
An in vitro model of the tumor–lymphatic microenvironment with simultaneous transendothelial and luminal flows reveals mechanisms of flow enhanced invasion†
M.
Pisano
a,
V.
Triacca
a,
K. A.
Barbee
b and
M. A.
Swartz
*ac
aInstitute of Bioengineering and Swiss Institute for Experimental Cancer Research (ISREC), School of Life Sciences, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland. E-mail: melody.swartz@epfl.ch
bSchool of Biomedical Engineering, Science, and Health Systems, Drexel University, Philadelphia, PA, USA
cInstitute for Molecular Engineering, University of Chicago, Chicago, IL, USA
First published on 10th April 2015
Abstract
The most common cancers, including breast and skin, disseminate initially through the lymphatic system, yet the mechanisms by which tumor cells home towards, enter and interact with the lymphatic endothelium remain poorly understood. Transmural and luminal flows are important biophysical cues of the lymphatic microenvironment that can affect adhesion molecules, growth factors and chemokine expression as well as matrix remodeling, among others. Although microfluidic models are suitable for in vitro reconstruction of highly complex biological systems, the difficult assembly and operation of these systems often only allows a limited throughput. Here we present and characterize a novel flow chamber which recapitulates the lymphatic capillary microenvironment by coupling a standard Boyden chamber setup with a micro-channel and a controlled fluidic environment. The inclusion of luminal and transmural flow renders the model more biologically relevant, combining standard 3D culture techniques with advanced control of mechanical forces that are naturally present within the lymphatic microenvironment. The system can be monitored in real-time, allowing continuous quantification of different parameters of interest, such as cell intravasation and detachment from the endothelium, under varied biomechanical conditions. Moreover, the easy setup permits a medium-high throughput, thereby enabling downstream quantitative analyses. Using this model, we examined the kinetics of tumor cell (MDA-MB-231) invasion and transmigration dynamics across lymphatic endothelium under varying flow conditions. We found that luminal flow indirectly upregulates tumor cell transmigration rate via its effect on lymphatic endothelial cells. Moreover, we showed that the addition of transmural flow further increases intravasation, suggesting that distinct flow-mediated mechanisms regulate tumor cell invasion.
Insight, innovation, integrationLymphatic vessels regulate flow in the tumor microenvironment and secrete factors that attract and facilitate tumor cell invasion, a critical step in metastasis. Their integrated mechanical–biological effects are complex yet important, since interstitial, transmural, and luminal flows can each affect tumor and lymphatic endothelial cells (LECs) to modulate invasion. Here, we develop a robust in vitro model that integrates these flows with image analysis tools to quantitatively assess tumor cell invasion and transmigration across LECs. We demonstrate that MDA-MB-231 tumor cell intravasation is differentially sensitive to interstitial and transmural vs. luminal flows, and when combined, these biomechanical cues additively increase tumor cell invasion. These data provide new mechanistic insight into the mechanobiology of tumor cell invasion into lymphatics. |
Introduction
An extensive network of lymphatic vessels drain fluid, cells, solutes and antigens from the periphery and, after filtering through the lymph nodes for immune surveillance, deposit their contents in the blood via the thoracic duct. Recent research is increasing our appreciation for the active role that lymphatic endothelium plays in regulating immune and tumor cell transport,1–5 and recent studies from our lab and others suggest that lymphatic endothelial cells (LECs) may play important and direct roles in immune tolerance.6–9 However, the mechanisms by which LECs regulate cell transport from the peripheral tissues to the lymph nodes remain unclear. While reports have shown that flow shear stress influences a wide range of blood endothelial cell functions, including leukocyte adhesion and extravasation,10–13 the effects of fluid forces on LEC behaviours – and in particular on its interactions with trafficking leukocytes and tumor cells – has been poorly understood, at least partially due to the lack of biomechanically relevant model systems.Therefore, the design of an in vitro tunable model of the lymphatic microenvironment is a critical step to further explore regulatory mechanisms of the lymphatic network. Such models have the potential to provide useful information about the role of lymphatics in pathological conditions such as cancer or chronic inflammation, and may further support the development of novel and improved therapeutic strategies.
Biomechanically, the lymphatic microenvironment is quite different from that of the blood, and can be characterized by the presence of three types of fluid forces:14 interstitial flow on interstitial cells, transmural flow on the LECs (basal to apical or abluminal to luminal), and luminal flow parallel to the apical surface of LECs (Fig. 1a–c). Physiologically, we assume that LECs in the absorbing or capillary vessels experience both transmural and luminal flow simultaneously, resulting in a complex pattern of biomechanical cues that may regulate immune or tumor cell motility in addition to interstitial flow (Fig. 1d). Interestingly, it has been observed that the velocity of lymph flow rapidly increases during inflammation and around tumors,3,15,16 though the downstream implications of these altered fluid forces on the lymphatic endothelium are not fully understood.
 | ||
| Fig. 1 Modeling the biomechanical microenvironment of the tumor–lymphatic microenvironment in vitro. (a) Tumor cells in the tumor margin experience interstitial flow (wiggly arrows) directed towards the nearest draining lymphatic capillary. This interstitial flow may induce mechanical signals on the cell as well as drive autologous chemotaxis. (b) Lymphatic capillaries drain fluid from the interstitium, and thus interstitial flow becomes transmural flow across the LECs as lymph forms. (c) Inside the lymphatic capillary, luminal flow (straight arrows) induces shear stress on the apical surface of the LECs. (d) These three forces are inherently coupled in the interstitial–lymphatic interface and may influence tumor cell transmigration into the lymphatic vessel lumen. (e) Schematic of the flow chamber unit, where tumor cells within a 3D extracellular matrix are cultured above a membrane and LEC monolayer. The tumor cells experience interstitial flow, while the LECs experience both transmural flow as well as luminal flow that are each finely controlled. (f) Photograph of the flow chamber, which hosts five independent channels. (g–j) The experimental steps to set up the chamber are represented. (g) First, in a culture insert, LECs are cultured for 24 h in the downward-facing side of the insert membrane. Then, the solution containing fluorescently labeled tumor cells and extracellular matrix is placed in the upper chamber of the insert and allowed to polymerize. (h) Meanwhile, the chamber is mounted and primed with cell medium. (i) The inserts are placed in the dedicated housings and sealed with the caps. (j) Peristaltic pumps connected to the device by pre-primed tubing allows fine control of the desired interstitial and luminal flow rates. | ||
Our group has recently demonstrated that transmural flow modulates dendritic cell and fluid transport across LECs, with higher transmural flow velocities causing LECs to increase their expression of cytokines and adhesion molecules that attract and facilitate leukocyte transmigration such as CCL21, ICAM-1, E-selectin, and VCAM-1.17 ICAM-1 upregulation by luminal shear stress on LECs has also been suggested to play a role in establishing a pre-metastatic niche in the draining lymph node.18 Luminal shear stress on LECs can also upregulate endothelial nitric oxide synthase (eNOS), which in turn regulates lymphatic pump function in the collecting vessels.19–22 Finally, luminal shear stress has also been shown to modulate the lymphatic endothelial barrier function by a mechanism dependent upon Rac1-mediated actin dynamics.23 In these examples, luminal flow was induced across a LEC surface without the ability to recapitulate or observe cell transmigration from the interstitial space into the vessel under relevant flow forces. As such, the role of luminal shear stress on regulating cell transport into lymphatics, as well as its differential effects compared to those of transmural flow, are poorly understood.
As mentioned, conventional in vitro experiments do not fully recapitulate the multiple factors that likely contribute strongly to lymphatic function. Migration and transmigration assays have typically been conducted using Boyden chambers24 or parallel plate flow chambers25 for qualitative and quantitative assessment of cellular responses to soluble factors. While these assays are well-established, reliable and easy to setup, they only examine one aspect of the lymphatic microenvironment at a time. In contrast, microfluidic models of the endothelial–interstitial environment have been developed.26–32 Microfluidic systems closely match the size dimensions of biological systems, and are thus potentially useful tools for the study of the lymphatic microenvironment. In principle, they allow recapitulation of complex 3D microenvironments and stimulation with precisely defined mechanical forces. However, such setups are typically difficult to use routinely, require skilled expertise, and are less amenable to simultaneously applying different conditions or output parameters. Furthermore, while micro-scale systems allow for direct observation of complex biological processes on the single-cell level, they yield data with small cell numbers, making statistical analyses problematic.
We were interested in developing a ‘meso-scale’ in vitro model that could recapitulate the fluid forces in the lymphatic microenvironment on interstitial cells that enter the lymphatic vessel as well as on the lymphatic endothelium itself, since both of these work in concert during cell trafficking from peripheral tissues such as when tumor cells invade before metastasis (Fig. 1a–d). To pursue this objective, we designed a culture chamber that allows simultaneous stimulation of LECs with both transmural and luminal flows in a standard cell transmigration assay. Previous work from our group established a simpler model of the tumor–lymphatic microenvironment containing only transmural flow,36 although it was easy to set up and analyze, and amenable to high-throughput experimentation. The new model was designed to keep the simplicity of the earlier model, where the co-cultures could be first established in standard incubator conditions, outside the fluidic device, to ensure LEC confluence and desired morphology before imposing fluid stresses. It was also designed to allow both interstitial and luminal flows to be imposed and controlled individually. We characterized fluid flow patterns in the device, verified cell viability and developed a protocol for high-throughput automated data analyses. Using this new model, we demonstrate how luminal and transmural flows each enhance the invasion and transmigration of MDA-MB-231 tumor cells through the extracellular matrix and across a LEC monolayer.
Materials and methods
Cell lines
Human dermal LECs were isolated from neonatal foreskin and cultured as described previously.33 MDA-MB-231 human mammary adenocarcinoma cells were purchased from ATCC and maintained in DMEM with 10% FBS (Gibco, Invitrogen Corporation, Carlsbad, CA USA) until 70–80% confluence before being detached and used for the experiments.Chamber design, fabrication and mounting
To mimic the lymphatic microenvironment (Fig. 1d), the flow chamber was designed to be easy to use and handle, to guaranty cell viability for a time frame of at least 24 h, to avoid liquid leakages and to allow time lapse imaging with an inverted fluorescent microscope.The chamber design was drafted using commercially available CAD software (SolidWorks Corp., Waltham, MA USA). The chamber is composed of three components (Fig. 1f–j): a base, comprising 5 microfluidic channels (500 μm high, 8 mm wide, 40 mm long); a top, which includes the fluidic inlets and outlets, and five regions to house the 6.4 mm cell culture inserts (BD Biosciences, San Jose, CA USA); and caps for each housing, each including a transmural flow inlet. The components were produced by high resolution stereolithography (PROFORM AG, Marly, CH) using U.V. curable resin Somos® Watershed XC11122 (DSM, Heerlen, NL), which is optically transparent and certified to be biocompatible (which we independently verified).
O-rings were placed in the dedicated grooves on the top component and on each cap, in order to seal the microfluidic channels and the cell culture insert, respectively. Before each experiment, the chambers were mounted by placing the top component on the base and tightening with bolts. The devices were then sterilized with ethylene oxide gas for 12 h and placed under vacuum for 48 h.
Flow control and measurement
Separate peristaltic pumps (IDEX GmbH, Wertheim, DE) were used to generate transmural and luminal flows, which were kept constant throughout the experiments. The transmural flow rate was set at either 0 or 2 μL min−1, leading to a transmural flow velocity of 0 or 1 μm s−1 across the LEC monolayer. For luminal flow, the flow rates at the luminal inlet were fixed in order to provide the desired shear stress to cells on the membrane (0, 0.01 or 1 dyn cm−2), according to the Poiseuille approximation for rectangular section channels: τ = (6μ·Q)/(w·h2), where τ is the shear stress (dyn cm−2), μ is the dynamic viscosity of the culture medium (P), Q is the inlet flow rate (μL), and w and h are the width and the height of the channel, respectively (mm). Starting from this first estimate, a more detailed calculation was done by finite element analyses, as described below. The luminal flow velocity in the channel was also validated with a particle tracking method34 using 5 μm diameter fluorescent beads (Spherotech Inc., Lake Forest, IL), and long exposure times (0.15 s) to calculate their track lengths and thus estimate the average luminal velocity within the channel.Finite element analysis of the luminal fluid flow distribution
To calculate flow profiles in the chamber, the fluidic channel and the gel compartment were discretized in 3D using COMSOL Multiphysics numerical simulation software (COMSOL Inc., Burlington, MA USA). The geometry of the channel was imported from the original CAD design and a tetrahedral mesh was created. The flow field in the channel was computed using the Navier–Stokes equations, with a fixed inlet flow rate and the outlet pressure set to zero. The above-mentioned equations were coupled with the Darcy equation, accounting for flux in the porous matrix: ∇p = −μ/k·v, where ∇p is the pressure gradient across the transmural portion, v is the fluid velocity; μ is the fluid viscosity; k is permeability of the 3D matrix, assumed to be 10−6 cm2 as previously determined.35LEC culture under flow conditions and transmigration assay
LECs were cultured to confluence on the underside (e.g., facing down) of collagen-coated, opaque Fluoroblok™ culture inserts (BD Biosciences, San Jose, CA, USA), which contain a 18 μm-thick membrane with pore diameters of 8 μm and which block light transmission from the upper well when visualized from below. Cell tracker green (1 μM, Invitrogen, Carlsbad, CA, USA) was used to label the MDA-MB-231 cells, which were suspended in 1.5 mg mL−1 type I collagen with 10% Matrigel (both from BD Biosciences) at 106 cells per mL and added to the inserts. The matrix was allowed to polymerize for 1 h in a humidified incubator at 37 °C and 5% CO2, during which time the chamber and tubings were primed with Endothelial Basal medium (EBM with 2% FBS and 0.1%BSA; all from Gibco). In some experiments, 5 μl ml−1 mouse anti-human CCR7 neutralizing antibody (MAB197, R&D System, Minneapolis, MN) was added to the medium and to the tumor cell–matrix solution. Following polymerization, the inserts were placed in the flow chamber and sealed with the dedicated caps. Tubing was connected via luer-lock adaptors and fluid flow was introduced at the desired rate.For transmigration experiments, the chamber was placed on an inverted fluorescence microscope (Axiovert 200 M, Carl Zeiss AG, Feldbach, CH) maintained at 37 °C. Images, focused on the LEC monolayer, were acquired every 15 min for 12 h (5 regions per membrane, 5 membranes per experiment). Flowing cell culture medium was saturated in 5% CO2 and supplemented with HEPES buffer (Gibco).
Cell viability assay
LECs were cultured on the underside of the insert membranes as described above, and subsequently exposed for 24 h under different flow combinations (0, 0.01 or 1 dyn cm−2 luminal flow and 0 or 1 μm s−1 transmural flow) for 24 h. The inserts were then removed from the flow chamber, placed in a standard 24-well plate, rinsed with PBS, and treated with a cell viability assay kit for mammalian cells (Invitrogen) following the manufacturer's protocol. Fluorescence images were acquired and the relative fractions of live vs. dead cells were computed.Immunostaining
Immunofluorescence staining on the culture inserts was performed with FITC-conjugated mouse anti-human CD31 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, Ancell, Stillwater, MN), mouse anti-human ICAM-1 (1:50, R&D Systems,) and goat anti-human CCL21 (1:25, R&D Systems). Cells were fixed in 2% paraformaldehyde in PBS for 15 min, rinsed with PBS, and immunostained according to standard protocols. Cell nuclei were labelled with 4′6′-diamidino-2-phenylidole (DAPI, Vector Laboratories, Burlingame, CA USA) and imaged using a LSM 700 confocal microscope (Zeiss). Imaris (Bitplane AG, Zurich, CH) was used for the z-stack reconstruction.
:50, Ancell, Stillwater, MN), mouse anti-human ICAM-1 (1:50, R&D Systems,) and goat anti-human CCL21 (1:25, R&D Systems). Cells were fixed in 2% paraformaldehyde in PBS for 15 min, rinsed with PBS, and immunostained according to standard protocols. Cell nuclei were labelled with 4′6′-diamidino-2-phenylidole (DAPI, Vector Laboratories, Burlingame, CA USA) and imaged using a LSM 700 confocal microscope (Zeiss). Imaris (Bitplane AG, Zurich, CH) was used for the z-stack reconstruction.
Quantification of cell transmigration
During the imaging acquisition process, the focal plane was maintained on the luminal side of the LEC monolayer. Due to the light-blocking insert membranes, fluorescence from the labeled tumor cells could only be detected from the luminal side. We could ensure that the fluorescent objects that we tracked represented transmigrated tumor cells (rather than, for example, abluminally positioned cells that might, extend cell projections across the LECs) by their size and by tracking in the x–y plane their movements across the LEC surface until they detached and disappeared from the focal plane.The images acquired during the transmigration experiments were processed with ImageJ (NIH, Bethesda, MD USA) using a custom macro to identify the cells. Imaris (Bitplane AG, Zurich, CH) was used with its tracking algorithm to automatically extrapolate the appearance, trajectory, and disappearance of each tumor cell from the membrane. An in-house Matlab (Mathworks, Natick, MA) code was used to obtain the transmigration dynamics, transmigration rates and adhesion times of each cell.
Results and discussion
Characterization of luminal flow in the chamber
To characterize the flows induced on the LECs cultured on the insert membranes, we measured flow velocities in the channel for each fixed inlet flow rate using fluorescent beads and time lapse fluorescent imaging as previously described.34 The measured average velocities were consistent with Poiseuille flow profiles (Fig. 2a), confirming the correct positioning of the insert membrane at 500 μm from the chamber bottom. The computationally estimated shear stress profiles on the luminal surface of the LECs further demonstrated that when both transmural and luminal flows were present, the contribution from transmural flow to the overall average luminal shear stress was negligible (Fig. 2b). Even with the lowest average luminal flow rate used, the transmural flow increased the average luminal shear stress by roughly 5%, and the maximum shear stress (i.e., at the downstream end of the membrane, relative to the luminal flow direction) by ∼20%. | ||
| Fig. 2 Characterization of luminal shear stress in the flow chamber. (a) Computational estimate of flow velocity (red line) in the luminal flow channel compared to experimental measurements (black points), which were calculated from trajectories of fluorescent beads as tracing particles. The consistency between measured and estimated velocity profiles confirms the correct positioning of the insert in the device. (b) Computational estimate of luminal shear stress on the LEC monolayer when the inlet luminal flow velocity was set at 25 μL min−1, with and without interstitial flow. With this fixed inlet velocity, the average shear stress on the insert membrane is 0.010 ± 0.001 dyn cm−2, while adding the interstitial flow this value increases of only 5%. | ||
LECs form and maintain a viable monolayer under different flow conditions
The integrity of the LEC monolayer after 24 h culture in the device was confirmed by fluorescence microscopy. The LECs remained confluent and viable, with no visible differences in expression patterns of cell–cell junctions (PECAM-1, Fig. 3a) or in cell viability (Fig. 3b and c) between static and flow conditions. Of course, many features of the in vivo lymphatic vessel cannot be recapitulated in this in vitro system, and it is unknown how the more complex microenvironment around a lymphatic vessels affects cell transmigration; however, the system allows us to carefully control the biomechanical environment and monitor subtle changes in cell migration, which would be difficult to achieve in vivo. | ||
| Fig. 3 Lymphatic endothelial cells maintain confluence, viability, and junctional integrity under different flow conditions. Lymphatic endothelial cell (LEC) monolayer was evaluated by fluorescence microscopy after 24 h culture under different flow conditions (TF, transmural flow; LF, luminal flow). (a) Intercellular junctions were maintained under all flow conditions (green, PECAM-1; gray, DAPI). (b) Live (green, calcein AM) and dead (red, propidium iodide) staining of the LECs on −1, the membrane. (c) Quantification of cell viability; no significant differences were observed. Scale bars: 50 μm. | ||
Quantification of cell transmigration dynamics
In our previous work,36 we assessed transmigrated tumor cells at a single time point and could not differentiate between cells that had transmigrated early on (i.e., those that were positioned near the abluminal side of the LECs at the start) from those that had transmigrated later (i.e., those that also had to invade through the ECM). Furthermore, because transmigrated tumor cells could either stay attached to the LEC monolayer, detach and float, or settle and attach to the plastic bottom of the well, three compartments had to be analyzed for each sample: the apical side of the LEC monolayer, the bottom of the culture dish, and the medium in between. To avoid these issues and also record the kinetics of each transmigrated cell, we performed live imaging on multiple regions of the apical side of each sample over 6 h.We used opaque insert membranes and imaged from the focal plane fixed on the LEC luminal surface so that fully transmigrated, fluorescently labelled tumor cells could be clearly distinguished by their size and their movement along the x–y plane. Representative confocal images in the z-plane confirmed that LECs remained confluent and firmly adhered to the membrane and that transmigrating tumor cells were only seen on the luminal, not the abluminal, surface of the LECs (Fig. S1, ESI†).
We developed an algorithm to identify and track the trajectories of each cell appearing on the membrane over time (Fig. 4a and b), since we found that simply counting the number of transmigrated cells present on the membrane at each timepoint was not accurate in reflecting the true transmigration rate due to the inability to account for those cells that have detached in the meantime (Fig. 4c and d). By tracking each cell individually and recording when it both appears and disappears from the luminal surface, we can obtain a more accurate picture of the transmigration dynamics.
 | ||
| Fig. 4 Quantification of cell intravasation dynamics. Data shown are representative of the average responses observed. (a) Tumor cells migrating across lymphatic endothelial cells (LECs) were imaged over 6 h; scale bar: 120 μm. (b) After image processing, cell movements on the membrane were tracked. (c) Identification of the time of appearance of each tumor cell on the apical side of the LEC monolayer (green dots) as well as its detachment from the LECs (red dots). (d) Tumor cell transmigration rate (cells per time) was calculated by considering both the appearance and disappearance of cells on the membrane, which is different from the standard method of counting only the cells present on the membrane at an endpoint. Δ reflects the number of cells leaving the membrane, as indicated by the red dots in panel (c). | ||
Luminal and transmural flows increase MDA-MB-231 transmigration rate and modulate their adhesion to the lymphatic endothelium
Cell intravasation in the lymphatic capillaries is a critical step in tumor cell dissemination and metastasis. Most carcinomas sprout from the primary tumor into the surrounding interstitial space, invade the surrounding tissues, and eventually intravasate into lymphatic vessels to disseminate.37 To explore the effects of various fluid forces on tumor cell transmigration, we used our new model to examine first the role of luminal shear flow. Based on previous estimations, the velocity of the lymph in the lymphatic capillaries in a mouse tail model is in the range of 5–30 μm s−1,38 leading to wall shear stresses between 0.01–0.06 dyn cm−2. In a rat mesenteric pre-nodal lymphatic model, the intraluminal flow has been shown to lead to an average shear stress of about 0.6 dyn cm−2, with peaks of 4–12 dyn cm−2.39 Lymph flow in the lymphatic capillaries is highly variable, depending on the location in the body. Moreover, it is increased during inflammatory events.17,40 Fischer and collaborators measured resting average lymph flow velocities in human skin lymphatic capillaries and found values in the range of 7–14 μm s−1.41 Assuming a viscosity of 1.5 cP42 and a Hagen–Poiseuille flow, the relative shear stress would be between 0.01 and 0.04 dyn cm−2.Based on these data, we decided to examine the effect of two different shear stress values, 0.01 (low shear) and 1 (high shear) dyn cm−2, modeling the shear forces present in the lymphatic capillaries during physiological conditions and inflammation, respectively.
Interestingly, we observed a shear flow dependent increase in MDA-MB-231 breast cancer cell transmigration rate across the lymphatic wall (Fig. 5a and b).
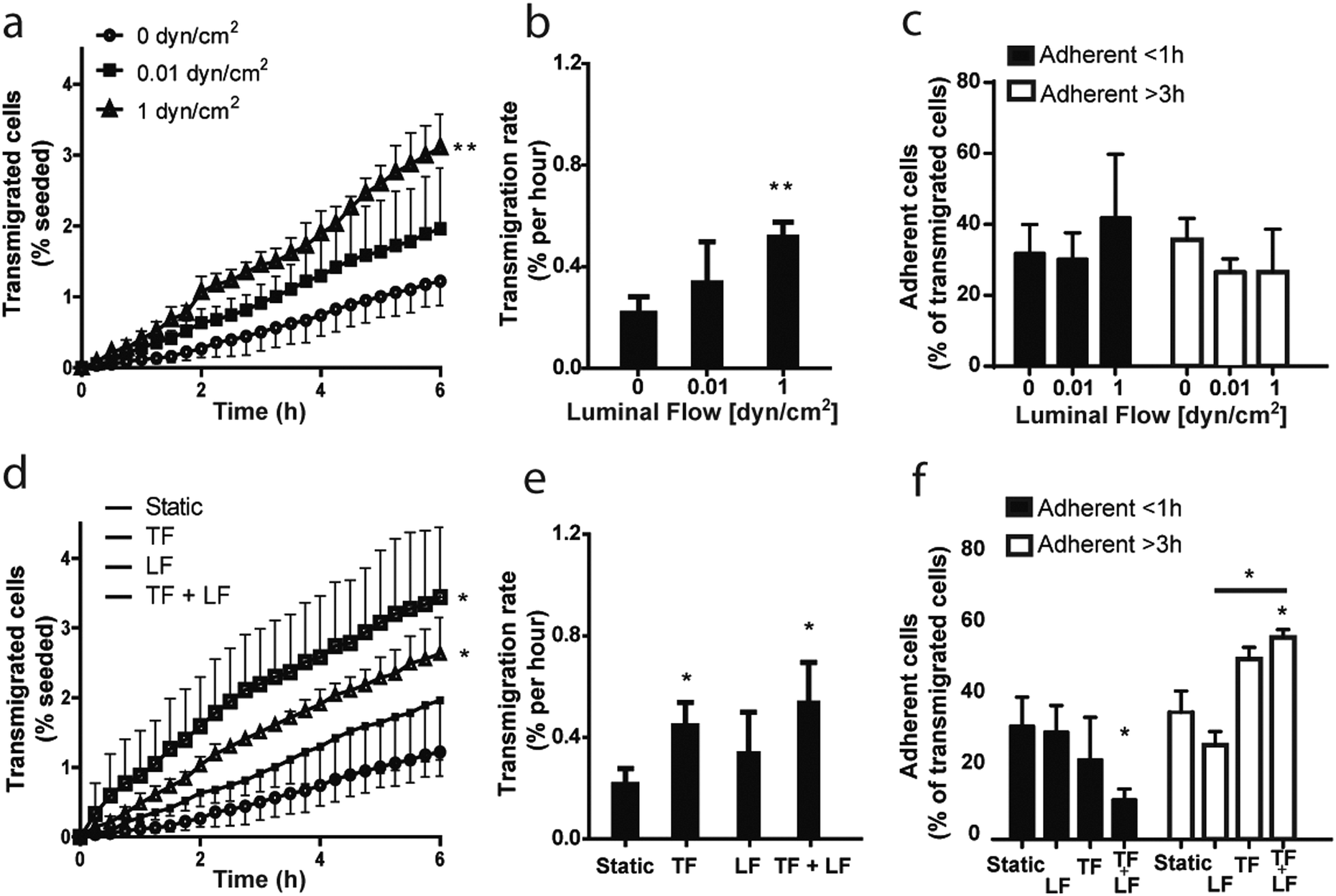 | ||
| Fig. 5 Luminal flow enhances tumor cell transmigration across lymphatic endothelium, while interstitial flow further drives tumor cell intravasation. (a) Dynamics of invasion and intravasation of MDA-MB-231 breast cancer cells from a 3D matrix across a lymphatic endothelial cell (LEC) monolayer under luminal flow alone, as measured by the appearance of newly transmigrated cells on the luminal side of the LECs as a function of time. (b) Transmigration rates for luminal flow conditions as quantified from 3–4 independent experiments. (c) Percentage of transmigrated tumor cells that adhere to the lymphatic endothelium under luminal flow conditions for less than 1 h (black bars) or more than 3 h (white bars). (d) Dynamics of intravasation of MDA-MB-231 tumor cells under various combinations of luminal and transmural flow. (e, f) Quantification of tumor cell (e) transmigration rates and (f) adhesion to the LECs. *p ≤ 0.05; **p ≤ 0.01 compared to static conditions. TF = transmural flow (1 μm s−1); LF = luminal flow (0.01 dyn cm−2). Data shown are pooled from 3–4 independent experiments. | ||
These data show for the first time that luminal shear stress indirectly affects tumor cells in the interstitial space through its action on the lymphatic endothelium, which in turn actively modifies the surrounding microenvironment and modulates tumor cell migration dynamics.
Moreover, we examined MDA-MB-231 adhesion time on the endothelial monolayer after transmigration, but we couldn't observe any significant effect of luminal flow (Fig. 5c).
We then investigated whether the combination of luminal and transmural flow has an effect on tumor cell transmigration. Indeed, simultaneous shear stress and transmural mechanical stimuli provoked an upregulation of intravasation higher than when the single flows were applied (Fig. 5d), as indicated by a significant increase in the transmigration rate (Fig. 5e).
It is important to note that transmural flow alone, set at a velocity of 1 μm s−1, caused a significant increase of cell transmigration (Fig. 5e). These findings are consistent with what has been previously reported using the standard transwell system.43 Nevertheless, combining transmural and luminal flows doubled the number of transmigrated cells with respect to luminal flow alone (Fig. 5d). In addition, we observed that transmural flow promotes cell adhesion to the endothelium when compared to luminal flow, suggesting that these distinct biomechanical cues have opposite effects in modulating cell adhesion to lymphatics (Fig. 5f). Interestingly, when we combined the two stimuli we noticed a significant increase in the adhesion time of the tumor cells on the endothelium, as there are significantly less cells detaching after less than 1 hour of adhesion respect to the static condition and significantly more cells still adherent after 3 hours respect to the luminal flow condition (Fig. 5f).
These results show that our in vitro model is capable of detecting differences in tumor cell transmigration across a lymphatic monolayer in response to different biophysical stimuli. Previous studies had shown and justified the effect of transmural flow on tumor cell intravasation in vitro, but the effect of luminal or luminal plus transmural flows could not be investigated because of the lack of reliable model systems. Here, we have confirmed previously published data obtained with well-established systems, and we have given evidences for a new mechanism of regulation of tumor cell migration, not observable with any other existing system.
The flow-mediated change in tumor cell transmigration observed here could be attributed to multiple factors. We previously demonstrated that transmural flow strongly induces CCL21 expression by LECs,17 which engages with ICAM-1 to facilitate cell transmigration. Here, we found that luminal shear stress also induces CCL21 upregulation by LECs (Fig. 6a and b). Flow-enhanced CCL21 upregulation was required for the flow-enhanced tumor cell transmigration, since blocking antibodies against CCR7 signaling prevented this effect (Fig. 6c).
 | ||
| Fig. 6 Luminal shear stress alters LEC expression of CCL21 to enhance tumor cell transmigration. (a) Immunostaining in LECs for CCL21 (red) and PECAM-1 (green) after 12 h stimulation with luminal flow (0, 0.01 or 1 dyn cm−2); cell nuclei (DAPI) shown in grey. Scale bar: 10 μm. (b) Quantification of CCL21 staining, from 3 independent experiments. (c) Transmigration rate of MDA-MB-231 cell across a LEC monolayer, pre-conditioned with different levels of luminal shear stress and in the presence or absence of CCR7 blocking antibody, as quantified from 3–4 independent experiments. *p ≤ 0.05 compared to static conditions. | ||
CCL21 is known to strongly bind extracellular matrix proteoglycans, and we previously measured its binding constant KD (koff/kon) to the Matrigel–collagen matrix used here as KD = 7 nM.43 This strong binding should counteract convection-driven forces into the lymphatic lumen. Such low binding constant allows the chemokine not to be transported away from the endothelium by the convective flows. To further confirm this, we perform a z-stack confocal imaging of an exogenous CCL21 gradient in a Matrigel–collagen matrix, both in static and flow condition, revealing no difference between the two (Fig. S2, ESI†).
Moreover, the effect of an overexpression of a non-matrix-binding chemokine by the lymphatic endothelium would also affect the tumor cell in the extracellular space, since the Peclet number in our system is in the order of 0.1, indicating that diffusion dominates over convection in the matrix (although the effect of convection cannot be ignored).
Interstitial flow on tumor cells has been shown to cause counterbalanced directional clues upstream (CCR7 independent) or downstream (CCR7 dependent) with respect to interstitial flow; this balance was found to be dependent on the cell density in the matrix.44 In particular, our group has recently demonstrated that it is possible to identify distinct tumor subpopulations, one of which responds with a high directedness towards the flow stream.43
While interstitial flow effect on tumor cell migration has been already examined, here we show for the first time that luminal flow is a potential regulator of the lymphatic function through its action on CCL21 expression. To further investigate this hypothesis, we performed the transmigration experiments with different level of luminal flow, in presence or absence of a CCR7 blocking antibody (Fig. 6c). We observed that blocking CCR7 signalling abolished luminal flow – driven transmigration of MDA-MB-231 cells, thus confirming that luminal flow can drive CCR7-expressing tumor cell transmigration across a lymphatic endothelium through the regulation of CCL21 in LECs.
However, luminal and transmural flow may have a much broader impact on lymphatic function, and further studies are needed to clarify new regulation pathways dependent on such biomechanical stimuli.
Conclusions
We have presented and characterized a novel flow chamber, which combines a standard Boyden chamber assay with controlled microfluidics to simulate in vivo-like biomechanics. Indeed, we were able to model the different compartments of the microenvironment of a lymphatic capillary (vessel lumen, vessel wall and extracellular space) and its specific biomechanical cues (transmural and luminal flow). Moreover, the presented system allows for the study of cell transmigration across a LEC monolayer; it permits the quantification of cell transmigration dynamics, transmigration rates and cell detachment from the endothelium.Our platform was applied to study the role of transmural and luminal flow on intravasation of a breast cancer cell line (MDA-MB-231) across a LEC monolayer. We demonstrated that luminal flow indirectly increases tumor cell transmigration by its action on LECs, specifically by upregulating CCL21 expression. We have also demonstrated that luminal and transmural flow increase tumor cell transmigration more than luminal flow alone. Our study provides new insights regarding flow-mediated regulation of tumor migration in the lymphatic system, and presents a novel in vitro tool for further research on potential targets for cancer therapy.
Acknowledgements
The authors are grateful to Samuel Gex, Olivier Burri, and Cara Buchanan for helpful advice and assistance. This study was funded by grants from the Swiss National Science Foundation (31-135756), the Swiss Cancer League (2696-08-2010), and the European Research Commission (AdG – 323053).Notes and references
- M. L. Kahn and D. J. Rader, Cell Metab., 2013, 17, 627–628 CrossRef CAS PubMed.
- G. J. Randolph, V. Angeli and M. A. Swartz, Nat. Rev. Immunol., 2005, 5, 617–628 CrossRef CAS PubMed.
- H. Wiig and M. A. Swartz, Physiol. Rev., 2012, 92, 1005–1060 CrossRef CAS PubMed.
- J. D. McAllaster and M. S. Cohen, Adv. Drug Delivery Rev., 2011, 63, 867–875 CrossRef CAS PubMed.
- A. Gogineni, M. Caunt, A. Crow, C. V. Lee, G. Fuh, N. van Bruggen, W. L. Ye and R. M. Weimer, PLoS One, 2013, 8, e68755 CAS.
- F. V. D. A. W. Lund, S. Hirosue, V. R. Raghavan, C. Nembrini, S. N. Thomas, A. Issa, S. Hugues and M. A. Swartz, Cell Rep., 2012, 1, 1–9 CrossRef PubMed.
- E. F. Tewalt, J. N. Cohen, S. J. Rouhani, C. J. Guidi, H. Qiao, S. P. Fahl, M. R. Conaway, T. P. Bender, K. S. Tung, A. T. Vella, A. J. Adler, L. Chen and V. H. Engelhard, Blood, 2012, 120, 4772–4782 CrossRef CAS PubMed.
- M. A. Swartz and A. W. Lund, Nat. Rev. Cancer, 2012, 12, 210–219 CrossRef CAS PubMed.
- S. Hirosue, E. Vokali, V. R. Raghavan, M. Rincon-Restrepo, A. W. Lund, P. Corthesy-Henrioud, F. Capotosti, C. Halin Winter, S. Hugues and M. A. Swartz, J. Immunol., 2014, 192, 5002–5011 CrossRef CAS PubMed.
- N. Resnick, H. Yahav, A. Shay-Salit, M. Shushy, S. Schubert, L. C. Zilberman and E. Wofovitz, Prog. Biophys. Mol. Biol., 2003, 81, 177–199 CrossRef.
- B. D. Johnson, K. J. Mather and J. P. Wallace, Vasc. Med., 2011, 16, 365–377 CrossRef PubMed.
- K. Yamamoto and J. Ando, J. Pharmacol. Sci., 2011, 116, 323–331 CrossRef CAS.
- S. Liang, M. J. Slattery, D. Wagner, S. I. Simon and C. Dong, Ann. Biomed. Eng., 2008, 36, 661–671 CrossRef PubMed.
- M. A. Swartz, Adv. Drug Delivery Rev., 2001, 50, 3–20 CrossRef CAS.
- A. M. Tchafa, A. D. Shah, S. Wang, M. T. Duong and A. C. Shieh, J. Visualized Exp., 2012, 65, 4159 Search PubMed.
- P. Koumoutsakos, I. Pivkin and F. Milde, Annu. Rev. Fluid Mech., 2013, 45, 325–355 CrossRef PubMed.
- D. O. Miteva, J. M. Rutkowski, J. B. Dixon, W. Kilarski, J. D. Shields and M. A. Swartz, Circ. Res., 2010, 106, 920–931 CrossRef CAS PubMed.
- Y. Kawai, M. Kaidoh, Y. Yokoyama and T. Ohhashi, Cancer Sci., 2012, 103, 1245–1252 CrossRef CAS PubMed.
- Y. Kawai, Y. Yokoyama, M. Kaidoh and T. Ohhashi, Am. J. Physiol.: Cell Physiol., 2010, 298, C647–C655 CrossRef CAS PubMed.
- H. G. Bohlen, O. Y. Gasheva and D. C. Zawieja, Am. J. Physiol.: Heart Circ. Physiol., 2011, 301, H1897–H1906 CrossRef CAS PubMed.
- G. W. Schmid-Schonbein, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3–4 CrossRef CAS PubMed.
- A. M. Andrews, D. Jaron, D. G. Buerk, P. L. Kirby and K. A. Barbee, Nitric oxide, 2010, 23, 335–342 CrossRef CAS PubMed.
- J. W. Breslin and K. M. Kurtz, Lymphatic Res. Biol., 2009, 7, 229–237 CrossRef PubMed.
- Z. Pujic, D. Mortimer, J. Feldner and G. J. Goodhill, Comb. Chem. High Throughput Screening, 2009, 12, 580–588 CrossRef CAS.
- S. A. Parsons, C. Jurzinsky, S. L. Cuvelier and K. D. Patel, Methods Mol. Biol., 2013, 946, 285–300 CAS.
- S. Chung, R. Sudo, P. J. Mack, C. R. Wan, V. Vickerman and R. D. Kamm, Lab Chip, 2009, 9, 269–275 RSC.
- M. K. Shin, S. K. Kim and H. Jung, Lab Chip, 2011, 22, 3880–3887 RSC.
- J. S. Jeon, I. K. Zervantonakis, S. Chung, R. D. Kamm and J. L. Charest, PLoS One, 2013, 8, e56910 CAS.
- K. H. Wong, J. M. Chan, R. D. Kamm and J. Tien, Annu. Rev. Biomed. Eng., 2012, 14, 205–230 CrossRef CAS PubMed.
- E. Bianchi, R. Molteni, R. Pardi and G. Dubini, J. Biomech. Eng., 2013, 46, 276–283 CrossRef PubMed.
- C. Buchanan and M. N. Rylander, Biotechnol. Bioeng., 2013, 110, 2063–2072 CrossRef CAS PubMed.
- M. B. Chen, J. A. Whisler, J. S. Jeon and R. D. Kamm, Integr. Biol., 2013, 5, 1262–1271 RSC.
- S. Podgrabinska, P. Braun, P. Velasco, B. Kloos, M. S. Pepper and M. Skobe, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16069–16074 CrossRef CAS PubMed.
- C. K. Tung, O. Krupa, E. Apaydin, J. J. Liou, A. Diaz-Santana, B. J. Kim and M. Wu, Lab Chip, 2013, 13, 3876–3885 RSC.
- C. P. Ng and M. A. Swartz, Am. J. Physiol.: Heart Circ. Physiol., 2003, 284, H1771–H1777 CrossRef CAS PubMed.
- J. D. Shields, M. E. Fleury, C. Yong, A. A. Tomei, G. J. Randolph and M. A. Swartz, Cancer Cell, 2007, 11, 526–538 CrossRef CAS PubMed.
- J. P. Sleeman and W. Thiele, Int. J. Cancer, 2009, 125, 2747–2756 CrossRef CAS PubMed.
- D. A. Berk, M. A. Swartz, A. J. Leu and R. K. Jain, Am. J. Physiol., 1996, 270, H330–H337 CAS.
- J. B. Dixon, S. T. Greiner, A. A. Gashev, G. L. Cote, J. E. Moore and D. C. Zawieja, Microcirculation, 2006, 13, 597–610 CrossRef PubMed.
- Z. Yuan, H. Rodela, J. B. Hay, D. Oreopoulos and M. G. Johnston, Lymphology, 1994, 27, 114–128 CAS.
- M. Fischer, U. K. Franzeck, I. Herrig, U. Costanzo, S. Wen, M. Schiesser, U. Hoffmann and A. Bollinger, Am. J. Physiol., 1996, 270, H358–H363 CAS.
- V. T. Turitto, Prog. Hemostasis Thromb., 1982, 6, 139–177 CAS.
- U. Haessler, J. C. Teo, D. Foretay, P. Renaud and M. A. Swartz, Integr. Biol., 2012, 4, 401–409 RSC.
- W. J. Polacheck, J. L. Charest and R. D. Kamm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11115–11120 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00085h |
| This journal is © The Royal Society of Chemistry 2015 |