
Ultraviolet radiation combined with microwave-assisted wet digestion of Antarctic seaweeds for further determination of toxic elements by ICP-MS
Marcia F.
Mesko
*a,
Rochele S.
Picoloto
b,
Lizângela R.
Ferreira
a,
Vanize C.
Costa
a,
Claudio M. P.
Pereira
a,
Pio
Colepicolo
c,
Edson I.
Muller
b and
Erico M. M.
Flores
b
aCentro de Ciências Químicas, Farmacêuticas e de Alimentos, Universidade Federal de Pelotas, 96010-610, Pelotas, RS, Brazil. E-mail: marciamesko@yahoo.com.br; Fax: +55-53-3275-7387; Tel: +55-53-3275-7387
bDepartamento de Química, Universidade Federal de Santa Maria, 97105-90, Santa Maria, RS, Brazil
cInstituto de Química, Departamento de Bioquímica, Universidade de São Paulo, 05508-000, São Paulo, SP, Brazil
First published on 26th September 2014
Abstract
In this work, the use of ultraviolet radiation combined with microwave-assisted wet digestion (MW-UV) was applied for digestion of seaweed samples and further determination of As, Cd and Pb by inductively coupled plasma mass spectrometry (ICP-MS). In the proposed method, UV radiation was generated in situ by electrodeless Cd discharge lamps inserted into digestion quartz vessels. This approach increased the digestion efficiency allowing lower consumption of acids. The feasibility of using diluted acid solution (0.5 to 7 mol L−1 HNO3) was evaluated for relatively higher sample masses (up to 800 mg). The efficiency of digestion was evaluated taking into account the residual carbon content and residual acidity in digests. Under the selected conditions, it was possible to digest up to 700 mg of sample using a nitric acid solution as diluted as 2 mol L−1 HNO3 allowing an efficiency of digestion higher than 77% (considering the total C content in the sample) and acidity as low as 0.19 mol L−1 that is convenient for ICP-MS measurements. The accuracy of the proposed MW-UV method was evaluated by the digestion of two certified reference materials of aquatic plant (BCR 060 and BCR 670) and by comparison with the results obtained after digestion by microwave-assisted wet digestion in a high pressure system using concentrated HNO3 (MW-AD). The results obtained by the proposed method did not present difference (t-test, 95% confidence level) with the certified values and with results obtained after seaweed sample digestion using the MW-AD method. Using 700 mg of sample, the limit of detection was 0.005, 0.001, and 0.012 μg g−1 for As, Cd, and Pb, respectively. A clear advantage of the proposed method over classical approaches is that only diluted solution was necessary and it is possible to digest a relatively high sample mass, that is important to minimize the generation of laboratory residues and to improve the limits of detection, respectively.
1. Introduction
In recent studies, the feasibility of using marine organisms as bioindicators for the evaluation of trace element pollution has been pointed out. In this sense, algae are among the most used matrices for this purpose.1,2 Macro-algae are able to accumulate toxic elements, generally reaching concentration values that are thousands of times higher than the corresponding concentrations in seawater.3 This ability to accumulate toxic elements can be related to the presence of polysaccharides and proteins in the algae structure.4 However, even under similar environmental conditions, different families, genera, species and geographical origin of macro-algae, among other parameters, can interfere with the capacity of trace element accumulation. The development of reliable analytical methods for the determination of trace elements in seaweed is an important task to fulfill this requirement5,6 and, in this sense, As, Cd and Pb have been studied as bioindicators in many studies.1–10Many analytical techniques have been used for the determination of As, Cd and Pb, in seaweed including spectrometric techniques, such as atomic absorption spectrometry,7,8 inductively coupled plasma optical emission spectrometry (ICP OES)9–12 and inductively coupled plasma mass spectrometry (ICP-MS).13–16 Taking into account some characteristics, such as sensitivity and multielement capability, ICP-MS has been the most recommended technique for the determination of As, Cd and Pb in seaweed samples.15,17,18 Despite the advantages related to the use of ICP-MS for trace element determination, some drawbacks have been observed in case of matrices with high content of sodium (e.g., seaweed collected from the Antarctic region). Sodium presents a low ionization potential, which changes plasma characteristics and can interfere during analyte determination by ICP-MS. In this sense, some strategies have been used such as high sample dilution and standard addition calibration.19 Moreover, this technique usually requires that samples are completely decomposed in order to obtain a suitable solution with low residual carbon content (RCC) for further analysis. In this context, it is important to highlight that RCC could interfere with the determination of some analytes by ICP-MS as reported in the literature.20,21 Especially, for As determination the high concentration of carbon in digests could enhance the As signal intensity in ICP-MS and the sample preparation step is considered as essential to ensure the accuracy of results.21
Microwave-assisted wet digestion (MW-AD) using concentrated acids and closed vessels has been successfully applied for seaweed sample digestion for further determination of As, Cd and Pb by several analytical techniques.4,11,15,22–24 The use of microwave radiation combined with closed vessels allows minimizing the risk of analyte loss, contamination and also generally ensures a more complete sample digestion in a relatively short time. However, the use of concentrated reagents is always hazardous, due to the risk of contamination, high blank values and generation of laboratory residues. In addition, digests with high acid content could not be suitable for analyte determination by some analytical techniques, such as ICP-MS and ICP OES.25–27 Consequently, it is necessary to perform a dilution of digests before the determination step to remove or dilute the acid excess. However, excessive dilution could result in a poor limit of detection (LOD) making difficult the determination of elements at low concentration, in the case of seaweeds.
Sample preparation methods using diluted acids have been applied to minimize some drawbacks related to the MW-AD method presenting good suitability for many matrices.28–31 However, it generally includes the use of concentrated acids and relatively high digestion time to achieve efficient matrix decomposition.30 In this sense, the combination of ultraviolet radiation with microwave-assisted wet digestion (MW-UV) can be a suitable alternative to the digestion of seaweed samples. In this method, electrodeless discharge lamps (Hg or Cd with main emission lines of 253.7 or 228.8 nm, respectively) can be used for UV generation. However, Cd lamps are preferred due to the low wavelength that is more effective for matrix digestion. In contrast to conventional systems, these electrodeless lamps can be directly immersed into digestion vessels generally used for MW-AD. With this approach, a minimum amount of reagents compared to conventional acid digestion procedures is required.32 Based on these advantages, this method can be an effective alternative to achieve good digestion efficiency and also to minimize the use of concentrated reagents. This system has been applied to the digestion of biological samples with suitable digestion efficiency. However, the maximum sample mass was 75 mg that impairs the achievement of lower LODs.32
In the present work, the feasibility of MW-UV digestion in closed vessels was evaluated for the first time for digestion of several types of seaweeds from the Antarctic region for further determination of As, Cd and Pb by ICP-MS. A systematic study was performed by evaluation of HNO3 concentration (0.5 to 7 mol L−1) and relatively high sample mass (500 to 800 mg). The efficiency of digestion was evaluated taking into account the RCC. The accuracy of the proposed method was evaluated by the digestion of two certified reference materials of aquatic plant (BCR CRM 060 and CRM 670), and also by the comparison of results with those obtained using conventional wet digestion MW-AD with concentrated HNO3 in closed vessels and subsequent determination of As, Cd and Pb by ICP-MS.
2. Experimental
2.1. Instrumentation
A microwave oven (Multiwave 3000 microwave sample preparation system, Anton Paar, Graz, Austria) equipped with eight high-pressure quartz vessels was used for the development of the proposed digestion method. Using this system, it is possible to carry out the operation at maximum pressure and temperature of 80 bar and 280 °C, respectively. Pressure and temperature were monitored in each vessel for all runs. The internal volume of the vessels was 80 mL. UV radiation was produced by using a Cd low-pressure discharge microwave lamp (Anton Paar) that was placed inside each quartz vessel. The emission of the UV radiation lamp is mainly located at 228 nm, and it was fully initiated and maintained by the microwave field within the microwave oven cavity. The same microwave oven was also employed for the conventional MW-AD digestion procedure that was used for comparison of results.An inductively coupled plasma mass spectrometer (PerkinElmer SCIEX, Model Elan DRC II, Thornhill, Canada) equipped with a concentric nebulizer (Meinhard Associates, Golden, USA), a cyclonic spray chamber (Glass Expansion, Inc., West Melbourne, Australia) and a quartz torch with a quartz injector tube (2 mm i.d.) was used for the determination of As, Cd and Pb in seaweed digests. Argon 99.996% (White Martins-Praxair, São Paulo, Brazil) was used for plasma generation, nebulization and auxiliary gas. Operating conditions are described in Table 1.
| Parameter | ICP-MS |
|---|---|
| Radio-frequency power (W) | 1300 |
| Plasma gas flow rate (L min−1) | 15.0 |
| Auxiliary gas flow rate (L min−1) | 1.2 |
| Nebulizer gas flow rate (L min−1) | 1.08 |
| Spray chamber | Cyclonic |
| Nebulizer | Concentric |
| Sampler and skimmer cones | Pt |
| Ion lens | Auto lens “on” |
| Analytes | Isotope (m/z) |
| As | 75 |
| Cd | 111 |
| Pb | 208 |
For carbon content measurements, an inductively coupled plasma optical emission spectrometer with axial view configuration (PerkinElmer, Model Optima 4300 DV, Shelton, CT, USA) equipped with a concentric nebulizer coupled to a cyclonic nebulization spray chamber was used. Operating conditions are used according to those described in ref. 33. In order to remove volatile carbon compounds in digests, sample aliquots were previously bubbled with argon for 2 minutes.34 The results of RCC were expressed as the percentage of total carbon originally present in the sample that remained in solution after each digestion procedure. Results for residual acidity were obtained using a titration system (Titrando 836, Metrohm, Herisau, Switzerland) equipped with a module of automatic stirring (module 803 Ti Stand, Metrohm) and a pH electrode (LL Electrode plus, model 6.0262.100).
Sodium was determined in seaweed digests obtained by the MW-UV method. Sodium measurements were performed using an inductively coupled plasma optical emission spectrometer with radial view configuration equipped with a concentric nebulizer coupled to a cyclonic nebulization spray chamber. The selected wavelength for Na determination was 589.592 nm and the operating conditions were used according to previous work.35
2.2. Reagents, standards and samples
Ultra pure water (18.2 MΩ cm) obtained from a Milli-Q water purification system (Millipore Corp., Bedford, MA, USA) and nitric acid (Merck, Darmstadt, Germany) purified using a sub-boiling system (Milestone, Model Duopur, Bergamo, Italy) were used in this work. Multielement stock standard solution (SCP33MS, SCP Science, Quebec, Canada) containing 10 mg L−1 of As, Cd and Pb was used to prepare analytical standards for the determination by ICP-MS using external calibration as well as for standard addition calibration, used for comparison of results. Analytical standards were prepared by sequential dilution of the multielement stock standard solution in 5% (v/v) HNO3, in the range of 0.025 to 10 μg L−1. Reference solutions used for the determination of residual carbon content by ICP OES were prepared by the dissolution of citric acid (Merck) in 5% (v/v) HNO3 and yttrium (1.0 mg L−1 solutions, Spex CertPrep, Metuchen, New Jersey, USA) was used as the internal standard in all samples, blank and reference solutions. A solution of 0.1 mol L−1 KOH (Merck) was used for residual acidity determination (this solution was previously standardized using potassium hydrogen phthalate). It is important to mention that all results presented for the residual acidity were determined in digests with final volume made up to 25 mL. Glass, plastic and quartz materials were soaked in 10% (v/v) HNO3 for 24 h and further washed with water before use.Four species of seaweeds (Desmarestia anceps, Iridaea cordata, Palmaria decipiens and Pyropia endiviifolia) from the Antarctic region, collected between November 2011 and January 2012, in Demay on King George Island (62°13′19.30′′S, 58°26′36.59′′W; 62°13′13.93′′S, 58°26′17.85′′W; 62°13′13.93′′S, 58°26′17.85′′W and 62°13′21.01′′S, 58°26′36.59′′W for Desmarestia anceps, Iridaea cordata, Palmaria decipiens and Pyropia endiviifolia, respectively) were used in this work. Collected samples were rinsed with ultra-pure water, dried in a lyophilizer (LIOTOP®, Model L101, Liobras, Brazil), ground in a cryogenic mill (Spex CertiPrep, model 6750 Freezer Mill, Metuchen, USA) and stored at −10 °C before analysis.
The accuracy of the proposed (MW-UV) and reference methods (MW-AD) was evaluated using two certified reference materials (CRMs) of aquatic plant (BCR 060 and BCR 670) purchased from the Community Bureau of Reference. Moreover, in order to evaluate the stability of the plasma conditions during analysis, a standard solution and CRM digested solutions (using the MW-AD method) were analyzed each five times.
2.3. Conventional microwave-assisted digestion method (MW-AD)
The conventional MW-AD method was performed for seaweed sample digestion for comparison of results with those obtained by MW-UV. In this procedure, 500 mg of Desmarestia anceps sample were digested with 6 mL of concentrated HNO3 (14.5 mol L−1). The microwave heating program was carried out as follows: 1000 W for 15 min (ramp of 10 min), and 0 W for 20 min (cooling step). For routine operation, the maximum temperature achieved during digestion ranged from 220 to 240 °C. After cooling, digests were diluted with water to 25 mL and the determination of As, Cd and Pb was carried out by ICP-MS. Taking into account the final acid concentration and NaCl content in digests, further dilutions (ranging from 10 to 50 times) were performed in order to obtain a suitable solution for ICP-MS analyses. The cleaning procedure of quartz vessels was performed by using 6 mL of concentrated HNO3 and applying the microwave program to 1000 W for 10 min and 0 W for 20 min (cooling step).2.4. Microwave-assisted UV-digestion method (MW-UV)
Seaweed samples (500 mg to 800 mg) were directly inserted into the quartz vessels. The digestion efficiency using nitric acid solutions (10 mL) was evaluated in the following concentrations: 0.5, 1, 2, 3, and 7 mol L−1. Before sample weighing, a polytetrafluoroethylene (PTFE) device was introduced into the quartz vessels in order to maintain the UV lamp in the vertical position and to avoid consequent damage to the lamps. The same digestion procedure was carried out without UV lamps. In both cases (digestion with or without UV lamps), the microwave-heating program was 900 W for 10 min and 0 W for 20 min (cooling step). The maximum temperature and pressure were set at 250 °C and 80 bar, respectively. After digestion, the pressure of each vessel was carefully released and the resultant solution was transferred to a volumetric flask and the final volume was completed with water to 25 mL. After each run, vessels, UV lamps and PTFE devices were cleaned using 6 mL of concentrated HNO3 in the microwave oven at 1000 W for 10 min and 0 W for 20 min for cooling. Further, all devices were rinsed with ultrapure water and dried in a class 100 laminar bench (CSLH-12, Veco, Brazil). Final digests were analyzed by ICP-MS (As, Cd and Pb). The residual carbon content and Na concentration were determined in the digests by ICP OES and residual acidity was determined by titration.All statistical calculations were performed using GraphPad InStat (GraphPad InStat Software Inc, Version 3.06, 2003) software. A significance level of P < 0.05 was selected for all the statistical evaluation. Sample preparation methods used in this work are summarized in Fig. 1.
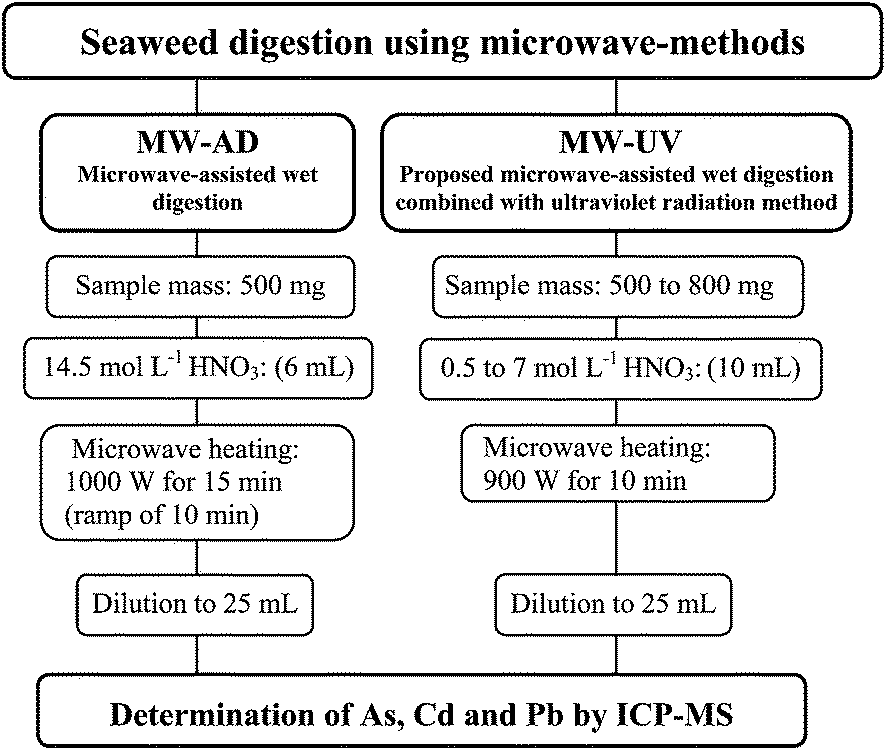 | ||
| Fig. 1 Sample preparation methods evaluated for the digestion of seaweed samples. | ||
3. Results and discussion
3.1. MW-AD reference method – preliminary studies
Initially, Desmarestia anceps seaweed sample (500 mg) was digested using 6 mL of 14.5 mol L−1 nitric acid by the MW-AD method. This procedure was performed according to that described by the instrument manufacturer for plant digestion.36 In addition, the MW-AD method was applied for the digestion of aquatic plant certified reference materials (BCR 060 and 670). Using this method, the results for all analytes were in agreement with the certified values (t-test, 95% confidence level) for both CRMs (BCR 060 and 670). It is important to mention that other isotopes for Cd (112 and 114) and Pb (207) were also evaluated and the respective results did not present significant difference. Then, the results obtained for the Desmarestia anceps sample (As: 28.0 ± 0.6 μg g−1, Cd: 0.582 ± 0.022 μg g−1 and Pb: 0.431 ± 0.021 μg g−1) were considered as reference values for the subsequent optimization of the proposed MW-UV method.3.2. MW-UV digestion method – evaluation of HNO3 concentration and sample mass for seaweed digestion
Preliminary experiments were performed in order to evaluate the influence of nitric acid concentration on seaweed sample digestion. Nitric acid concentrations of 0.5, 1, 2, 3 or 7 mol L−1 were used in order to achieve a suitable condition related to efficient organic matter digestion using a minimum nitric acid amount. These initial tests were carried out using 500 mg of seaweed sample. The determination of RCC and acidity was performed after each digestion step. All studies were performed using a total volume of 10 mL of nitric acid solution in order to cover the surface of the UV lamp. The results obtained for As, Cd and Pb determination by ICP-MS using (0.5 to 7 mol L−1) HNO3 solution are shown in Fig. 2. Using 0.5 mol L−1 HNO3 solution, the digestion was not effective (RCC > 70%, respective to the total C content in the sample) and solid residues remained as suspended particles with a deep colored aspect. Digests using 0.5 mol L−1 HNO3 were not analyzed by ICP-MS for As, Cd and Pb determination. Using 1 mol L−1 HNO3 solution, the RCC and acidity were 37.5 ± 5.2% and 0.015 ± 0.006 mol L−1, respectively. Digests obtained using 1 mol L−1 HNO3 by MW-UV were not considered suitable for direct analysis and a dilution step was necessary before determination by ICP-MS. This dilution step was necessary taking into account that high RCC could cause interference during ICP-MS determination, especially in the case of As.21 On the other hand, using 2, 3 or 7 mol L−1 HNO3 solutions, final digests presented a colorless aspect and the RCC was always below 3% (2.82, 2.50 and 2.20%, respectively). In this case, direct determination of the analytes by ICP-MS was possible using external calibration. These values correspond to 120, 136 and 88 mg L−1 of C in digests diluted to 25 mL that cannot interfere with ICP-MS analysis. The final acidity of digests was 0.21, 0.57 and 1.79 mol L−1, respectively. As expected, it was observed that the acidity increased with the use of higher HNO3 concentration for seaweed sample digestion by MW-UV although the RCC was practically the same (below 3%). The results could be considered as an indication that it is possible to use HNO3 solution as diluted as 2 mol L−1 for seaweed digestion still keeping a suitable decomposition efficiency. This fact, not observed in previous studies,37–39 was attributed to the additional effect of UV radiation combined with microwave heating.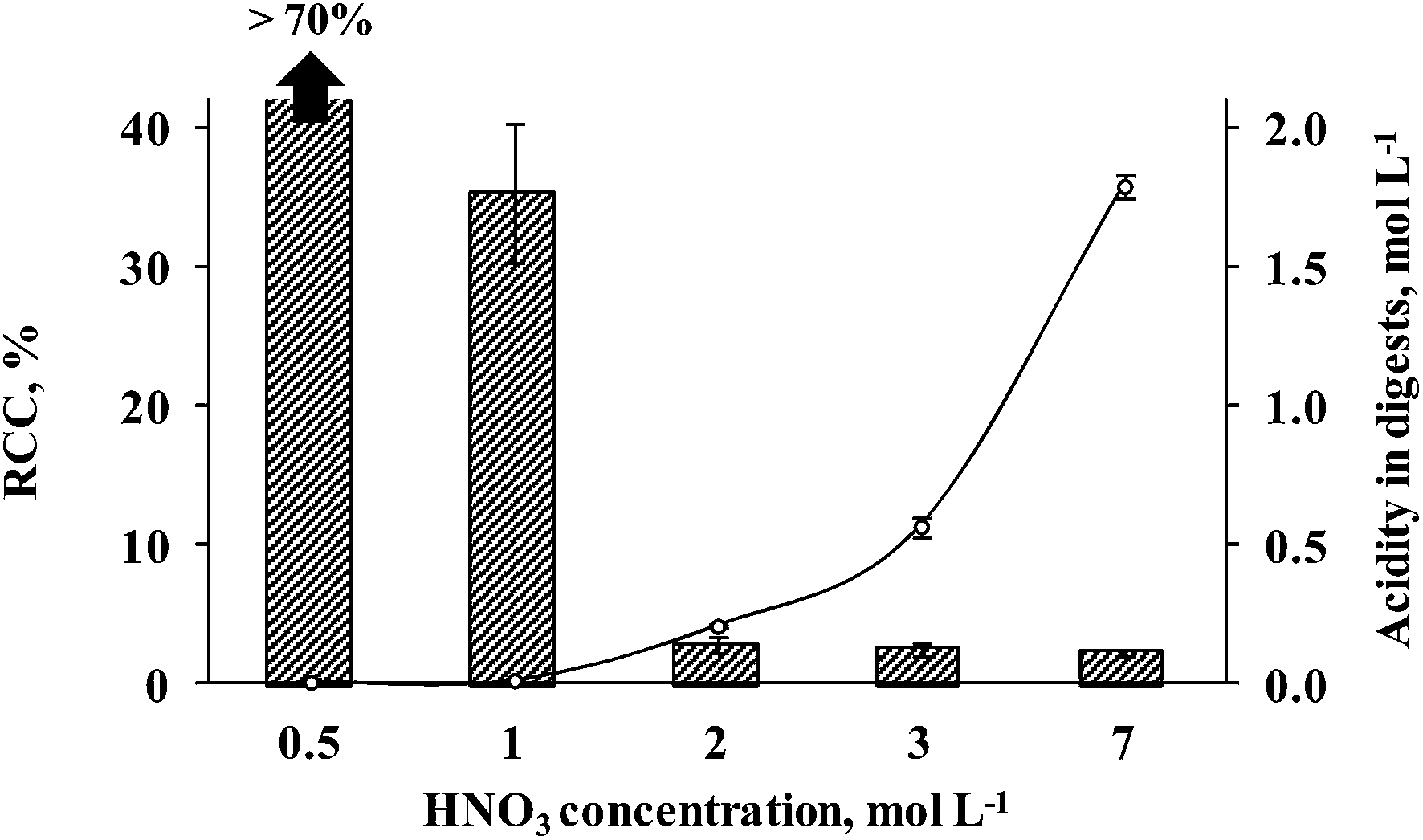 | ||
Fig. 2 Effect of HNO3 concentration (0.5 to 7 mol L−1) on the digestion of 500 mg of seaweed: effectiveness of organic matter digestion ( ) and residual acidity ( ) and residual acidity ( ) performed using the MW-UV method. Error bars are the respective standard deviation (n = 3). ) performed using the MW-UV method. Error bars are the respective standard deviation (n = 3). | ||
Arsenic, Cd and Pb concentrations were determined by ICP-MS in solutions obtained using 1 to 7 mol L−1 HNO3 concentrations. The results indicated that using 2, 3 or 7 mol L−1 HNO3, an agreement with reference values better than 98% was achieved for all analytes (Fig. 3). By using 1 mol L−1 HNO3, similar results were obtained only for As (agreement better than 95%), but the relative standard deviation (RSD) was relatively high (RSD = 14%). For Cd and Pb, the agreement was lower than 85 and 35%, respectively, using 1 mol L−1 HNO3 solution. These results can be explained due to low digestion efficiency when using 1 mol L−1 HNO3 solution (RCC > 36%, respective to the total C content in the sample) and the RSD was higher than 10% for all analytes. On the other hand, using 2, 3 or 7 mol L−1 HNO3 solutions, the RSD was lower than 8% for all analytes.
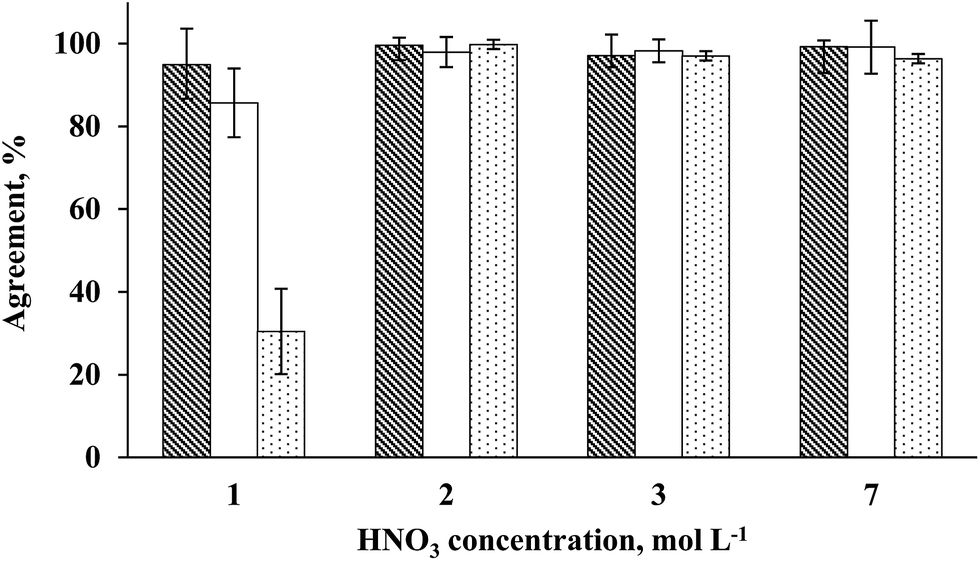 | ||
Fig. 3 Influence of HNO3 concentration (1 to 7 mol L−1) on the digestion of 500 mg of seaweed sample (Desmarestia anceps) using the MW-UV method. Determination of As, Cd and Pb ( , ,  , ,  , respectively) by ICP-MS. Error bars are the respective standard deviation (n = 3). , respectively) by ICP-MS. Error bars are the respective standard deviation (n = 3). | ||
It is important to mention that the presence of As, Cd and Pb in seaweed samples, especially in the Antarctic region, can occur in low concentrations. In this sense, a study of using higher sample mass was performed in order to achieve LODs as low as possible. Then, the proposed method was investigated for the digestion of 600, 700 and 800 mg of seaweed sample, under the previously optimized conditions (10 mL of 2 mol L−1 HNO3, microwave heating time = 10 min). Digestion of 800 mg of sample using the proposed MW-UV method was considered unsuitable for subsequent analyte determination by ICP-MS, mainly due to the presence of solid residues in the final solution and relatively high RCC (>75%). On the other hand, the MW-UV method was suitable to digest up to 700 mg of sample, without solid residues in digests (RCC lower than 22.7%) making solutions suitable for further ICP-MS analysis. The acidity in solutions after digestion of 700 mg was 0.24 mol L−1. The results obtained for As, Cd and Pb by ICP-MS after digestion using 700 mg of seaweed sample were respectively 27.7 ± 0.5 μg g−1 for As, 0.558 ± 0.016 μg g−1 for Cd and 0.441 ± 0.020 μg g−1 for Pb. These results were compared with the results obtained after digestion of the seaweed sample using the MW-AD method and the results presented no statistical difference (confidence level of 95%).
Additional experiments were performed in order to evaluate the influence of UV lamps in digestion of seaweed samples. In this sense, the same microwave heating program and digestion solution were applied without the insertion of the UV lamp inside the quartz vessels. Sample masses of 500, 600 or 700 mg were digested with 10 mL of 2 mol L−1 HNO3 solution without the use of a UV-lamp. The residual carbon content was determined in final digests. Final digests for 500 and 600 mg of sample presented a yellow color and the RCC was 16.3 and 30.3%, respectively. However, using 700 mg of sample (without UV-lamp) the aspect of solution was dark yellow and the RCC was higher than 47.5%. Residual carbon content values obtained after digestion of the seaweed sample without the UV-lamp were always higher than those obtained using the MW-UV method. It shows that the effectiveness of matrix digestion was favored by the use of the UV-lamp during the digestion of seaweed using the MW-UV method. In this sense, subsequent experiments to digest seaweed samples by MW-UV were performed using 700 mg of sample and 10 mL of 2 mol L−1 HNO3 using the Cd UV-lamp.
3.3. Determination of As, Cd and Pb in seaweed and CRM samples after MW-AD and MW-UV methods
The accuracy of the proposed MW-UV method was evaluated by using two CRMs of aquatic plant (BCR 060 and BCR 670). In this way, 700 mg of BCR 060 or BCR 670 were weighed and transferred to a quartz vessel with the UV-lamp. Digestion was performed under optimized conditions (10 mL of 2 mol L−1 HNO3 solution) and a heating program was applied for 10 min at 900 W. The results obtained for As, Cd and Pb in both CRMs presented an agreement better than 95% with reference values (except for As in BCR 060) as it can be seen in Table 2. Certified reference materials BCR 670 and 060 were also digested using the reference MW-AD method (Table 2). The agreement for As determination by ICP-MS in BCR 060 after MW-AD and MW-UV digestions was 87 and 81%, respectively. Similar results for As were reported in another work,40 where sample digestion procedures were evaluated using several types of acid mixtures, and even using a mixture of concentrated HNO3, H2O2 and HF, the maximum agreement with reference certified values was 85%. On the other hand, for BCR 670, the agreement with the As certified value was better than 95% for both digestion methods (MW-AD and MW-UV). It can be an indication that reference values (only for As) in BCR 060 must be carefully evaluated for further studies.| Analyte | Sample | MW-UV | MW-AD | Certified values |
|---|---|---|---|---|
| a Informed values. | ||||
| As | Iridaea cordata | 11.7 ± 0.6 | 11.0 ± 0.8 | — |
| Palmaria decipiens | 5.68 ± 0.11 | 5.85 ± 0.10 | — | |
| Pyropia endiviifolia | 25.8 ± 0.7 | 26.1 ± 0.5 | — | |
| BCR 060 | 6.52 ± 0.35 | 6.96 ± 0.50 | 8a | |
| BCR 670 | 1.89 ± 0.02 | 1.91 ± 0.05 | 1.98 ± 0.19a | |
| Cd | Iridaea cordata | 0.656 ± 0.013 | 0.646 ± 0.051 | — |
| Palmaria decipiens | 0.307 ± 0.018 | 0.314 ± 0.002 | — | |
| Pyropia endiviifolia | 3.49 ± 0.03 | 3.46 ± 0.08 | — | |
| BCR 060 | 2.21 ± 0.06 | 2.33 ± 0.10 | 2.20 ± 0.10 | |
| BCR 670 | 0.073 ± 0.005 | 0.075 ± 0.016 | 0.0755 ± 0.0025 | |
| Pb | Iridaea cordata | 0.104 ± 0.003 | 0.105 ± 0.006 | — |
| Palmaria decipiens | 0.324 ± 0.007 | 0.330 ± 0.007 | — | |
| Pyropia endiviifolia | 0.222 ± 0.014 | 0.227 ± 0.010 | — | |
| BCR 060 | 67.8 ± 1.2 | 67.3 ± 3.8 | 64 ± 4 | |
| BCR 670 | 1.94 ± 0.10 | 1.98 ± 0.13 | 2.06 ± 0.12a | |
Additionally, in order to evaluate the suitability of the MW-UV method for seaweed digestion and further As, Cd and Pb determination, the proposed method was applied for other three seaweed samples (Iridaea cordata, Palmaria decipiens and Pyropia endiviifolia) and CRMs (Table 2). For all the analytes, the agreement with certified values and with the reference method (MW-AD) was better than 94%. It is important to mention that the results presented for the MW-UV digestion method were obtained when 700 mg of sample and CRM were digested. However, for the MW-AD method, 500 mg of sample and CRM were digested.
It is important to mention that the seaweed samples used in this work presented a Na content ranging from 0.173 ± 0.006 to 6.49 ± 0.18% (Na determination was performed by ICP OES). Therefore, in order to avoid Na interference of plasma during determination of As, Cd and Pb by ICP-MS mainly for Desmarestia anceps samples that present a high Na concentration, digests were diluted up to 50 times before the determination step. In addition, these solutions were also analyzed by ICP-MS using calibration by the standard addition method. In this case, the results obtained by standard addition calibration were in agreement with those obtained using external calibration. Additionally, due to the high Na content (and consequently high Cl content) arsenic was also determined by ICP OES in all samples used in this work.41 The results obtained for As by ICP-MS and ICP OES presented no statistical difference (confidence level of 95%).
4. Conclusions
The proposed MW-UV method was suitable for the digestion of seaweed samples for further simultaneous determination of As, Cd and Pb by ICP-MS. It was possible to use a solution as diluted as 2 mol L−1 HNO3 to digest up to 700 mg of sample. In this aspect, the MW-UV method avoids the use of concentrated acids, significantly reducing interference with the determination step and the laboratory wastes, which is in agreement with the recommendations of green chemistry.42 Using the proposed method, an agreement better than 94% (t-test, 95% confidence level) to reference values (after MW-AD digestion and determination by ICP-MS) was obtained. The accuracy was also evaluated by comparison with CRM values, and an agreement better than 95% was obtained (with the exception of As for BCR 060). In addition, LODs obtained by MW-UV were always lower in comparison with those obtained by MW-AD. The use of MW-UV showed good performance for seaweed digestion with an efficiency higher than 77% (considering the total C content in the sample) and a convenient throughput for routine analysis (up to eight samples can be digested in less than 40 min).Acknowledgements
The authors are grateful to Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS) for supporting this study. We are also grateful to Programa Antártico Brasileiro (PROANTAR) for supplying seaweed samples.References
- T. Tin, Z. L. Fleming, K. A. Hughes, D. G. Ainley, P. Convey, C. A. Moreno, S. Pfeiffer, J. Scott and I. Snape, Antarct. Sci., 2009, 1, 3–33 CrossRef.
- J. E. A. Moreno, M. S. Gerpe, V. J. Moreno and C. Vodopivez, Polar Biol., 1997, 17, 131–140 CrossRef.
- G. W. Bryan and W. J. Langston, Environ. Pollut., 1992, 76, 89–131 CrossRef CAS.
- S. Farías, S. P. Arisnabarreta, C. Vodopivez and P. Smichowski, Spectrochim. Acta, Part B, 2002, 57, 2133–2140 CrossRef.
- F. Riget, P. Johasen and G. Asmund, Mar. Pollut. Bull., 1997, 34, 171–176 CrossRef CAS.
- E. Marinho-Soriano, P. C. Fonseca, M. A. A. Carneiro and W. S. C. Moreira, Bioresour. Technol., 2006, 97, 2402–2406 CrossRef CAS PubMed.
- I. Akcali and F. A. Kucuksezgin, Mar. Pollut. Bull., 2011, 62, 637–645 CrossRef CAS PubMed.
- V. Besada, J. M. Andrade, F. Schultze and J. J. González, J. Mar. Syst., 2009, 75, 305–313 CrossRef PubMed.
- A. Pérez, S. S. Farías, A. M. Strobl, L. B. Pérez, C. M. López, A. Piñeiro, O. Roses and M. A. Fajardo, Sci. Total Environ., 2007, 376, 51–59 CrossRef PubMed.
- R. Domínguez-González, A. Moreda-Piñeiro, A. Bermejo-Barrera and P. Bermejo-Barrera, Talanta, 2005, 66, 937–942 CrossRef PubMed.
- S. Farías, P. Smichowski, D. Vélez, R. Montoro, A. Curtosi and C. Vodopívez, Chemosphere, 2007, 69, 1017–1024 CrossRef PubMed.
- J. F. Gonçalves, F. G. Antes, J. Maldaner, L. B. Pereira, L. A. Tabaldi, R. Rauber, L. V. Rossato, D. A. Bisogninc, V. L. Dressler, E. M. M. Flores and F. T. Nicoloso, Plant Physiol. Biochem., 2009, 47, 814–821 CrossRef PubMed.
- R. Rocha, F. J. Sánchez-Muniz, M. Gómez-Juaristi and M. T. L. Marín, J. Food Compos. Anal., 2009, 22, 330–336 CrossRef PubMed.
- R. J. C. Brown and M. J. T. Milton, TrAC, Trends Anal. Chem., 2005, 3, 266–274 CrossRef PubMed.
- G. B. Brito, T. L. Souza, F. C. Bressy, C. W. N. Moura and M. G. A. Korn, Mar. Pollut. Bull., 2012, 64, 2238–2244 CrossRef CAS PubMed.
- J. W. Runcie, A. T. Townsend and A. J. Seen, Mar. Pollut. Bull., 2009, 58, 961–966 CrossRef CAS PubMed.
- R. Domínguez-González, V. Romarís-Hortas, C. García-Sartal, A. Moreda-Piñeiro, M. C. Barciela-Alonso and P. Bermejo-Barrera, Talanta, 2010, 82, 1668–1673 CrossRef PubMed.
- S. Ródenas de la Rocha, F. J. Sánchez-Muniz, M. Gómez-Juaristi and M. T. Larrea Marín, J. Food Compos. Anal., 2009, 22, 330–336 CrossRef PubMed.
- P. Abbyad, J. Tromp, J. Lamb and E. Salin, J. Anal. At. Spectrom., 2001, 16, 464–469 RSC.
- F. A. Duarte, J. S. F. Pereira, J. S. Barin, M. F. Mesko, V. L. Dressler, E. M. M. Flores and G. Knapp, J. Anal. At. Spectrom., 2009, 24, 224–227 RSC.
- G. Grindlay, J. Mora, M. Loos-Vollebregt and F. Vanhaecke, Spectrochim. Acta, Part B, 2013, 86, 42–49 CrossRef CAS PubMed.
- M. E. Contia and G. Cecchett, Environ. Res., 2003, 93, 99–112 CrossRef.
- M. T. Larrea-Marín, M. S. Pomares-Alfonso, M. Gómez-Juaristi, F. J. Sánchez-Muniz and S. Ródenas de la Rocha, J. Food Compos. Anal., 2010, 23, 814–820 CrossRef PubMed.
- F. G. Antes, F. A. Duarte, M. F. Mesko, M. A. G. Nunes, V. A. Pereira, E. I. Muller, V. L. Dressler and E. M. M. Flores, Talanta, 2010, 82, 364–369 CrossRef PubMed.
- J.-L. Todoli and J.-M. Mermet, Spectrochim. Acta, Part B, 1999, 54, 895–929 CrossRef.
- J. S. F. Pereira, P. A. Mello, F. A. Duarte, M. F. P. Santos, R. C. L. Guimarães, G. Knapp, V. L. Dressler and E. M. M. Flores, Energy Fuels, 2009, 23, 6015–6019 CrossRef CAS.
- A. L. H. Muller, C. C. Muller, F. Lyra, P. A. Mello, M. F. Mesko, E. I. Muller and E. M. M. Flores, Food Anal. Methods, 2013, 6, 258–264 CrossRef.
- C. A. Bizzi, E. M. M. Flores, J. S. Barin, E. E. Garcia and J. A. Nóbrega, Microchem. J., 2011, 99, 193–196 CrossRef CAS PubMed.
- C. A. Bizzi, J. S. Barin, E. E. Garcia, J. A. Nóbrega, V. L. Dressler and E. M. M. Flores, Spectrochim. Acta, Part B, 2011, 66, 394–398 CrossRef CAS PubMed.
- C. A. Bizzi, E. M. M. Flores, R. S. Picoloto, J. S. Barin and J. A. Nóbrega, Anal. Methods, 2010, 2, 734–738 RSC.
- E. I. Muller, M. F. Mesko, D. P. Moraes, M. G. A. Korn and E. M. M. Flores, in Microwave-Assisted Sample Preparation for Trace Element Determination, ed. E. M. M. Flores, Elsevier, Waltham, 2014, vol. 1, pp. 100–142 Search PubMed.
- D. Florian and G. Knapp, Anal. Chem., 2001, 73, 1515–1520 CrossRef CAS.
- A. L. H. Muller, P. A. Mello, M. F. Mesko, F. A. Duarte, V. L. Dressler, E. I. Muller and E. M. M. Flores, J. Anal. At. Spectrom., 2012, 27, 1889–1894 RSC.
- C. A. Bizzi, E. L. M. Flores, J. A. Nóbrega, J. S. S. Oliveira, L. Schmidt and S. R. Mortari, J. Anal. At. Spectrom., 2014, 29, 332–338 RSC.
- J. S. F. Pereira, L. S. F. Pereira, L. Schmidt, C. M. Moreira, J. S. Barin and E. M. M. Flores, Microchem. J., 2013, 109, 29–35 CrossRef CAS PubMed.
- Anton Paar GmbH, Microwave Sample Preparation System, Software version v1.27-Synt, Graz, Austria, 2003 Search PubMed.
- J. S. F. Pereira, R. S. Picoloto, L. S. F. Pereira, R. C. L. Guimarães, R. A. Guarnieri and E. M. M. Flores, Anal. Chem., 2013, 85, 11034–11040 CrossRef CAS PubMed.
- A. Limbeck, Anal. Chim. Acta, 2006, 575, 114–119 CrossRef CAS PubMed.
- K. Dash, L. Rastogi and J. Arunachalam, Analyst, 2012, 137, 668–674 RSC.
- J. Sucharová and I. Suchara, Anal. Chim. Acta, 2006, 576, 163–176 CrossRef PubMed.
- F. A. Duarte, J. S. F. Pereira, M. F. Mesko, F. Goldschmidt, E. M. M. Flores and V. L. Dressler, Spectrochim. Acta, Part B, 2007, 62, 978–984 CrossRef PubMed.
- D. L. Rocha, A. D. Batista, F. R. P. Rocha, G. L. Donati and J. A. Nóbrega, TrAC, Trends Anal. Chem., 2013, 45, 79–92 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |