
Combined experimental and bioinformatics analysis for the prediction and identification of VHR/DUSP3 nuclear targets related to DNA damage and repair†
Fabio Luis
Forti
Laboratory of Signalling in Biomolecular Systems, Department of Biochemistry, Institute of Chemistry, University of Sao Paulo, Av. Prof. Lineu Prestes, 748 - Bl.09i, Sl.922, CEP: 05508-900 - Cidade Universitária, Sao Paulo-SP, Brazil. E-mail: flforti@iq.usp.br; Fax: +55-11-3091-2186; Tel: +55-11-3091-9905
First published on 28th October 2014
Abstract
The atypical dual-specificity phosphatases (aDUSPs) are a group of protein tyrosine phosphatases (PTPs) that have been increasingly studied recently, but little is known about their substrates or their roles and regulation. aDUSPs are typically low-molecular-weight enzymes that are distinct from the mitogen-activated protein kinase phosphatases (MKPs) but that still function in the regulation of the MAPK signalling cascade. aDUSPs may also have non-MAPK substrates, based on homologies observed in the sequences flanking potential phosphotyrosine target sites of other proteins and the cell type-specific characteristics of certain aDUSPs. Here, we combined experimental and computational tools to identify new substrates and protein partners of VHR (DUSP3) phosphatase in HeLa cells exposed to genotoxic stress. Experimental approaches confirmed the good stability of VHR and its nuclear co-localisation with classical MAPK substrates. The bioinformatics analysis of 4539 human nuclear proteins to identify a subset with functions related to DNA damage response and repair or to checkpoints and cell cycle control, that contain the phosphorylatable Thr–X–Tyr motif of MAPK with a high probability of dual phosphorylation, and that have structural homology to the MAPK activation loop resulted in a list of 57 putative VHR substrates. Fluorescence confocal microscopy and pull-down experiments followed by immunoblots revealed that VHR co-localised and interacted with components of the MRN complex and pH2AX, a DNA double-strand break sensor. Our platform, which combines experimental data from structure–function and bioinformatics analyses based on MAPK substrate similarities, provides a low-cost and rapid approach for the identification of novel aDUSP-interacting proteins with unknown roles in genotoxic stress response and genome stability maintenance.
Introduction
Tyrosine phosphorylation is a crucial process in signal transduction pathways. The steady state of this post-translational modification is finely regulated by the coordinated actions of protein-tyrosine kinases (PTKs) and protein-tyrosine phosphatases (PTPs). Drugs, peptides, growth factors, hormones, cytokines, antigens, and microorganism infection can shift this balance by rapidly stimulating PTK activity, inducing tyrosine phosphorylation and consequently activating signal transduction pathways.1 Conversely, tyrosine phosphatase inhibition has been shown to mimic signal transduction that is typically activated by tyrosine kinase activation.Cell fate decisions require several kinase- and phosphatase-regulated signalling pathways, and their deregulation is often found in pathological conditions.2 For instance, mitosis is primarily regulated by serine/threonine kinases (CDKs, PLKs, AURKs, NEKs) and fine-tuned by specific serine/threonine phosphatases (PPs 1, 2 and 4). However, nuclear mitotic events dependent on tyrosine phosphorylation also require tyrosine kinases (WEEs) and tyrosine phosphatases (CDC25s).3 Recently, after two reports of its activity in S. cerevisiae,4 the dual-specificity phosphatase CDC14 was found in the nucleus of mammalian cells and shown to be extremely important for efficient DNA repair and early aging after gamma radiation-induced damage.5,6
Similarly, the dual-specificity phosphatases (DUSPs or DSPs), which dephosphorylate mitogen-activated kinases (MAPKs) and mitogen kinase phosphatases (MKPs), are being increasingly recognised for their roles in human cancer development and progression.7 These ten enzymes can be classified into three groups; they are cell type-specific, they have sub-cellular compartment-dependent activity, and some are rapidly induced early response genes. However, they have different specificities for MAPK isoforms. These classical MKPs dephosphorylate threonine and/or tyrosine residues within the signature sequence pT–X–pY in the activation loop (A-loop or AL) of all MAPKs.7,8 Non-classical MKPs known as atypical DUSPs (aDUSPs) do not harbour the N-terminal (NT) Cdc25 homology (CH2) domain or the MAP kinase-binding (MKB) domain (also known as kinase-interacting motif, KIM) but can specifically dephosphorylate MAPKs with different affinities.8 The aDUSP prototype is VHR (DUSP3), which is over- or under-expressed in various human cancer cells. Prior to this report, the only known substrates of VHR were ERK1/2 and JNK1/2 (ERK ≫ JNK ∼ p38) phospho-enzymes.9,10
VHR plays a role in JNK dephosphorylation during 12-o-tetradecanoyl-phorbol-13-acetate (TPA)- or thapsigargin (TA)-induced apoptosis in LNCaP cells.11 In cervical carcinoma cells, VHR downregulates ERK1/2 and JNK1/2 to promote cell proliferation, and VHR knockdown by siRNA causes cell cycle arrest at G1/S and G2/M with consequent cellular senescence.12 Additionally, in human cervical carcinoma cell lines and patient tumour samples, VHR is considered as a new marker for cancer progression and a potential new target for cancer therapies. Not only is VHR up-regulated in these cells, it is also strongly localised to the nucleus.13 In contrast, VHR is expressed at very low levels in MCF7 and SKBR3 breast cancer cells, which allows phosphorylated JNK levels to accumulate and consequently promotes CYCLIN D1 and BRCA1-IRIS over-expression to establish a malignant phonotype.14 Similarly, VHR is downregulated in non-small cell lung cancer (NSCLC) cells, leading to a tumorigenic phenotype, whereas its over-expression inhibits cell growth and proliferation via ERBB2 receptor dephosphorylation.15
Because the factors that determine aDUSP substrate specificity are not completely known, recent efforts have been made to identify new VHR substrates and to elucidate its biological functions.16 We have contributed to these efforts by extensively immunoprecipitating proteins and/or protein complexes including VHR from cellular lysates, subjecting them to mass spectrometry identification, and generating interactome maps.17 However, here we developed a new strategy to quickly scan putative nuclear VHR targets in human protein databases by combining bioinformatics with biochemical experiments based on the functional and structural properties of MAPK targets. We combined theoretical analysis with validation analysis in HeLa and other human tumorigenic cell lines in the presence or absence of gamma radiation to identify novel VHR substrates and/or protein partners specifically involved in genomic stability/integrity. Taken together, our analysis generated an extensive list of putative VHR substrates involved in DNA damage response and repair and cell cycle progression. The proteins pH2AX, NBS1, CHK2, pATM, pATR, CENP-F, RAD50, CYCLIN A, and p53 strongly co-localised with VHR in a stress radiation-dependent manner, and pull-down experiments revealed that NBS1 in the MRN complex physically interacts with VHR.
Materials and methods
The flowchart below provides a logical, graphical representation of all materials and methods used in this work (described subsequently) and correlates the procedures with the two parts of the Results section. Part I (in grey) shows the experiments performed to test the hypothesis that VHR has other, non-MAPK nuclear targets. These experiments were performed in parallel with Part II (in green), which is a detailed description of the prediction, identification and validation method proposed in this work that is driven and substantiated by the results obtained in Part I.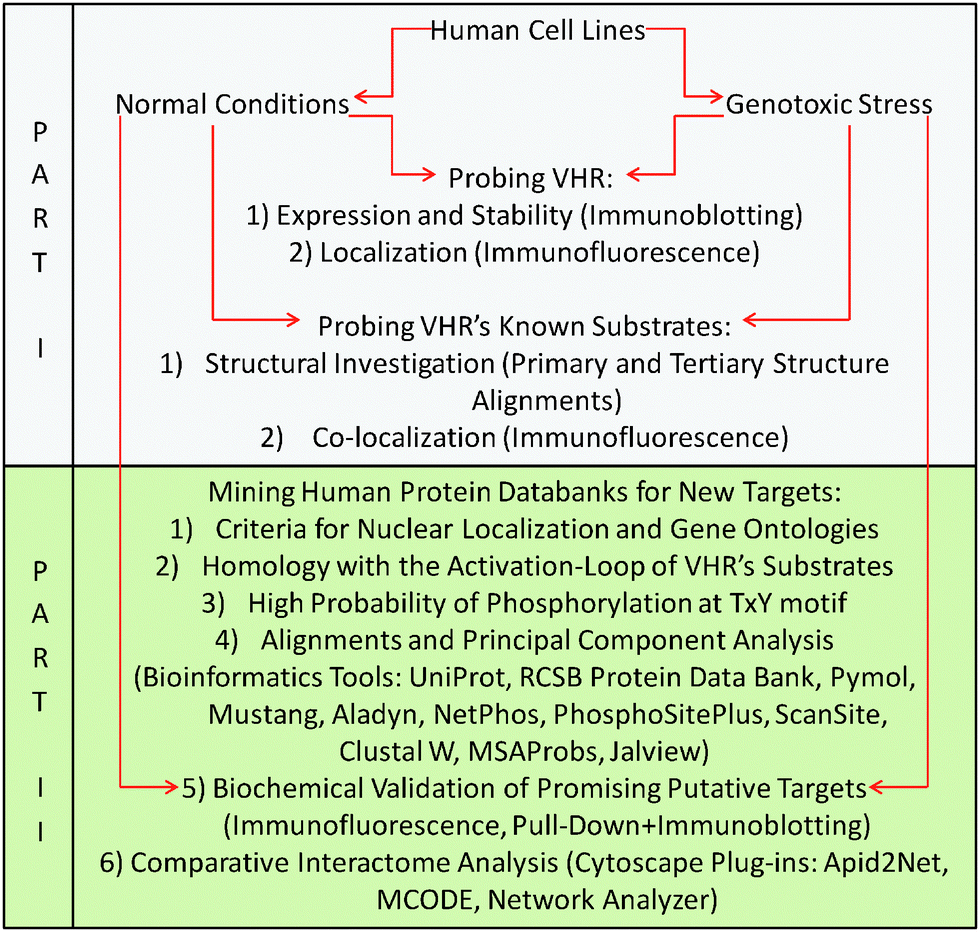
Cell culture and radiation treatments
The HeLa (CCL-2; American Type Culture Collection), HaCaT (kindly donated by Prof. Hugo A. Armelin, Butantan Institute, Sao Paulo-SP-Brazil), HEK-293 (CRL-1573; American Type Culture Collection), and MeWo (HTB-65; American Type Culture Collection) cell lines were cultured in DMEM or RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with streptomycin and ampicillin (Invitrogen, Carlsbad, CA) and 10% foetal bovine serum (FBS; Cultilab, Campinas, SP, Brazil) in a 37 °C MCO-19AICUV-PA incubator (Sanyo, Tokyo, Japan) with a humidified 5% CO2 atmosphere. Cells were maintained in culture at early passages (up to ten), sub-cultured every three days at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 or 1:5 dilutions, and plated in dishes of varying diameter according to the experimental protocols. For radiation treatments, the cells were subjected to 5 Gy ionising gamma radiation from a cobalt-60 source (Co60-GammaCel 220; Atomic Energy of Canada Limited (AECL), Ontario, Canada) at the Institute of Energy and Nuclear Research (IPEN, São Paulo-SP, Brazil).
:3 or 1:5 dilutions, and plated in dishes of varying diameter according to the experimental protocols. For radiation treatments, the cells were subjected to 5 Gy ionising gamma radiation from a cobalt-60 source (Co60-GammaCel 220; Atomic Energy of Canada Limited (AECL), Ontario, Canada) at the Institute of Energy and Nuclear Research (IPEN, São Paulo-SP, Brazil).
Cell synchronisation
Unsynchronised HeLa cells at approximately 60–70% confluence were arrested in G1/S by double thymidine blocking (DTB) for 18 h using 2 mM thymidine; the medium was changed, 2 mM thymidine was added to the medium 9 h later, and cells were incubated for an additional 12 h. Finally, cells were released into fresh medium containing 10% FBS for up to 24 h. For pulse-chase experiments, HeLa cells arrested at G1/S by DTB were cultured in the presence of 50 μg mL−1 cycloheximide (CHX) added 0.5 h before each time point, after which cells were released into fresh medium for up to 24 h. Unsynchronised HeLa cells at approximately 50% confluence were arrested at G2/M for 18 h by 300 nM nocodazole (NOCO) with or without a 10 μg mL−1 CHX pulse at time zero, followed by their release into fresh medium for up to 12 h. HeLa cells subjected to G2/M arrest by NOCO were sometimes also pre-treated with 10 μM of the proteasome inhibitor MG132 added at time zero and released into fresh medium for up to 12 h. HeLa cells were arrested at G0/G1 using serum-free medium (SFM) for 48 or 72 h and were released by adding fresh medium containing 20% FBS for up to 10 h. VHR protein expression was assessed in unsynchronised HeLa cells subjected to different doses of γ-radiation (0.5, 2, 5, 10, 15 and 20 Gy) and collected at several time points, 1 to 24 h after damage.Cell lysates
HeLa or MeWo cell suspensions were seeded in 100 mm-diameter dishes at 50–75% confluence 24 h before gamma radiation treatment. At specific times after radiation treatments, cells were lysed in RIPA buffer (50 mM Tris pH 7.2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mM NaCl and 10 mM MgCl2; Sigma) containing a protease inhibitor cocktail (2 μg mL−1 aprotinin, 2 μg mL−1 leupeptin, 1 mM DTT, 1 mM PMSF, 2 μg mL−1 pepstatin; Sigma).Pull-down assay and immunoblotting
Pull-downs and immunoblotting were performed as described previously.17 Briefly, E. coli cultures (strain BL-21) were transformed with the empty pGEX-4T1 vector (GE Healthcare) or pGEX-4T1 containing wild-type DUSP3 cDNA (DUSP3-WT) and grown in LB (15% Bacto-tryptone, 5% yeast extract and 15% NaCl; Sigma) culture medium. Cells were induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 2 h at 37 °C to obtain bacterial cell lysates overexpressing the VHR(WT)-GST fusion protein or GST protein alone as a control. Bacterial cell suspensions were sonicated on ice using eight 2 min cycles (ten pulses of 15 s at 50% amplitude with intervals of 30 s) in a Vibra-Cell model CVX-130 sonicator (Sonics & Material Inc.). After clearing the lysates by centrifugation for 30 min at 10000 rpm at 4 °C, the supernatants containing soluble bacterial proteins were incubated with glutathione-sepharose beads (4B; GE Healthcare) to pull down GST-fused proteins, which were then resolved by SDS-PAGE and quantified using the Bradford (Bio-Rad, Hercules, CA) method. Beads were washed twice in ice-cold PBS and centrifuged for 3 min at 3000 rpm at 4 °C and subsequently incubated with 250 μL bacterial lysates containing either the GST or GST-DUSP3 fusion protein for 1 h with stirring at 4 °C. After washing with RIPA buffer under the same conditions as above, the beads were incubated with HeLa/MeWo cell lysates (0.5 mg of total protein) for 1 h and washed again with RIPA buffer under the same conditions as described previously (adapted from ref. 18). The purified protein and protein complexes were resolved on 10 or 13% SDS-PAGE gels, transferred onto Hybond-C nitrocellulose membranes (GE Healthcare) in semi-dry transfer buffer (25 mM Tris pH 7.5, 0.2 M glycine and 20% methanol) in a trans-blot TE77 PWR semi-dry transfer system (Amersham Biosciences), and then blocked for 1 h at room temperature with 5% non-fat milk in Tris-buffered saline (0.01 g mL−1) with Tween-20 (0.1%; TBST). The following specific antibodies were used for immunoblots at 1:1000 dilutions: mouse anti-VHR (#610547; BD Biosciences), rabbit anti-actin (sc-10731 H-300; Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-NBS1 (#610870; BD Biosciences), mouse anti-phospho-JNK1/2/3 (sc-6254 G-7; Santa Cruz Biotechnology, Santa Cruz, CA), and rabbit anti-phospho-H2AX (#2577; Cell Signalling Technology Inc.). Incubations were all performed in TBST. Incubation times varied from 2 h at room temperature to overnight (18 h) at 4 °C. After three 5-min washes in TBST, the nitrocellulose membranes were incubated for 1 h at room temperature with anti-mouse, anti-rabbit or anti-goat antibodies conjugated to IRDye (Li-Cor, Lincoln, NE, USA) or Alexa-Fluor (Invitrogen, Carlsbad, CA) at 1:15000 dilutions for detection at 800 nm or 680 nm, respectively. The membranes were scanned with an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA) and quantified using Odyssey software (Version 3.0).
Immunofluorescence
HeLa, HaCaT, HEK-293 and MeWo cells seeded on round glass coverslips the day before treatment were subjected to 5 Gy gamma radiation and fixed at specific time points in fixing solution (3% formaldehyde, 37% solution, 3% paraformaldehyde, 2% sucrose in PBS; Synth, Sao Paulo, Brazil) for 10 min at room temperature and permeabilised (0.5% Triton X-100, 6.84% sucrose, 3 mM MgCl2 in PBS; Synth, Sao Paulo, Brazil) for 5 min on ice. Cells were incubated with 1:250 dilutions of the following primary antibodies in PBS/3% Triton X-100 for 1–2 h at room temperature in a humidified chamber: mouse anti-VHR (BD Biosciences) or goat anti-VHR (Santa Cruz), mouse anti-pATM-Ser1981 (Cell Signalling), rabbit anti-pATR-Ser428 (Cell Signalling), rabbit anti-pBRCA1-Ser1423 (Santa Cruz), rabbit anti-BRCA2 (Santa Cruz), rabbit anti-CENP-F (Santa Cruz), rabbit anti-cyclin A (Santa Cruz), mouse anti-NBS1 (BD Biosciences), rabbit anti-APE1 (Cell Signalling), rabbit anti-pATF2-Ser490/498 (Santa Cruz), rabbit anti-MRE11 (Cell Signalling), rabbit anti-RAD50 (Santa Cruz), rabbit anti-pCHK2-Thr68 (Cell Signalling), rabbit anti-pP53-Ser15 (Cell Signalling), rabbit anti-pERK1/2-Thr185/Ty187 (Cell Signalling), mouse anti-JNK1/2/3-Thr183/Ty185 (Santa Cruz), mouse anti-p-p38-Thr180/Tyr182 (Santa Cruz), rabbit anti-pH2AX-Ser139 (Cell Signalling), rabbit anti-pSMC1-Ser360 (Cell Signalling), goat anti-KU86 (Santa Cruz), and rabbit anti-MSH2 (Santa Cruz). The following secondary antibodies were diluted 1:500 in PBS/Triton X-100 and incubated for 1 h at room temperature: donkey anti-goat (Alexa Fluor 488 nm), donkey anti-mouse (Alexa Fluor 568 nm), goat anti-mouse (Alexa Fluor 488 nm), and goat anti-rabbit (Alexa Fluor 568 nm). Coverslips were mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA) fixing agent plus DAPI, and cells were observed with a confocal microscope (Zeiss LSM 510-Meta; Germany). The image acquisition and analyses were performed using the Start LSM Image Browser software (Zeiss, Germany).
Co-localisation analyses
After systematic image acquisition, analyses were performed in fields containing several nuclei using the Start LSM Image Browser software (Zeiss, Germany). Co-localisation coefficients were obtained by plotting the fluorescence intensity from channel 1 (red) versus the fluorescence intensity from channel 2 (green). The weighted co-localisation coefficient was obtained for each channel, and the overlap coefficient was extracted from the merge field, representing the co-localisation percentage. The correlation R and R2 were obtained for the merge field between the red and green channels. High co-localisation was assumed between two fluorophores (in our case, labelled in green with Alexa Fluor 488 and labelled in red with Alexa Fluor 555) when overlap coefficients were > 0.5.Protein data mining
All information about proteins, sequences, biological functions, and other annotation data was mined from UniProt (http://www.uniprot.org/) databases.Protein structure visualisation and modification
Structures acquired from the Protein Data Bank (PDB; http://www.rcsb.org/pdb/) were rotated, coloured, and/or cut in specific and desired sequences with the Pymol software (http://www.pymol.org/) and subsequently saved.Structural alignments
Solved 3D structures deposited in the PDB for human ERK1 (1ERK), ERK2 (1TVO), JNK1 (3ELJ), JNK2 (3E7O), JNK3 (2WAJ), p38a (1P38), ErbB2 (1OVC), phospho-ERK1 (2ZOQ), phospho-ERK2 (2ERK) and p-p38a (3PY3) proteins were structurally aligned using the Mustang (http://pxgrid.med.monash.edu.au:8080/mustangserver/)19,20 or Aladyn (http://aladyn.escience-lab.org/)21 software on freely available online web servers. Alignments shown in Fig. 3 and Fig. S3 (ESI†) were obtained with the Mustang web server after loading the .pdb files and superimposing them onto the ERK2 structure as a template. The results represent the optimal RMSD (root-mean-square deviation of atomic positions) with the minimal distances (in ångströms) for all the aligned structures shown in the figures.Phosphorylation site predictions
Phosphorylation sites of threonine and tyrosine residues were predicted in protein sequences of interest using three different freely accessible web software packages that generated similar values: (1) NetPhos 2.0 (http://www.cbs.dtu.dk/services/NetPhos/),22,23 (2) PhosphoSitePlus (http://www.phosphosite.org/homeAction.do),24 and (3) ScanSite (http://scansite.mit.edu/).25 The prediction of phosphorylation sites shown in Fig. 3A and 5B was obtained from the NetPhos 2.0 Server.Amino acid alignment
The primary sequence or amino acid sequence of proteins of interest in the FASTA format (http://https://www.ncbi.nlm.nih.gov/BLAST/blastcgihelp.shtml) was analysed using the Clustal W/Omega26 and MSAProbs27 software on web servers available from the Bioinformatics Toolkit (http://toolkit.tuebingen.mpg.de/).Principal component analysis
The free program Jalview (http://www.jalview.org/)28,29 was used to view and edit multiple sequence alignments (from the MSAProbs software), which were analysed using phylogenetic trees and principal component analysis (PCA) plots.Construction of physical protein–protein interaction (PPPI) networks and clustering analyses
The 57 protein candidate hits obtained from the bioinformatics approach described in Fig. 5A were used to construct a potential VHR interaction network in the context of physical protein–protein interactions (PPPIs). Data mining and network design were performed with the Cytoscape version 2.6.3 software (http://www.cytoscape.org)30 in different protein databases using the APID2NET Cytoscape plugin (http://bioinfow.dep.usal.es/apid/apid2net.html).31 The obtained network was analysed with the Molecular Complex Detection (MCODE) Cytoscape plugin (http://baderlab.org/Software/MCODE)32 to detect protein clusters and/or complexes that were representative of biological processes that involve VHR and that are related to cell cycle regulation, senescence and/or DNA repair.Statistical analysis of general and simplest network parameters
The resulting global network (Fig. 8A) and clusters of interest (Fig. 8B and C) were subjected to the Network Analyzer Cytoscape plugin (http://med.bioinf.mpi-inf.mpg.de/networkanalyser/)33 to assay robustness and topological and centrality parameters, such as the clustering coefficient, diameter, centralisation, shortest paths, average number of neighbours, density, heterogeneity, number of nodes, number of self-loops, path length and analysis time in seconds. These results are listed in ESI,† Table S4.Results
Part I: probing the true substrates of VHR: are these substrates adequate to support the hypothesis that VHR has other nuclear targets in cells under genotoxic stress conditions?
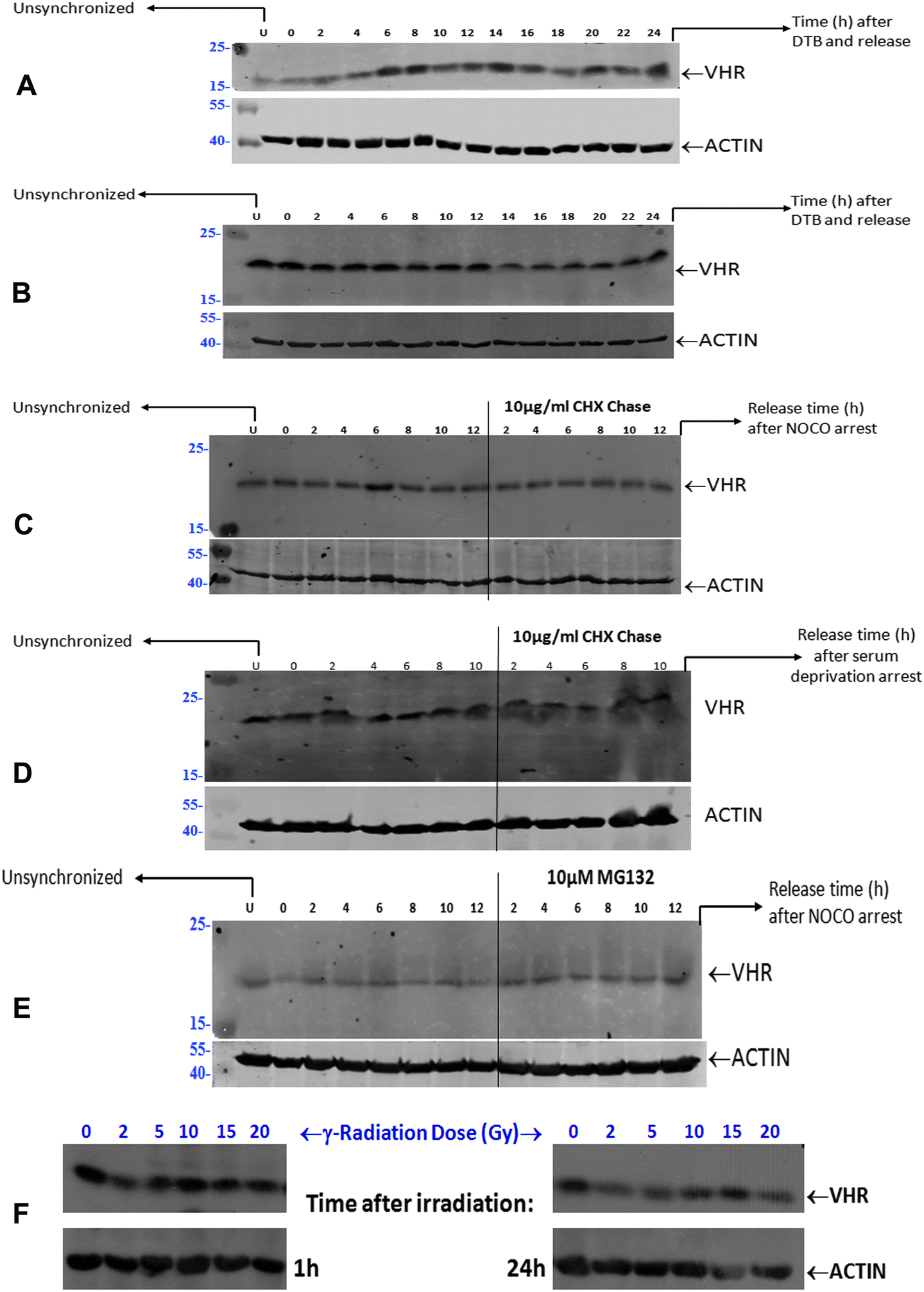 | ||
| Fig. 1 VHR protein expression and stability are unchanged in HeLa cells throughout the cell cycle and under stress conditions. (A) Unsynchronised HeLa cells subjected to G1/S arrest by double thymidine blocking (DTB) + FBS release up to 24 h. (B) Unsynchronised HeLa cells subjected to G1/S arrest by DTB + 50 μg mL−1 cycloheximide (CHX) pulse chase added 0.5 h before each time point + FBS release up to 24 h. (C) Unsynchronised HeLa cells subjected to G2/M arrest by 300 nM nocodazole (NOCO) + 10 μg mL−1 CHX pulse at time zero + FBS release up to 12 h. (D) Unsynchronised HeLa cells subjected to G2/M arrest by 300 nM nocodazole (NOCO) + pre-treatment with 10 μM proteasome inhibitor MG132 added at time zero + FBS release up to 12 h. (E) Unsynchronised HeLa cells subjected to G0/G1 arrest by serum-free medium (SFM) for 72 h + SBF release up to 10 h. (F) VHR protein expression in unsynchronised HeLa cells subjected to different doses of γ-radiation and collected at different time points from 1 h to 24 h after damage. | ||
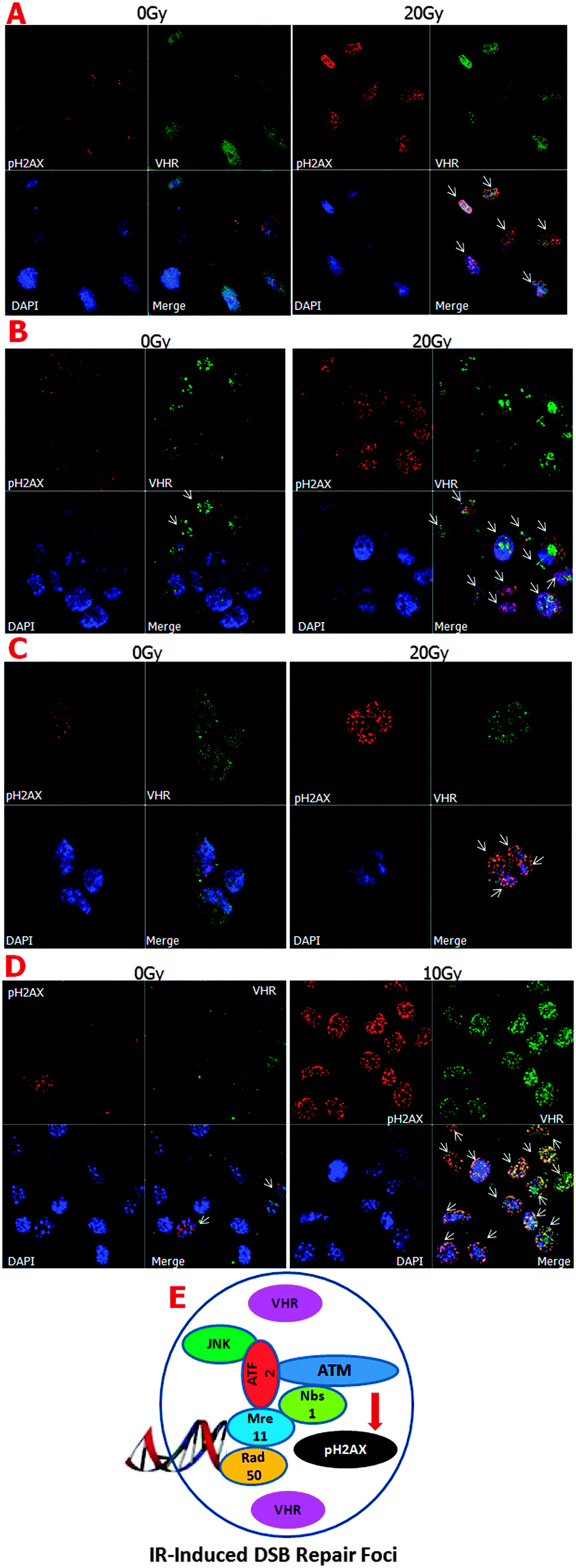 | ||
| Fig. 2 VHR and pH2AX proteins localise at ionising radiation-induced foci (IRIF) in multiple cell lines. Expression and localisation of VHR (green) and pH2AX (red) proteins in unsynchronised HeLa (A), HaCaT (B), HEK-293 (C), and MeWo (D) cells subjected to high doses of gamma radiation (10 or 20 Gy) and collected 1 h after damage. (E) Schematic showing pH2AX localisation at the IRIF, where other proteins and complexes are known to participate in DNA repair processes. VHR also localises at the IRIF, where it may play an unknown role in these stress conditions. | ||
It is well known that the MRE11–RAD50–NBS1 complex (MRN complex) is important for detecting single- and double-strand DNA breaks. ATM kinase is then recruited to the breaks, where it phosphorylates histone H2AX at serine 139, which in turn promotes the rapid recruitment of the repair machinery to the damage sites.34 It was recently shown that the ATF2 protein, which is phosphorylated by active (phosphorylated) JNK, is necessary for ATM recruitment to ionising radiation-induced foci (IRIF).35 As shown in Fig. 2E, we hypothesised a preliminary scheme in which VHR molecules in close proximity to the protein complexes formed after DNA damage at IRIF function in the repair processes. We confirmed part of this hypothesis by investigating VHR and pATF2 (Ser 490/498) co-localisation using confocal fluorescence microscopy (ESI,† Fig. S5). Thus, our results show that high VHR levels are maintained for at least 1 h after 20 Gy gamma radiation exposure and specifically co-localise with pH2AX at IRIF. These results support our hypothesis that new roles and targets of VHR have not yet been identified in DNA damage recognition and/or repair mechanisms.
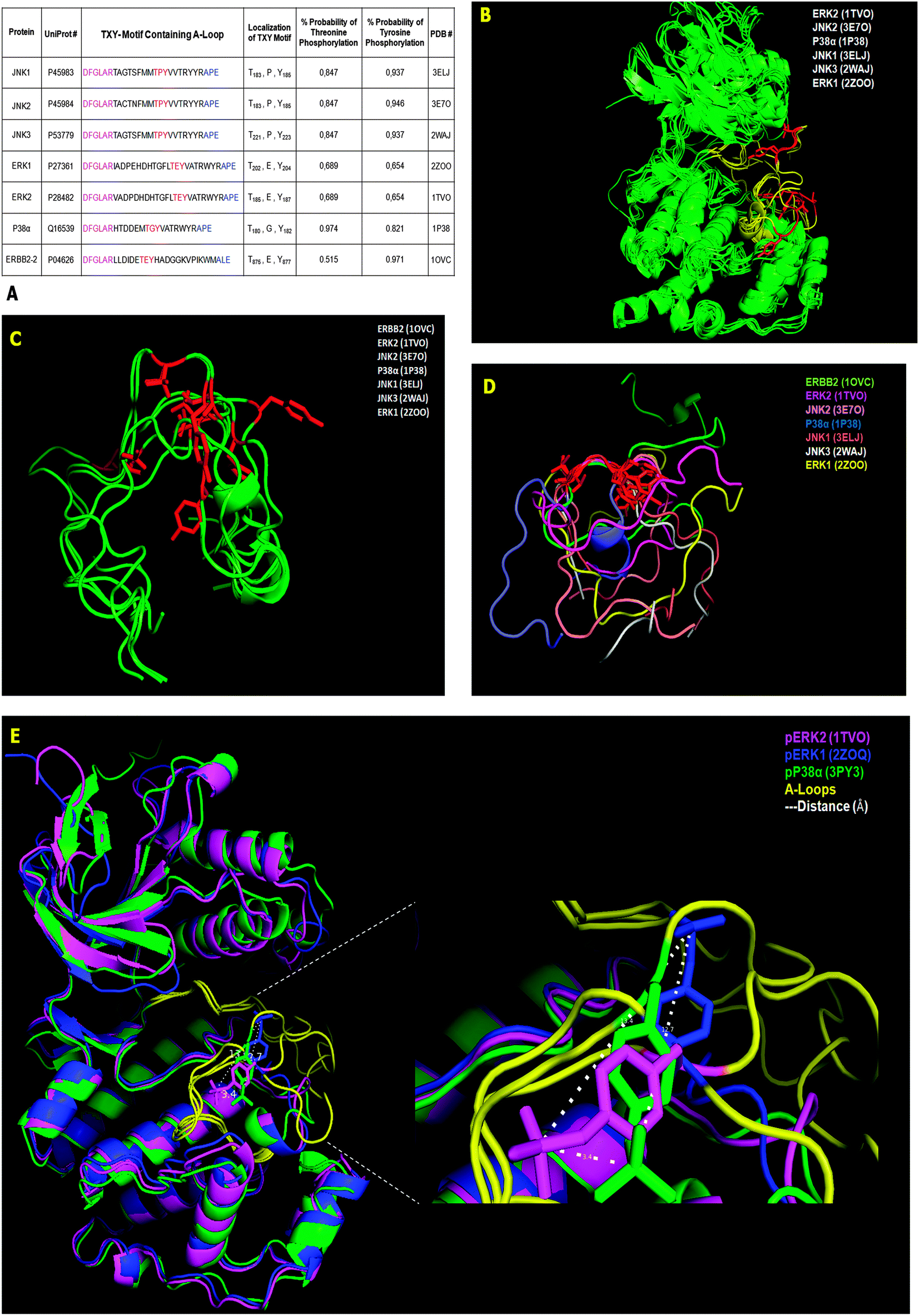 | ||
| Fig. 3 MAPKs, the VHR classical substrates: sequence and structural characteristics. (A) Seven known VHR substrates’ activation loop (AL or A-loop) primary sequence and phosphorylation predictions of the TXY motif. (B) Structural alignments of full length MAPK isoforms known as VHR substrates in vitro and in vivo. The activation A-loop region is marked in yellow and the TXY motif is in red. (C) Structural alignments of the A-loop from the seven VHR substrates shown in (A) highlighting only the Thr and Tyr (red) residues of the TXY motif. (D) Structural alignment of A-loops from the seven VHR substrates by fixing the spatial coordinates of only the T and Y residues. (E) The full-length structures of three tyrosine-phosphorylated MAPK isoforms (ERK1 = blue, ERK2 = magenta and p38 = green), with crystal structures solved and deposited in PBD, were structurally aligned with a resolution of 2.3 Å. The spatial positioning of phosphotyrosyl residues in atomic distances is shown (dotted white lines) in the activation loop (yellow). | ||
The ERBB2 protein is a transmembrane receptor for tyrosine-phosphorylated EGF whose tyrosine kinase catalytic domain is phosphorylated at the cytosolic C-terminus.15 This phosphorylation creates a distinct folding pattern that does not overlap with classical MAPKs. However, the structural alignment of the remaining six substrates using their solved 3-D structures in the non-phosphorylated state (see the PDB # shown in Fig. 3A) shows high overlap along the entire protein α-C backbones (green), medium overlap in the A-loop (yellow), and low overlap in the TXY motifs (red) (Fig. 3B). Alternatively and complementarily, by directly performing the structural alignment with only the A-loop, with similar N- and C-terminal structural constraints of the full-length proteins, and maintaining their structural coordinates from the PDB structure, the seven VHR substrates from Fig. 3A (green) did not improve their overlap even in their TXY motifs (red) (Fig. 3C). Furthermore, by fixing the Thr and Tyr residue coordinates to overlap and performing structural alignments for the seven A-loop regions, we found that the substrates have different rotational positions and a complete non-overlapping pattern along the N- and C-terminal flanking sequences (coloured) (Fig. 3D). Finally, the structural alignment (2.3 Å resolution) of the three available phosphorylated MAPK structures deposited in the PDB (pERK1, pERK2, and p-p38) shows that the A-loop is the least overlapping region, and by comparing the three phospho-tyrosine residues (magenta phosphate of ERK2 is 3.4 Å away from the green phosphate of p38, which is 12.7 Å away from the blue phosphate of ERK1, etc.), we found that they each occupy different spatial positions and distances ranging from 3.4 Å to 13.4 Å between the phosphorus atoms of the phosphate groups (dashed lines in Fig. 3E).
These different structural investigations using deposited data gave us a better idea of how VHR might interact with MAPK to remove the phosphate groups from the tyrosine and threonine residues in such a flexible region. Even for these very specific substrates, there is no absolute consensus sequence flanking the TXY motif that precludes other proteins as VHR substrates. Consequently, these findings support our screen of protein databanks for novel VHR interaction partners based on recent substrate-driven homology approaches.40
Despite the apparent structural flexibility of the TXY motifs independent of their phosphorylation status and the directionality of the activation loops, the three classical MAPKs remain as important VHR substrates. Thus, we investigated the effects of ionising radiation on the sub-cellular co-localisation of MAPK and VHR (Fig. 4). We found that ERK, JNK and p38 (to a lesser extent) were all highly phosphorylated 10 min after 5 Gy gamma irradiation and strongly localised to the nuclei of HeLa cells. We examined a similar strong nuclear localisation of VHR, and more surprisingly, it was highly co-localised (overlap coefficients ≥0.5, vs. ≤0.3 in untreated cells, Table S1, ESI†) with each protein examined after DNA damage (Fig. 4A, B and C, respectively). VHR and p-p38 MAPK colocalisation was less pronounced compared to VHR colocalisation with p-ERK or p-JNK. These experiments show that phosphorylated MAPKs are potential VHR targets, even under stress conditions, which allows us to use their biological and structural properties as driving models for our screening approach. Additionally, VHR was diffusely distributed throughout the nucleus and in nucleolar sub-compartments and was heterogeneously distributed in more and less compacted DNA regions, where it may have additional unknown substrates and functions.
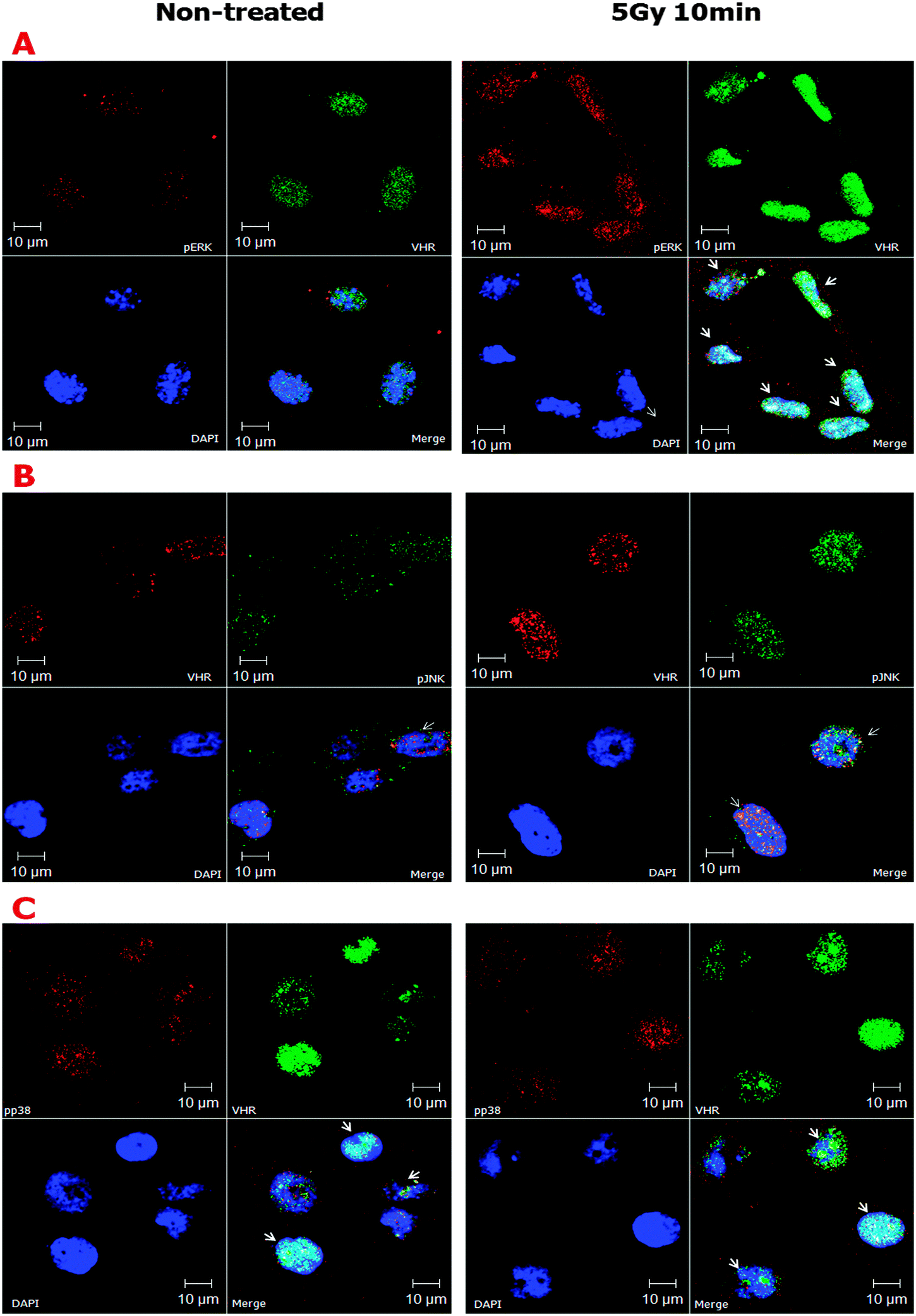 | ||
| Fig. 4 Confocal fluorescence microscopy showing co-localisation of VHR with pERK (A), pJNK (B), and p-p38 (C) in HeLa cells treated with gamma radiation. HeLa cells grown asynchronously were subjected to 5 Gy gamma irradiation, collected after 10 min, fixed, and permeabilised. Double labelling with anti-VHR and anti-phospho-MAPK antibodies shows high levels of co-localisation after irradiation, especially for VHR-pERK and VHR-pJNK. p-p38 levels did not increase after gamma radiation treatment, but p-p38 partially co-localised with VHR independent of damage. | ||
Part II: identifying and validating new nuclear protein targets of VHR using the substrate structure-based approach proposed in this work
000 deposited human proteins (Fig. 5A). The second filter we used was to include proteins involved in the following seven specific biological functions (or gene ontologies, GO): (1) MAPK pathway (positive control for VHR substrates); (2) mitosis; (3) cell cycle; (4) telomerase; (5) DNA damage; (6) DNA repair; and (7) genomic stability. The selected ontologies were based on substrates and biological functions already described for this DUSP (Torres TEP & Forti FL, unpublished results).9–12,14,15,36,41 The resulting ∼800 proteins were subsequently filtered according to the presence of at least one T–X–Y motif in the primary sequence. We identified 520 TXY-containing sequences in 323 proteins (Fig. 5A) and analysed their TXY motifs for the probability of both tyrosine and threonine phosphorylation using three different freely available software packages [NetPhos (http://www.cbs.dtu.dk/services/NetPhos/); ScanSite (http://scansite.mit.edu/motifscan_id.phtml); and PhosphoSitePlus (http://www.phosphosite.org/siteSearchAction.do)]. We required the predicted phosphorylation of both sites to be greater than 50% for all three software analyses, and this criterion culled our list to 121 sequences in 101 proteins (ESI,† Table S2) (Fig. 5A and B). The distributions of the best-predicted sequences according to their gene ontologies are shown in Fig. 5B (right superior quadrant) along with the classical VHR substrates (MAPK pathway in red squares).
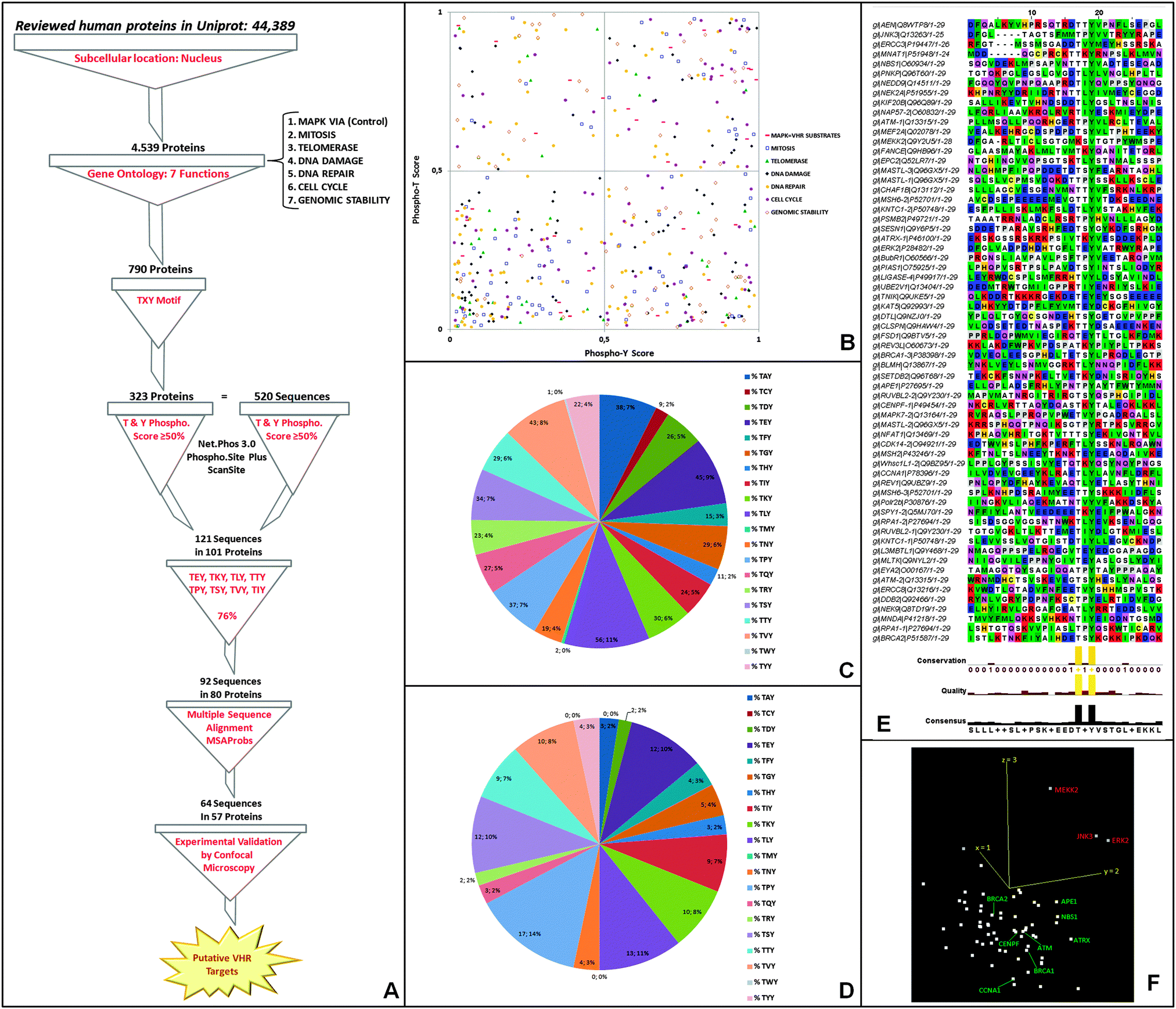 | ||
| Fig. 5 Bioinformatics approaches based on MAPK substrate-driven similarities for identifying putative novel VHR targets involved in genome integrity maintenance. (A) Workflow of the seven filtering steps used to generate a list of 64 putative VHR targets. (B) Distribution of candidates according to the seven gene ontology processes showing the probability of phosphorylation at Tyr versus Thr residues. (C) Percentage of the twenty amino acids in the X position of 520 TXY-containing sequences. (D) Percentage of the twenty amino acids in the X position of 121 TXY-containing sequences with the phospho-Y/T scores > 0.5. (E) Resulting alignment of 64 A-loop-like sequences of 29 aa showing the final consensus sequence flanking the TXY motif. (F) Principal component analysis calculation of the 64 sequences aligned. The classical VHR substrates are shown in red, and the eight randomly chosen targets for experimental validation are shown in green. | ||
An additional filtering step was performed based on the frequency at which the twenty amino acids appear in the 2nd or X position in the TXY motif in the 520 sequences (Fig. 5C); this quantification showed relative conservation of certain amino acids in the highest scoring 121 phospho-sequences (Fig. 5D), suggesting that some amino acids are evolutionarily preferential in that position. We then selected the eight most frequent residues (E, K, L, T, P, S, V, I), which represented the majority (76%) and limited our analysis to 92 sequences from 80 proteins. Intriguingly and consistent with our data (Fig. 3A), these residues include glutamate and proline, which are the preferred X residues in the best in vitro and in vivo VHR substrates, ERK and JNK,9,10 and in the ERBB2 receptor catalytic domain.15
Following our approach, we performed pairwise alignments of the 92 sequences based on hidden Markov models (HMMs) combined with partition functions to calculate posterior probabilities of multiple matching (MSAProbs). We only considered a 31-aa sequence similar to the largest A-loop in VHR substrates (as in ERK proteins, see Fig. 3A), including the TXY motif with 10 downstream and 18 upstream amino acids [(AA)18–TXY–(AA)10]. In the 64 sequences with the highest scores (best scored alignments without inserted residues) found in 57 proteins [Fig. 5E, visualised with Alignment Viewer on the JalviewLite applet tool (http://www.jalview.org/)], in addition to Thr and Tyr conservation, other consensus residues were in specific positions flanking the TXY motif in a significant number of aligned sequences, comprising a putative novel consensus sequence for VHR recognition and binding that might exist in other DUSP substrates. Finally, these 64 sequences (which include, for instance, some MAPK VHR substrates) were submitted to a principal component analysis (PCA) calculation using a BLOSUM62 sequence similarity protein matrix (http://www.jalview.org/help/html/calculations/pca.html). These calculations showed that ERK2, JNK3 and MEKK2 kinases had the most significant sequence similarities (in red), were distinct from the others (in green), and localised in the same similarity space (Fig. 5F). The last result demonstrates that, in principle, all these proteins are putative VHR targets. Thus, we randomly selected hits for experimental investigation based solely on their spatial proximity to ERK2, JNK3, and MEKK2 in the PCA results (Fig. 5F).
Before experimentally validating potential VHR protein targets, we performed a structural alignment of several hits that had structural data deposited at the PDB (full length or partial) containing one or more regions of approximately 31 amino acids of the T–X–Y motif. Of the 64 hits, only 15 had .pdb structures (ESI,† Table S3 and Fig. S6). We performed structural alignments of these sequences with the A-loop of ERK2 as a control (as in Fig. 3, shown here in magenta) by fixing the Thr and Tyr coordinates. The majority of alignments overlapped poorly, with only one or two amino acids flanking the T–X–Y motif (red), as in APE1 (ESI,† Fig. S6D). In general, their sequences had no obvious structural homology with the ERK2 A-loop. This analysis may (or not) exclude these 15 hits, but the others require experimental validation.
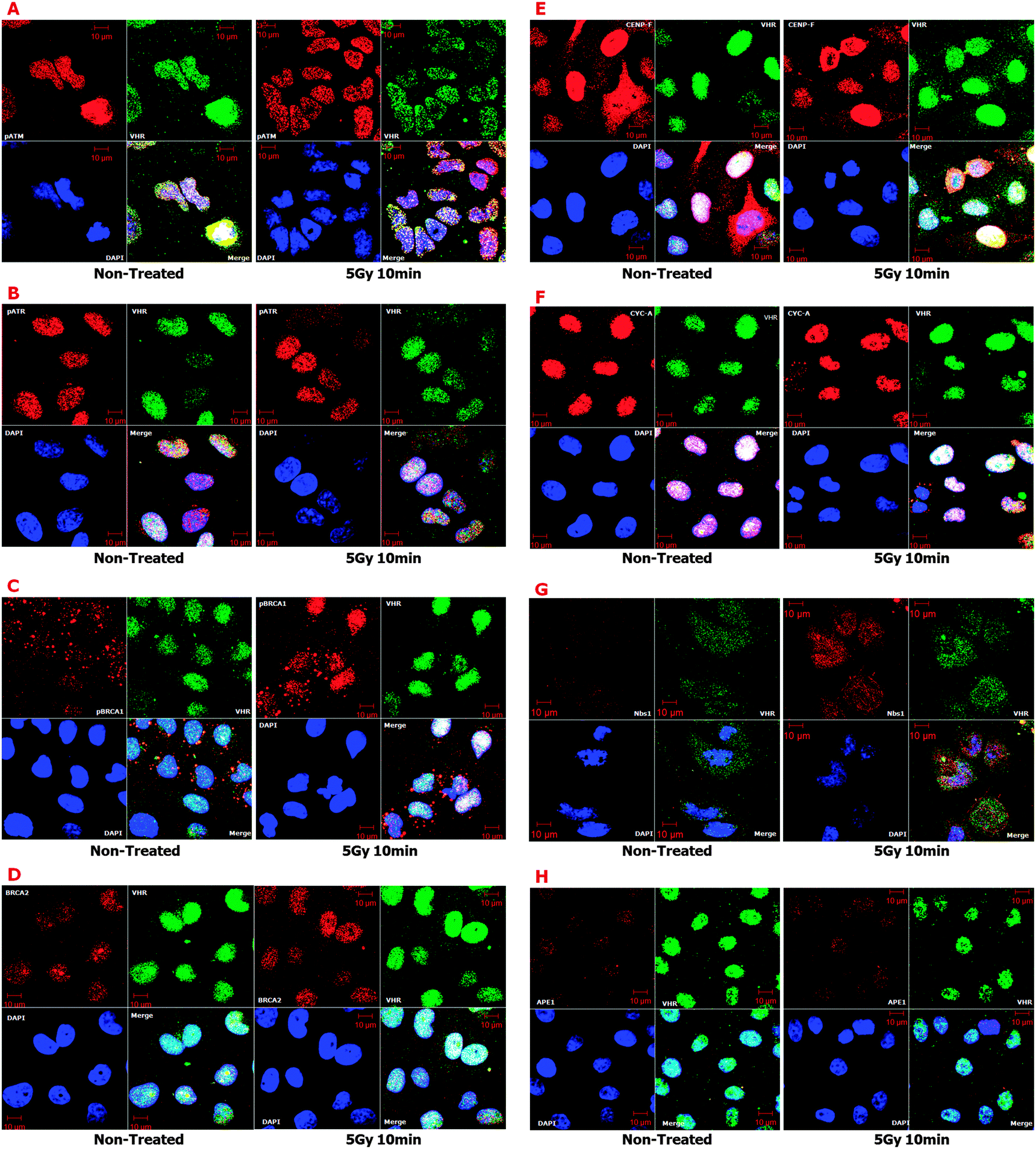 | ||
| Fig. 6 Confocal fluorescence microscopy showing co-localisation of VHR with (A) pATM (Ser1981); (B) pATR (Ser428); (C) pBRCA1 (Ser1423); (D) BRCA2; (E) CENP-F; (F) Cyclin A; (G) NBS1, and (H) APE1 in HeLa cells 10 min after 5 Gy gamma radiation treatment. Double labelling of VHR (labelled in green by Alexa Fluor 488) with the eight protein hits (labelled in red by Alexa Fluor 555) was performed by incubating with a primary antibody for 1 h and then for 1 h with a secondary antibody at room temperature, and DAPI was used to stain nuclear DNA. Micrographs were obtained on a Zeiss LSM confocal microscope in triplicate and analysed using the Zeiss LSM-Meta software. | ||
To investigate potential physical interactions of VHR with the eight randomly chosen proteins, we used VHR-GST as bait to pull down interacting proteins or complexes from cell lysates.17 HeLa and MeWo cell lines were untreated or exposed to 5 Gy gamma radiation, lysed 30 min after irradiation, and incubated with recombinant VHR-GST. Glutathione-Sepharose beads retained VHR-GST and its interacting partners, as assessed by immunoaffinity chromatography. We performed immunoblotting experiments with specific antibodies against the classical ERK1/2 (not shown) and JNK substrates (Fig. 7B) as controls. Of the eight candidate protein targets, VHR was only able to pull down Nibrin (NBS1 or NBN), one of the three components of the DNA repair MRN complex. Whether it interacts directly or indirectly with VHR is still unknown (Fig. 7A).
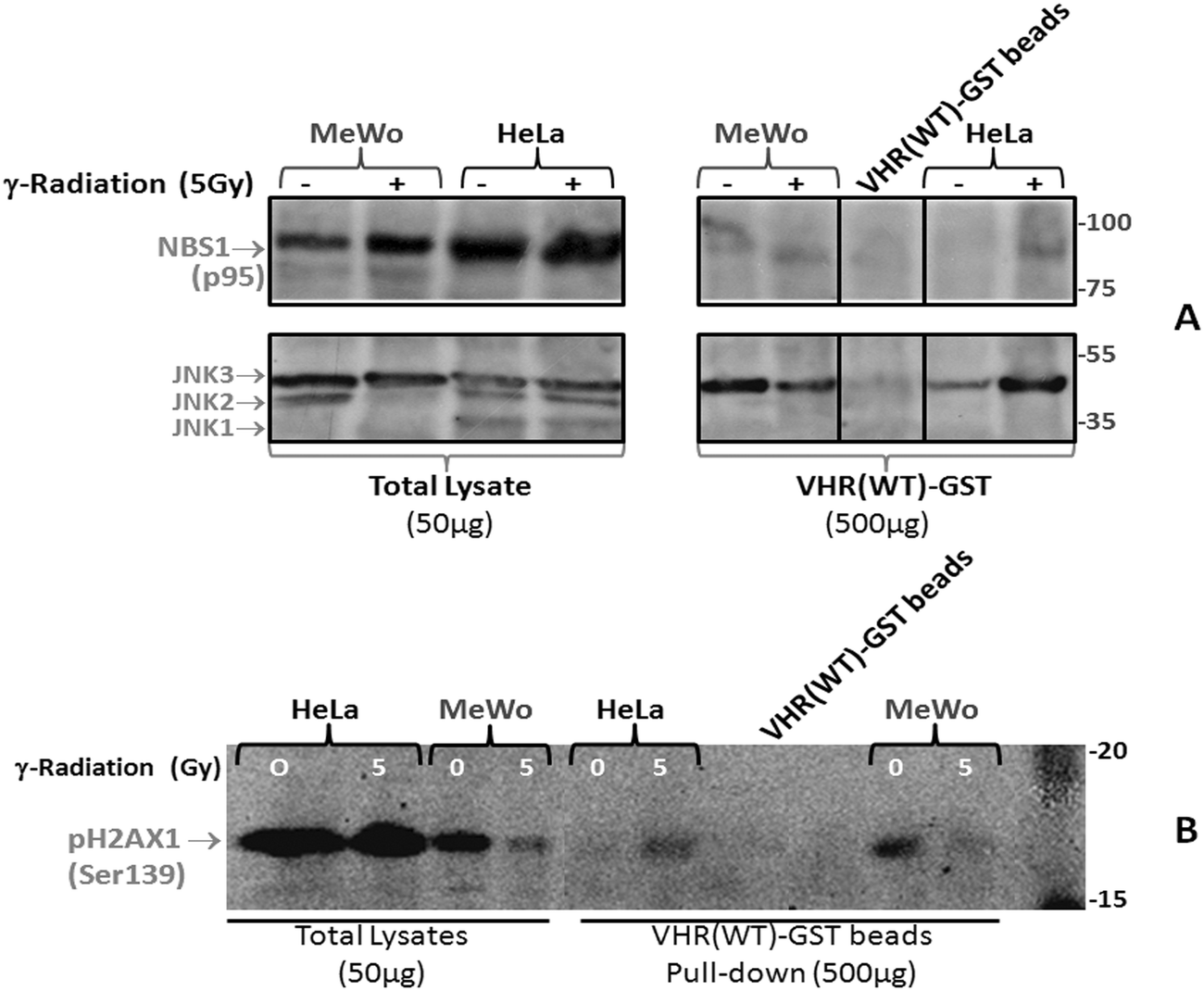 | ||
| Fig. 7 Immunoprecipitation of VHR can pull down NBS1 and p-H2AX (Ser139) from HeLa and MeWo cells. VHR-GST was used as bait to precipitate interacting protein partners from 500 μg of lysates from 5 Gy gamma-irradiated HeLa or MeWo cells. A total of 50 μg cell lysates were compared. The pGEX-4T1 vector produced bacterial GST recombinant proteins that were incubated with cellular lysates as a control for nonspecific binding to the tag protein, and the VHR(WT)-GST beads were used alone as a control for nonspecific binding to the glutathione-sepharose beads. | ||
Finally, we performed pull-down experiments to determine whether the Ser-139 phosphorylation of H2AX or γ-H2AX by the PIKK kinases (ATM/ATR) occurs after MRN complex recruitment to DNA strand break points and whether VHR could simultaneously localise at these foci. This result would confirm the results from Fig. 2 that showed co-localisation between VHR and p-H2AX, but it would also show evidence of an indirect physical interaction via the NBS1 protein (Fig. 7A and B). Our results indicate that pulling down VHR in HeLa or MeWo cells also pulled down phospho-H2AX and MRN complex components, which are recruited to DNA strand breaks in an ionising radiation-dependent or -independent manner. Phospho-H2AX detection by immunoprecipitation is difficult because the interaction between VHR and the MRN complex is likely transient and very rapid, and the interaction between MRN and pH2AX is likely to be similarly transient, thus diminishing the probability of γ-H2AX precipitation. However, we can now propose many functions for VHR in DNA damage and repair.
Finally, we performed confocal immunofluorescence experiments on HeLa cells irradiated with 5 Gy gamma radiation to probe for other key proteins directly involved in DNA damage response and repair signalling (ESI,† Fig. S7), including MRE11, RAD50, pCHK2 (Thr68), and p-p53 (Ser15). We found strong co-localisation of VHR with these proteins (ESI,† Table S1) that did not depend on ionising radiation treatment and thus reinforced the pull-down results from Fig. 7. These results combined with those from Fig. 6 suggest that VHR likely co-localises with important complexes at IRIF. Similar results were found for other proteins involved in classical DNA repair pathways triggered by ionising gamma radiation, such as HR, NHEJ and MMR, which were not identified by our VHR substrate-oriented bioinformatics approach because they do not meet the criteria used in Fig. 5A (ESI,† Fig. S8).
To further test our bioinformatics prediction platform based on substrate structure similarities and to gain insight into our identification of new VHR nuclear targets involved in DNA repair, we generated physical protein–protein interaction (PPPI) networks of the 57 identified protein hits (Fig. 5E). We mined PPPI databases using a seed list composed of these proteins and generated a very complex and huge network (1242 nodes and 19055 edges) of interactions that have been experimentally validated through at least one biochemical method (Fig. 8A). Although our input protein list (green circle nodes in Fig. 8A) contained samples of already known gene ontology according to the previously mentioned criteria (Fig. 5A), the rationale behind interactome construction and analysis was to identify clusters or complexes of functionally related proteins to confirm the results shown in this work and to direct future experimental investigation and validation. Clustering procedures for molecular complex detection (the adopted cut-off score was 2) were applied to the network from Fig. 8A, and we identified two clusters: one cluster contained H2AX (cluster 1, score 44.585; Fig. 8B) and the other contained NBS1 (cluster 9, score 2.041; Fig. 8C) proteins (the red rounded square nodes in Fig. 8A), which exhibit strong and close interactions with many predicted (and some validated) proteins that participate in DNA repair mechanisms. Clusters 1 and 9 (Fig. 8B and C) are represented according to a force-directed layout; the differences in node colour and size and edge colour and thickness represent a score based on the betweenness centrality scales of this topological parameter.
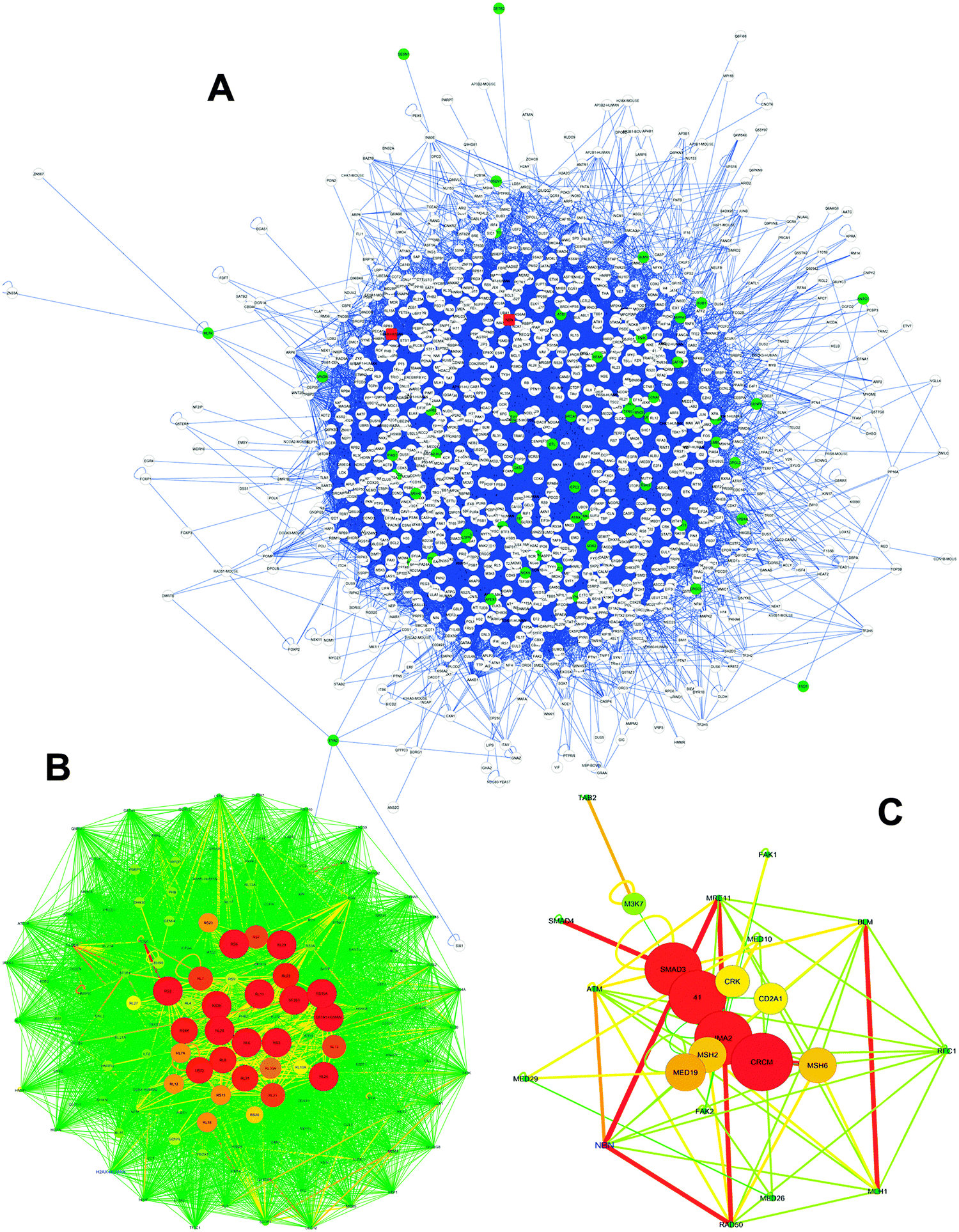 | ||
| Fig. 8 VHR PPPI network analysis by topological and statistical parameters. (A) The global VHR network built from our bioinformatics approach using 57 protein hits. This network was subjected to molecular complex detection procedures using MCODE software, and the 10 highest scoring clusters (density × number of nodes) were isolated, but only clusters 01 (B) and 09 (C) included experimental protein targets identified in this work. H2AX is marked in blue in (B), and NBN1 (NBS1) is marked in blue in (C). The three networks were analysed using the Network Analyzer software to identify general statistical and topological parameters, as shown in Table S4 (ESI†). Colour, thickness and size of nodes and edges in B and C networks are based on the betweenness centrality parameter. Networks are represented by the force-directed layout. | ||
Discussion
VHR (DUSP3) is the smallest member of the aDUSP subclass, and two non-MAPK substrates of this enzyme, STAT5 and the ERBB receptor, have been identified15,36 in addition to ERK2 and JNK.9,10 Additionally, very recent studies suggest that two classes of VHR substrates exist, such that class II substrates bind to VHR in an opposite orientation relative to the canonical binding mode of class I substrates, consisting of the classical MAPK substrates, and are good yet unidentified p-Tyr substrates of VHR in vitro.16 All these studies suggest that understanding the new structure–function properties and the physiologic substrates of typical or atypical DUSPs is necessary for DUSPs to be realised as cancer therapy targets.42,43VHR is over-expressed11,13 or under-expressed14,15 in some human tumour cells, and the consequences of these changes suggest that its altered expression results in strong phenotypic perturbations, including its preferential nuclear localisation13 and interactions with binding partners in the nucleolus.17 These results suggest that VHR may play a role in mediating DNA damage and/or repair and genomic stability processes. We first showed that VHR is constitutively and stably expressed through all phase transitions of the cell cycle, and its expression was not affected by gamma radiation-induced cellular and DNA damage. We observed this result in four different cell lines (HeLa, HaCaT, HEK293 and MeWo), suggesting that VHR regulates biochemical processes or pathways (such as the MAPK cascade) that need to be activated quickly by a gene that is not a MAPK-regulated early-response gene.44
Parallel to our hypothesis that VHR regulates genomic stability are reports that an effective nuclear regulation of MAPK activity would improve DNA repair machinery activities. For example, JNK and p38 MAPK have been shown to mediate cellular responses to ionising radiation in HeLa cells,45 and ERK1/2 positively regulates the ATM/γ-H2AX pathway to control homologous recombination (HR) repair in gamma radiation-induced DNA damage.46 Additionally, the positive regulation of JNK activity by UV light can induce H2AX phosphorylation and apoptosis.47 These previous reports are consistent with our results that VHR strongly co-localises with these three MAPK isoforms in the nucleus upon gamma radiation-induced damage. We also detected strong and DNA damage-dependent co-localisation of VHR with phospho-H2AX (Ser139) and phospho-ATF2 (Ser 490/498), suggesting that this aDUSP localises to the areas surrounding ionising-radiation induced foci (IRIF) where many other DNA repair systems are known to be recruited.34,35 Altogether, these data reinforced our preliminary hypothesis illustrated in Fig. 2E.
We cannot discount the possibility that VHR down-regulates MAPKs in the nucleus of cells with DNA damage; however, the principal aim of this manuscript was to suggest and identify new VHR protein substrates in radiation stress conditions. Considering the well-known structure of MAPKs and specifically their A-loop region as a template for putative VHR substrates, we screened human protein databanks using a substrate-driven strategy40,48 as described in Fig. 5. We identified proteins that fulfilled prerequisites established in a seven-step bioinformatics approach. We isolated 64 sequences from 57 protein hits for subsequent experimental validation. Among these putative VHR targets, eight (pATM-Ser1981, pATR-Ser428, pBRCA1-Ser1423, BRCA2, CENP-F, CYCLIN A, NBS1, and APE1) co-localised with VHR in control and gamma-radiation stress conditions. Additionally, four targets that were not identified through our bioinformatics approach (MRE11, RAD50, pCHK2-Thr68, and p53-Ser15) also strongly co-localised with VHR, consistent with the idea that protein complexes and pathways that participate in and/or regulate DNA damage repair may interact with VHR at different times. Our confocal microscopy results were consistent with immunoprecipitation experiments in which VHR directly pulled down NBS1 and indirectly pH2AX-Se139 proteins. These results strongly suggest that VHR is at least proximal to proteins surrounding and interacting with classical complexes involved with DNA repair.
For instance, the MRN (MRE11–RAD50–NBS1) complex, which is exclusively dedicated to DNA damage detection and repair initiation,34 physically interacts with VHR through NBS1 (Fig. 6G, 7A and ESI,† Table S2), and all three proteins in the complex co-localise with VHR, especially after gamma irradiation. Under genotoxic stress and after recruitment to DNA damage sites, MRN can trigger canonical DNA repair pathways, as in the following scenarios: (i) pATM-Ser1981/pATR-Ser428 → pCHK2-Thr68; (ii) pATM-Ser1981/pATR-Ser428 → p-p53-Ser15; (iii) pATM-Ser1981/pATR-Ser428 → pH2AX-Ser139; (iv) pATM-Ser1981 → pATF2-Ser490/498; and (v) pATM-Ser1981/pATR-Ser428 → pBRCA1-Ser1423; all these phosphorylated active proteins co-localise with VHR. Other putative VHR substrates identified in our bioinformatics analysis also co-localised with VHR (ESI,† Fig. S8), and each represented a different DNA repair pathway known to be activated by gamma radiation, such as (i) homologous recombination (HR), pSMC1-Ser360; (ii) non-homologous end joining (NHEJ), KU86; and (iii) mismatch repair (MMR), MSH2.
According to our data17,49,50 and reports by others,51–53 systems biology approaches based on the construction and analysis of protein–protein interaction networks (interactomes) have been useful prediction tools for the validation of bioinformatics and experimental datasets. By analysing our results from Fig. 8 for sub-networks composed of putative VHR-targeted protein complexes, we found proteins in cluster 1 (Fig. 8B) that we had previously identified using proteomics approaches, such as NPM, HNRPC, PHB, PABP1 and KU70.17 These VHR-interacting partners directly regulate biological processes related to DNA/RNA, such as repair (in agreement with our unpublished experimental data), senescence12 and apoptosis.11 Furthermore, many ribosomal proteins from PL and PS (RPL, RPS and MRPS) families belong to this cluster, which are ribonucleoprotein components of ribosomes with biological functions related to ribosome assembly, rDNA transcription, rRNA stability and processing, etc., and many of these contain a T–X–Y motif. Additionally, these ribonucleoproteins are key components of cluster 1 and are intimately related to the main nucleolar proteins NPM and NUCL, which were previously identified as VHR-interacting partners.17
In summary, the high expression and preferential nuclear localisation of VHR in cervical carcinomas and other human tumour cell lines, its involvement in MAPK down-regulation, and its interactions with other non-canonical or distinct nuclear protein partners suggest potentially novel substrates of this aDUSP and reveal unknown roles in the genotoxic stress response and genome stability maintenance.
Author contributions
FLF designed and performed all the experiments, analysed and interpreted the data and wrote the manuscript.Conflict of interest
The author declares that there is no conflict of interest.Acknowledgements
This research was supported by the Brazilian agency FAPESP through a Young Investigator fellowship, #2008/58264-5, to FLF. This work was also supported by the federal Brazilian agencies CNPq and CAPES, which supported our graduate programmes in Biochemistry and Molecular Biology. We thank Andressa P. Costa, Juliana R. Domingos, Benedita Oliveira and Viviane Q. Machtura for technical assistance. Additionally, we thank the AJE staff and editors for their editing services.References
- T. Hunter, Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling, Cell, 1995, 80, 225–236 CrossRef.
- A. Bononi, C. Agnoletto, E. De Marchi, S. Marchi, S. Patergnani, M. Bonora, C. Giorgi, S. Missiroli, F. Poletti, A. Rimessi and P. Pinton, Protein kinases and phosphatases in the control of cell fate, Enzyme Res., 2011, 2011, 329098, DOI:10.4061/2011/329098.
- C. Wurzenberger and D. W. Gerlich, Phosphatases: providing safe passage through mitotic exit, Nat. Rev. Mol. Cell Biol., 2011, 12, 469–482, DOI:10.1038/nrm3149.
- B. Akiyoshi and S. Biggins, Cdc14-dependent dephosphorylation of a kinetochore protein prior to anaphase in Saccharomyces cerevisiae, Genetics, 2010, 186, 1487–1491, DOI:10.1534/genetics.110.123653.
- A. Mocciaro and E. Schiebel, Cdc14: a highly conserved family of phosphatases with non-conserved functions, J. Cell Sci., 2010, 123, 2867–2876, DOI:10.1242/jcs.074815.
- Z. Wei, S. Peddibhotla, H. Lin, X. Fang, M. Li, J. M. Rosen and P. Zhang, Early-onset aging and defective DNA damage response in Cdc14b-deficient mice, Mol. Cell. Biol., 2011, 31, 1470–1477, DOI:10.1128/MCB.01330-10.
- S. M. Keyse, Dual-specificity MAP kinase phosphatases (MKPs) and cancer, Cancer Metastasis Rev., 2008, 27, 253–261, DOI:10.1007/s10555-008-9123-1.
- A. Farooq and M. M. Zhou, Structure and regulation of MAPK phosphatases, Cell Signalling, 2004, 16, 769–779, DOI:10.1016/j.cellsig.2003.12.008.
- J. L. Todd, K. G. Tanner and J. M. Denu, Extracellular regulated kinases (ERK) 1 and ERK2 are authentic substrates for the dual-specificity protein-tyrosine phosphatase VHR. A novel role in down-regulating the ERK pathway, J. Biol. Chem., 1999, 274, 13271–13280 CrossRef CAS PubMed.
- J. L. Todd, J. D. Rigas, L. A. Rafty and J. M. Denu, Dual-specificity protein tyrosine phosphatase VHR down-regulates c-Jun N-terminal kinase (JNK), Oncogene, 2002, 21, 2573–2583, DOI:10.1038/sj.onc.1205344.
- Y. J. Arnoldussen, P. I. Lorenzo, M. E. Pretorius, H. Waehre, B. Risberg, G. M. Maelandsmo, H. E. Danielsen and F. Saatcioglu, The mitogen-activated protein kinase phosphatase vaccinia H1-related protein inhibits apoptosis in prostate cancer cells and is overexpressed in prostate cancer, Cancer Res., 2008, 68, 9255–9264, DOI:10.1158/0008-5472.CAN-08-1224.
- S. Rahmouni, F. Cerignoli, A. Alonso, T. Tsutji, R. Henkens, C. Zhu, C. Louis-dit-Sully, M. Moutschen, W. Jiang and T. Mustelin, Loss of the VHR dual-specific phosphatase causes cell-cycle arrest and senescence, Nat. Cell Biol., 2006, 8, 524–531, DOI:10.1038/ncb1398.
- R. Henkens, P. Delvenne, M. Arafa, M. Moutschen, M. Zeddou, L. Tautz, J. Boniver, T. Mustelin and S. Rahmouni, Cervix carcinoma is associated with an up-regulation and nuclear localization of the dual-specificity protein phosphatase VHR, BMC Cancer, 2008, 8, 147, DOI:10.1186/1471-2407-8-147.
- L. Hao and W. M. ElShamy, BRCA1-IRIS activates cyclin D1 expression in breast cancer cells by downregulating the JNK phosphatase DUSP3/VHR, Int. J. Cancer, 2007, 121, 39–46, DOI:10.1002/ijc.22597.
- J. Y. Wang, C. L. Yeh, H. C. Chou, C. H. Yang, Y. N. Fu, Y. T. Chen, H. W. Cheng, C. Y. Huang, H. P. Liu, S. F. Huang and Y. R. Chen, Vaccinia H1-related phosphatase is a phosphatase of ErbB receptors and is down-regulated in non-small cell lung cancer, J. Biol. Chem., 2011, 286, 10177–10184, DOI:10.1074/jbc.M110.163295.
- R. Luechapanichkul, X. Chen, H. A. Taha, S. Vyas, X. Guan, M. A. Freitas, C. M. Hadad and D. Pei, Specificity profiling of dual specificity phosphatase vaccinia VH1-related (VHR) reveals two distinct substrate binding modes, J. Biol. Chem., 2013, 288, 6498–6510, DOI:10.1074/jbc.M112.449611.
- K. Panico and F. L. Forti, Proteomic, cellular, and network analyses reveal new DUSP3 interactions with nucleolar proteins in HeLa cells, J. Proteome Res., 2013, 12, 5851–5866, DOI:10.1021/pr400867j.
- F. L. Forti and H. A. Armelin, Vasopressin triggers senescence in K-ras transformed cell, via RhoA-dependent downregulation of cyclin D1, Endocr.-Relat. Cancer, 2007, 14, 1117–1125, DOI:10.1677/ERC-07-0154.
- A. S. Konagurthu, C. F. Reboul, J. W. Schmidberger, J. A. Irving, A. M. Lesk, P. J. Stuckey, J. C. Whisstock and A. M. Buckle, MUSTANG-MR structural sieving server: applications in protein structural analysis and crystallography, PLoS One, 2010, 5, e10048, DOI:10.1371/journal.pone.0010048.
- A. S. Konagurthu, J. C. Whisstock, P. J. Stuckey and A. M. Lesk, MUSTANG: a multiple structural alignment algorithm, Proteins, 2006, 64, 559–574, DOI:10.1002/prot.20921.
- R. Potestio, T. Aleksiev, F. Pontiggia, S. Cozzini and C. Micheletti, ALADYN: a web server for aligning proteins by matching their large-scale motion, Nucleic Acids Res., 2010, 38, W41–W45, DOI:10.1093/nar/gkq293.
- N. Blom, S. Gammeltoft and S. Brunak, Sequence and structure-based prediction of eukaryotic protein phosphorylation sites, J. Mol. Biol., 1999, 294, 1351–1362, DOI:10.1006/jmbi.1999.3310.
- N. Blom, T. Sicheritz-Ponten, R. Gupta, S. Gammeltoft and S. Brunak, Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence, Proteomics, 2004, 4, 1633–1649, DOI:10.1002/pmic.200300771.
- P. V. Hornbeck, J. M. Kornhauser, S. Tkachev, B. Zhang, E. Skrzypek, B. Murray, V. Latham and M. Sullivan, PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse, Nucleic Acids Res., 2012, 40, D261–D270, DOI:10.1093/nar/gkr1122.
- J. C. Obenauer, L. C. Cantley and M. B. Yaffe, Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs, Nucleic Acids Res., 2003, 31, 3635–3641 CrossRef CAS PubMed.
- J. D. Thompson, D. G. Higgins and T. J. Gibson, CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice, Nucleic Acids Res., 1994, 22, 4673–4680 CrossRef CAS PubMed.
- Y. Liu, B. Schmidt and D. L. Maskell, MSAProbs: multiple sequence alignment based on pair hidden Markov models and partition function posterior probabilities, Bioinformatics, 2010, 26, 1958–1964, DOI:10.1093/bioinformatics/btq338.
- M. Clamp, J. Cuff, S. M. Searle and G. J. Barton, The Jalview Java alignment editor, Bioinformatics, 2004, 20, 426–427, DOI:10.1093/bioinformatics/btg430.
- A. M. Waterhouse, J. B. Procter, D. M. Martin, M. Clamp and G. J. Barton, Jalview Version 2—a multiple sequence alignment editor and analysis workbench, Bioinformatics, 2009, 25, 1189–1191, DOI:10.1093/bioinformatics/btp033.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Res., 2003, 13, 2498–2504, DOI:10.1101/gr.1239303.
- J. Hernandez-Toro, C. Prieto and J. De las Rivas, APID2NET: unified interactome graphic analyzer, Bioinformatics, 2007, 23, 2495–2497, DOI:10.1093/bioinformatics/btm373.
- G. D. Bader and C. W. Hogue, An automated method for finding molecular complexes in large protein interaction networks, BMC Bioinf., 2003, 4, 2 CrossRef PubMed.
- Y. Assenov, F. Ramirez, S. E. Schelhorn, T. Lengauer and M. Albrecht, Computing topological parameters of biological networks, Bioinformatics, 2008, 24, 282–284, DOI:10.1093/bioinformatics/btm554.
- R. S. Williams, J. S. Williams and J. A. Tainer, Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template, Biochem. Cell Biol., 2007, 85, 509–520, DOI:10.1139/O07-069.
- A. Bhoumik, S. Takahashi, W. Breitweiser, Y. Shiloh, N. Jones and Z. Ronai, ATM-dependent phosphorylation of ATF2 is required for the DNA damage response, Mol. Cell, 2005, 18, 577–587, DOI:10.1016/j.molcel.2005.04.015.
- R. Hoyt, W. Zhu, F. Cerignoli, A. Alonso, T. Mustelin and M. David, Cutting edge: selective tyrosine dephosphorylation of interferon-activated nuclear STAT5 by the VHR phosphatase, J. Immunol., 2007, 179, 3402–3406 CrossRef CAS.
- J. H. Her, S. Lakhani, K. Zu, J. Vila, P. Dent, T. W. Sturgill and M. J. Weber, Dual phosphorylation and autophosphorylation in mitogen-activated protein (MAP) kinase activation, Biochem. J., 1993, 296(Pt 1), 25–31 CAS.
- Y. Miyata and E. Nishida, Distantly related cousins of MAP kinase: biochemical properties and possible physiological functions, Biochem. Biophys. Res. Commun., 1999, 266, 291–295, DOI:10.1006/bbrc.1999.1705.
- C. Tarrega and R. Pulido, A one-step method to identify MAP kinase residues involved in inactivation by tyrosine- and dual-specificity protein phosphatases, Anal. Biochem., 2009, 394, 81–86, DOI:10.1016/j.ab.2009.07.006.
- J. E. Fuchs, S. von Grafenstein, R. G. Huber, C. Kramer and K. R. Liedl, Substrate-driven mapping of the degradome by comparison of sequence logos, PLoS Comput. Biol., 2013, 9, e1003353, DOI:10.1371/journal.pcbi.1003353.
- F. L. Forti, M. H. M. Nascimento and T. E. P. Torres, Investigating roles of dual tyrosine phosphatases in DNA damage responses, Int. J. Low Radiat., 2010, 4, 8, DOI:10.1504/IJLR.2010.034914.
- O. Bermudez, G. Pages and C. Gimond, The dual-specificity MAP kinase phosphatases: critical roles in development and cancer, Am. J. Physiol.: Cell Physiol., 2010, 299, C189–C202, DOI:10.1152/ajpcell.00347.2009.
- P. Rios, C. E. Nunes-Xavier, L. Tabernero, M. Kohn and R. Pulido, Dual-specificity phosphatases as molecular targets for inhibition in human disease, Antioxid. Redox Signaling, 2014, 20, 2251–2273, DOI:10.1089/ars.2013.5709.
- J. M. Brondello, A. Brunet, J. Pouyssegur and F. R. McKenzie, The dual specificity mitogen-activated protein kinase phosphatase-1 and -2 are induced by the p42/p44MAPK cascade, J. Biol. Chem., 1997, 272, 1368–1376 CrossRef CAS PubMed.
- M. J. Kim, S. Y. Choi, I. C. Park, S. G. Hwang, C. Kim, Y. H. Choi, H. Kim, K. H. Lee and S. J. Lee, Opposing roles of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase in the cellular response to ionizing radiation in human cervical cancer cells, Mol. Cancer Res., 2008, 6, 1718–1731, DOI:10.1158/1541-7786.MCR-08-0032.
- S. E. Golding, E. Rosenberg, S. Neill, P. Dent, L. F. Povirk and K. Valerie, Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response, Cancer Res., 2007, 67, 1046–1053, DOI:10.1158/0008-5472.CAN-06-2371.
- C. Lu, F. Zhu, Y. Y. Cho, F. Tang, T. Zykova, W. Y. Ma, A. M. Bode and Z. Dong, Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3, Mol. Cell, 2006, 23, 121–132, DOI:10.1016/j.molcel.2006.05.023.
- G. Tasnadi, C. K. Winkler, D. Clay, N. Sultana, W. M. Fabian, M. Hall, K. Ditrich and K. Faber, A substrate-driven approach to determine reactivities of alpha,beta-unsaturated carboxylic esters towards asymmetric bioreduction, Chemistry, 2012, 18, 10362–10367, DOI:10.1002/chem.201200990.
- L. A. Pescatore, D. Bonatto, F. L. Forti, A. Sadok, H. Kovacic and F. R. Laurindo, Protein disulfide isomerase is required for platelet-derived growth factor-induced vascular smooth muscle cell migration, Nox1 NADPH oxidase expression, and RhoGTPase activation, J. Biol. Chem., 2012, 287, 29290–29300, DOI:10.1074/jbc.M112.394551.
- N. M. Garavello, D. A. Pena, J. M. Bonatto, M. L. Duarte, H. M. Costa-Junior, R. I. Schumacher, F. L. Forti and D. Schechtman, Activation of protein kinase C delta by psideltaRACK peptide promotes embryonic stem cell proliferation through ERK 1/2, J. Proteomics, 2013, 94, 497–512, DOI:10.1016/j.jprot.2013.10.021.
- P. Braun and A. C. Gingras, History of protein-protein interactions: from egg-white to complex networks, Proteomics, 2012, 12, 1478–1498, DOI:10.1002/pmic.201100563.
- A. C. Gingras and A. Nesvizhskii, Protein complexes and interaction networks, Proteomics, 2012, 12, 1475–1477, DOI:10.1002/pmic.201270054.
- K. Raman, Construction and analysis of protein-protein interaction networks, Automated Experimentation, 2010, 2, 2, DOI:10.1186/1759-4499-2-2.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ib00186a |
| This journal is © The Royal Society of Chemistry 2015 |