
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic analysis of extracellular matrix-bFGF crosstalk on primary human myoblast chemoproliferation, chemokinesis, and chemotaxis†
Meghaan M.
Ferreira
a,
Ruby E.
Dewi
b and
Sarah C.
Heilshorn
*b
aDepartment of Chemical Engineering, Stanford University, Stanford, CA 94305-4045, USA
bDepartment of Materials Science and Engineering, Stanford University, 476 Lomita Mall, McCullough 246, Stanford, CA 94305-4045, USA. E-mail: heilshorn@stanford.edu; Fax: +1-650-498-5596; Tel: +1-650-723-3763
First published on 14th April 2015
Abstract
Exposing myoblasts to basic fibroblast growth factor (bFGF), which is released after muscle injury, results in receptor phosphorylation, faster migration, and increased proliferation. These effects occur on time scales that extend across three orders of magnitude (100–103 minutes). Finite element modeling of Transwell assays, which are traditionally used to assess chemotaxis, revealed that the bFGF gradient formed across the membrane pore is short-lived and diminishes 45% within the first minute. Thus, to evaluate bFGF-induced migration over 102 minutes, we employed a microfluidic assay capable of producing a stable, linear concentration gradient to perform single-cell analyses of chemokinesis and chemotaxis. We hypothesized that the composition of the underlying extracellular matrix (ECM) may affect the behavioral response of myoblasts to soluble bFGF, as previous work with other cell types has suggested crosstalk between integrin and fibroblast growth factor (FGF) receptors. Consistent with this notion, we found that bFGF significantly reduced the doubling time of myoblasts cultured on laminin but not fibronectin or collagen. Laminin also promoted significantly faster migration speeds (13.4 μm h−1) than either fibronectin (10.6 μm h−1) or collagen (7.6 μm h−1) without bFGF stimulation. Chemokinesis driven by bFGF further increased migration speed in a strictly additive manner, resulting in an average increase of 2.3 μm h−1 across all ECMs tested. We observed relatively mild chemoattraction (∼67% of myoblast population) in response to bFGF gradients of 3.2 ng mL−1 mm−1 regardless of ECM identity. Thus, while ECM-bFGF crosstalk did impact chemoproliferation, it did not have a significant effect on chemokinesis or chemotaxis. These data suggest that the main physiological effect of bFGF on myoblast migration is chemokinesis and that changes in the surrounding ECM, resulting from aging and/or disease may impact muscle regeneration by altering myoblast migration and proliferation.
Insight, innovation, integrationWe present a quantitative analysis of how insoluble cues from the extracellular matrix (ECM) alter the response of primary human myoblasts to soluble cues, specifically basic fibroblast growth factor (bFGF). As a technological innovation, we employed a microfluidic device that enabled quantitative, single-cell measurements of migration speed and direction, thus distinguishing between chemoproliferation, chemokinesis, and chemotaxis, which cannot be performed in traditional Transwell assays. These studies led to the biological insight that while ECM identity impacts bFGF-induced chemoproliferation, it does not modify bFGF-induced chemokinesis or chemotaxis. We discovered that bFGF is only weakly chemoattractive, and that its dominant effect on myoblast migration is chemokinesis. These discoveries would not have been possible without integrating an engineered microfluidic device with cell biology. |
Introduction
Muscle regeneration is mediated by both soluble and insoluble cues in the muscle microenvironment, which direct the behavior of myoblasts following injury. One such soluble cue, basic fibroblast growth factor (bFGF), is released following muscle injury;1–3 bFGF levels are elevated in dystrophic muscle, as it is more susceptible to injury.4,5 Subsequent binding and phosphorylation of the high-affinity fibroblast growth factor receptor-1 (FGFR1) occurs within minutes,6,7 but the short-lived receptor activation is thought to result in long-term downstream effects including enhanced myoblast migration (i.e., chemokinesis8 and chemotaxis8–13) and proliferation14–16 that require hours and days to observe, respectively. Insoluble cues, such as cell-adhesion sites in the extracellular matrix (ECM), are recognized by specific integrin receptors and can have pronounced effects on myoblast behavior. For example, laminin promotes myoblast adhesion, migration, and proliferation.17,18 Laminin, fibronectin, and collagen (IV and V) are all components of the muscle ECM in vivo,19 the composition of which changes over time as a result of aging and disease.20The signaling pathways downstream of integrin and growth factor receptors have many constituents in common; as a result, there are many opportunities for interactions between integrin and growth factor signals to further modulate cell behavior.21 These interactions can result in a synergy that enhances growth factor activity. For example, previous studies have demonstrated that laminin and epidermal growth factor (EGF) interact synergistically to enhance myoblast migration and proliferation.22 The increased chemoproliferation was hypothesized to be a consequence of the faster migration rate, since the daughter cells were observed to migrate away from one another much faster following division. Previous work with other cell types has suggested that crosstalk between integrin receptors and bFGF may exist.23 For example, bFGF has been found to act as a ligand and bind directly to the fibronectin-associated ανβ3 integrin receptor.24,25 Based on these earlier studies, we hypothesized that insoluble ECM cues may synergistically interact with soluble bFGF to alter the chemokinesis, chemotaxis, and chemoproliferation of primary human myoblasts.
To evaluate this hypothesis, we utilized a microfluidic technology that enables longitudinal, quantitative, single-cell analysis of cell movement. Previous observations of myoblast migration in response to bFGF have been performed almost exclusively with Transwell assays,8–13 which were developed as a tool to assess cell chemotaxis.26 However, Transwells fail to produce stable concentration gradients, which are necessary for quantitative analysis of chemotaxis. To determine the variation in the concentration gradient produced in a Transwell chamber at the cellular level (i.e., along the pore depth), we performed a longitudinal analysis of the soluble concentration gradient using finite element modeling (FEM). Our analysis revealed that the concentration gradient diminishes by 45% within the first minute of the assay. On average, we measured migration rates of around 0.2 μm min−1, which would require a minimum of 50 minutes for a cell to migrate through the 10 μm deep pores in a Transwell membrane, assuming the cell moved in a straight path. Another common challenge in the interpretation of data from Transwell experiments is that chemokinesis and chemoproliferation may be incorrectly interpreted as chemotaxis.27 Thus, to independently evaluate bFGF-induced chemokinesis (an increase in random motility) and chemotaxis (an increase in directional migration) in myoblasts, we utilized a microfluidic assay capable of producing a stable, linear concentration gradient.28,29 In contrast to Transwell assays, which preclude direct visualization of cell migration, this microfluidic assay allows quantitative, single-cell measurements of migration speed and direction through the use of time-lapse microscopy.
Although previous studies using Transwell assays reported bFGF as a chemoattractant,8–13 our studies revealed that bFGF promoted only weak chemotaxis in primary human myoblasts. In contrast, bFGF stimulated relatively strong chemokinesis, indicating that the dominant effect of bFGF on myoblast migration is chemokinesis, not chemotaxis. These results, combined with the FEM analysis of the instability of the concentration gradient produced by Transwell assays, indicate that Transwells are not an appropriate method for evaluating chemotaxis. Our microfluidic approach allowed us to not only decouple chemokinesis from chemotaxis, but also to study how the ECM modified those behaviors. We found that ECM identity had significant crosstalk effects on bFGF-induced chemoproliferation. In contrast, ECM identity and bFGF stimulation effects were strictly additive for chemokinesis and chemotaxis, suggesting that crosstalk does not impact changes in migratory behavior that occur in response to bFGF stimulation.
Materials and methods
Primary human myoblast isolation and culture
De-identified muscle samples were collected from patients during surgical treatment at Stanford University Hospital in accordance with the Human Subjects Committee of Stanford University guidelines. Primary human myoblasts were isolated from these samples as previously described.30 Myoblasts were plated onto 15 cm polystyrene dishes coated overnight at 37 °C with a 22 μg mL−1 collagen solution (Sigma, C-8919). The cells were cultured in skeletal muscle cell growth medium (SkGM) supplemented with human epidermal growth factor (EGF), dexamethasone, gentamicin/amphotericin-B, fetuin, insulin, and bovine serum albumin provided in the SkGM Bullet Kit (Lonza, CC-3160); cell medium was changed every other day. HyQtase (Thermo Scientific, SV3003001) was used to dissociate cells from polystyrene dishes. All migration studies were performed in a modified version of SkGM where EGF was removed. Myoblasts were not used for experiments beyond passage six.Western blot
Myoblasts were exposed to bFGF for 0 (not exposed), 1, 5, 10, 15, or 30 min. After exposure, whole-cell protein lysates were obtained by lysing cells in RIPA buffer (Cell Signaling Technology) containing protease inhibitors. Samples were centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200g for 10 min at 4 °C. Protein concentrations were measured using the DC Protein Assay kit (Bio-Rad), and 15–20 μg of protein was resolved in each lane of a 10% SDS-PAGE gel before transferring to PVDF membranes. Membranes were blocked in TBST (1× Tris buffered saline with 1% v/v Tween-20) and 5% nonfat dry milk. Primary antibodies for β-tubulin (1:1000, Cell Signaling Technology, 2146), and phospho-FGF receptor (1:1000, Cell Signaling Technology, 3476) were diluted in the same buffer, and the membranes were incubated in this solution overnight. Horseradish peroxidase-conjugated secondary antibodies against rabbit (711-035-152) and mouse (715-035-150) were purchased from Jackson ImmunoResearch and used at a 1:10000 dilution. Protein bands were visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce, 34080) in a Chemi-Doc MP system (Bio-Rad). The intensities of the protein bands were quantified by densitometry using Image J software (NIH freeware).
200g for 10 min at 4 °C. Protein concentrations were measured using the DC Protein Assay kit (Bio-Rad), and 15–20 μg of protein was resolved in each lane of a 10% SDS-PAGE gel before transferring to PVDF membranes. Membranes were blocked in TBST (1× Tris buffered saline with 1% v/v Tween-20) and 5% nonfat dry milk. Primary antibodies for β-tubulin (1:1000, Cell Signaling Technology, 2146), and phospho-FGF receptor (1:1000, Cell Signaling Technology, 3476) were diluted in the same buffer, and the membranes were incubated in this solution overnight. Horseradish peroxidase-conjugated secondary antibodies against rabbit (711-035-152) and mouse (715-035-150) were purchased from Jackson ImmunoResearch and used at a 1:10000 dilution. Protein bands were visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce, 34080) in a Chemi-Doc MP system (Bio-Rad). The intensities of the protein bands were quantified by densitometry using Image J software (NIH freeware).
Transwell assay
Transwell migration assays were conducted with polycarbonate membrane inserts with 8 μm diameter pores (Corning, 3422). Cells in SkGM at a concentration of 1.0 × 106 cells per mL were seeded onto Transwell membranes using a volume of 0.1 mL. The Transwell inserts were placed into 24-well plates containing 0.65 mL per well modified SkGM and incubated at 37 °C and 5% CO2 for 30 min before transferring to wells containing 0, 1.25, 5, 10, or 100 ng mL−1 recombinant human bFGF (Life Technologies, PHG0264) diluted in SkGM. Myoblasts were incubated for 6 h before collecting cells from the bottom chamber and counting with a hemacytometer. A minimum of three independent trials was performed at each concentration.Proliferation assay
Adsorbed protein substrates were prepared by incubating glass coverslips (Bellco Glass, 1943-10015) in mouse laminin from Engelbreth-Holm-Swarm (EHS) sarcoma (10 μg mL−1, Life Technologies, 23017-015), fibronectin from human plasma (10 μg mL−1, Sigma, F2006), or Type I collagen from calf skin (21.7 μg mL−1, Sigma, C-8919) dissolved in phosphate buffered saline (PBS) overnight at 37 °C. Coverslips were rinsed in warm PBS and placed in 24-well plates. Myoblasts in SkGM (4000 cells per mL, 0.5 mL per well) were cultured overnight at 37 °C and 5% CO2. Cells were collected from half of the wells for DNA quantification (PicoGreen dsDNA quantitation assay kit, Life Technologies, P7589). The remaining cells were cultured for an additional 96 h in SkGM supplemented with 100 ng mL−1 heparin sodium salt (Sigma-Aldrich, H3149) with or without 25 ng mL−1 bFGF (Life Technologies, PHG0264). Media was changed every other day, and cells were harvested 96 h after the initial time point. Cells were collected in lysis buffer comprised of 10 mM Tris (pH 7.4), 1 mM MgCl2, 20 μM ZnCl2 and 0.02% Triton X-100 dissolved in deionized water. Samples were kept on ice and stored at −80 °C. Thawed samples were sonicated prior to DNA quantification with PicoGreen dsDNA assay.Finite element modeling and gradient simulation
Gradient formation and steady-state concentration in both Transwell assays and the microfluidic device were simulated using the commercial finite element modeling (FEM) software, COMSOL Multiphysics (Burlington, MA). The diffusion coefficient for bFGF (17.2 kDa) in water at 37 °C was estimated to be D = 1.16 × 10−10 m2 s−1 using the Stokes–Einstein equation and a reported hydrodynamic radius (RH) of 28 Å [ref. 31]. To create the geometry for Transwell assay simulations, pore dimensions of an 8 μm diameter and a 10 μm height (the thickness of a polyester or polycarbonate membrane) were used. Both the number of pores per insert and the distance between pores were estimated using a known pore density of 1 × 105 pores per cm and a membrane growth area of 0.33 cm2. The heights of the fluid columns above and below the membrane were estimated by dividing the total volume of the upper and lower chambers (0.1 mL and 0.6 mL, respectively) by the total number of pores.To create the geometry for microfluidic device simulations, a two-dimensional, top-down projection of the device dimensions was employed. Verification of gradient formation within the microfluidic device was performed by perfusing the source channel with 1 μM solutions of fluorescently labeled dextran (10 kDa, RH = 23 Å, Sigma, D1828) and imaging the cell culture chamber (CCC) at the end of each 14 h experiment. Gradient formation was verified for each chemotaxis experiment, and only data collected from studies with a gradient matching the 3.2 ng mL−1 mm−1 slope predicted by FEM were included in the overall analyses.
Microfluidic device fabrication
The microfluidic device was designed using AutoCAD software (Autodesk, Inc., San Rafael, CA) and fabricated according to standard soft lithography methods32 by the Stanford Microfluidics Foundry as previously reported.28 Briefly, the device design was printed onto mylar masks and transferred to a 4-inch silicon wafer with positive relief features by crosslinking a negative photoresist (SU-8, MicroChem Corp.) with ultraviolet light exposure. The surface of the SU-8 master was coated with 3-aminopropyl trimethoxysilane (Sigma, St. Louis, MO). A 10:1 mixture of Sylgard 184 monomer and hardener (polydimethylsiloxane (PDMS), Dow Corning, Corning, NY) was poured over the SU-8 master and degassed under vacuum for 30 min. The PDMS was then cured at 65 °C for a minimum of 2 h before releasing and creating inlet and outlet holes using a biopsy punch (Syneo Corp., Angleton, TX). Devices were bonded to the glass surface of Lab-Tek chamber slides (Thermo Scientific, 154461) using oxygen plasma (SPI Plasma Prep III Plasma Etcher, West Chester, PA) and heated at 60 °C for 20 min to strengthen and accelerate bonding.
Chemotaxis and chemokinesis assays
Microfluidic devices were prepared by rinsing first with 70% ethanol and then with PBS. Devices were coated with Type I collagen, laminin, or fibronectin overnight at 37 °C before rinsing with PBS and filling with modified SkGM. Primary human myoblasts at a concentration of 1.0 × 106 cells per mL were suspended in SkGM and gently seeded into the cell culture chamber (CCC) through cell-seeding ports using a 10 μL pipetter. Devices were placed in an incubator at 37 °C and 5% CO2 for 2 h to allow cells to adhere before sealing the cell-seeding ports with plugs (Instech Laboratories, Inc., SP20/12) and connecting tubing to inlets. For chemotaxis experiments, modified SkGM was introduced into the sink inlets using 100 μL syringes (Hamilton, 201797), while modified SkGM plus bFGF (11 ng mL−1) and 10 kD Texas Red dextran (1 μM) were introduced into the source inlets. For chemokinesis experiments, source and sink channels were both filled with modified SkGM with or without bFGF (5 ng mL−1). Syringes were driven at a flow rate of 0.6 μL h−1 using a syringe pump (World Precision Instruments, SP220i). Phase contrast images were taken every 15 min for 16 h using an inverted fluorescence microscope (Zeiss, Axiovert 200M) with an environmental control chamber operating at 37 °C and 5% CO2. Images from the first 2 h were not included in the data analysis to ensure stable gradient formation, which was confirmed by imaging fluorescently-labeled dextran as described above. Myoblast migration was analyzed using MTrackJ,33 a manual cell-tracking plugin for the NIH ImageJ software. Cell speed and chemotactic index were quantified from position data using the Chemotaxis Tool plugin (Ibidi, Martinsried, Germany), which was also used to create compass plots of cell tracks and angular histograms.Results and discussion
Cellular responses to bFGF stimulation occur across various time scales
Basic FGF stimulation has been reported to mediate myoblast behavior with regards to differentiation, migration, and proliferation.1,8–16 The time scales over which cellular responses to bFGF stimulation are observed range from minutes, to hours, and even days. Within minutes of bFGF stimulation and binding, fibroblast growth factor receptor-1 (FGFR1), a receptor tyrosine kinase, is known to dimerize and autophosphorylate.7,31,34 Consistent with previous reports,7 we observed receptor phosphorylation at short time scales after exposing myoblasts to bFGF, with maximum phosphorylation occurring as early as 5 min post-exposure (Fig. 1A, Fig. S1, ESI†). Despite the short-lived effects of bFGF on FGFR1 phosphorylation, bFGF stimulation has long-lived consequences for cell behavior. For example, changes in myoblast migration are observed hours after stimulation. To assess the effect of bFGF stimulation on cell migration, we first employed the traditionally used Transwell assay, which consists of an insert with a porous membrane culture substrate. After seeding cells onto the membrane, the insert was placed into a well containing bFGF for 6 h. The number of cells that migrated across the membrane towards the bFGF source was enumerated at the end of the study. The Transwell assays indicated that bFGF enhanced migration, with peak migration occurring when a source concentration of 5 ng mL−1 bFGF was used to stimulate cells (Fig. 1B). Other studies have also revealed enhanced migration in response to bFGF using the Transwell platform, although the reported peak concentration varied between 1–100 ng mL−1 [ref. 8,10–13,15]. In contrast to the hours-long migration studies, the mitogenic (i.e., proliferative) effect of bFGF occurs on a time scale of days, as shown by a decrease in doubling time for myoblasts exposed to 25 ng mL−1 bFGF compared to unstimulated cells (Fig. 1C). Previous studies have reported a 10-fold increase in the number of murine skeletal myoblasts after 7 days of bFGF stimulation compared to untreated cells.15 Although chemoproliferative effects can confound the results of Transwell assays, the doubling times observed (38–62 h) were much longer than our 6 h Transwell study, and therefore should not have affected our interpretation of the migration data.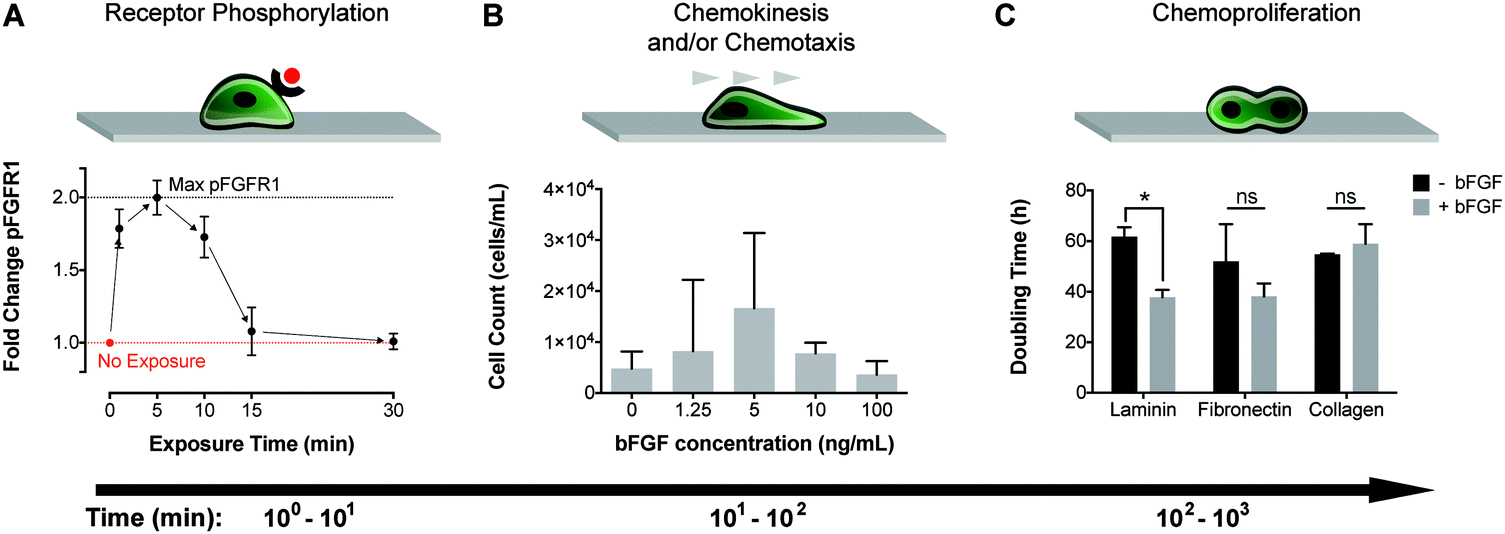 | ||
| Fig. 1 Primary human myoblast exposure to bFGF results in cellular responses that occur over a range of time scales from 100–103 min. (A) Phosphorylation of FGFR1 (pFGFR1) in primary myoblasts peaks 5 min after bFGF stimulation. (B) Stimulation with 5 ng mL−1 bFGF promoted the greatest amount of migration in myoblasts as measured in Transwell assays over 6 h. Error bars represent the standard deviation between three independent studies. (C) The doubling time for myoblasts with or without bFGF stimulation was determined on laminin, fibronectin, and collagen. Error bars represent standard deviation; *p < 0.05 and ns = not significant (p > 0.05). | ||
While the mitogenic effect of bFGF on myoblasts has been reported,14–16 it is unknown how the underlying matrix might affect bFGF-induced mitogenesis. Previous research has suggested that bFGF may interact or crosstalk with specific integrin receptors.23–25 These data led us to hypothesize that the composition of the underlying ECM, and hence the types of integrin receptors engaged, might arbitrate how cells respond to bFGF stimulation. We examined how the underlying matrix affected mitogenesis by culturing myoblasts on glass coverslips coated with laminin, fibronectin, or Type I collagen. We found that the ECM identity had a significant effect on chemoproliferation. Myoblasts cultured on laminin exhibited a statistically significant reduction in doubling time with the addition of bFGF, whereas myoblasts cultured on fibronectin and collagen did not. This finding confirmed our notion that matrix-bFGF crosstalk impacted chemoproliferation, and it encouraged us to look for additional matrix-dependent responses to bFGF in the form of chemokinesis and chemotaxis.
Microfluidic device to quantify chemokinetic and chemotactic responses to bFGF
To assess the impact of the underlying substrate on bFGF-induced myoblast migration, we employed a microfluidic device comprised of a polydimethylsiloxane (PDMS) layer bonded to thin coverslip glass (Fig. 2A and B). In contrast to the Transwell assay, which prevents visualization of cell migration across the membrane, the microfluidic device allows direct imaging of myoblast migration over time. As a result, the device enables individual cell tracking, which makes it possible to quantify migration speed and directionality at both the population and individual-cell levels. Cells are seeded in a 1 mm wide cell-culture chamber (CCC) at the center of the device. The CCC is flanked by two arrays of micro-capillaries, which increase fluidic resistance and prevent fluid flow and the generation of shear forces within the CCC.28 The absence of fluid flow within the CCC makes it easy to adapt the device to observe the behavior of loosely adherent cells29,35 or cells in 3D matrices.36,37 Channels on either side of the arrays are perfused with medium, such that the direction of fluid flow runs perpendicular to the micro-capillaries. These channels are fed through their two inlets simultaneously by the same syringe pump to ensure equal flow rates.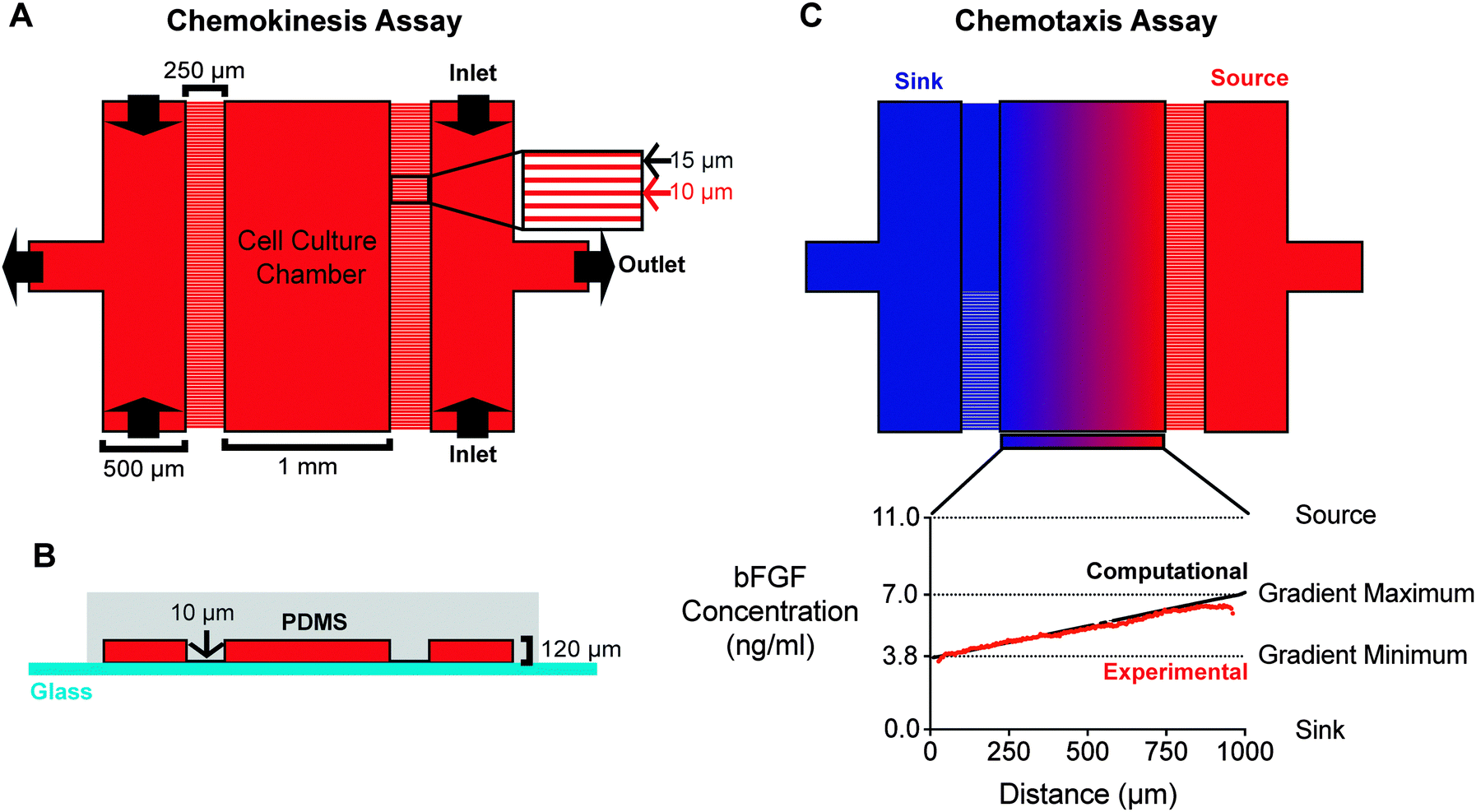 | ||
| Fig. 2 A microfluidic device for chemokinesis and chemotaxis assays. (A) The microfluidic device viewed from the top-down. The outermost channels (500 μm wide) are separated from the cell-culture chamber (CCC, 1 mm wide) by a micro-capillary array with capillaries (250 μm long and 10 μm wide) spaced 15 μm apart. For chemokinesis assays, the same medium is perfused into both outer channels to create a uniform concentration across the device and replenish growth factors. (B) A cross-sectional view of the microfluidic device. The medium-carrying channels and the CCC are 120 μm high. The difference in height between the CCC and the micro-capillaries (10 μm) increases fluidic resistance and, consequently, minimizes flow within the CCC. (C) For chemotaxis assays, the outermost channels (“source” and “sink”) are perfused with media containing different bFGF concentrations to create a stable, linear concentration gradient. Computational transport models (black line) of the predicted concentration profile were validated with experimental studies (red line). | ||
For chemokinesis assays, both outermost channels were filled with media containing either no bFGF or media containing 5 ng mL−1 bFGF (Fig. 2A), where the bFGF concentration was chosen based on results obtained from Transwell assays. For chemotaxis assays, media containing no bFGF was perfused into the “sink” channel, while media containing 11 ng mL−1 bFGF was perfused into the “source” channel, thus creating a stable, linear concentration gradient that ranged from 3.8–7.0 ng mL−1 bFGF across the CCC (Fig. 2C). The concentration gradient was determined computationally using a finite element modeling software package (COMSOL) and validated experimentally using a 10 kDa fluorescent dextran (Fig. 2C, lower panel). Unlike Transwell assays, which are commonly used to assess the chemotactic response of mammalian cells to a chemokine, the microfluidic chamber creates predictable concentration gradients that are stable over long time periods; the gradient generated by the microfluidic device remained stable for >14 h.37
For comparison, we performed an engineering analysis of the gradient produced by the traditional Transwell assay. Finite element simulations revealed that the gradient across the Transwell membrane diminishes by 45% within the first minute of the assay and is almost completely absent after 6 h (Fig. 3). Both the transiency and the variability of the Transwell gradient may be problematic for chemotaxis studies, as previous work has shown that gradient steepness can affect the chemotactic response of mammalian cells.38–40 Measurements of bulk concentrations in Transwell chambers give a deceptive indication of gradient stabilization, as the large volumes used in the upper and lower chambers (0.1 and 0.6 mL, respectively) dilute the flux of growth factor across the membrane making changes difficult to detect (Fig. S2, ESI†). These measurements do not accurately represent the concentrations presented to cells, which are located at the membrane. Furthermore, because Transwell assays prohibit direct imaging of cell migration, they can only provide a qualitative, population-based measurement of cell migration that does not distinguish between chemoproliferation, chemokinesis, and chemotaxis. While the ‘checkerboard’ method (i.e., reversing the media concentrations in the upper and lower chambers as an experimental control in Transwell studies), effectively reverses the gradient direction, the distinctly different volumes in the upper and lower chambers can result in different average growth factor concentrations across the membrane. For example, when the source is supplemented into the upper vs. the lower chamber the steady state concentration is 14% of the original source concentration, compared to 86% when the lower chamber is supplemented. The loss of gradient across the Transwell membrane indicates that differences in migration occurring after 6 h are a result of concentration-dependent chemokinesis and not chemotaxis. These limitations can confound results when attempting to quantify chemokinesis or chemotaxis independently, and they may obscure any behavior that primarily occurs in only a subset of the population. In contrast, the microfluidic device presented here provides quantitative, single-cell measurements for both chemokinesis and chemotaxis evaluation.
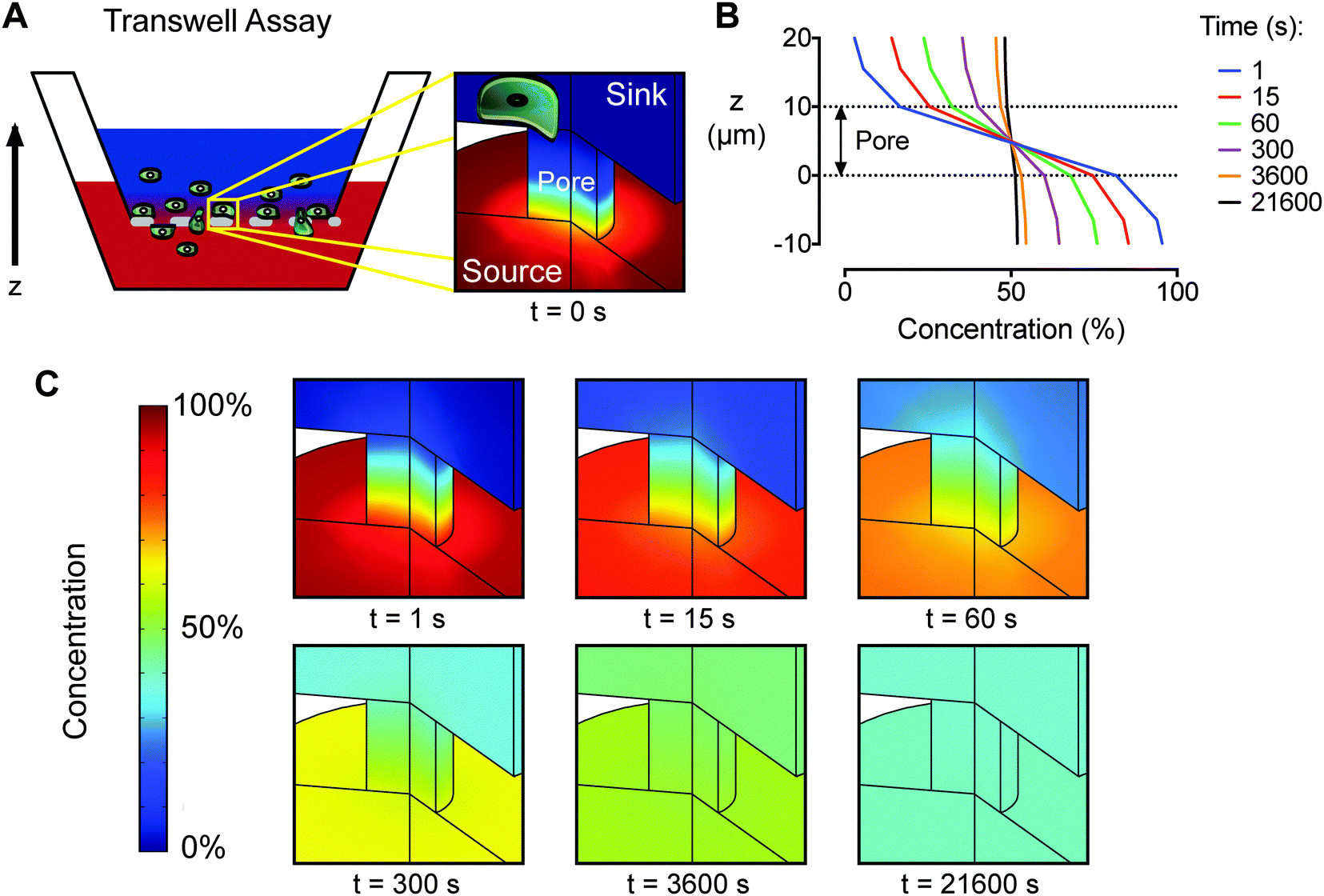 | ||
| Fig. 3 An engineering analysis of diffusion reveals significant instability of the concentration gradient within a Transwell assay. (A) Schematic diagram of a Transwell assay with the sink above the membrane and the growth factor source below the membrane. (B) Concentration gradient profiles across a membrane pore are displayed as a percentage of the initial source concentration for times between 1–21600 s (i.e., 1 s–6 h). (C) Finite element simulations of the concentration gradient across a membrane pore as a function of time. Red is 100% of the initial source concentration and dark blue is 0%. | ||
Basic FGF has an additive effect on myoblast migration rates, which are regulated by the extracellular matrix
To assess the chemokinetic activity of bFGF, we initially seeded myoblasts in static-culture, chamber-slides coated with fibronectin. Time-lapse microscopy revealed no significant difference between the migration speeds of myoblasts cultured in media with and without bFGF supplementation (5 ng mL−1) (Fig. 4A). However, when a media exchange was used to expose the cells to fresh bFGF immediately prior to imaging, a significant increase in migration speed was observed. These results suggest that the concentration of active bFGF decreases over time as a result of degradation41–43 or cell uptake.34,42,44–47 To circumvent these issues, we repeated the chemokinesis assays utilizing the microfluidic device, which enables constant perfusion of fresh bFGF-containing medium into the CCC. The results were similar to those obtained by refreshing the bFGF-supplemented media in the chamber-slide, demonstrating that the microfluidic device provided a suitable cell culture platform for chemokinesis studies.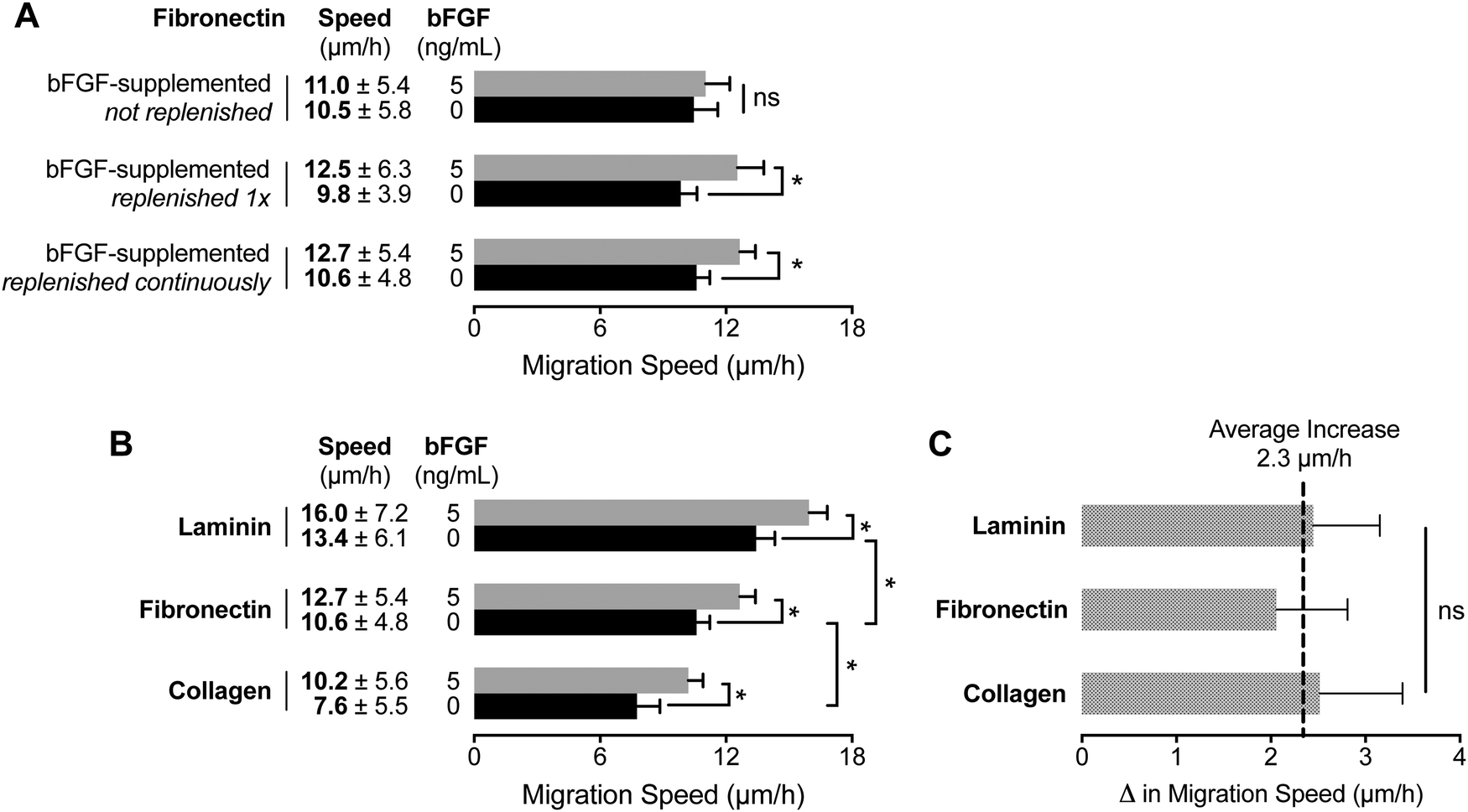 | ||
| Fig. 4 Migration speed of primary human myoblasts is mediated through interactions with the extracellular matrix and soluble bFGF. (A) Migration speeds for myoblasts cultured on fibronectin were quantified for media with (5 ng mL−1) and without bFGF supplementation using time-lapse microscopy. The bFGF-supplemented medium was delivered using three different methods: (1) without any replenishment, (2) replenishment immediately before imaging, and (3) continuous delivery of fresh supplemented medium using a microfluidic device. (B) Differences in myoblast migration speed as a result of substrate composition and bFGF stimulation with continuous medium replenishment. (C) The increased migration speed resulting from bFGF stimulation is not affected by the underlying substrate, indicating that the effect is strictly additive. Error bars represent 95% confidence intervals; *p < 0.01 and ns = not significant (p > 0.05). | ||
Next, we compared bFGF-induced chemokinesis on three different ECM substrates (laminin, fibronectin, and Type I collagen) using the microfluidic device. Although Transwells are sometimes coated with ECM proteins for what are commonly referred to as invasion assays,48,49 the combinatorial effect of matrix and growth factor on cell migration is rarely explored. Unlike Transwells, the device is amenable to quantification of protein adsorption on the surface of the CCC as a function of protein concentration per area (Fig. S2, ESI†). In the absence of bFGF, myoblasts exhibited significantly higher migration speeds on laminin (13.4 μm h−1) compared to fibronectin (10.6 μm h−1) (Fig. 4B). This observed trend is consistent with a previous study of MM14 cells (a myogenic cell line derived from mouse skeletal muscle), which exhibited faster migration speeds on laminin (20 μm h−1) compared to fibronectin.18 In contrast, migration on Type I collagen exhibited significantly slower speeds (7.6 μm h−1) than laminin or fibronectin. These differences are presumably a result of the engagement of different integrins by the unique cell-adhesive domains presented by each matrix.50–54
The addition of a uniform concentration of 5 ng mL−1 bFGF prompted significantly higher migration speeds for each of the different matrices (Fig. 4B). However, the net increase in myoblast migration speed with bFGF stimulation did not change significantly between the three matrices tested (Fig. 4C). Therefore, the chemokinetic effect of bFGF stimulation is strictly additive, and matrix identity does not influence bFGF-induced increases in migration speed.
Basic FGF produces mild chemoattraction above a minimum concentration threshold
Microfluidic devices were employed to assess the potential bFGF-induced chemotaxis of myoblasts, independent of the potentially confounding effects of chemokinesis or chemoproliferation. Myoblasts were seeded in the CCC of the microfluidic device, and the source and sink channels were perfused with media containing 11 and 0 ng mL−1 bFGF, respectively, to create a stable, linear gradient across the CCC (Fig. 2). The glass surface of the CCC was coated with laminin, fibronectin, or Type I collagen to explore the effects of any potential interactions between the ECM and bFGF on chemotaxis. Time-lapse microscopy was used to track cell positions over time; images of cell position were collected every 15 min over a 14 h time period after allowing time (∼2 h) to establish a stable gradient. Images of the 1 mm wide CCC were discretized into 250 μm regions referred to as G1, G2, G3, or G4, which correlate with gradients of 3.8–4.6, 4.6–5.4, 5.4–6.2, and 6.2–7.0 ng mL−1, respectively. Thus, each 250 μm region has a different concentration range but an identical gradient slope of 3.2 ng mL−1 mm−1. Individual cell tracks were categorized as belonging to either G1, G2, G3, or G4 based on their initial position within the CCC. The chemotactic index is defined as the net distance the cell migrated in the direction of the gradient, divided by the total accumulated distance. Single-cell level measurements were calculated for individual cells and are depicted in Fig. 5A as individual black dots, while population averages are indicated by red lines. For each ECM substrate, two control experiments were performed: (1) cell migration in the absence of bFGF, denoted as (-) and (2) cell migration within a uniform bFGF concentration of 5 ng mL−1, denoted as NG for no gradient. A chemotactic index of zero implies random migration, whereas a positive chemotactic index indicates the presence of a chemoattractive response. The maximum chemotactic index possible is 1 and would indicate that the cell traveled in a direct line towards the chemokine source: a typical maximal chemotactic index for a cell population is around 0.7.55 Conditions with population means statistically different from zero (one-sample t-test, p < 0.01) were considered chemotactic. For myoblasts migrating along a laminin-coated substrate, a mild chemotactic response was observed in G4 only, with a mean chemotactic index of 0.11. Similar results were observed for myoblasts on fibronectin, with a mean chemotactic index in G4 of 0.08. The collagen substrate also exhibited similar trends, however, statistically significant chemoattraction was observed in both G3 and G4 with means of 0.08 and 0.11, respectively.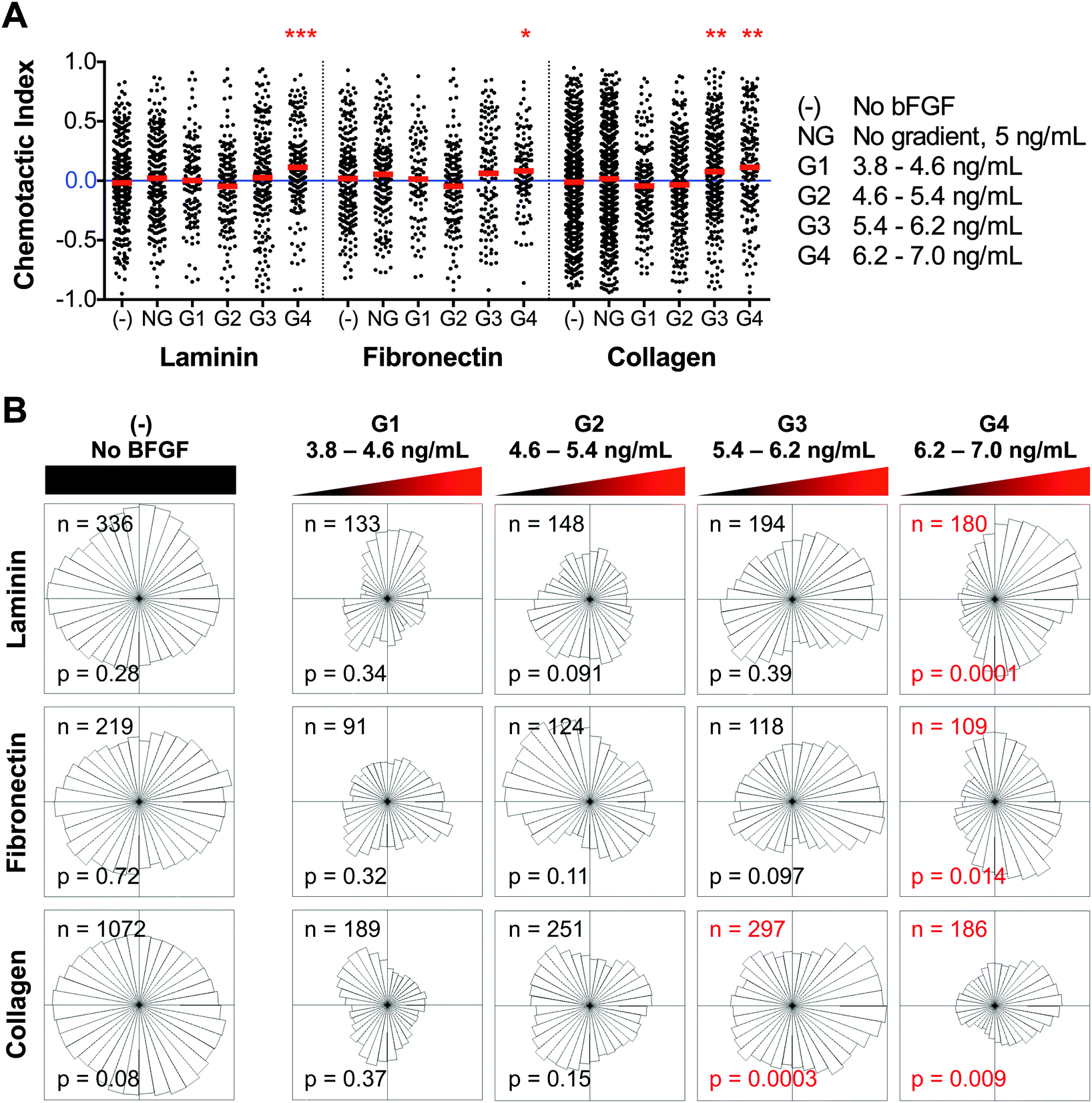 | ||
| Fig. 5 Myoblasts subjected to a bFGF gradient exhibit mild chemoattraction above a threshold concentration regardless of the underlying substrate. (A) The chemotactic index for individual cells (black dots) and population averages (red lines) are plotted for each condition tested (-, NG, G1, G2, G3, and G4). Red asterisks are used to indicate population averages with a chemotactic index significantly different from zero (*p < 0.01, **p < 0.001, ***p < 0.0001). (B) Angular histograms of myoblast positions, relative to their starting points, after 14 h under various conditions. The number of individual cells tracked (n) and the p-value of the Rayleigh Test for Vector Data (p), which tests histogram asymmetry, are also shown. Red labels denote histograms with statistically significant asymmetry (p < 0.05). | ||
The distribution of cell migratory directions is illustrated by an angular histogram (Fig. 5B). Here chemotaxis is evaluated using the Rayleigh Test for Vector Data (RTVD),56,57 which examines the uniformity of a circular distribution taking into account distance from the origin, where p < 0.05 indicates an asymmetric distribution of cells. Thus, a circular distribution would indicate a lack of response, (i.e. random migration direction) whereas a non-circular distribution oriented towards the source would indicate chemoattraction. Consistent with the chemotactic index analysis, chemoattraction is observed at bFGF concentrations greater than 6.2 ng mL−1 on laminin and fibronectin, and greater than 5.4 ng mL−1 on collagen. Thus, these data suggest that the minimum bFGF concentration to induce myoblast chemotaxis is around 6.2 ng mL−1, which is higher than the concentration required for myoblast chemokinesis (∼5 ng mL−1).
To confirm this finding in an independent set of experiments, we altered the bFGF concentration in the source and sink channels to increase the concentration range while maintaining the slope of the gradient at 3.2 ng mL−1 mm−1. Angular histograms illustrate the distribution of migration direction for myoblasts in laminin-coated devices with source and sink concentrations of 11 and 0 ng mL−1 bFGF (Fig. 6, top panel) and 16 and 5 ng mL−1 (Fig. 6, bottom panel), respectively. The new gradients, G1′, G2′, G3′, and G4′ correspond to concentrations of 5.3–6.1 ng mL−1, 6.1–6.9 ng mL−1, 6.9–7.7 ng mL−1, and 7.7–8.5 ng mL−1, respectively. We expected that shifting the concentration range to slightly higher concentrations would result in chemotaxis across more of the device. As expected, we observed statistically significant chemotaxis in regions G3′ and G4′. These data confirm that while significant chemokinesis occurs at a bFGF concentration of 5 ng mL−1, a higher threshold concentration is required to initiate chemotaxis.
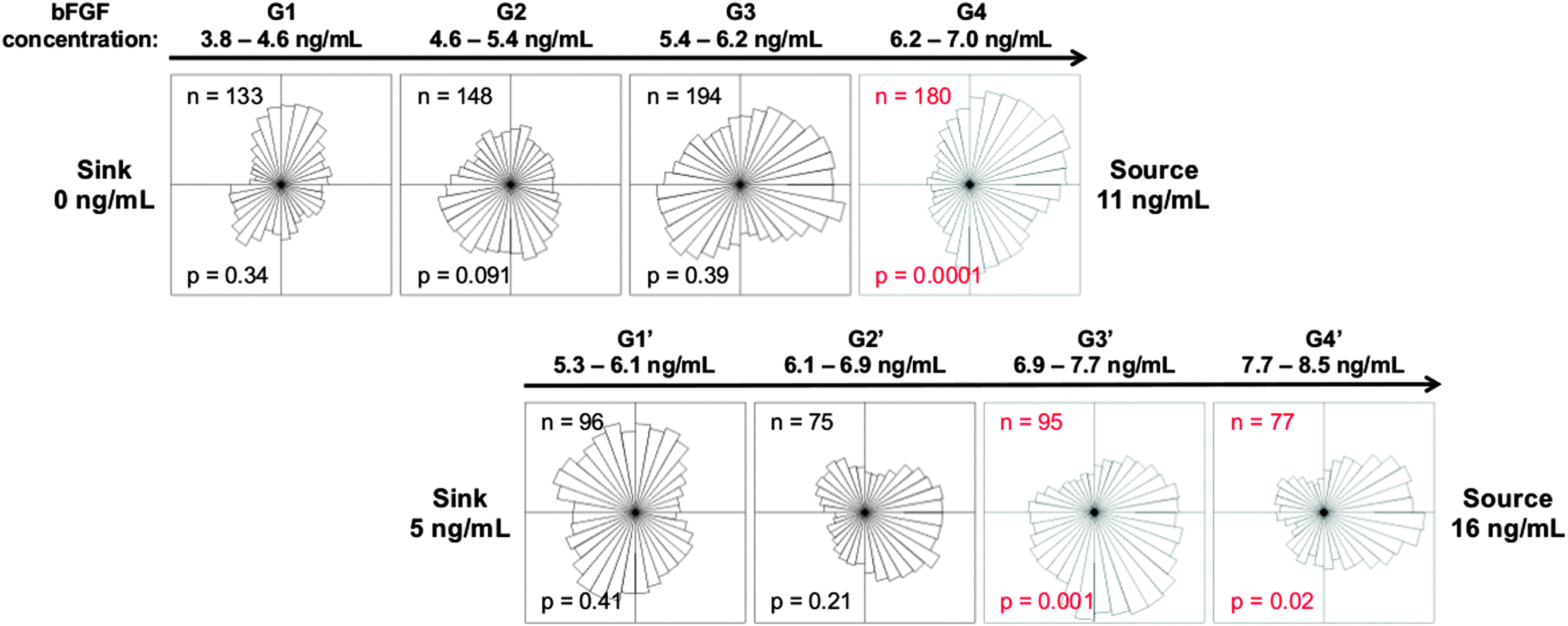 | ||
| Fig. 6 Increasing the concentration range, while maintaining the gradient slope, reveals similar chemotactic behavior in myoblasts above a threshold concentration. The top panel of angular histograms illustrates the distribution of myoblasts after exposure to the corresponding bFGF gradient for 14 h when the device source and sink are perfused with 11 and 0 ng mL−1 bFGF, respectively. The bottom panel illustrates the distribution of myoblasts after 14 h when the device source and sink are perfused with 16 and 5 ng mL−1 bFGF, respectively. The gradient slopes are identical between the top and bottom panels. The number of individual cells tracked (n), and the p-value calculated using the Rayleigh Test for Vector Data (p) are shown. Red labels denote histograms with statistically significant asymmetry (p <0.05). | ||
The chemotactic response occurs on a time scale of hours
Another advantage of using a microfluidic device to analyze cell migration is the ability to perform time-based, single-cell analyses that would be difficult with an end-point assay such as the Transwell. Tracks for individual myoblasts migrating on a laminin-coated surface in the G4 gradient (6.2–7.0 ng mL−1 bFGF) are displayed over time (Fig. 7, top panel) with corresponding angular histograms (Fig. 7, bottom panel). At short time scales (i.e., 0.25 h), the distribution of cell migration is random as expected. As time progresses, the distribution slowly shifts towards the bFGF source, until chemoattraction becomes statistically significant at 6 h. Thus, a stable gradient must be present for at least 6 h to accurately observe bFGF-induced myoblast chemotaxis. Longer time periods (10 and 14 h) appear to slightly enhance the chemotactic response, as the asymmetry in the angular histogram becomes more pronounced. However, even at 14 h a subset of myoblasts (33%) does not respond to the bFGF gradient, as evidenced by examination of the traces of individual cell paths (Fig. 7, top right panel). These data demonstrate the importance of longitudinal, single-cell analysis when quantifying chemotactic behavior and further emphasize that the short-term concentration gradients formed by Transwell assays are insufficient for identifying chemotactic behavior.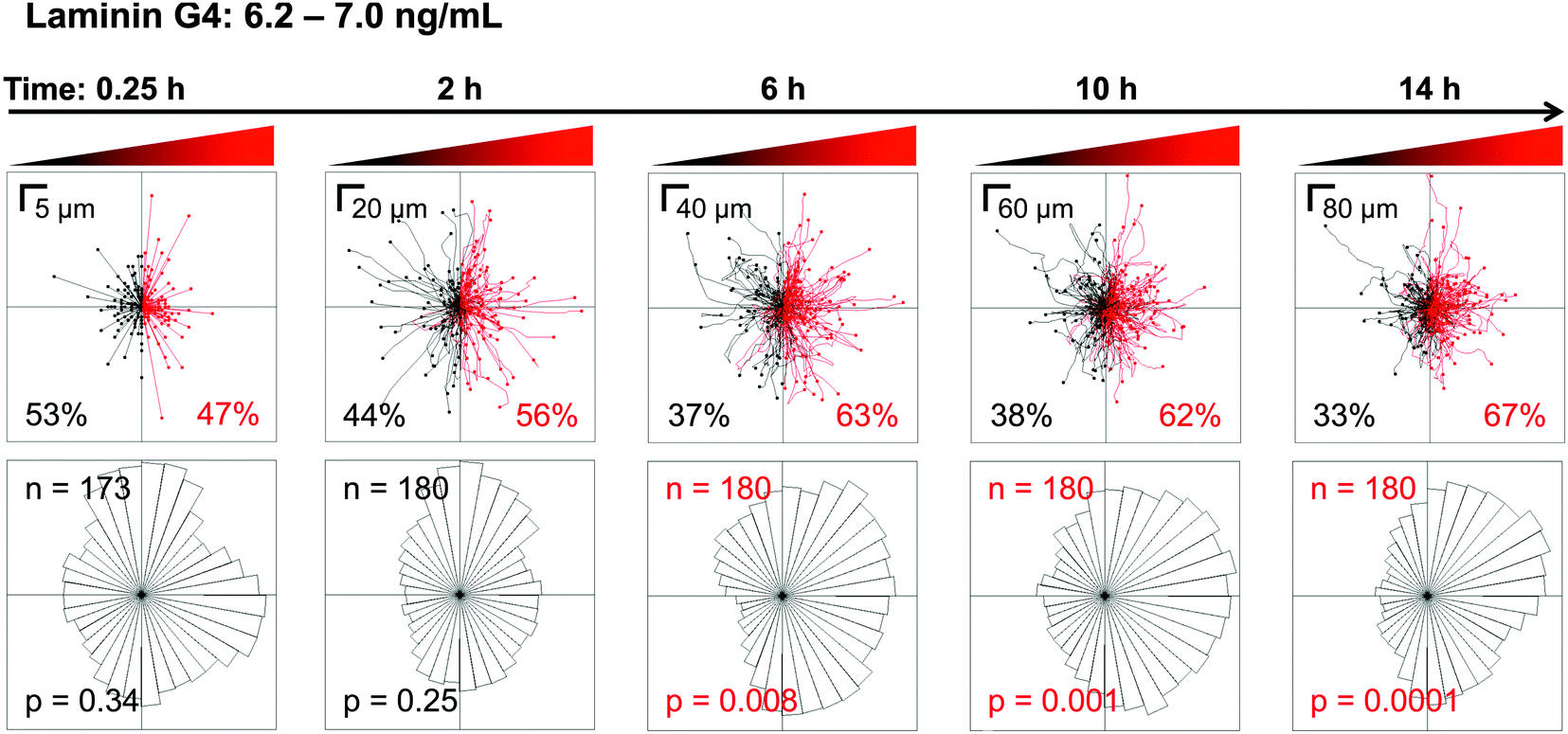 | ||
| Fig. 7 Myoblasts establish a biased directional response towards the bFGF source after 6 h. Myoblast migration in the G4 bFGF gradient on laminin was observed for 14 h. The top panels show the paths taken by individual myoblasts, where all initial positions (t = 0) are plotted at the origin. Red indicates paths with a positive net displacement towards the bFGF source, and black indicates paths moving away from the source. The percent of cells exhibiting movement towards and away from the bFGF source are shown in red and black fonts, respectively. The bottom panels show the corresponding angular histograms, where n is the number of individual cells tracked, and p is the p-value calculated using the Rayleigh Test for Vector Data. Red labels denote statistically significant asymmetry of histograms. | ||
Conclusion
Despite short-term receptor activation, which occurs within minutes, bFGF exposure has long-term effects on myoblast behavior, including chemotaxis, chemokinesis, and chemoproliferation. These latter effects require observation over hours and even days after initial bFGF exposure to accurately quantify. It is important to consider the time scale on which these behaviors occur when choosing an appropriate analysis method. While the commonly used Transwell insert provides a useful, simple assay that allows researchers to test for enhanced migration in response to a chemokine, the rapid decline of the concentration gradient at the cellular length scale (Fig. 3) makes it an inappropriate method to evaluate chemotaxis. This is especially true for slow-moving cells like myoblasts, which required 6 h before significant bFGF-induced chemoattraction could be observed (Fig. 7), and for assays using growth factors with activity that diminishes over time (Fig. 4A). While Transwell assays may not be suited for chemotaxis measurements, they offer a valuable tool for quickly assessing the dose-dependent chemokinetic response of cells over a wide range of concentrations, as chemokinesis does not require the presence of a concentration gradient (Fig. 1).We used a microfluidic device to create a stable gradient that was compatible with the long time-scales required to observe bFGF-induced chemotaxis in primary human myoblasts. The chemotactic response that we observed was relatively mild; 67% of cells exhibited net movement towards the bFGF source, and it required a minimum threshold concentration around 6.2 ng mL−1. In contrast, we observed robust chemokinesis at a bFGF concentration of 5 ng mL−1, which is below the observed threshold concentration needed to initiate chemotaxis. While the underlying ECM substrate did alter the migration speed of myoblasts, it did not significantly affect bFGF-induced chemotaxis or chemokinesis. Conversely, bFGF-induced chemoproliferation did exhibit crosstalk with ECM signaling, as the doubling time of myoblasts cultured on laminin, but not fibronectin or collagen, was significantly reduced upon bFGF treatment.
Together, these data indicate that changes in the surrounding ECM as a result of aging and disease may impact muscle regeneration by affecting the proliferative and migratory potential of myoblasts. It also suggests that chemotaxis is not the main physiological role of bFGF and that other growth factors, either alone or in combination with bFGF, are required to initiate directed migration following muscle injury. Deconvolution of the individual contributions that soluble (e.g., bFGF) and insoluble (e.g., ECM) cues within the microenvironment make to muscle regeneration will help to inform future therapies for treating conditions such as sarcopenia and muscular dystrophy. Importantly, these types of quantitative biological discoveries require experimental methods that are compatible with the time-scales of the cellular behavior of interest. Our engineering analysis has shown that, despite their widespread use, Transwell assays create an ever-changing gradient that becomes too shallow to invoke directional migration after 6 h. Coupled with our longitudinal studies, we discovered that it may take as long as 6 h to observe significant chemotaxis in slow-moving cells, such as myoblasts. Together, these analyses suggest that the gradient time-scale within a Transwell assay is insufficient to accurately evaluate chemotaxis in some mammalian cell types. In addition to providing a stable concentration gradient, our studies demonstrated that microfluidic devices could provide longitudinal, quantitative, single-cell analyses of mammalian cell migration in response to soluble and insoluble cues that would not have been possible using traditional methods. These studies highlight the importance of selecting experimental methods that are compatible with the time scales of the cellular behavior of interest and applying technological innovation to develop new experimental methods when necessary.
Acknowledgements
The authors thank Drs. Helen Blau, Penney Gilbert, and Stephane Corbel for their kind gift of the primary human myoblasts. We thank Dr. Debanti Sengupta for helpful discussions. We acknowledge funding support from the National Institutes of Health grants DP2-OD006477-01 NIH R21-AR062359. MF was a recipient of the Diversifying Academia, Recruiting Excellence fellowship from the Office of the Vice Provost of Graduate Education at Stanford University and the National Science Foundation Graduate Research Fellowship.References
- J. G. Tidball, Compr. Physiol., 2011, 1, 2029–2062 Search PubMed.
- M. S. Clarke, R. Khakee and P. L. McNeil, J. Cell Sci., 1993, 106(Pt 1), 121–133 CAS.
- M. S. Clarke and D. L. Feeback, FASEB J., 1996, 10, 502–509 CAS.
- P. A. D'Amore, R. H. Brown, P. T. Ku, E. P. Hoffman, H. Watanabe, K. Arahata, T. Ishihara and J. Folkman, Ann. Neurol., 1994, 35, 362–365 CrossRef PubMed.
- J. DiMario, N. Buffinger, S. Yamada and R. Strohman, Science, 1989, 244, 688–690 CAS.
- T. J. Templeton and S. D. Hauschka, Dev. Biol., 1992, 154, 169–181 CrossRef CAS PubMed.
- R. L. Panek, G. H. Lu, T. K. Dahring, B. L. Batley, C. Connolly, J. M. Hamby and K. J. Brown, J. Pharmacol. Exp. Ther., 1998, 286, 569–577 CAS.
- S. E. Webb, K. K. Lee, M. K. Tang and D. A. Ede, Dev. Dyn., 1997, 209, 206–216 CrossRef CAS PubMed.
- M. D. Grounds and M. J. Davies, Basic Appl. Myol., 2009, 6, 469–484 Search PubMed.
- S. Corti, Exp. Cell Res., 2001, 268, 36–44 CrossRef CAS PubMed.
- J. Suzuki, Y. Yamazaki, G. Li, Y. Kaziro, H. Koide and L. Guang, Mol. Cell. Biol., 2000, 20, 4658–4665 CrossRef CAS PubMed.
- R. Bischoff, Dev. Dyn., 1997, 208, 505–515 CrossRef CAS PubMed.
- T. A. Robertson, M. A. Maley, M. D. Grounds and J. M. Papadimitriou, Exp. Cell Res., 1993, 207, 321–331 CrossRef CAS PubMed.
- S. M. Sheehan and R. E. Allen, J. Cell. Physiol., 1999, 181, 499–506 CrossRef CAS PubMed.
- A. Yanagiuchi, H. Miyake, M. Nomi, A. Takenaka and M. Fujisawa, BJU Int., 2009, 103, 1569–1573 CrossRef CAS PubMed.
- A. Bikfalvi, S. Klein, G. Pintucci and D. B. Rifkin, Endocr. Rev., 1997, 18, 26–45 CAS.
- S. Crawley, E. M. Farrell, W. Wang, M. Gu, H. Y. Huang, V. Huynh, B. L. Hodges, D. N. Cooper and S. J. Kaufman, Exp. Cell Res., 1997, 235, 274–286 CrossRef CAS PubMed.
- M. Ocalan, S. L. Goodman, U. Kühl, S. D. Hauschka and K. von der Mark, Dev. Biol., 1988, 125, 158–167 CrossRef CAS PubMed.
- J. R. Sanes, J. Cell Biol., 1982, 93, 442–451 CrossRef CAS PubMed.
- C. J. Mann, E. Perdiguero, Y. Kharraz, S. Aguilar, P. Pessina, A. L. Serrano and P. Muñoz-Cánoves, Skeletal Muscle, 2011, 1, 21 CrossRef PubMed.
- M. A. Schwartz and M. H. Ginsberg, Nat. Cell Biol., 2002, 4, E65–E68 CrossRef CAS PubMed.
- S. R. Chowdhury, Y. Muneyuki, Y. Takezawa, M. Kino-oka, A. Saito, Y. Sawa and M. Taya, J. Biosci. Bioeng., 2009, 108, 174–177 CrossRef CAS PubMed.
- S. Mori and Y. Takada, Med. Sci., 2013, 1, 20–36 CAS.
- M. Rusnati, E. Tanghetti, P. Dell'Era, A. Gualandris and M. Presta, Mol. Biol. Cell, 1997, 8, 2449–2461 CrossRef CAS PubMed.
- E. Tanghetti, R. Ria, P. Dell'Era, C. Urbinati, M. Rusnati, M. G. Ennas and M. Presta, Oncogene, 2002, 21, 3889–3897 CrossRef CAS PubMed.
- S. Boyden, J. Exp. Med., 1962, 115, 453–466 CrossRef CAS PubMed.
- C. Zhang, S. Jang, O. C. Amadi, K. Shimizu, R. T. Lee and R. N. Mitchell, BioMed Res. Int., 2013, 2013, 373569 Search PubMed.
- A. Shamloo, N. Ma, M.-M. Poo, L. L. Sohn and S. C. Heilshorn, Lab Chip, 2008, 8, 1292–1299 RSC.
- A. Shamloo, M. Manchandia, M. Ferreira, M. Mani, C. Nguyen, T. Jahn, K. Weinberg and S. Heilshorn, Integr. Biol., 2013, 5, 1076–1085 RSC.
- H. M. Blau and C. Webster, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 5623–5627 CrossRef CAS.
- M. Mohammadi, I. Dikic, A. Sorokin, W. H. Burgess, M. Jaye and J. Schlessinger, Mol. Cell. Biol., 1996, 16, 977–989 CrossRef CAS PubMed.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- E. Meijering, O. Dzyubachyk and I. Smal, Methods Enzymol., 2012, 504, 183–200 Search PubMed.
- P. A. Maher, J. Cell Biol., 1996, 134, 529–536 CrossRef CAS PubMed.
- L. M. McLaughlin, H. Xu, S. E. Carden, S. Fisher, M. Reyes, S. C. Heilshorn and D. M. Monack, Integr. Biol., 2014, 6, 438–449 RSC.
- A. Shamloo and S. C. Heilshorn, Lab Chip, 2010, 10, 3061–3068 RSC.
- N. H. Romano, K. J. Lampe, H. Xu, M. M. Ferreira and S. C. Heilshorn, Small, 2015, 11, 722–730 CrossRef CAS PubMed.
- D. Fuller, W. Chen, M. Adler, A. Groisman, H. Levine, W.-J. Rappel and W. F. Loomis, PNAS, 2010, 107, 9656–9659 CrossRef CAS PubMed.
- C. M. Isbister, P. J. Mackenzie, K. C. W. To and T. P. O'Connor, J. Neurosci., 2003, 23, 193–202 CAS.
- L. Song, S. M. Nadkarni, H. U. Bödeker, C. Beta, A. Bae, C. Franck, W.-J. Rappel, W. F. Loomis and E. Bodenschatz, Eur. J. Cell Biol., 2006, 85, 981–989 CrossRef CAS PubMed.
- A. Sommer and D. B. Rifkin, J. Cell. Physiol., 1989, 138, 215–220 CrossRef CAS PubMed.
- M. A. Nugent and R. V. Iozzo, Int. J. Biochem. Cell Biol., 2000, 32, 115–120 CrossRef CAS PubMed.
- T. Gauthier, M. Maftouh and C. Picard, Biochem. Biophys. Res. Commun., 1987, 145, 775–781 CrossRef CAS PubMed.
- M. Rusnati, C. Urbinati and M. Presta, J. Cell. Physiol., 1993, 154, 152–161 CrossRef CAS PubMed.
- V. Baldin, A. M. Roman, I. Bosc-Bierne, F. Amalric and G. Bouche, EMBO J., 1990, 9, 1511–1517 CAS.
- F. Amalric, G. Bouche, H. Bonnet, P. Brethenou, A. M. Roman, I. Truchet and N. Quarto, Biochem. Pharmacol., 1994, 47, 111–115 CrossRef CAS PubMed.
- I. A. Ferguson, J. B. Schweitzer and E. M. Johnson, J. Neurosci., 1990, 10, 2176–2189 CAS.
- N. Kramer, A. Walzl, C. Unger, M. Rosner, G. Krupitza, M. Hengstschläger and H. Dolznig, Mutat. Res., Rev. Mutat. Res., 2013, 752, 10–24 CrossRef CAS PubMed.
- K. A. Moutasim, M. L. Nystrom and G. J. Thomas, Methods in Molecular Biology, Humana Press, Totowa, NJ, 2011, vol. 731, pp. 333–343 Search PubMed.
- E. A. Cox and A. Huttenlocher, Microsc. Res. Tech., 1998, 43, 412–419 CrossRef CAS PubMed.
- J. D. Hood and D. A. Cheresh, Nat. Rev. Cancer, 2002, 2, 91–100 CrossRef PubMed.
- F. G. Giancotti and E. Ruoslahti, Science, 1999, 285, 1028–1032 CrossRef CAS PubMed.
- A. J. Ridley, Science, 2003, 302, 1704–1709 CrossRef CAS PubMed.
- A. Huttenlocher and A. R. Horwitz, Cold Spring Harbor Perspect. Biol., 2011, 3, a005074 Search PubMed.
- H. Levine and W.-J. Rappel, Phys. Today, 2013, 66, 24 CrossRef CAS PubMed.
- B. R. Moore, Biometrika, 1980, 67(1), 175–180 CrossRef.
- N. I. Fisher, Statistical Analysis of Circular Data, Cambridge University Press, 1995 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00060b |
| This journal is © The Royal Society of Chemistry 2015 |