
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Parallel feedback loops control the basal activity of the HOG MAPK signaling cascade†
Hoda
Sharifian‡
a,
Fabienne
Lampert
a,
Klement
Stojanovski
b,
Sergi
Regot§
b,
Stefania
Vaga¶
c,
Raymond
Buser
a,
Sung Sik
Lee‡
a,
Heinz
Koeppl
d,
Francesc
Posas
b,
Serge
Pelet
*e and
Matthias
Peter‡
*a
aInstitute of Biochemistry, Department of Biology, ETH Zürich, Zürich, Switzerland. E-mail: matthias.peter@bc.biol.ethz.ch
bDepartment of Experimental and Health Sciences, Universitat Pompeu Fabra, Barcelona, Spain
cInstitute of Molecular Systems Biology, Department of Biology, ETH Zürich, Switzerland
dDepartment of Electrical Engineering and Information Technology, Technische Universität Darmstadt, Darmstadt, Germany
eDepartment of Fundamental Microbiology, University of Lausanne, Lausanne, Switzerland. E-mail: serge.pelet@unil.ch
First published on 23rd February 2015
Abstract
Tight regulation of the MAP kinase Hog1 is crucial for survival under changing osmotic conditions. Interestingly, we found that Hog1 phosphorylates multiple upstream components, implying feedback regulation within the signaling cascade. Taking advantage of an unexpected link between glucose availability and Hog1 activity, we used quantitative single cell measurements and computational modeling to unravel feedback regulation operating in addition to the well-known adaptation feedback triggered by glycerol accumulation. Indeed, we found that Hog1 phosphorylates its activating kinase Ssk2 on several sites, and cells expressing a non-phosphorylatable Ssk2 mutant are partially defective for feedback regulation and proper control of basal Hog1 activity. Together, our data suggest that Hog1 activity is controlled by intertwined regulatory mechanisms operating with varying kinetics, which together tune the Hog1 response to balance basal Hog1 activity and its steady-state level after adaptation to high osmolarity.
Insight, innovation, integrationMAPK signaling pathways play a key role in transducing extra-cellular stimuli into specific cellular responses tuned by positive and negative feedback loops. The p38/Hog1 MAPK in yeast is essential for cell survival in high osmolarity environments. The pathway activity under hyperosmotic stress is controlled by intracellular glycerol accumulation, however, less is known about feedback mechanisms within the signaling cascade regulating Hog1 basal activity. Here, we applied an interdisciplinary approach integrating data from in vivo and in vitro phosphorylation assays and dynamics single cell measurements using microfluidic devices in a mathematical model. This strategy allowed us to identify Hog1 upstream activators that are direct targets of the MAPK as novel rapid-response regulators of Hog1 basal activity. |
Introduction
In order to rapidly adapt to the changing conditions, cells have established complex signaling networks that sense their intra- and extracellular environment. In particular, conserved mitogen-activated protein kinase (MAPK) pathways are pivotal to transmit extracellular stimuli and stress conditions to orchestrate appropriate cellular responses. Genetic and biochemical approaches have identified the core signaling components that transduce the activation signal through a conserved MAPK cascade. Recently quantitative and dynamic single cell measurements coupled with mathematical modeling have provided new insights into feedback regulation and signaling kinetics to describe the complex cellular signaling outputs.1,2In yeast, the high osmolarity glycerol (HOG) pathway is required to re-establish the balance between internal and external osmotic pressures upon hyper-osmotic shock by increasing the intra-cellular concentration of glycerol.3 A sudden increase in the osmolarity of the medium causes cells to shrink by losing water, which triggers activation of the osmosensors Sln1, Sho1, Msb2 and Hkr1.4 While Sln1 transmits the signal via a two-component phosphorelay mechanism comprised of Ypd1 and Ssk1 to the MAPK kinase kinases (MAPKKK) Ssk2 and its functionally redundant homologue Ssk22,5 the other sensors activate the MAPKKK Ste11. All three MAPKKKs subsequently converge on the MAPK kinase (MAPKK) Pbs2,5–9 which in turn phosphorylates the MAPK Hog1 on Thr174 and Tyr176,10 thereby triggering its activation and rapid translocation to the nucleus.11,12 Adaptation to high osmolarity is mostly achieved by cytoplasmic glycerol accumulation through altering the metabolic fluxes towards increased glycerol synthesis1,13 and closure of the glycerol channel Fps1, thereby preventing glycerol efflux under osmotic stress.14,15 As cells increase glycerol production, the osmotic pressure on the cell wall decreases. Consequently Hog1 activation declines and Hog1 becomes dephosphorylated due to the combined action of multiple phosphatases.10,16–19 Mathematical modeling suggests that cellular adaption by increasing cytoplasmic glycerol concentration is sufficient to explain the dynamics of Hog1 activity during stress.1,20
Although the HOG pathway is acutely induced by high osmolarity, basal activity of Hog1 exists under isosmotic conditions. This basal signaling is mediated by the Sln1 branch of the pathway, thereby enabling a fast and sensitive Hog1 response upon stress.21 Interestingly, basal Hog1 phosphorylation further increases when Hog1 kinase activity is inhibited, implying that a Hog1-dependent negative feedback mechanism must exist to control basal signaling.21 However, the targets and mechanism of this Hog1-dependent negative feedback loop remain unclear.
Identification and characterization of signaling feedback loops within the HOG pathway can be facilitated using quantitative single cell assays that accurately report on Hog1 activity with high temporal resolution, and experimental conditions that minimize the influence of the glycerol adaptation feedback. Indeed, a computational study suggests that the increase in Hog1 phosphorylation upon kinase inhibition can be explained by a drop in the internal glycerol level.20 Here, by virtue of an unexpected interruption of signal transduction in the HOG pathway in absence of glucose, we have established conditions that allow uncoupling of Hog1 activity from glycerol accumulation.
We employed quantitative live cell microscopy coupled to microfluidic devices to monitor Hog1 dynamics in response to glucose availability and salt stress. Interestingly, our experimental data in combination with mathematical simulations suggest that Hog1-dependent feedback loops specifically target the Sln1 branch by phosphorylating multiple targets, including Ssk2 and components of the sensory module, which includes the phosphorelay proteins. These together modulate basal Hog1 activity and rapidly adjust steady-state levels following a high osmolarity response.
Results
Hog1 phosphorylates its upstream activators in vivo and in vitro
In order to identify novel Hog1 substrates, we compared the phospho-proteome of wild-type cells exposed or not to osmostress by the addition of 0.4 M NaCl. In addition to known phosphorylation sites including Thr 174 and Tyr176 on Hog1, we detected other proteins of the pathway whose phosphorylation sites are mapped to serine residues followed by a proline, suggesting that they may be directly phosphorylated by Hog1 (Table S1, ESI†). The Hog1-dependence of specific phospho-sites was subsequently confirmed by inhibiting the Hog1 kinase using the hog1-as allele, which allows rapid and specific inhibition of Hog1 activity by addition of 1-NA-PP1. Interestingly, this analysis revealed that Ssk1 (S193 and S195) and Ssk2 (S54, 57, 74, 78) are phosphorylated in vivo in a Hog1-dependent manner (Fig. 1A and Fig. S1A, ESI†). As Ssk1 and Ssk2 function in the osmostress signaling cascade upstream of Hog1, these results suggest that Hog1 may regulate its own activity via a positive or negative feedback mechanism.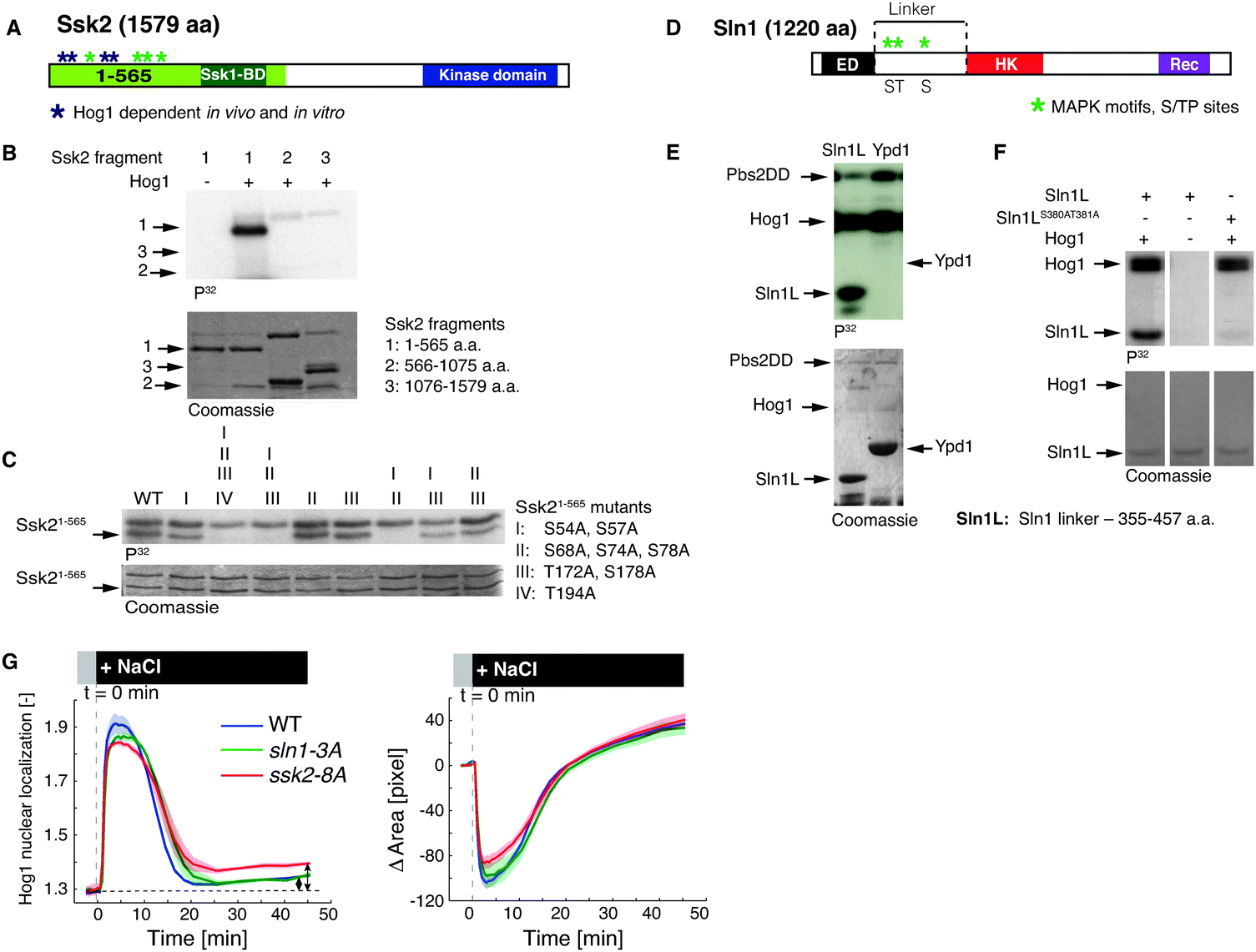 | ||
| Fig. 1 Hog1 phosphorylates multiple sites on Ssk2 and Sln1 in vitro. (A) Schematic diagram of Ssk2, S/TP sites at the N-terminus are marked with *, dark blue asterisks (S54, 57, 74, 78) are phosphorylated in vitro and in vivo (Table S1, ESI†). The binding domain of Ssk1 (Ssk1-BD) and the kinase domain are indicated. (B) In vitro phosphorylation assay in which purified fragments of Ssk2 were incubated with pre-activated Hog1 (see Material and methods) in the presence of γ-32P-ATP. The upper panels show autoradiographs to visualize phosphorylation of the different proteins, while Coomassie staining (lower panels) control for protein amounts. (C) In vitro phosphorylation by pre-activated Hog1 as in (B) of the Ssk21-565 fragment containing as indicated alanine mutations of S54, S57, S68, S74, S78, T172, T178 and T194. (D) Schematic diagram highlighting the domain structure of Sln1. ED: extracellular domain, HK: histidine kinase, Rec: receiver domain. MAPK motifs, S/TP sites in the linker are marked with *. (E) In vitro phosphorylation by pre-activated Hog1 as in (B) of Sln1 linker and full length Ypd1. (F) Sln1L (linker) fragment containing mutations of S380 and T381 to alanine were analysed by the in vitro phosphorylation assay as in (B) in the presence of pre-activated Hog1. (G) Quantification of Hog1 nuclear relocation and cells area upon hyper-osmotic stress in wild-type (WT) cells, sln1-3A and ssk2-8A mutants where the serine or threonine residues are mutated to non-phosphorylatable alanine. Cells were treated by 0.4 M NaCl at time 0, and the Hog1 activity (left) was quantified by plotting the mean of nuclear vs. rim (see Material and methods and Fig. S2, ESI†) Hog1-mCherry fluorescence intensity. In addition, adaptation was followed by measuring the cell area using cytoplasmic Hog1-mCherry signal as a function of time (right). The data are averaged from 3 independent experiments (2 replicates for sln1-3A), each measuring at least 200 cells and the shaded areas show ±SEM from 3, or 2 for sln1-3A, independent experiments. Arrows indicate the difference between the steady-state values in ssk2-8A cells and the wild-type (WT) control. | ||
In order to test whether Hog1 directly phosphorylates Ssk2 and possibly other upstream components, we set up in vitro kinase assays using bacterially expressed and activated GST-Hog1 and purified substrates (see Material and methods). While Ssk1 was a poor substrate (Fig. S1A and B, ESI†), Ssk2 and in particular its amino-terminal domain was readily phosphorylated by Hog1 under these conditions (amino acids 1–565, Fig. 1A and B). Consistent with the in vivo analysis, mutagenesis revealed that the MAPK consensus sites S54, S57, S74 and S78 were among the major phospho-acceptors in vitro (Fig. 1C). In addition, we found that the Sln1 linker between the second transmembrane segment and the histidine kinase motif was strongly phosphorylated by Hog1 (Fig. 1D and E and Fig. S1C and D, ESI†). Indeed, mutating the MAPK consensus sites serine 380 and threonine 381 to non-phosphorylatable alanine residues abolished Sln1 phosphorylation in vitro (Fig. 1F). In contrast, no phosphorylation of Ypd1 could be detected (Fig. 1E), consistent with the fact that the sequence of Ypd1 does not contain any serine or threonine residues followed by a proline, the minimal requirement for MAPK consensus sites. Taken together, these results suggest that Hog1 may regulate its activity by novel positive or negative feedback loops targeting upstream components of the signaling cascade.
In order to test this hypothesis we first measured the Hog1 response to hyper-osmotic stress in cells expressing non-phosphorylatable Sln1 or Ssk2 mutants (sln1-3A and ssk2-8A, respectively), using Hog1 nuclear shuttling as a readout for its activity.1,22,23 We followed Hog1 activity in a microfluidic chip using an established assay that takes advantage of the rapid translocation of active Hog1 into the nucleus23,24 (Fig. S2, ESI† and Material and methods). As shown in Fig. 1G, Hog1 activation in ssk2-8A and sln1-3A cells exposed to 0.4 M NaCl was comparable to wild-type controls, while inactivation of Hog1 was slightly delayed. The adaptation time was not prolonged under these conditions indicating that glycerol accumulation was unaltered, consistent with previous mathematical modeling.20 However, only in ssk2-8A cells the steady-state level of active Hog1 after adaptation to osmostress was about 57% higher compared to wild-type controls (Fig. 1G, arrows and Fig. S3, ESI†). Together, these data suggest that a Hog1-dependent internal feedback loop targeting Ssk2 may help to fine-tune steady state levels of Hog1 activity, while osmoadaptation is mainly achieved by glycerol feedback.
In addition, these data exemplify the limitation of the conventional osmostress stimulation assay for dissecting internal feedback loops in the signaling cascade.
Hog1-dependent negative feedback in the Sln1 branch
In order to characterize the internal signaling feedback, we established experimental conditions that allow measuring feedback regulation in the signaling cascade independent of the adaptation feedback through glycerol synthesis. This can be achieved by transiently starving cells for glucose, the main precursor for glycerol production. Strikingly, we found that Hog1 nuclear accumulation decreased during glucose starvation and returned to initial levels following a transient peak after glucose re-addition (Fig. 2A). Monitoring Hog1 phosphorylation by western blot confirmed that double-phosphorylation of Hog1 followed similar dynamics as Hog1 nuclear relocation under these conditions (Fig. 2B). A comparable peak of nuclear Hog1 was detected after re-addition of glucose when glucose starvation was induced by replacing glucose for the non-metabolizable glucose analog 2-deoxy-D-glucose25 in the starvation medium (Fig. 2C), excluding the possibility that changes in osmolarity rather than signaling feedback cause this behavior. Interestingly, extending the starvation period before glucose re-addition slightly increased the amplitude of the transient peak of nuclear Hog1 (Fig. S4, ESI†). These results suggest that an upstream activating signal may accumulate during glucose starvation, which leads to rapid Hog1 activation upon relief of the starvation switch, causing an overshoot in Hog1 activity. This transient activation of Hog1 is dependent on the presence of its activator, Pbs2, since no Hog1 activity peak was detectable after switching pbs2Δ cells back to glucose-containing media (Fig. 2C and Fig. S5A, ESI†), and instead we observed a slow and minor increase of nuclear Hog1 which might reflect Hog1 autophosphorylation.26 Importantly, the transient peak in nuclear accumulation and phosphorylation of Hog1 did not occur in cells expressing a kinase-dead mutant of Hog1 (hog1KN, Fig. 2D and Fig. S5B and S6, ESI†). Since Hog1 can be activated by two parallel inputs, we next asked whether this Hog1-dependent negative feedback is confined to a single branch or targets components in both branches. Interestingly, while ste11Δ cells displayed a transient peak in Hog1 localization following glucose re-addition that was comparable to wild-type cells, overshoot of Hog1 activity was lost in ssk1Δ cells where only the Sho1 branch remains functional (Fig. 2E and Fig. S5C, ESI†). As there are two redundant MAPKKK in the Sln1 branch (Ssk2 and Ssk22), we tested the Hog1 response in the presence of only one of the MAPKK kinases. The phenotype of ste11Δ ssk22Δ double mutants is similar to ste11Δ cells, while the presence of Ssk22 in ste11Δ ssk2Δ double mutants is not sufficient for the transient peak in nuclear Hog1 after glucose re-addition (Fig. S7, ESI†), indicating that the feedback loops are routed through Ssk2 but not its redundant homolog Ssk22. Moreover, the overshoot of Hog1 activity in fps1Δ1 cells (an open channel mutant)15 was comparable to cells expressing wild-type Fps1 (Fig. S8, ESI†), demonstrating that re-establishing basal Hog1 activity occurs independently of the adaptation feedback and thus most likely operates within the signaling cascade. To better compare the dynamics of the Hog1 response, we defined a parameter termed “signaling feedback efficiency” (SFE, see Fig. 2F and Material and methods for details). In order to calculate the SFE, we identified the time window of the overshoot in Hog1 activity based on the average behavior of wild-type cells. Before data processing, individual single cell traces were smoothened to reduce measurement noise by removing random fluctuations. Within this defined time window, for each single cell trace of Hog1 activation signal, we identified the maximum value and the minimum value reached after this maximum. The SFE is calculated as the ratio of the maximum minus the minimum reached after overshoot (y2) over the maximum minus the basal value before glucose addition (y1). Obviously, for traces without overshoot, the maximum can be reached at the end of the time window resulting in an SFE of zero (Fig. S9, ESI†). Indeed, experimental analysis revealed strong feedback regulation for wild-type and ste11Δ cells, while the SFE was zero for most of pbs2Δ, hog1KN and ssk1Δ single cell traces, suggesting that this value allows to quantitatively compare feedback strengths in different backgrounds. Plotting the SFE distributions separates two distinct classes of strains either displaying an overshoot or a gradual increase (Fig. 2G). Together, these data suggest that a Hog1-dependent negative feedback mechanism targeting components in the Sln1 but not the Sho1 branch of the HOG signaling pathway limits Hog1 activation (Fig. 2H). | ||
| Fig. 2 Hog1-dependent negative feedback in the Sln1 branch. (A) Wild-type cells expressing Hog1-mCherry were imaged in a microfluidic chamber, and the media was exchanged as indicated in the gray bar. Hog1 nuclear localization over time was quantified from images processed as in Fig. 1G, Number of cells (nc) = 196. Unless stated otherwise, the solid line represent the average of the single cell traces and the shaded area the standard deviation. (B) Hog1 phosphorylation was examined in cells expressing Hog1-mCherry, which were cultured in SC-full, washed and grown for 10 minutes in media with (Glc 2%) or without (Glc 0%) glucose. Glucose was then added (2% final concentration) and aliquots were fixed with 10% TCA at the indicated times (min). Hog1 activity was additionally monitored by immunoblotting using a phospho-specific Hog1-antibody (αP-p38). Cells treated with 0.4 NaCl for 5 min were included as control (left lane). Total Hog1 protein levels were controlled by probing the samples against RFP (αRFP). (C)–(E) Hog1 nuclear localization was quantified over time from images processed as in Fig. 1G during the glucose-switch in wild-type (WT, C, nc = 314), pbs2Δ (C, nc = 201), catalytically inactive Hog1 (hog1KN, D, nc = 424), ste11Δ (E, nc = 438) and ssk1Δ (E, nc = 134) cells. Cells were cultured in a microfluidic chamber and starved in medium lacking glucose but containing 2% 2-deoxy-D-glucose (2DG) to keep the osmolarity of the switching media constant. (F) Quantification of “signaling feedback efficiency” (SFE) for comparing transient overshoot of Hog1 activity. The light green area indicates a fixed time window based on the average signal of Hog1 in wild-type cells, which covers the expected overshoot response. SFE values (y2/y1) were calculated as described in Material and methods for each smoothened cell trace and the distributions are shown in panel G. (G) The SFE distributions calculated on single cell traces from wild-type (WT) and mutants presented in panels C to E. (H) Schematic view of the HOG pathway illustrating the potential feedback loops. | ||
Computational model of the Sln1 branch including multiple feedback loops to upstream proteins
In order to understand the feedback mechanisms acting in the Sln1 branch, we developed a minimal computational model that describes Hog1 dynamics upon glucose starvation and re-addition. Based on our in vitro phosphorylation analysis (Fig. 1A–F and Fig. S1A and B, ESI†), we included three feedback loops targeting Sln1, Ssk1 or Ssk2 (Fig. 3A). The Ssk1 and Ssk2 feedback mechanisms were modeled such that phosphorylation by Hog1 prevents their activation, while phosphorylation of Sln1 increases the transfer rate of the phosphoryl group between Sln1-His576, Sln1-Asp1144, and Ypd1-His64, thereby leading to reduced Ssk1 activity (see ESI†). All reactions are listed in Table S4 (ESI†) and were solved by assuming mass action kinetics. | ||
| Fig. 3 Computational model of the Sln1 branch including multiple feedback loops to upstream proteins. (A) Schematic view of the model depicting feedback loops (FBLs) to Ssk2 (red), Ssk1 (green) and Sln1 (blue). Proteins in their active states are shown in gray. (B) Experimental data for wild-type (WT, nc = 314) and cells expressing the Ypd1-AID fusion protein (nc = 325), which is rapidly degraded upon addition of auxin. Degradation of Ypd1-AID was induced 30 min before the glucose-switch was implemented as in Fig. 2C. (C) Simulation of Hog1 activation over time of the model with wild-type (black, all feedback loops, FBLs) and conditions where Ypd1 is degraded (red to yellow curves for varying Ssk2 feedback parameters). The ratio between phosphorylated Hog1 (Hog1-PP) and total Hog1 is plotted versus time (min). The glucose starvation condition is simulated in the model for 10 min and the normalized Hog1 activity is followed upon implementation of the glucose switch in the model (see computational model description in the ESI†). The concentration of Ypd1 normalized to the initial value was decreased from 1 to 0.0068 in the simulated degradation process before implementing the glucose switch. (D) Simulation results for Hog1 activity with all feedback loops (black, all FBLs) and an Ssk1 mutant (purple, phosphorelay disruption), where the Ssk1 phosphorylation rate was decreased by 100 fold. (E) Experimental data showing Hog1 nuclear localization over time from images processed as in Fig. 1G of wild-type (WT, black, nc = 314), ssk2ΔN (gray, nc = 98) and ssk1-D554S cells (purple, nc = 223), which cannot be phosphorylated by Ypd1. Expression of the dominant-active ssk2ΔN and ssk1-D554S proteins were induced by addition of 100 nM β-estradiol, and the glucose-switch was implemented as in Fig. 2C. | ||
In order to assess Hog1 response dynamics, we systematically varied feedback loop strength and simulated the model for different sets of feedback parameters (see ESI†). Interestingly, in the explored parameter space, individual negative feedback to Sln1 was not sufficient to describe the observed Hog1 dynamics. Conversely, including feedback loops to either Ssk1 or Ssk2 mimicked Hog1 inactivation kinetics, and the feedback behavior was enhanced when both mechanisms were combined (Fig. S10, ESI†).
Interestingly, varying feedback strength confirmed that regulation of Sln1 has a minor effect on the simulated SFE, while the SFE is more tunable by combining the Ssk1 and Ssk2 feedback loops (Fig. S11A, ESI†, compare to B and C). Thus, our modeling approach favored topologies with multiple feedback loops, suggesting that such systems allow for more adjustable regulation of Hog1 activity. Changing the feedback parameters for one feedback loop mostly alters the inactivation time following the overshoot but not its amplitude, implying that each loop has a given capacity to adjust Hog1 activity. Parallel feedback loops have additive effects and can thereby control the amplitude of the overshoot by increasing the feedback capacity.
In order to calibrate the feedback mechanism in our model, we developed experimental conditions allowing to measure Ssk2 regulation and estimate its parameters (and strength) relative to Ssk1. Since Ypd1 is a negative regulator of the phosphorelay, the pathway can be activated by degrading Ypd1, which bypasses Ssk1 regulatory mechanisms by either the phosphorelay or the Hog1-dependent feedback. Thus, Ypd1 was fused to an auxin-binding domain, which allows auxin-dependent degradation of the fusion protein by the ubiquitin–proteasome system.27 Indeed, Ypd1 was degraded within minutes after exogenous auxin addition resulting in activation of the HOG pathway as measured by nuclear translocation of Hog1 (Fig. S12, ESI†). Interestingly, although Hog1 activity rapidly increased upon glucose readdition, Hog1 deactivation was significantly slower compared to wild-type controls (Fig. 3B), implying that feedback regulation of Hog1 activity was defective in Ypd1-depleted cells. Mathematical simulation of this perturbation revealed that Hog1-dependent feedback is less efficient and also less affected by feedback to the sensory module (Fig. S13, ESI†). Importantly, the best match between simulation and experimental data is obtained under conditions where only moderate feedback to Ssk2 is implemented (Fig. 3C). Therefore we set the Ssk2 feedback rate constants to a third of the Ssk1 feedback parameters.
We further validated these feedback parameters by measuring Hog1 activation after triggering the pathway by expressing hyperactive Ssk1. In the absence of salt stress, Ypd1-dependent phosphorylation at Asp554 of Ssk1 keeps Ssk1 inactive. Consequently, mutating Ssk1-Asp554 to serine prevents its phosphorylation by Ypd1 and thus hyperactivates Ssk1.28 Simulating the effect of this mutation in our mathematical model by decreasing the phosphorylation rate of Ssk1 by 100 fold predicted strongly reduced feedback efficiency (83% reduction), which is apparent in the smaller overshoot of Hog1 activity (Fig. 3D). In order to test this prediction, we followed Hog1 activity in cells expressing ssk1-D554S from the β-estradiol-inducible promoter upon glucose re-addition. In accordance with the model, we found that Hog1 activity rapidly increased but then declined very slowly (Fig. 3E, purple curve), demonstrating that multiple feedback loops are impaired under these conditions.
In order to test the simulated mechanism of Ssk2 feedback, we profited from a dominant-active allele of Ssk2, ssk2ΔN, which lacks the Ssk1 binding domain8 and all phosphorylation sites targeted by Hog1 in vitro. Since our model assumes that Ssk2 activity is controlled by Hog1-dependent feedback, cells expressing ssk2ΔN should be devoid of intrinsic feedback similar to hog1KN cells. Thus, we stimulated Hog1 activity by expressing ssk2ΔN from the β-estradiol-inducible promoter. As expected, nuclear Hog1 reached the final steady state value without any transient peak (Fig. 3E, gray curve). The levels of Hog1 activation in individual cells varied considerably probably due to different induction levels of ssk2ΔN. However, Hog1 dynamics were comparable in all cells irrespective of the initial Hog1 activity, thus excluding the possibility that the lack of overshoot is caused by saturating Hog1 activation (Fig. S14, ESI†). Taken together, these experimental and computational results suggest that the feedback regulation is more efficient by including both Ssk1 and Ssk2 feedback loops, while Sln1 feedback cannot explain the overshoot dynamics of Hog1 activity.
Parallel feedback loops ensure controlled basal activity of Hog1
In order to corroborate these findings, we next simulated Hog1 activation in sln1-3A and ssk2-8A mutants, where feedback regulation to Sln1 and Ssk2 is specifically abolished. In contrast to Sln1, model simulations predicted that removal of Ssk2 feedback regulation increases basal Hog1 activity and decreases the amplitude of the feedback overshoot (Fig. 4A). The experimental data match these predictions (Fig. 4B). In comparison to sln1-3A cells and wild-type controls, the amplitude of the decay after the Hog1 peak was smaller in ssk2-8A cells and Hog1 activity settled at higher steady-state levels (Fig. 4B). Indeed, SFE analysis revealed that sln1-3A cells exhibit normal feedback control, while the feedback efficiency in ssk2-8A cells decreased on average by 25% with a subpopulation of non-overshooting cells (SFE = 0) (Fig. 4C). According to the simulation and experimental data, the partial Hog1 control is most likely due to additional feedback targeting Ssk1. Therefore we tested whether the phosphosites detected by the phospho-proteomic analysis in vivo are involved in this feedback regulation. We mutated serines 193 and 195 together with two adjacent serines (110 and 351) to non-phosphorylatable alanine residues (ssk1-4A). However, no feedback defect was detected in ssk1-4A cells alone or in combination with the ssk2-8A allele (Fig. S15, ESI†), implying that phosphorylation of these sites is not sufficient to block Hog1-dependent phosphorylation on Ssk1 in vivo. Thus, based on the model predictions, Hog1 is likely to phosphorylate additional sites on Ssk1 that are required for feedback inhibition (note that Ssk1 has a total of 16 candidate SP/TP sites for Hog1 phosphorylation). In summary, our simulations and experimental data suggest that in addition to the adaptive glycerol feedback regulation, Hog1-dependent negative feedback mechanisms target Ssk2 and most likely Ssk1 of the Sln1 branch, thereby controlling Hog1 basal activity in isosmotic conditions and after adaptation to osmostress. | ||
| Fig. 4 Parallel feedback loops ensure controlled basal activity of Hog1. (A) Simulations of Hog1 activity for different model topologies, including all feedback loops (All FBLs, black), no Sln1 feedback (FB, blue) or no Ssk2 FB (red). (B) Experimental data showing Hog1 nuclear localization over time from images processed as in Fig. 1G of wild-type (WT, black), sln1-3A where Sln1 feedback is inhibited (blue) and ssk2-8A where Ssk2 feedback is prevented (red). The mutants were integrated in the deletion backgrounds with their endogenous promoters. The glucose-switch was implemented as in Fig. 2C. The data are averaged from 3 independent experiments each measuring at least 200 cells, the dark shaded areas show ±SEM from 3 independent experiments and the light areas show the standard deviation. (C) The SFE distribution for the data presented in B. The mean values of SFE for wild-type (WT) and sln1-3A are 0.53 and 0.52 respectively while for ssk2-8A cells it is reduced by 25% to 0.4. The statistical significance is evaluated by the Student's t-test and the Kolmogorov–Smirnov test where no assumption for normality is required. (D) Schematic representation of the negative feedback loops controlling the HOG pathway. Glycerol accumulation balances the turgor pressure, negative feedback loops in the signaling cascade control the basal activity, the solid line is the feedback to Ssk2 and the dashed line suggests a feedback loop to the phosphorelay. For detailed explanations see the text. | ||
Discussion
In this study, we integrated quantitative single-cell data and simulation techniques to investigate feedback regulations in the HOG signaling cascade. Together with previous findings, our data demonstrate that the fidelity of the HOG pathway is ensured by multiple regulatory mechanisms (Fig. 4D) that act at different time-scales.Transcriptional up-regulation of stress response genes such as GPD1 and GPP229 contribute to the long-term adaptation of cells to high-osmolarity environment. It is a slow process where first proteins are synthesized 5–10 minutes after stress30 but can remain in the cell for multiple divisions. Protein expression in the HOG pathway has been shown to be dispensable for the immediate response to mild hyper-osmotic stresses31 but contributes to the resistance of the cells to future stress events.32,33 The so-called integral feedback acting via glycerol accumulation by closure of the Fps1 channel14,15 and increased synthesis rates through glycolysis13 drives the adaptation of the cells to its new environment and takes between 5 to 30 minutes to reestablish osmotic balance. Finally, the signaling feedback described in this study acts on the minute time scale and contributes to attenuate Hog1 activity under basal conditions and during acute osmotic stress.
Hog1 targets Ssk1 and Ssk2 to regulate its basal activity
It has been shown previously that basal signaling in the Sln1 branch enables higher efficiency, faster response times and higher sensitivity to signal variations compared to the Sho1 branch.21 However, the source of this basal signaling and the control mechanism remained unclear. Our model suggests that Hog1 dependent feedback control is achieved by targeting Ssk2 and the sensory module in the Sln1 branch (Fig. 4D). Assessing the feedback efficiency by simultaneously varying all feedback parameters suggests that a combination of feedback loops impinging on Ssk2 and Ssk1 can best explain the Hog1 activity overshoot. Unfortunately, purified Ssk1 is only weakly phosphorylated by Hog1 in vitro, and additional factors may thus be required in vivo such as priming of Ssk1 by Ypd1. In contrast, Ssk2 is readily phosphorylated by Hog1 in vitro on multiple MAPK consensus sites clustered within its amino-terminal domain, and several of these sites are also phosphorylated in vivo in a Hog1-dependent manner. Cells expressing a non-phosphorylatable Ssk2 mutant exhibit higher basal levels of active Hog1, and slightly slower kinetics of Hog1 inactivation after salt stress. These defects are independent of the well-studied glycerol feedback, but part of an internal signaling feedback loop that restricts Ssk2 activity. Interestingly, the amino-terminal domain of Ssk2 is known to inhibit its activity in vivo and mediates the interaction with Ssk1,7,8 providing a plausible molecular mechanism for how feedback phosphorylation of this domain by Hog1 may interfere with Ssk2 activity. We found Hog1-dependent phosphorylation sites on Sln1 in vitro, in particular in the linker segment between the second transmembrane domain and the histidine kinase motif.34 However these sites did not play a role in controlling basal Hog1 activity and thus their molecular function needs to be studied further.Crosstalk between Hog1 activation and glucose availability
We eliminated the glycerol feedback by starving the cells for glucose, which is rapidly metabolized through glycolysis to produce glycerol. Surprisingly, we found that cells are unable to activate Hog1 under these conditions, although the cells shrink and thus activate the membrane receptors sensing turgor pressure. While we do not yet understand the mechanism of this rapid glucose-switch, our preliminary data show that Hog1 activity upon osmostress is tuned by glucose availability, ranging from no activity under complete starvation to full activity under saturated glucose levels (2%). This suggests that an unknown factor tunes Hog1 activity by rapidly and reversibly gauging glucose availability. Preliminary experiments suggest that that this factor is independent of the Snf1 and Ras/PKA pathways. A possible explanation for this behavior is that cells control Hog1 activation to limit the transformation of glucose into glycerol when glucose is scarce and required for growth. Note that the closure of the Fps1 channel is reported to occur at least partially by a Hog1-independent mechanism,14,15,35 suggesting that cells may be able to retain intracellular glycerol even when Hog1 activity is low. In any case, further studies are necessary to understand the molecular crosstalk between glucose availability and Hog1 signaling.Feedback loops operating at different scales coordinate the MAPK cascades
Undeniably, positive and negative feedback regulation in signaling pathways are ubiquitous among different organisms. In addition to a well-conserved MAPKKK-MAPKK-MAPK cascade, a feedback topology linking the MAPK to the most upstream components seems to be recurrent.36–39 Moreover parallel control systems have been proposed as a general strategy for suppressing noise to improve information capacity in signaling pathways.40 Here we found evidence for parallel regulatory mechanisms adjusting Hog1 activity through the adaptation and negative feedback loops in the signaling cascade targeting upstream components, MAPKKK and the sensory module. It will be interesting to examine whether similar feedback loops also regulate basal signaling and adaptation of other signaling cascades.The benefits of feedback control in engineering systems to increase robustness to parameters variation, information capacity and bandwidth are well characterized,41 and have been observed in diverse biological systems.22,36,42,43 As the HOG pathway performs with high fidelity and robust signaling both in iso- and hyper-osmotic situations, the contributions of multiple feedback loops with distinct kinetics acting at different phases of the cellular responses are important to ensure cell survival, adaptation and recovery.
Conclusions
Using quantitative single cell microscopy combined with mathematical modeling, we uncovered a rapid negative feedback mechanism in the HOG signaling cascade targeting Ssk2 and most likely Ssk1. This new control mechanism regulates basal Hog1 activity under isosmotic conditions and is also important for tuning steady-state level of active Hog1 after adaptation to high osmolarity. Interestingly, our experimental and simulation data suggest that a single feedback loop has a limited capacity for regulating Hog1 activity, while double additive (not redundant) feedback loops in parallel are able to adjust Hog1 activity more efficiently.Material and methods
Plasmids and yeast strains construction
Plasmids and yeast strains are listed in Tables S2 and S3 (ESI†), respectively. Standard methods were used for yeast strain construction and molecular biology. For details see (ESI†).Growth conditions and antibodies
Cells were grown in synthetic medium (SC: 0.17% yeast nitrogen base, 0.5% ammonium sulfate, 2% glucose, amino acids as required). For glucose starvation, cells of 0.5–0.8 OD600 were washed and grown in glucose starvation medium without glucose (SC-Glc: 0.17% yeast nitrogen base, 0.5% ammonium sulfate, amino acids as required) or with 2-deoxy-D-glucose (2DG) instead of glucose (SC + 2DG: 0.17% yeast nitrogen base, 0.5% ammonium sulfate, 2% 2-deoxy-D-glucose, amino acids as required). Hog1 activation by osmostress was induced by the addition of 0.4 M NaCl.Commercially available antibodies against RFP (ChromoTek), Hog1 (Santa Cruz yC-20), and phospho-p38 (9216L, Bioconcepts or Cell Signaling 3D7) were used in this study.
NaCl-stimulation and label-free phospho-proteomics
Cells were grown in 50 ml standard medium to OD600 0.6, at which time they were stimulated with 0.4 M NaCl. Cells were harvested by 6.25% trichloroacetic acid (TCA) 0, 1, 5, 10, 20 and 45 minutes after NaCl stimulation. For each time-point, 3 biological replicates were generated. Sample preparation and mass spectrometry was performed as described.44 The acquired data was searched against the SGD database using Sorcerer Sequest version 4.2.0 search algorithm,45 and the OpenMS version 1.846 was used both to detect MS1 features and to align them between the different experimental conditions. Based on a decoy search,47 the maximum false discovery rate was set to 1%. Phosphopeptides features with identical sequence and phosphorylation state but different charge states were merged.GST-protein expression and extraction
Escherichia coli (BL21) cultures transformed with appropriate vectors were grown at 37 °C to an OD600 of 0.4. 1 mM isopropyl thiogalactopyranoside (IPTG) was added to the culture to induce expression of GST-tagged proteins and cells were incubated at 25 °C for 5 h. Cells were pelleted and resuspended in STET buffer (100 mM NaCl, 10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 5% Triton X-100 supplemented with 2 mM dithiothreitol (DTT), 1 mM phenylmethylsulphonyl fluoride (PMSF), 2 mM benzamidine, 200 μg ml−1 leupeptin and 2 μg ml−1 pepstatin). Cells were lysed by sonication at 4 °C and cell debris was removed by centrifugation at 7000 × g. GST-fused proteins were pulled down from supernatants with 300 μl of glutathione-sepharose resin (GE Healthcare, 50% slurry equilibrated with STET buffer) by mixing for 90 min at 4 °C. The glutathione-sepharose beads were collected by brief centrifugation and washed four times in 50 mM Tris-HCl pH 8.0 buffer supplemented with 2 mM DTT. The GST-fused proteins were then eluted in 300 μl of 50 mM Tris-HCl pH 8.0 buffer with 2 mM DTT and 10 mM reduced glutathione (Sigma) by rotating for 30 min at 4 °C.In vitro kinase assays
Eluted proteins were mixed with activated purified GST-Hog1 (pre-activation reaction: GST-Pbs2EE, 100 μM cold ATP, 1× kinase buffer, 30 min at 30 °C) in 1× kinase buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 2 mM DTT) with ATP32 (5 μM). The mix was incubated for 20 min at 30 °C, after which samples were boiled at 95 °C and resolved on SDS-PAGE. The proteins were visualized by Coomassie staining, while phosphorylation was detected using BioMax XAR (Kodak) films.Protein extraction and western blotting
For western blotting, cells were grown in SC with 2% glucose to OD600 ≈ 1, washed, and resuspended either in SC or SC-Glc for 10 min before addition of 2% glucose (time 0). Aliquots were removed at the indicated time, the cells were fixed with 10% TCA (Merck) and extracts were prepared for western blotting with appropriate antibodies.Microfluidic devices and quantitative microscopy
Live cell images were acquired using a fully automated inverted epi-fluorescence microscope (Ti-Eclipse, Nikon) in an incubation chamber set at 30 °C with 60× oil objective (N.A. 1.4). Image acquisition was controlled using micro-manager open source software,48 and each frame was imaged with the CFP, RFP and Cy5 filter sets in a single Z-plane and various XY-positions at variable time intervals between 20 seconds to 5 minutes. Microfluidic chips (CellAsic Y4) were used to rapidly exchange the media, and operated by the CellAsic controller. The pressures were set to 3 psi for the “ON” state and 0.25 psi for the “OFF” state to avoid cross contamination between the media. The fluorescent dye Alexa Fluor® 680 (M.W. 3 kDa, Invitrogen) was used to visualize switching. We used a MATLAB® based routine established in YeastQuant to automatically analyze images.24 Nuclear segmentation and cell tracking were based on Hta2-CFP images, while cells were segmented by RFP images (Hog1-mCherry). We defined the “rim object” to normalize nuclear accumulation of Hog1 (Fig. S2, ESI†), and remove artifacts due to fluorescence bleaching, cell movement in the z-direction and mis-segmentation of cells. Briefly, a small sub-cytoplasmic region, 1 pixel away from the nucleus and 2 pixels wide, was defined as a “rim” object. To quantify Hog1 relocation, the ratio between the RFP signal intensity in the nucleus and rim objects was calculated. The cells area was obtained by the summation of pixels in the “cell object” using the RFP channel. The values were quantified for individual cells and the average value over indicated number of cells from a representative experiment has been shown in all figures, unless otherwise stated. The experiment shown in Fig. S8 (ESI†) was carried out in a 96-well glass bottom microwell plate (630 μl, 0.17 mm low glass, Matrical bioscience) coated with a filtered solution of Concanavalin A (GE Healthcare) in PBS (0.1 mg ml−1).SFE calculations and modeling
A fixed time window was defined based on the average signal of Hog1 in wild-type cells, which starts immediately after switching [t1] and extends for 6 minutes [t2] to cover the expected overshoot response. The following algorithm was applied to each single cell trace and used to calculate the signaling feedback efficiency (SFE):• Smooth the measured signal (local regression using weighted linear least squares and a 2nd degree polynomial model with a span of 20%)
• Find the maximum and the time of the maximum point in [t1,t2], respectively Max and tmax
• Find the minimum in [tmax,t2], Min
• Calculate y1 = Max − y0, where y0 is the value just before switching
• Calculate y2 = Max − Min
• Calculate SFE = y2/y1
The model based on ordinary differential equations (ODE) is described in detail in ESI,† and all reactions are listed in Table S4 (ESI†). The MATLAB SimBiology® toolbox was used to design and simulate the model.
Author's contribution
H.S. and S.S.L. performed the quantitative microscopy assays, F.L. and R.B. the western blotting and immunoprecipitation experiments, K.S. and S.R. the in vitro kinase assay, and S.V. the mass spectrometry analysis. H.S., S.P. and H.K. were involved in the modeling, and H.S., S.P., F.P. and M.P. conceived the study and wrote the paper.Acknowledgements
We thank Haruo Saito for sharing plasmids, Ingrid Stoffel-Studer and Janny Tilma for technical assistance, members of the Peter and Pelet laboratories for helpful discussions, and Reinhard Dechant and Alicia Smith for critical reading of the manuscript. Funding: F.L. is supported by a HFSPO post-doctoral fellowship. The Pelet and Peter laboratories are supported by grants of the Swiss National Science Foundation (SNF), and work in the Peter laboratory is funded by the European Research Council (ERC), the Swiss Initiative in Systems Biology SystemsX (RTD project YeastX) and the ETH Zurich. In addition grants from the Spanish Ministry of Economy and Competitiveness (BFU2012-33503 and FEDER), 2014 SGR (Generalitat de Catalunya) and the Fundación Marcelino Botín (FB). F.P. is a recipient of an ICREA Acadeèmia award (Generalitat de Catalunya).Notes and references
- D. Muzzey, C. A. Gomez-Uribe, J. T. Mettetal and A. van Oudenaarden, Cell, 2009, 138, 160–171 CrossRef CAS PubMed.
- A. S. Howell, M. Jin, C.-F. Wu, T. R. Zyla, T. C. Elston and D. J. Lew, Cell, 2012, 149, 322–333 CrossRef CAS PubMed.
- S. Hohmann, FEBS Lett., 2009, 583, 4025–4029 CrossRef CAS PubMed.
- K. Tanaka, K. Tatebayashi, A. Nishimura, K. Yamamoto, H.-Y. Yang and H. Saito, Sci. Signaling, 2014, 7, ra21 CrossRef PubMed.
- F. Posas, S. M. Wurgler-Murphy, T. Maeda, E. A. Witten, T. C. Thai and H. Saito, Cell, 1996, 86, 865–875 CrossRef CAS.
- J. L. Brewster, T. de Valoir, N. D. Dwyer, E. Winter and M. C. Gustin, Science, 1993, 259, 1760–1763 CAS.
- F. Posas and H. Saito, Science, 1997, 276, 1702–1705 CrossRef CAS.
- F. Posas and H. Saito, EMBO J., 1998, 17, 1385–1394 CrossRef CAS PubMed.
- K. K. Tatebayashi, M. M. Takekawa and H. H. Saito, EMBO J., 2003, 22, 3624–3634 CrossRef CAS PubMed.
- S. M. Wurgler-Murphy, T. Maeda, E. A. Witten and H. Saito, Mol. Cell. Biol., 1997, 17, 1289–1297 CAS.
- P. Ferrigno, F. Posas, D. Koepp, H. Saito and P. ASilver, EMBO J., 1998, 17, 5606–5614 CrossRef CAS PubMed.
- V. Reiser, H. Ruis and G. Ammerer, Mol. Biol. Cell, 1999, 10, 1147–1161 CrossRef CAS.
- J. Bouwman, J. Kiewiet, A. Lindenbergh, K. van Eunen, M. Siderius and B. M. Bakker, Yeast, 2011, 28, 43–53 CrossRef CAS PubMed.
- K. Luyten, J. Albertyn, W. F. Skibbe, B. A. Prior, J. Ramos, J. M. Thevelein and S. Hohmann, EMBO J., 1995, 14, 1360–1371 CAS.
- M. Tamas, K. Luyten, F. C. W. Sutherland, A. Hernandez, J. Albertyn, H. Valadi, H. Li, B. A. Prior, S. G. Kilian, J. Ramos, L. Gustafsson, J. M. Thevelein and S. Hohmann, Mol. Microbiol., 1999, 31, 1087–1104 CrossRef CAS.
- T. Jacoby, H. Flanagan, A. Faykin, A. G. Seto, C. Mattison and I. Ota, J. Biol. Chem., 1997, 272, 17749–17755 CrossRef CAS PubMed.
- C. P. Mattison and I. M. Ota, Genes Dev., 2000, 14, 1229–1235 CAS.
- J. Warmka, J. Hanneman, J. Lee, D. Amin and I. Ota, Mol. Cell. Biol., 2001, 21, 51–60 CrossRef CAS PubMed.
- C. Young, J. Mapes, J. Hanneman, S. Al-Zarban and I. Ota, Eukaryotic Cell, 2002, 1, 1032–1040 CrossRef CAS.
- J. O. R. Schaber, R. Baltanas, A. Bush, E. Klipp and A. Colman-Lerner, Mol. Syst. Biol., 2012, 8, 1–17 CrossRef PubMed.
- J. Macia, S. Regot, T. Peeters, N. Conde, R. Solé and F. Posas, Sci. Signaling, 2009, 2, ra13, DOI:10.1126/scisignal.2000056.
- P. Hersen, M. N. McClean, L. Mahadevan and S. Ramanathan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7165–7170 CrossRef CAS PubMed.
- S. Pelet, F. Rudolf, M. Nadal-Ribelles, E. de Nadal, F. Posas and M. Peter, Science, 2011, 332, 732–735 CrossRef CAS PubMed.
- S. Pelet, R. Dechant, S. S. Lee, F. van Drogen and M. Peter, Integr. Biol., 2012, 4, 1274–1282 RSC.
- A. N. Wick, D. R. Drury, H. I. Nakada and J. B. Wolfe, J. Biol. Chem., 1957, 224, 963–969 CAS.
- I. Maayan, J. Beenstock, I. Marbach, S. Tabachnick, O. Livnah and D. Engelberg, PLoS One, 2012, 7, e44749 CAS.
- K. Nishimura, T. Fukagawa, H. Takisawa, T. Kakimoto and M. Kanemaki, Nat. Methods, 2009, 6, 917–922 CrossRef CAS PubMed.
- T. Horie, K. Tatebayashi, R. Yamada and H. Saito, Mol. Cell. Biol., 2008, 28, 5172–5183 CrossRef CAS PubMed.
- M. Rep, M. Krantz, J. M. Thevelein and S. Hohmann, J. Biol. Chem., 2000, 275, 8290–8300 CrossRef CAS PubMed.
- G. Mas, E. de Nadal, R. Dechant, M. L. Rodríguez de la Concepción, C. Logie, S. Jimeno-González, S. Chávez, G. Ammerer and F. Posas, EMBO J., 2009, 28, 326–336 CrossRef CAS PubMed.
- P. J. Westfall, J. C. Patterson, R. E. Chen and J. Thorner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12212–12217 CrossRef CAS PubMed.
- J. T. Mettetal, D. Muzzey, C. Gomez-Uribe and A. van Oudenaarden, Science, 2008, 319, 482–484 CrossRef CAS PubMed.
- D. B. Berry and A. P. Gasch, Mol. Biol. Cell, 2008, 19, 4580–4587 CrossRef CAS PubMed.
- M. Krantz, E. Becit and S. Hohmann, Curr. Genet., 2006, 49, 152–165 CrossRef CAS PubMed.
- R. Babazadeh, T. Furukawa, S. Hohmann and K. Furukawa, Sci. Rep., 2014, 4, 4697 Search PubMed.
- R. C. Yu, C. G. Pesce, A. Colman-Lerner, L. Lok, D. Pincus, E. Serra, M. Holl, K. Benjamin, A. Gordon and R. Brent, Nature, 2008, 456, 755–761 CrossRef CAS PubMed.
- B. Klinger, A. Sieber, R. Fritsche-Guenther, F. Witzel, L. Berry, D. Schumacher, Y. Yan, P. Durek, M. Merchant, R. Schäfer, C. Sers and N. Blüthgen, Mol. Syst. Biol., 2013, 9, 673 CrossRef PubMed.
- H. Shinohara, M. Behar, K. Inoue, M. Hiroshima, T. Yasuda, T. Nagashima, S. Kimura, H. Sanjo, S. Maeda, N. Yumoto, S. Ki, S. Akira, Y. Sako, A. Hoffmann, T. Kurosaki and M. Okada-Hatakeyama, Science, 2014, 344, 760–764 CrossRef CAS PubMed.
- K. Yamamoto, K. Tatebayashi, K. Tanaka and H. Saito, Mol. Cell, 2010, 40, 87–98 CrossRef CAS PubMed.
- I. Lestas, G. Vinnicombe and J. Paulsson, Nature, 2010, 467, 174–178 CrossRef CAS PubMed.
- H. S. Black, Bell Syst. Tech. J., 1934, 13, 1–18 CrossRef.
- S. Tay, J. J. Hughey, T. K. Lee, T. Lipniacki, S. R. Quake and M. W. Covert, Nature, 2010, 466, 267–271 CrossRef CAS PubMed.
- Y. Lin, Y. Li, S. Crosson, A. R. Dinner and N. F. Scherer, PLoS Comput. Biol., 2012, 8, e1002778 CAS.
- B. Bodenmiller and R. Aebersold, Methods Enzymol., 2010, 470, 317–334 CAS.
- J. K. Eng, A. L. McCormack and J. R. Yates, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef CAS.
- M. Sturm, A. Bertsch, C. Gröpl, A. Hildebrandt, R. Hussong, E. Lange, N. Pfeifer, O. Schulz-Trieglaff, A. Zerck, K. Reinert and O. Kohlbacher, BMC Bioinf., 2008, 9, 163 CrossRef PubMed.
- L. Käll, J. D. Storey, M. J. MacCoss and W. S. Noble, J. Proteome Res., 2008, 7, 29–34 CrossRef PubMed.
- A. Edelstein, N. Amodaj, K. Hoover, R. Vale and N. Stuurman, Curr. Protoc. Mol. Biol., 2010, 92, 14.20.1–14.20.17 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary text, 5 supplementary tables and 15 supplementary figures. See DOI: 10.1039/c4ib00299g |
| ‡ Competence Center for Systems Physiology and Metabolic Diseases, ETH Zürich, Switzerland. |
| § Current address: Bioengineering Department, Stanford University, USA. |
| ¶ Current address: MRC National Institute for Medical Research, London, UK. |
| This journal is © The Royal Society of Chemistry 2015 |