
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionic liquids assisted processing of renewable resources for the fabrication of biodegradable composite materials
Hamayoun
Mahmood
 a,
Muhammad
Moniruzzaman
*ab,
Suzana
Yusup
a and
Tom
Welton
c
a,
Muhammad
Moniruzzaman
*ab,
Suzana
Yusup
a and
Tom
Welton
c
aDepartment of Chemical Engineering, Universiti Teknologi PETRONAS, 32610 Bandar Seri Iskandar, Perak Darul Ridzuan, Malaysia. E-mail: m.moniruzzaman@utp.edu.my
bCenter of Research in Ionic Liquids, Universiti Teknologi PETRONAS, 32610 Bandar Seri Iskandar, Perak Darul Ridzuan, Malaysia
cDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK
First published on 13th April 2017
Abstract
In recent years, the utilization of renewable resources, particularly lignocellulosic biomass based raw materials, to replace synthetic materials/polymers for the manufacture of green materials has gained increased worldwide interest due to growing global environmental awareness, concepts of sustainability and the absence of conflict between food and chemical/materials production. However, structural heterogeneity and the presence of networks of inter- and intra-molecular interactions in biopolymer matrices remain unsolved challenges to clean pretreatment for biocomposite processing. A number of techniques including physical, physico-chemical and chemical methods have been investigated for the pretreatment of renewable resources. Most of these methods require high temperatures and pressures, as well as highly concentrated chemicals for the pretreatment process. Fortunately, ionic liquids (ILs) – potentially attractive “green” recyclable alternatives to environmentally harmful organic solvents – have been increasingly exploited as solvents and/or (co)solvents and/or reagents for biopolymer processing. Compared to conventional approaches, ILs in processing biodegradable composites exhibit many advantages such as being noncorrosive and nonvolatile, having excellent dissolution power under relatively mild conditions and high thermal stability. Presently, a wide range of different approaches have been explored to further improve the performance of ILs processing of biobased polymers for composites manufacturing. The main goal of this review is to present recent technological developments in which the advantages of ILs as processing solvents for biopolymers for the production of a plethora of green composites have been gradually realized. It is hoped that the present article will inspire new ideas and new approaches in ILs-assisted processing of renewable resources for green composite production.
 Hamayoun Mahmood | Hamayoun Mahmood received his B.Sc. (2007) and M.Sc. (2011) in Chemical Engineering from the University of Engineering & Technology (UET), Lahore, Pakistan. He served as a faculty member in the Department of Chemical Engineering, University of Gujrat, Pakistan and King Khalid University, Saudi Arabia. He then moved to Malaysia where he got his Ph.D. (2017) under supervision of Dr. Muhammad Moniruzzaman from Universiti Teknologi PETRONAS. During his Ph.D work, he explored the use of ionic liquids for pretreatment of lignocellulosic biomass to fabricate green composite materials. He is currently interested in the optimization, comparison and environmental impact assessment for use of ionic liquids in the biorefinery applications. |
 Muhammad Moniruzzaman | Muhammad Moniruzzaman received his B.Sc. in Chemical Engineering from the Bangladesh University of Engineering and Technology (BUET), Bangladesh. He then moved to Japan where he got his M.Sc. (2004) and Ph.D (2007) in biochemical engineering from Kanazawa University. In 2007, he moved to Kyushu University as a JSPS (Japan Society for the Promotion of Science) Post-doctoral fellow. Currently, M. Moniruzzaman serves as an Associate Professor in the Department of Chemical Engineering at the Universiti Teknologi PETRONAS, Malaysia. His current research interests focus on the application of ionic liquids as alternative “green” solvents for the design of bioconversion processes and novel drug delivery systems. |
 Suzana Yusup | Suzana Yusup currently serves as an Associate Professor in Department of Chemical Engineering at Universiti Teknologi PETRONAS, Malaysia. She received her B.Sc. in Chemical Engineering from the University of Leeds, United Kingdom (1992), an M.Sc. in Chemical Engineering (Adsorption) from the University of Wales, United Kingdom (1995), and PhD in Chemical Engineering (Powder Technology) from the University of Bradford, United Kingdom (1998). She has published numerous research articles and book chapters and has lead several research grants at both national and international level. Dr Suzana Yusup is currently affiliated with the Board of Engineers Malaysia. |
 Tom Welton | Tom Welton is the Dean of the Faculty of Natural Sciences and Professor of Sustainable Chemistry at Imperial College London. He has been working with ionic liquids for over 30 years and is interested in all aspects of the science of ionic liquids, form their fundamental physical properties to their applications in biomass conversions. He received his BSc and DPhil from the University of Sussex, UK. |
1. Introduction
Ionic liquids (ILs), liquids that are entirely composed of ions that can be liquids at ambient or below ambient temperatures, have been widely utilized as promising alternatives to hazardous, toxic, volatile and highly flammable organic solvents.1,2 In fact, various attractive and unique physicochemical properties of ILs such as extremely low vapor pressures,3,4 high solvation interactions with inorganic and organic compounds,5,6 excellent thermal and chemical stabilities,7 good ionic conductivities and broad electrochemical windows make ILs attractive candidates for the replacement of volatile organic compounds (VOCs) for polysaccharides processing.8 Furthermore, the physicochemical properties, such as densities, viscosities, hydrophobicities and solute solubilities of ILs can be tailored by appropriate selection of different combinations of anions and cations as well as attached substituent groups to manipulate these for particular demands. Hence, ILs have been called “designer solvents”.9 The combination of all these unique properties opens new avenues to an extensive range of applications, including, organic synthesis and catalysis,10–12 extraction,13,14 inorganic synthesis,15 nanomaterial synthesis,16 separation,17 biocatalysis18,19 pharmaceuticals20,21 and polysaccharide dissolution.22,23 Particularly, the utilization of environmentally benign ILs as dissolution media for lignocellulose and various biopolymers has received tremendous attention in the last few years.23–26The use of petroleum based non-biodegradable plastics and composites and their existing processing techniques have become recognized as a threat to the environment and consume much limited fossil resources.27 The UN conference on climate change in Copenhagen December 2009 evoked a fierce public debate about the future of the earth and the need for a transition towards a CO2 neutral biobased economy (bioeconomy) was emphasized.28 This situation has led people from both academia and industry to focus on the development of new eco-friendly materials based on biobased renewable resources. In contrast to the field of renewable energy, in which biobased energy resources are in competition with wind and solar energies, the only renewable alternative to materials based on carbon petrochemistry is biobased raw materials. To this end, polymeric carbohydrates, e.g., cellulose, starch, chitin, inulin, chitosan, lignin etc. are found abundantly in nature as structural building elements.29 Agricultural industries generate a substantial amount of lignocellulosic agricultural waste every year, which is mainly composed of cellulose, hemicellulose and lignin. The typical percentages of dry weight of these wastes are 35–50% cellulose, 20–35% hemicellulose and 5–30% lignin.30 Thus, the utilization of non-food lignocellulosic waste from various agro-industries for the production of high value composite materials could be an attractive approach instead of creating pollution problems.31
Sustainability in composites science depends upon not only the assortment of renewable, or environmentally benign raw materials for their manufacture, but also on the development of mild pretreatment methods that avoid the use and production of hazardous substances to minimize their environmental impact.32 The critical challenge to extend the novel applications of biopolymers and lignocellulose for manufacturing biocomposite materials is to overcome the strong inter- and intra-molecular hydrogen bonding which lead to its recalcitrant nature.26 These attributes assure the excellent chemical and physical stability of polysaccharides but also prevent their widespread utilization.33,34 Consequently, considerable efforts have been devoted to enhance the processability of these carbohydrate polymers and lignocellulosic materials. For example, the ‘Viscose process’ is a 100 year old technique for cellulose processing which involves derivatizing the cellulose with carbon disulphide to cellulose xanthate followed by dissolution in sodium hydroxide.35 Alternatively, the ‘Lyocell process’ employs the solvent N-methylmorpholine N-oxide (NMMO) for direct dissolution of cellulose in an industrial fiber-making process.36 Nevertheless, each of these technologies carries a significant drawback; considerable byproduct formation in cellulose/NMMO/water systems can cause detrimental effects such as degradation of cellulose, temporary or permanent discoloration of resulting fibers, pronounced decomposition of NMMO,37 and the Viscose process generates two kilograms of waste per kilogram of cellulose obtained.38 In addition, various pretreatment technologies have been developed for lignocelluloses which apply hydrothermal or chemical treatments after mechanical comminution. Hydrothermal techniques include steam explosion,39 carbon dioxide explosion,40 and hot water treatment,41 whereas chemical processes can be dilute-acid treatment,42 alkali treatment, the organosolv process using organic solvent,43 ammonia fiber explosion and ozonolysis.44 However, most of the above mentioned pretreatment methods feature several drawbacks. They have to be tailored to the specific biomaterial source and can cause significant decomposition of biopolymer components to side-products, which can severely inhibit further processing of the biocomposite.45 Additionally, some pretreatment methods require extreme conditions of temperatures and pressures or strong acids or bases for which special equipment are necessary. The perceived adverse effects of such processes combined with their safety and environmental issues, has resulted in increased pressure to minimize their use, particularly in the light of recent regulations that have aggressively targeted the reduction of the emission of industrial pollutants.46 Thus sustainability, green chemistry, and eco-efficiency are not just newly coined buzzwords, but are forming the principles that are directing the development of the next generation of industrial chemical operations. Subsequently, novel processing technologies for widespread potential applications of biobased polymeric carbohydrates and lignocellulosic agricultural waste are being extensively investigated.47,48
The technological utilization of natural polymers and lignocellulosic materials for biocomposite manufacturing can potentially be enhanced significantly by their dissolution in ILs rather than in traditional organic solvents because of ILs’ unusual solvent properties.26,49 Studies on the dissolution of different polysaccharides in ILs over the last 10–15 years have shown that by using these solvents efficient selective extraction of the components is possible.50 In addition, many biopolymers and lignocellulosic fibers after regeneration from their IL solutions exhibited high thermal and operational stability.51,52 Therefore, ILs are receiving increased interest as new and highly effective dissolution media for a wide range of biodegradable polymers and lignocellulosic materials. The main objective of this review is to present a brief overview of the current state-of-the-art on the role of ILs as dissolution media for various polysaccharides for engineered green materials applications.
2. Ionic liquids and their selective properties for dissolution of biopolymers
Typical ILs are comprised of an organic cation (most commonly an alkyl-substituted imidazolium, quaternary ammonium or pyridinium ion) with an inorganic or organic anion. While it is not a rigorous definition the term, ionic liquid is commonly used to infer a melting point below 100 °C. Many ILs are liquid at, or below room temperature and remain in the liquid state within a wide temperature window (typically <400 °C). The chemical, physical and biological characteristics of ILs generally depend on the cationic structure (e.g., class of cation, the length and the symmetry of substituent groups) as well as on the structure and extent of charge delocalization of the anion.53 The specific physicochemical properties of ILs can easily be tailored through suitable structural changes in the cation and anion. Thereby, ILs are a class of solvents that incorporate a wide range of properties and one should avoid over-generalized statements. Some comprehensive databases of physical properties of ILs such as viscosity, melting point, density etc. have been compiled.54–56Ionic liquids are generally considered to be capable of having a broad range of intermolecular interactions such as hydrogen bonding, dipolar, dispersive and ionic.57,58 Therefore, various compounds are remarkably soluble in ILs. Moreover, many compounds which are insoluble or only partially soluble in other organic solvents can be efficiently dissolved in ILs possessing coordinating anions such as [OAc]−, Cl−, [NO3]− that are strong hydrogen bond acceptors.59,60
Based on their solvation capabilities, ILs are classified generally as highly polar solvents. Various methods have been employed to understand the polarity of ILs such as partition,5 fluorescence probe methods61 and solvatochromic dyes.62 In addition, a number of empirical and semi-empirical polarity scales such as COSMO-RS,63 the Hansen solubility parameters,64 and the Kamlet–Taft polarity method5 have been applied to predict and correlate the solubility of biopolymers and biomolecules in ILs with their polarities. In the Kamlet–Taft system, the parameter describing the hydrogen-bond basicity is expressed by β, whereas α represents the hydrogen-bond acidity and π* is the measure of interactions through polarisability and dipolarity effects. In ILs having non-functionalised cations like dialkylimidazolium, the β value is primarily affected by the anion.65 For cellulose dissolution in ILs, it was found that dissolution capabilities of ILs are characterized by their high values of hydrogen-bond basicity parameters.24
Ionic liquids are usually miscible with polar solvents such as ketones, lower alcohols and dichloromethane, but immiscible with non-polar organic solvents including ethers and alkanes. Furthermore, on the basis of solubility of ionic liquids in water they can be classified into hydrophilic (water miscible) and hydrophobic (forming a biphasic system with water). It was found that water miscibility of an IL also generally depends on its anion.66
ILs have higher viscosities as compared to those of most common molecular solvents. In general, the viscosities of ILs depend on their interionic interactions, such as Coulomb forces, hydrogen bonding and van der Waals interactions. Therefore, viscosities of IL vary considerably with composition, temperature and chemical structure. The higher viscosities of ILs significantly hinder the dissolution of lignocellulose and other biopolymers in these. In order to reduce viscosities, organic co-solvents including dimethylsulfoxide, dimethylformamide and 1,3-dimethyl-2-imidazolidinone etc. have also been successfully used.67Fig. 1 provides structures of some ILs that have been used for processing of various biopolymers and lignocellulosic materials for biocomposite manufacturing.
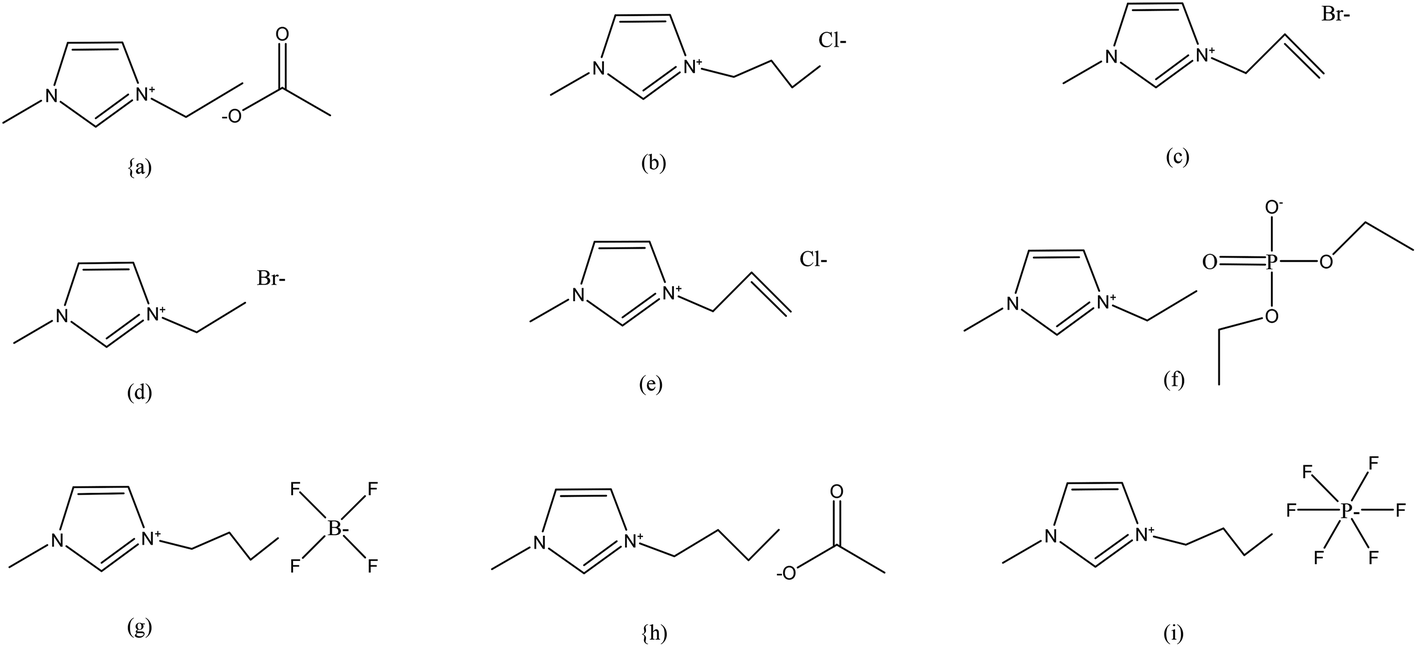 | ||
Fig. 1 Structures of the ILs used for dissolution of biopolymers and lignocellulosic materials for fabrication of biocomposites: (a) [C2C1im][OAc], (b) [C4C1im]Cl, (c) [(C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2)C1im]Br, (d) [C2C1im]Br, (e) [(C1C2)C1im]Cl, (f) [C2C1im][Et2PO4], (g) [C4C1im][BF4], (h) [C4C1im][OAc], (i) [C4C1im][PF6].24,93 C2)C1im]Br, (d) [C2C1im]Br, (e) [(C1C2)C1im]Cl, (f) [C2C1im][Et2PO4], (g) [C4C1im][BF4], (h) [C4C1im][OAc], (i) [C4C1im][PF6].24,93 | ||
2.1. Dissolution of biopolymers in ionic liquids: a comparison study with conventional solvents
In addition to the green aspects and the unique physicochemical properties of ILs, some important advantages of ILs that make these promising as potential solvents for cellulose and a wide range of other biopolymers are as follows.As ILs are tunable solvents, they can be designed by appropriate combination of cations and anions for a particular biopolymer, which is not possible using conventional organic solvents. In fact, since ILs can undergo a wider spectrum of intermolecular interactions, they are capable of dissolving a vast range of biopolymer compounds that are insoluble in organic solvents.68 For example, ILs can be used for dissolution of starch,69 chitin,70 chitosan,71 polylactic acid,72 lignin,73 cellulose,22 or for even complete dissolution of lignocellulosic materials.24,74 ILs can also be employed for selective dissolution and extraction of any desired or undesired component from a mixture or matrix solution.75
In comparison to molecular solvents, the remarkable benefit of using ILs for dissolution of lignocellulose and various biopolymers is that they can dissolve these under relatively mild operating conditions and at normal atmospheric pressure. Fukaya et al.76 reported the dissolution of cellulose at the temperature of 45 °C for 30 min by utilizing alkylimidazolium based ILs with a methyl phosphonate anion.
As well as acting as dissolution medium, recently many ILs, especially with methylimidazolium cations, were found to outperform other conventional plasticizers by significantly disrupting the inter- and intra-molecular hydrogen bonding within polysaccharides, such as starch, and thus can be employed as excellent media for polysaccharide modification and plasticization.77,78 Sankri et al.79 and Leroy et al.80 have performed pioneering work using the IL 1-butyl-3-methylimidazolium chloride ([C4C1im]Cl) as a novel plasticizer in melt processing of starch-based polymers with remarkable improvements in plasticization, hydrophobicity and electrical conductivity being reported.
Pretreatment is an essential unit operation in a polysaccharides based biorefinery and for sustainable material applications, but is considered to be of one the most cost intensive operations. The production of valuable products from lignocellulose or starch has a narrow profit margin. Various acidic, alkaline, hydrothermal and ammonia fiber expansion (AFEX) pretreatment technologies have been examined under high biomass loadings (>15%).81,82 Lately, ILs have been shown to be highly efficient dissolution media for lignocellulosic materials under solid biomass loads of as high as 50%.83 Recently, we have explored high solids loading, because of lower overall cost, improved efficiency and environmental benefits.51 Thus, the possibility of high-throughput pretreatment in a continuous process could improve the potential for the use of ILs as a cost-effective and extremely promising technology for manufacturing sustainable composites from various polysaccharide raw materials. The most important advantages of ILs as dissolution media for the fabrication of polysaccharides based biocomposites is the possibility of ILs recycling and product recovery schemes which are not viable with conventional organic solvent systems.74,84
It should be noted that biopolymer dissolution is often controlled by kinetics rather than by thermodynamics. Sescousse et al.85 performed a comparative study for the dissolution and regeneration of cellulose in the ILs 1-butyl-3-methylimidazolium acetate [C2C1im][OAc] and [C4C1im]Cl with conventional cellulose solvent systems, i.e., NaOH and N-methyl-morpholine N-oxide (NMMO). Kinetic study of the cellulose regeneration process from cellulose–IL solution revealed that this was a diffusion-controlled process. Gavillon and Budtova86 also predicted that the cellulose regeneration steps from cellulose–NaOH–water and cellulose–NMMO solutions were diffusion-controlled. For complete dissolution of a biopolymer, the solvent must diffuse into the structure of polymeric chain to affect both the amorphous and the crystalline regions. Therefore, high diffusivity and the capability of chain disentanglement are necessary attributes for an effective solvent.87 At a given cellulose concentration, the diffusion coefficient for 1-ethyl-3-methylimidazolium acetate [C2C1im][OAc] and [C4C1im]Cl were four to five times lower than the diffusion coefficient of NaOH and about twice lower than that of NMMO, which might be due to the larger size of the ILs.86–88 Moreover, compared with the NMMO process, the direct dissolution of cellulose in IL is more easily controlled and the process is inherently safer. It has been described that cellulose fibers prepared using ILs displayed similar properties in terms of elasticity and tenacity compared to the fibers manufactured by the Viscose and Lyocell processes. In addition, the process can be designed such that both fibrillating and non-fibrillating fibers can be fabricated specially for textile applications.89,126,127
Ionic liquids exhibit a far lower degradation potential compared to the NMMO. The study of Wendler et al. showed that a [C2C1im][OAc]–cellulose solution exhibited a Tonset (temperature corresponding to the point of intersection of tangents to two branches of the thermogravimetric curve) of 180 °C compared to the value of 146 °C for a NMMO–cellulose system. Apart from Tonset, numerical estimation of pressurization rates from pressure curves of cellulose–solvent mixture showed that compared to NMMO the pressure rise during the exothermic event for cellulose–IL solution was much less, which evidently suggested the ILs as solvents with much higher thermal stability.90
The dissolution of cellulose in acidic solvents is accompanied by a substantial hydrolysis of cellulose and causes depolymerization, which consequently limits its use in some applications.91 On the contrary, for cellulose dissolution in 1-allyl-3-methylimidazolium chloride [(C1C2)C1im]Cl at 60 °C, a well resolved 13C NMR spectrum of all the six anhydroglucose units was observed, indicating that the cellulose dissolved in a similar manner in [(C1C2)C1im]Cl as in other familiar solvents for cellulose including sodium hydroxide–carbon disulfide solution.92 Allyl- and n-alkyl-imidazolium based ILs with Cl−, Br− and [OAc]− anions showed 5–14.5% cellulose solubility over a temperature range of 80–110 °C.93 In comparison, an aqueous mixture of NaOH/urea/thiourea that could readily dissolve cellulose exhibited a maximum cellulose solubility of only 7.2%.94 Moreover, thermal gelation of cellulose in aqueous NaOH/urea/thiourea solution due to self-aggregation of the cellulose chains has also been reported.95
Although a great deal of experimental and computational advances have been made, there is no explicit understanding about the specific mechanism of biopolymer dissolution in ILs compared to other solvents. Currently, two main models for the ILs’ interactions with cellulose prevail: (1) the dissolution process is controlled by the interaction of the anion with the biopolymer with no particular role for the cation; and (2) the principal driving force for cellulose dissolution originates from the H-bond interactions of the cellulose hydroxyl group with both the cation and the anion of the IL.96 Conversely in the case of the NMMO solvent, the proposed mechanism presumes the cleavage of intermolecular bonds of cellulose via the creation of a soluble complex of stronger hydrogen bonds between the NO group of NMMO and the cellulosic hydroxyl groups.97 For mixed inorganic/organic solvents such as lithium chloride/dimethylacetamide (LiCl/DMAc), the dissolution mechanism is suggested to go through an intermediate involving the interaction of Cl− (due to its basicity) with cellulose.98 This type of interaction can be considered as a polyelectrolyte effect, in which polymer molecules are forced apart because of charge repulsion. A similar polyelectrolyte effect has been suggested for the tetrabutylammonium fluoride/dimethyl sulfoxide (TBAF/DMSO) solvent system.99
2.2. Ionic liquids as an effective tool for biopolymer processing
Certain ILs can dissolve lignocellulose by disrupting the hydrogen bonding network of the natural composite of cellulose, hemicellulose and lignin, as studied by 13C-NMR and 35/37Cl-NMR spectrometric results.100 Recently, cellobiose (the repeating unit of cellulose) solvation was investigated in [C2C1im][OAc] by in situ and variable-temperature NMR spectroscopy.101 The study suggested that the principal reason for cellobiose solvation was hydrogen bonds formed between the hydroxyl groups of cellobiose and both the anion and cation of [C2C1im][OAc]. The excellent dissolution capabilities of this IL for lignocellulosic materials under relatively mild conditions, its ease of synthesis and its potential for recycling have led to many researchers selecting this solvent to be a promising green processing medium for polysaccharide research in a future biorefinery. However, there are concerns regarding the reactivity of this and other closely related ionic liquids with cellulose (see below).3. Composites based on ionic liquid assisted processing of biopolymers
Composites are engineered materials fabricated from two types of constituents viz. reinforcement and matrix, which maintain their distinct characteristics within the finished structure.102 However, in the case of polysaccharide based composites, the final product properties could be achieved without keeping the strict classification for reinforcement and matrix.103 For conversion of various biopolymers and lignocellulosic materials into biodegradable composite products, biomaterial is typically dissolved in the IL in the temperature range of 65–110 °C with dissolution times of 10 min to 22 h.24,93 The resulting IL–polymer solution can be processed via different fabrication techniques such as electrospinning, casting, molding, gelation etc. to form numerous types of biocomposites. Conversely, the dissolved material in IL can be regenerated by addition of certain anti-solvents such as water, acetone, ethyl alcohol etc. The regenerated cellulosic material may be utilized in a broad range of applications including textiles, cosmetics, biomedical, fiberboard composites, fiber-reinforced polymeric composites, all-cellulose composites, wood-plastic composites etc. Fig. 2 provides a schematic view of manufacturing methodologies for numerous biocomposite products using ILs as the solvent. Here we describe ILs based manufacturing of various composites prepared solely from polysaccharides or their sources.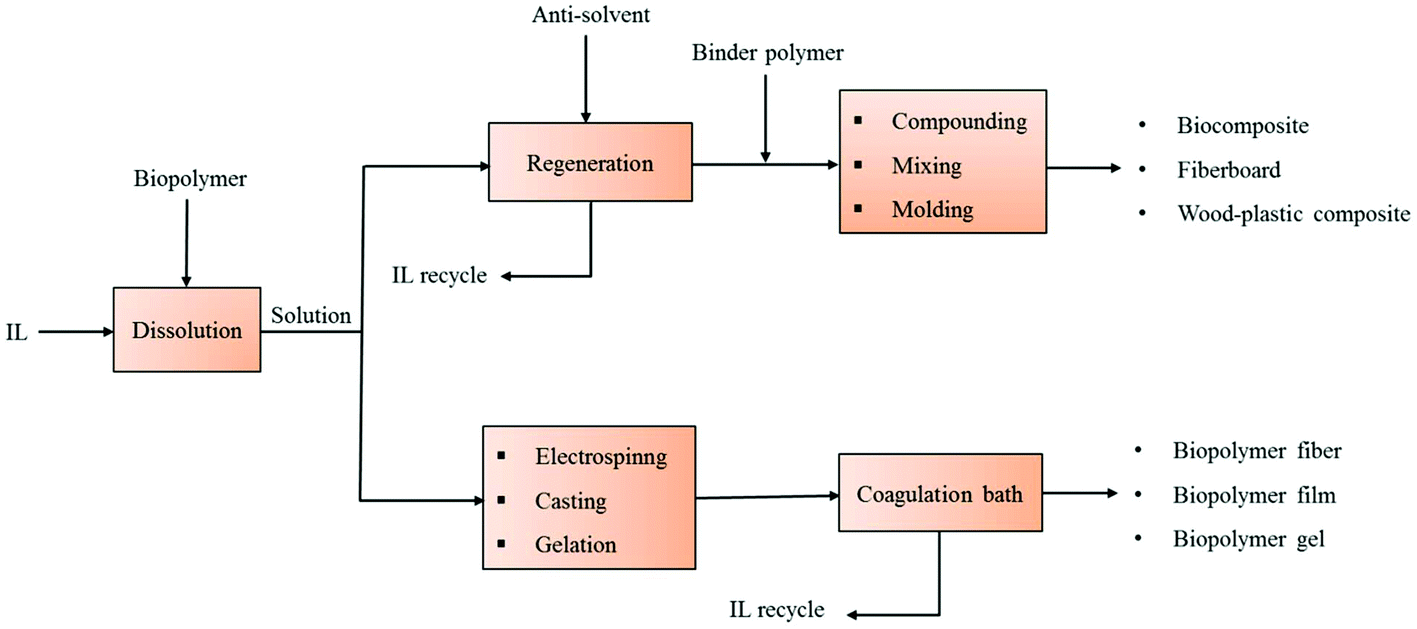 | ||
| Fig. 2 Schematic representation of the manufacture of various biodegradable composites based on ILs dissolution of biopolymers and lignocellulose. | ||
3.1. Biopolymer films
Currently, researchers from both academia and industry are increasingly focusing their interests onto the exploitation of natural biopolymers as potential alternatives to non-biodegradable plastic products. As the most important skeletal component in plants, the polysaccharide cellulose is an almost inexhaustible polymeric raw material with fascinating structure and properties.104 However, processing of cellulose is extremely difficult because, in general, it does not melt nor dissolve in common solvents. Recently, the dissolution capabilities of ILs for cellulose provoked a remarkable interest in cellulose processing for biobased polymer films.24,105The IL [(C1C2)C1im]Cl was used as a single component solvent for cellulose to fabricate the regenerated cellulose material with good mechanical properties by a solution casting method.106 The cellulose samples (cotton linters) were cut into smaller pieces and dried at 70 °C for 3 h in a vacuum oven. A specific mass of cellulose was dispersed into 20 mL of ([(C1C2)C1im]Cl) to get 4% polymer concentration, and the mixture was continuously heated and stirred until the material was completely dissolved. Finally, the solution was cast onto a glass plate to achieve a thickness of about 0.50 mm, air bubbles were removed in a vacuum oven and then a transparent regenerated cellulose film was formed in a water bath. The thickness of the film was controlled to within 0.5 mm to avoid curls in the cellulose films. The regenerated cellulose film was washed with plenty of distilled water and dried at 60 °C in a vacuum oven. Thus, [(C1C2)C1im]Cl based processing of cellulose was proposed to be a promising “green process” for the fabrication of regenerated cellulose films, which can solve the inherent environmental problems of the generation of waste toxic gases formed during the current industrial processes. Dissolution and regeneration of cellulosic biofilm from [C4C1im]Cl was also reported by Liu et al.,107 where cotton pulp was used as the raw cellulose source. The authors observed that [C4C1im]Cl was a direct solvent for cellulose and its solubility could reach up to 13 wt% at 90 °C in 7 h, but at the same time the degree of polymerization was remarkably reduced. Takegawa and coworkers108 prepared the bi-component biopolymer film containing cellulose and chitin each dissolved separately in [(C1C2)C1im]Br and [C4C1im]Cl respectively at 100 °C for 24 h with continuous stirring to give clear solutions of chitin and cellulose. The stress–strain curves were also measured under tensile mode and it was concluded that the biofilms became more elastic with decreasing the ratio of chitin to cellulose in the final product. Meng et al.109 successfully dissolved and regenerated the native skin collagen in [C4C1im]Cl (Fig. 3). However, the triple helical structure of the collagen had been partly destroyed during the dissolution and regeneration process. A possible mechanism of dissolution and regeneration of collagen in IL was also proposed and it was deduced that the IL dissolved the collagen fibers by mainly breaking the hydrogen bonds and the ionic bonds in collagen macromolecules. Regenerated wool keratin films were prepared from wool keratin/ionic liquid solutions through the addition of water, methanol or ethanol as coagulation solvents.110 It was suggested that [(C1C2)C1im]Cl had higher solubility for wool keratin fibers than that of [C4C1im]Cl. XRD data also confirmed that the regenerated films exhibited a β-sheet structure and the disappearance of the α-helix structure. Further, Byrne et al.111 reported the fabrication of regenerated film of three natural polymers, raw cotton, silk and wool using [(C1C2)C1im]Cl at 105 °C. The new biocomposite films showed enhanced thermal stability compared to single component films, which was attributed to the increase in intra molecular hydrogen bonds for the biofilms. Similarly, Rahatekar et al.112 successfully combined the biocompatible properties of chitin with the high electrical conductivity of carbon nanotubes (CNTs) by mixing these using an imidazolium-based ionic liquid as a common solvent/dispersion medium. The IL allowed the uniform dispersion of CNTs and dissolution of chitin to create a biocompatible, electrically conducting scaffold permissive for mesenchymal stem cell function. Recently, Byrne and co-workers113 presented an exciting example of using [(C1C2)C1im]Cl for the preparation of a novel regenerated cotton/duck feather composite film. The new blended films showed enhanced elastic properties as well as thermal stability in comparison to the single component films regenerated from the same IL. The amount of α-helix in the composite film was responsible for improvement in the elastic properties of the fabricated films.
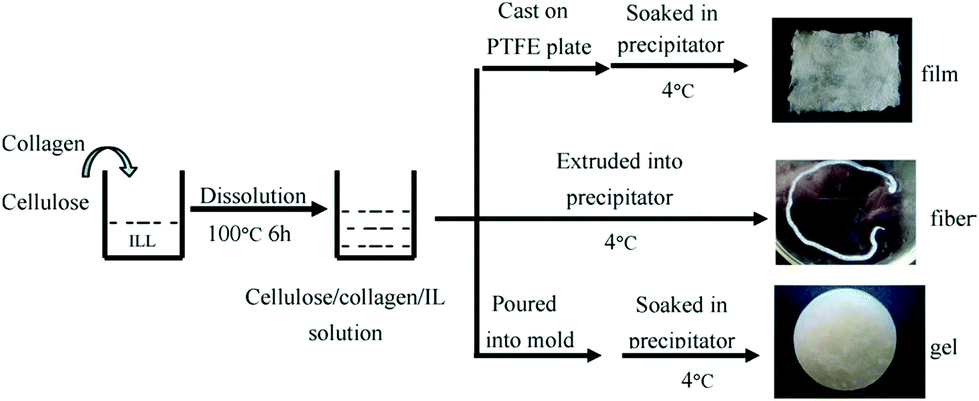 | ||
| Fig. 3 Schematic representation of collagen/cellulose composite materials preparation using IL [C4C1im]Cl.109 | ||
The novel hybrid green composite films comprising of cellulose, starch and lignin have been prepared by respective dissolution in [(C1C2)C1im]Cl.69 The experimental results showed that the relative contents of cellulose, starch and lignin had a significant impact on the mechanical properties of composite films. The composite films exhibited good thermal stability and high gas barrier capacity with a CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 permeability ratio close to 1.
:O2 permeability ratio close to 1.
3.2. Biopolymer fibers
Electrospinning of polymer materials has emerged as a powerful technology for the manufacture of fibrous materials with controllable compositions, high specific surface area and high porosities for a broad range of applications such as reinforcement in nano-biocomposites, as tissue engineering scaffolds, in wound dressing and as protective clothing.114,115 The electrospinning of natural polymers for biopolymer fiber fabrication is of particular interest, not only because of the renewable resources, but also due to the beneficial properties of these biomacromolecules, such as biodegradability, biocompatibility and excellent specificity.116 Electrospinning typically employs volatile organic solvents to prepare the polymeric spinning solution which evaporate in the course of fiber formation. This process can release high levels of harmful volatile compounds into the environment and it is unsuitable for effective recovery and recycling of the solvents.117 Electrospinning from ILs represents an interesting solution to this problem since these non-volatile solvents remain in the fibers as they are cast and are removed by means of a coagulation/washing bath, allowing their effective recovery using an array of strategies.118 A typical electrospinning scheme based on ILs assisted dissolution and regeneration of cellulose is illustrated in Fig. 4. The setup consists of a high-voltage power supply connected to a spinneret containing the IL–polymer solution and to a grounded conductive collector. The spinneret is often a syringe, controlled by a pump through which the IL–biopolymer solution can be fed with a specific flow rate to get a pendant droplet at the tip of the needle. An electrical charge is accumulated in the surface of the IL solution droplet after a voltage difference is created between the spinneret and the collector. The solution is electrospun under a certain threshold voltage to form a thin, charged IL–polymer jet which is ejected from the tip of the cone and is drawn towards the grounded collector. The residual IL in the fiber is removed using ethanol or water and the fiber is washed by deionized water and dried at 50–60 °C for a day.118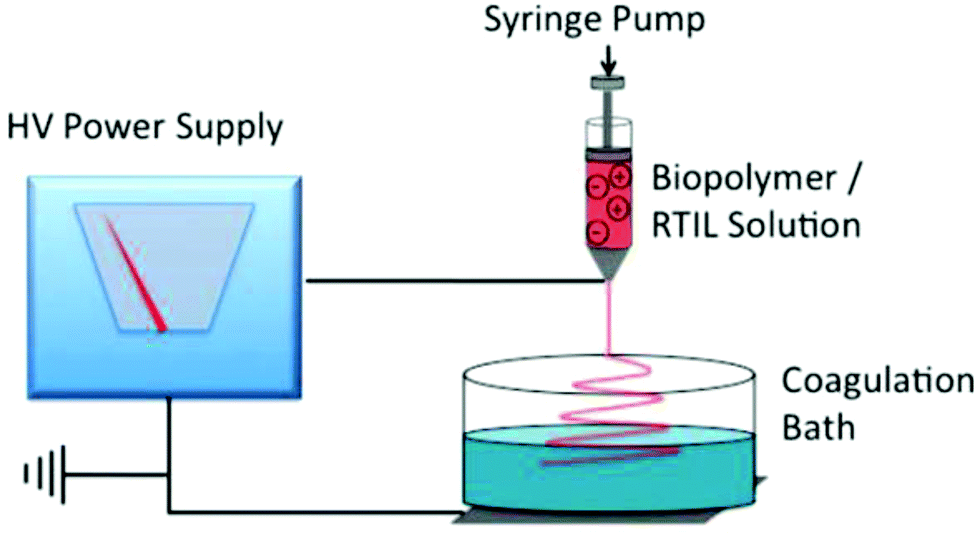 | ||
| Fig. 4 An illustration of a typical electrospinning apparatus for biofiber processing by ILs.118 | ||
Polaskova et al.119 used a wet electrospinning technique for the transformation of raw pine wood into microfiber (1–4 μm) by dissolution in [C2C1im][OAc] or [C2C1im][lactate]. It was reported that 5% wood concentration in the IL was most appropriate for electrospinning and an increase in the biomass loading up to 10% complicated the process due to drastic increase in the viscosities of the solutions. Raw delignified lignocellulose biomass (hemp) was successfully electrospun using ([C2C1im][OAc]) as the spinning solvent by Ahn et al.120 As expected, the spinning efficiency and fiber morphology strongly depended on the lignin contents of the raw biomass. It was observed that when the lignin content was higher than 6%, no fiber was formed and the solution was converted into large droplets at the end of the nozzle. Jiang et al.121 studied the microstructure and crystalline properties of the commercial cellulose fibers regenerated and processed with different solvents and technologies with synchrotron wide-angle X-ray diffraction (WAXD) and small-angle X-ray scattering (SAXS). On the basis of their findings, the IL was expected to be much more efficient and flexible in cellulose dissolution and fiber-forming processes.
Quan et al.122 prepared nonwoven nano-scale fibers of cellulose regenerated from [C4C1im]Cl using electrospinning and successfully obtained a minimum diameter of the continuous electrospun cellulose fibers in the range of 500–800 nm. Qin et al.123 observed that high molecular weight and high purity chitin powder could be recovered after complete dissolution of raw crustacean shells in [C2C1im][OAc] with the yield as high as 94% (Fig. 5). Furthermore, they reported the direct fabrication of chitin fibers and films from the extract solution. High tenacity chitosan fibers with excellent strength and initial modulus were generated from dissolved chitosan in binary IL mixtures of glycine hydrochloride [Gly·H]Cl and [C4C1im]Cl with a dry-wet spinning technology.124Fig. 6 shows that the same binary system of acidic and neutral IL ([Gly·H]Cl and [C4C1im]Cl) was used to fabricate chitosan-cellulose composite fiber with 9.4 wt% chitosan.125 The hybrid-type biopolymer fiber not only had good mechanical strength but also excellent thermal stability with Tonset of 305.1 °C.
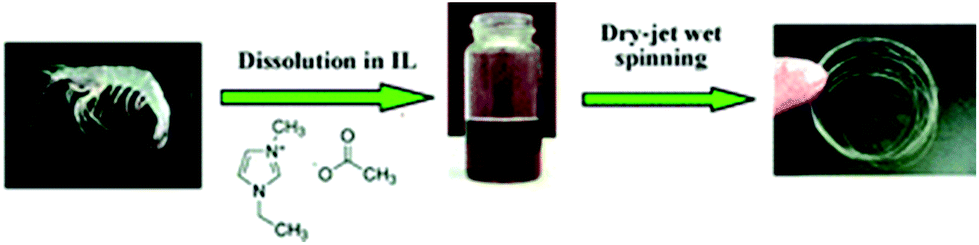 | ||
| Fig. 5 Fabrication of chitin fiber from an IL solution of crustacean shells.123 | ||
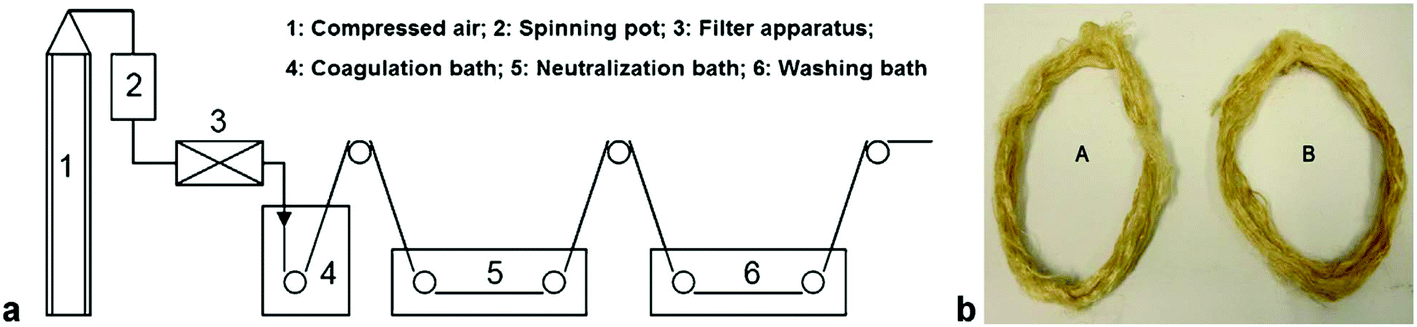 | ||
| Fig. 6 (a) Schematic representation of the dry-wet spinning technique (b) the obtained chitosan fibers.125 | ||
The suitability of several chloride containing ILs was tested for their cellulose dissolving and subsequent fiber-making properties by spinning the IL–cellulose solutions into water.126 The resulting fibers were asserted to belong to the class of Lyocell-fibers and showed the same or comparable characteristics to fibers obtained from NMMO solutions. Recently, cellulosic fibers were spun in a dry-jet wet spinning process from a solution in the IL 1,5-diazabicyclo[4.3.0]non-5-enium acetate ([DBNH][OAc]) into water, resulting in properties equal or better than that made from the conventional Lyocell process (tensile strength 37 cN per tex or 550 MPa).127 A spinneret aspect ratio L/D (length to diameter ratio of fiber) of 2.0 gave a better orientation than L/D of 0.2 and allowed higher draw ratios.
Another report describes the multiwall carbon nanotube (MWCNT)/cellulose composite fibers processed from solutions in [C2C1im][OAc].128 At 0.05 mass fraction of MWCNT, the fiber tensile strength increased by about 25%, strain to failure increased by 100% and modulus essentially remained unchanged. Later, Rahatekar and co-workers129 further extended the work by evaluating the effect of fiber extrusion speed and fiber winding speed on the degree of alignment and electrical properties of MWNTs/cellulose composite fiber processed by [C2C1im][OAc]. A decrease in degree of alignment was noted when fibers were spun with higher winding speed using a constant extrusion speed. More recently, the work of Rahatekar130 corroborated the manufacture of highly aligned cellulose fibers spun from optically anisotropic microcrystalline cellulose solutions in 1-ethyl-3-methylimidazolium diethylphosphate ([C2C1im][Et2PO4]). Fibers with an average diameter of ∼20 μm, a high Young's modulus up to ∼22 GPa and moderately high tensile strength of ∼305 MPa were successfully spun from an 18 wt% cellulose solution in the IL. Similarly, in the interesting study of Byrne et al.131 composite fiber comprising of cellulose and duck feather was extruded from a solution of the biopolymers in the IL [(C1C2)C1im]Cl via a wet spinning operation. The mechanical properties of the composite fiber was shown to be better than regenerated cellulose fibers alone with a 63.7% improvement in tensile strain. The hybrid composite fiber comprising regenerated raw lignocellulosic biomass (yellow pine and bagasse) from [C2mim][OAc] with a dry-jet wet spinning process under short dissolution times (10–30 min) and temperatures above the glass transition temperature of lignin was reported by Rogers et al.132 Fibers spun using the higher temperature/shorter time method were stronger than those obtained using the lower temperature/longer time method (Fig. 7).
 | ||
| Fig. 7 Cellulose–starch composite gel (a) and fiber (b).133 | ||
3.3. Aerogels/hydrogels
Aerogels are the class of highly porous synthetic solid materials with extremely low density and low thermal conductivity. Owing to their diverse physical and chemical properties, aerogels have a wide range of applications as engineering materials. These ultra-light materials can be manufactured from a variety of raw materials including silica, carbon, metal oxides etc.134,135 However, due to the rapid diminishment of petroleum reserves and serious environmental problems created by the non-biodegradable material products and their energy intensive syntheses, alternative polysaccharides based resources have attracted a great deal of attention for aerogels.136,137 Lignocellulose and various biopolymers based aerogels are new highly promising biomaterials that could offer a broad range of potential applications including cosmetics, bio-medical (e.g. scaffolds, delivery systems), electro-chemical, wastewater treatment and as insulation materials.138,139Li et al.140 reported a facile fabrication method of a lignocellulose aerogel by dissolution of wood in [(C1C2)C1im]Cl by a cyclic-freeze–thaw (FT) process, as shown schematically in Fig. 8. It was shown that the IL assisted FT treatment, when used as a physical cross-linking technique, can successfully form continuous 3D network structures. Later, Sescousse et al.141 fabricated an ultra-light and highly porous cellulose material which they called aerocellulose. The material was prepared via cellulose dissolution in [C2C1im][OAc] or [C4C1im]Cl and then regenerated and dried under supercritical CO2 conditions. The density of the aerocellulose was from 0.06 to 0.20 g cm−3. The “bead-like” morphology of aerocellulose from cellulose–ionic liquid solutions was similar to that of samples obtained from cellulose–NMMO monohydrate solutions. A facile preparation of a flexible gel material from a solution of cellulose in [C4C1im]Cl (15% w/w) by keeping it at room temperature for 7 days was reported (Fig. 9).142 Elemental analysis data showed that the fabricated gel was composed of cellulose, the IL, and water. Both XRD and TGA results indicated that the crystalline structure of cellulose was largely disrupted in the material.
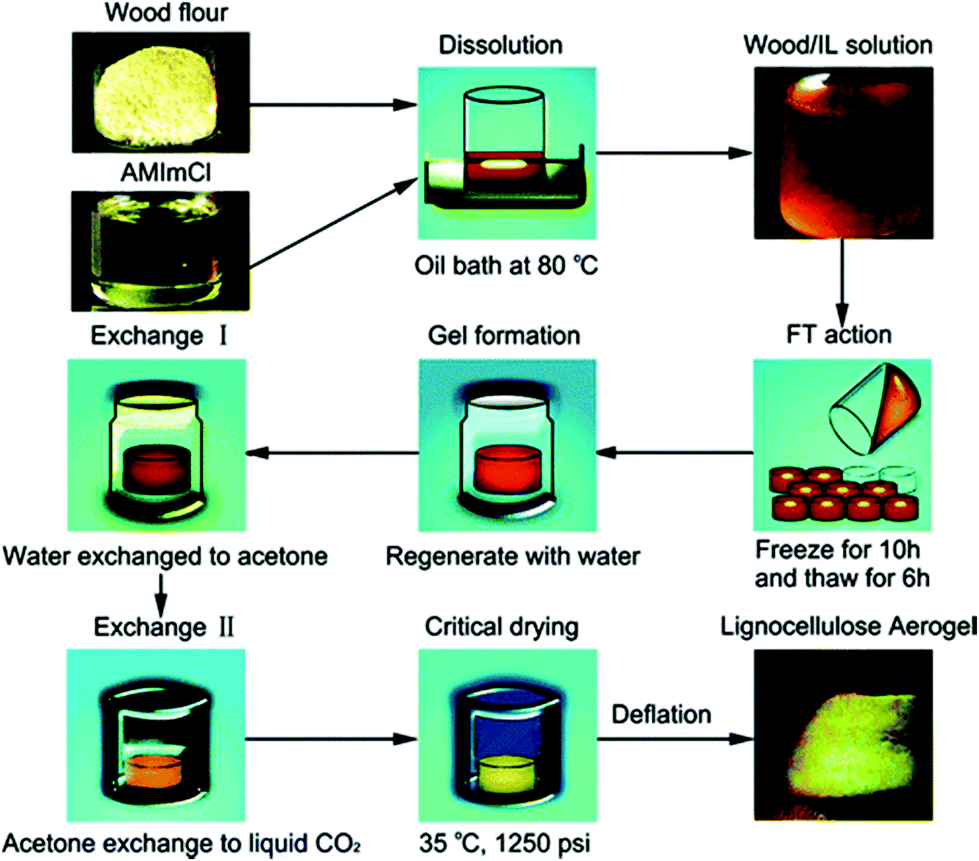 | ||
| Fig. 8 Schematic presentation of lignocellulose aerogel preparation.140 | ||
 | ||
| Fig. 9 Photographs of (a) a cellulose–IL solution gelation process, (b) the obtained gel product.142 | ||
Matrices based on silk fibroin show good applicability in the field of regenerative medicine, but the cocoons of Antheraea mylitta are underutilized because of the poor ability of traditional organic solvents to dissolve these. Consequently, Silva et al. investigated the solubilization and processing of degummed fibers extracted from the cocoons of silkworms into hydrogels using [C4C1im][OAc].143 The degummed silk fibers were dissolved in the IL at 95 °C with 10 wt% solid loading, as shown schematically in Fig. 10. The outcomes of the study suggested that the use of ILs for the dissolution/processing of degummed fibers derived from A. Mylitta cocoons into hydrogels could be interesting in cartilage regeneration repair strategies. Lately, a simple method for the preparation of cellulose/graphene composite hydrogels with high toughness was developed.144 The hydrogel was prepared by regeneration of the mixture of wood pulp and reduced graphene oxide from their IL solution using deionized water as an anti-solvent. This approach provides a simple and green method to compound the multifunctional properties of cellulose with the extraordinary performances of graphene based hydrogels (Fig. 11).
 | ||
| Fig. 10 The fabrication of silk hydrogels from mulberry and non-mulberry silk cocoons using the IL [C4C1im][OAc] as solvent. (A): Antheraea mylitta cocoons, (B): Bombyx mori cocoons.143 | ||
 | ||
| Fig. 11 (a) Images of cellulose and cellulose/graphene composite hydrogels (CGH); (b) the freeze-dried CGH; (c) the freeze-dried CGH with height of 1.50 cm and diameter of 1.40 cm supporting a 4 kg counterpoise which is more than 14800 times its own weight.144 | ||
Nanofibrillar aerogels were developed from cellulose, spruce wood and from mixtures of cellulose, lignin and xylan by first dissolving in [C4C1im]Cl and coagulating from IL solution using aqueous ethanol. These microporous aerogels could be readily disintegrated into fibrous or powder-like material by rubbing between one's fingers. Aerogels made from wood were much harder and possessed much more structural strength than the hybrid aerogels made from cellulose, lignin and xylan (Table 1).171
| Entry | IL | Raw material | Conditions | Loading (%) | Fabrication technique | Product | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | [(C1C2)C1im]Cl |
Cellulose | 100–130 °C | 4 | Solution casting | Film | 106 |
| 40–180 min | |||||||
| 2 | [C4C1im]Cl | Cotton pulp | 90 °C | — | Solution casting | Film | 107 |
| [C2C1im]Cl | 7 h | ||||||
| [C2C1im][OAc] | |||||||
| [C4C1im][OAc] | |||||||
| 3 | [C4C1im]Cl | Natural wool | 100 °C | 5 | Solution casting | Film | 145 |
| Cellulose | 10 h | ||||||
| 4 | [(C1C2)C1im]Cl |
Cotton | 100 °C | 10 | Solution casting | Film | 111 |
| Silk | |||||||
| Wool | |||||||
| 5 | [(C1C2)C1im]Cl |
Cornhusk | 80–120 °C | 4 | Solution casting | Film | 146 |
| [C2C1im][OAc] | Cellulose | 2–12 h | |||||
| 6 | [C4C1im]Cl | Silk fibroin | 90 °C | 2 | Molding | Film | 147 |
| Cellulose | 12 h | ||||||
| 7 | [C4C1im]Cl | Cellulose | 85 °C | 8 | Solution casting | Film | 148 |
| Montmorillonite | 28 h | ||||||
| 8 | [(C1C2)C1im]Cl |
Bamboo pulp | 80 °C | 6 | Solution casting | Film | 149 |
| Soy protein isolate | |||||||
| 9 | [C2C1im][OAc] | Crustacean shells | 100 °C | 10 | Dry-jet wet spinning | Film | 108 |
| 19 h | Fiber | ||||||
| 10 | [C4C1im]Cl | Cellulose | 100 °C | 10 | Solution casting | Film | 108 |
| [(C1C2)C1im]Br |
Chitin | 24 h | 5 | Gel | |||
| 11 | [C2C1im][OAc] | Chitin | 60–120 °C | 3 | Solution casting | Film | 112 |
| Carbon nanotubes | 2–12 h | ||||||
| 12 | [C2C1im][OAc] | Cellulose | 80 °C | 10 | Blending | Film | 150 |
| Silk | 1 h | Molding | |||||
| 13 | [C4C1im]Cl | Skin collagen | 100 °C | 5 | Solution casting | Film | 109 |
| Cellulose | 6 h | Extrusion | Fiber | ||||
| Molding | Gel | ||||||
| 14 | [(C1C2)C1im]Cl |
Wool keratin | 100 °C | — | Solution casting | Film | 110 |
| 15 | [(C1C2)C1im]Cl |
Cotton | 100 °C | — | Solution casting | Film | 113 |
| Duck feather | |||||||
| 16 | [(C1C2)C1im]Cl |
Cellulose | 80 °C | 6 | Solution casting | Film | 69 |
| Starch, lignin | |||||||
| 17 | [C4C1im][OAc] | Cellulose | 85–95 °C | 6 | Solution casting | Film | 151 |
| Chitosan | 6 h, 3–4 days | Vacuum freeze drying | |||||
| 18 | [C4C1im]Cl | Cellulose | 25–110 °C | — | Solution casting | Film | 152 |
| [C4C1im][OAc] | Chitosan | 4 h | |||||
| 19 | [C2C1im][OAc] | Hemp biomass | — | 14 | Electrospinning | Fiber | 120 |
| 20 | [(C1C2)C1im]Cl |
Cotton | 100 °C | — | Lab spinning set-up | Fiber | 131 |
| Duck feather | |||||||
| 21 | [C2C1im][OAc] | Pine wood | 80 °C | 5–10 | Wet electrospinning | Fiber | 119 |
| [C2C1im][lactate] | 24 h | ||||||
| 22 | [C4C1im]Cl | Cellulose pulp | — | — | Dry-jet-wet-spinning | Fiber | 121 |
| 23 | [C4C1im]Cl | Cellulose | 90 °C | 5 | Electrospinning | Fiber | 122 |
| 30 min | |||||||
| 24 | [C4C1im]Cl | Chitosan | 80 °C | 5 | Dry-wet spinning | Fiber | 124 |
| 1 h | |||||||
| 25 | [C4C1im]Cl | Chitosan | 80 °C | 5 | Dry–wet spinning | Fiber | 125 |
| Cellulose | 10 | ||||||
| 26 | [C2C1im][OAc] | Hemp biomass | 70 °C | 14 | Electrospinning | Fiber | 153 |
| 24 h | |||||||
| 27 | [C4C1im]Cl | Cellulose, starch | 100 °C | 4–10 | Dry-jet wet spinning | Fiber | 154 |
| Hindered amine light stabilizer | 20 h | ||||||
| 28 | [C2C1im]Cl | Silk fibroin | 95 °C | 10 | Extrusion | Fiber | 155 |
| 29 | [dbmim][BF4N2 ] | Quantum dots elastomer | 70 °C | 1% IL | Electrospinning | Fiber | 156 |
| 6 h | |||||||
| 30 | [C4C1im]Cl | Cellulose | Microwave heating | 10 | Electrospinning | Fiber | 157 |
| Heparin | 2 | ||||||
| 31 | [(C1C2)C1im]Cl |
Cellulose | 80 °C | 1–5 | Electrospinning | Fiber | 158 |
| 2 h | |||||||
| 32 | [C2C1im][OAc] | Cellulose | RT | 8 | Electrospinning | Fiber | 159 |
| [C1C1im][Cl] | |||||||
| 33 | [C4C1im]Cl | Wood pulp | 100 °C | 4 | Dry-jet wet spinning | Fiber | 160 |
| MWCNT | 45 min | ||||||
| 34 | [C2C1im][OAc] | Chitin | — | 1.5 | Electrospinning | Fiber | 161 |
| 35 | [C2C1im][OAc] | Cellulose | 80 °C | 4–8 | Dry-jet wet spinning | Fiber | 128 |
| MWCNT | 2–3 h | ||||||
| 36 | [C2C1im][OAc] | Cellulose | 80 °C | — | Lab-built spinning | Fiber | 129 |
| MWCNT | 2 h | equipment | |||||
| 37 | [C2C1im][Et2PO4] | Microcrystalline cellulose | 95 °C | 7.6–18 | Dry-jet wet spinning | Fiber | 130 |
| 24 h | |||||||
| 38 | [DBNH][OAc] | Birch kraft pulp | 80 °C | 13 | Multi-filaments piston-spinning | Fiber | 162 |
| 75 min | |||||||
| 39 | [C2C1im][OAc] | Yellow pine | 175 °C | 5 | Dry-jet wet spinning | Fiber | 132 |
| Bagasse | 30 min | ||||||
| 40 | [C4C1im]Cl | Cellulose | 100 °C | 3 | Spinning into water | Fiber | 126 |
| [(C1C2)C1im]Cl |
2–4 h | ||||||
| 41 | [DBNH][OAc] | Cellulose | 80 °C | 13 | Dry-jet wet spinning | Fiber | 127 |
| 3 h | |||||||
| 42 | [C4C1im]Cl | Cellulose | 100 °C | 10 | Solution sandwiched between glass plates, raising by spatula | Fiber | 133 |
| Starch | 24 h | Gel | |||||
| 43 | [C2C1im][OAc] | Cellulose | — | — | Electrospinning | Synthetic wood fiber | 163 |
| Xylan, lignin | |||||||
| 44 | [(C1C2)C1im]Cl |
Pineapple peel | 100 °C | 2.5–7.5 | Heating–cooling–freezing–thawing–washing process | Hydrogel | 164 |
| 12 h | |||||||
| 45 | [C4C1im]Cl | Microcrystalline cellulose | 80 °C | 1.5–5.5 | Chemical crosslinking | Hydrogel | 165 |
| 12 h | |||||||
| 46 | [C4C1im]Cl | Cellulose | 100 °C | 6 | Dropping solution from syringe needle | Hydrogel beads | 166 |
| Collagen | |||||||
| 47 | [C4C1im]Cl | Cellulose | 100 °C | 15 | Keeping in RT for 7 days | Hydrogel | 142 |
| 24 h | |||||||
| 48 | [C4C1im][OAc] | Silk fibroin from the silkworm | 95 °C | 10 | Molding followed by gelation overnight | Hydrogel | 143 |
| 8 h | |||||||
| 49 | [C4C1im]Cl | Cellulose | 80 °C | 3–5 | Degassing the solution by ultrasound | Hydrogel | 144 |
| Graphene | 24 h | ||||||
| 50 | [C4C1im]Cl | Cellulose | 70 °C | — | Solution casting | Hydrogel | 167 |
| 2 h | |||||||
| 51 | [C4C1im]Cl | Cotton cellulose | 100 °C | 8 | Solution casting | Aerogel | 168 |
| 24 h | |||||||
| 52 | [(C1C2)C1im]Cl |
Nalita wood | 80 °C, 4 h | 8 | Freeze thaw | Aerogel | 140 |
| 53 | [C2C1im][OAc] | Cellulose | 80 °C | 3 | Molding | Alcogels | 169 |
| 16 h | |||||||
| 54 | [C2C1im][OAc] | Cellulose | 80 °C | — | Regeneration/drying in supercritical CO2 | Aerocellulose | 141 |
| 48 h | |||||||
| [C4C1im]Cl | |||||||
| 55 | [(C1C2)C1im]Br |
Chitin | 100 °C | 7 | Dissolution + cooling | Gel | 170 |
| 48 h | |||||||
| 56 | [C4C1im]Cl | Spruce wood | 130 °C | 7 | Solution casting followed by coagulation and high pressure cell | Aerogel | 171 |
| 4 h | |||||||
| 57 | [C4C1im]Cl | Cellulose, starch | 95 °C | 3 | Molding | Aerogel | 172 |
| Zein protein, agar | |||||||
| 58 | [C4C1im]Cl | Agarose | 100 °C | 5 | Regeneration from IL solution | Ionogel | 173 |
| Chitosan |
3.4. Self-reinforced all-cellulose composites (ACCs)
Eco-friendly composites can be manufactured by compounding a variety of lignocellulosic fibers such as wood fiber, leaf fiber, bast fiber or regenerated cellulose with a thermoset or thermoplastic matrix.102,174 However, the mechanical strength of such biocomposites is often limited due to the poor chemical compatibility between the hydrophilic fiber and hydrophobic matrix, which results in weak interfacial stress transfer.45,84,175 Recently, in order to resolve the interfacial adhesion problem, a new class of composite materials with superior mechanical properties which entirely consist of non-derivatized cellulose have been developed – all-cellulose composites (ACCs).176,177A novel approach to prepare cellulose-based conductive hydrogels was demonstrated by dissolution of microcrystalline cellulose (MCC) in [C4C1im]Cl (Fig. 12).165 The obtained MCC composite hydrogel showed excellent electrical conductivity of 7.83 × 10−3 S cm−1, as measured using a four-probe method. Ma et al.178 utilized the excellent selective solubility of the IL 1-(2-hydroxyethyl)-3-methylimidazolium chloride ([(HO)2C2C1im]Cl) for microcrystalline cellulose (MCC) and nanocrystalline cellulose (NCC) to fabricate green all-cellulose composite films. The ACC film was prepared by adding NCC to the MCC–IL solution. The composite film was fully biobased, biodegradable and easily recyclable, and proposed to be useful as biomaterials and food ingredients (Fig. 13b). Huber et al.176 developed a fiber-reinforced all-cellulose composite (ACC) laminate on the basis of a conventional hand lay-up method (Fig. 13a). Four layers of each natural fiber textile (linen) and man-made cellulosic textile (Cordenka rayon) were impregnated with [C4C1im][OAc]. The layers were heated under pressure to partially dissolve in the IL so that compaction of the single laminate was achieved to form a composite. The formation of the matrix phase was obtained in situ by the regeneration of the dissolved fraction of the cellulose fiber, which resulted in a composite with strong fiber–matrix interfacial properties.
 | ||
| Fig. 12 (a) Cellulose hydrogel and (b) swollen hydrogel.165 | ||
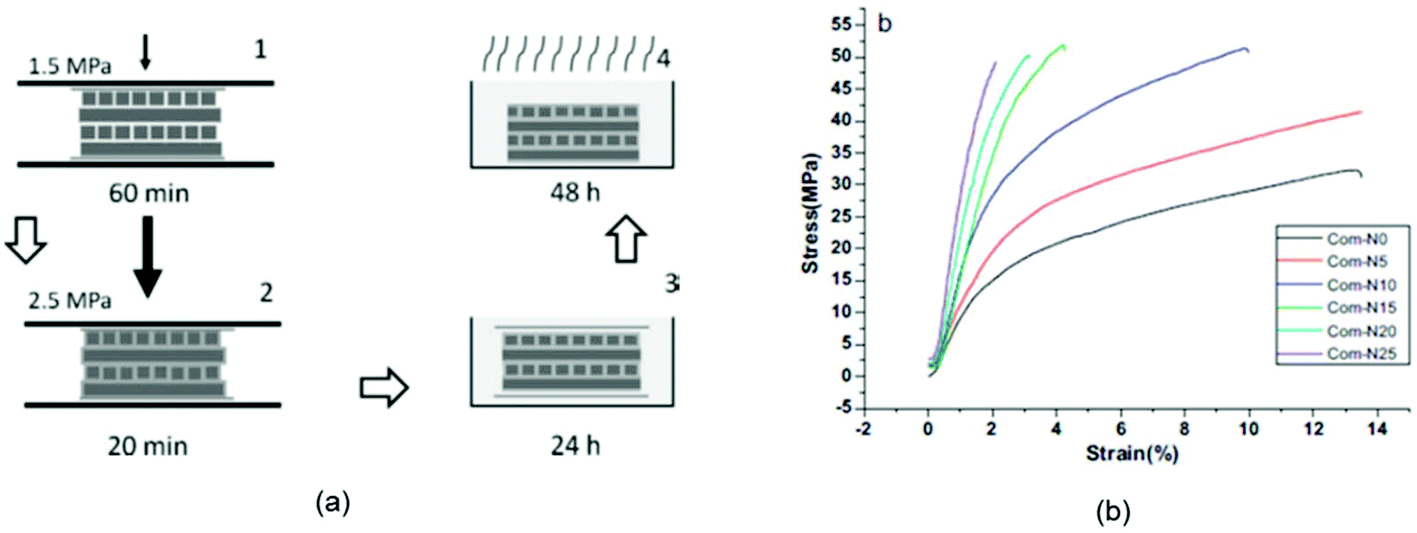 | ||
| Fig. 13 (a)The fabrication process for ACC laminate by hand lay-up method,176 (b) stress–strain curve of the all-cellulose composites.178 | ||
Recently, in the interesting work of Reddy et al.,179 the cellulose was extracted from the Agave plant and the extracted cellulose microfibrils were successfully dissolved in [(C1C2)C1im]Cl along with a small amount of microfibrils which remained undissolved as can clearly be observed in Fig. 14. The self-reinforced regenerated cellulose biocomposite films with an average strength as high as 135 MPa were prepared from this solution. Furthermore, a comparison of an IL based all-cellulose composite with an epoxy matrix composite was reported by partial dissolution of flax and Lyocell fiber in [C4C1im]Cl.180 It was observed that ACCs fabricated from Lyocell fiber showed similar strength and stiffness but superior extensibility as compared to Lyocell fiber–epoxy composites.
 | ||
| Fig. 14 (a) Photograph and (b) SEM images of regenerated all-cellulose composite film.179 | ||
 | ||
| Fig. 15 An example of fiber welding. (a) Raw silk thread, welded with [C2C1im][OAc] for 5 min at 60 °C to create a solid, yet flexible band structure, (b) and (c). SEM images of (untreated) desized silk and of the welded silk band are shown in (d) and (e), respectively.181 | ||
3.5. All-wood composites
Wood is widely utilized for furniture and building materials. However, most of the waste timbers recovered from demolished wooden buildings and massive deforestation are usually incinerated for energy purposes, which causes severe environmental problems.185,186 Furthermore, it has also been reported that a massive increase of damaged trees due to various diseases such as sugi-mizogusare, which can no more be useful as lumber, is becoming a critical problem in countries such as Chiba and Japan.187 On the other hand, the requirement for wood based composite materials for furniture and construction industries in the form particleboard, fiberboard, oriented strand board etc. is increasing every year.188 Therefore, there has been a substantial demand for novel technologies that could provide new routes for consumption of the untapped or waste lumber as well as for the production of wood composite materials in a more efficient and environmentally benign manner (Table 2).| Entry | Ionic liquid | Raw material | Conditions | Loading (%) | Fabrication technique | Ref. |
|---|---|---|---|---|---|---|
| 1 | [C4C1im][OAc] | Linen | 110 °C | 50 | Hot press | 176 |
| Rayon | 80 min | |||||
| 2 | [C4C1im]Cl | Microfibrillated cellulose | 80 °C | — | Solution casting | 191 |
| 20–160 min | ||||||
| 3 | [C2C1im]Cl | Microcrystalline cellulose | 85 °C | 3 | Solution casting | 178 |
| 2 h | ||||||
| 4 | [(C1C2)C1im]Cl |
Agave fibers | 80 °C | 4 | Solution casting | 179 |
| 5 | [C4C1im]Cl | Lyocell fibers | 130 °C | — | Solution casting | 180 |
| Flax fibers | 1 min impregnation | |||||
| 6 | [C4C1im]Cl | Rice husk | 100 °C | 5–10 | Solution casting | 192 |
| Filter paper | ||||||
| 7 | [C4C1im]Cl | Cedar wood flour | 100 °C | 60 | Compression molding followed by annealing | 189 |
| Bark flour | 10 min | |||||
| 8 | [C4C1im]Cl | Cotton fabric | 100 °C | — | Hot press followed by annealing | 190 |
| hinoki lumber | 30 min | |||||
| 9 | [C4C1im]Cl | Mulberry wood | 80 °C | — | Twin-screw kneader followed by injection-molding | 193 |
| [C4C1im][OAc] | 2–3 h | |||||
| 10 | [C2C1im][OAc] | Silk fiber | 60 °C | — | Natural fiber welding | 181 |
| Hemp fiber | 5 min | |||||
| 11 | [C2C1im][OAc] | Cotton cloth | 40–80 °C | — | Natural fiber welding | 182 |
| 0.5–24 h |
Shibata et al.189 developed a new method for waste wood processing based on the partial dissolution of waste wood flour in [C4C1im]Cl as depicted in Fig. 16. Wood flour (WF) and bark flour (BF) derived from cedar wood were mixed with 40 wt% [C4C1im]Cl at 100 °C and the resulting mixtures were compression-molded at 210 °C in a stainless steel mold. The tensile strength and thermal properties of the biocomposites were significantly improved by the extraction of the IL from the final product and further by the annealing process. In their further work,190 Shibata and co-workers tried to improve the tensile strength and modulus of all-wood based biocomposites from dipped hinoki lumber (HL) with dimensions 50 mm × 10 mm × 2 mm in the same IL at 100 °C and the IL-impregnated HL was hot-pressed at 210 °C for 30 min (Fig. 17a). Although the tensile strength of the fabricated biocomposite was found to be lower than that of original HL, the tensile modulus of the former was significantly improved. In-depth analysis of the structural and chemical changes of wood during IL interaction is needed to explore the opportunities for using ILs in wood and wood based composite materials.
 | ||
| Fig. 16 Photographs of the all-wood biocomposites compression-molded at various temperatures.189 | ||
 | ||
| Fig. 17 (a) Original hinoki lumber (HL), (b) HL composite after IL pretreatment with [C4C1im]Cl,190 (c) synthetic wood films.194 | ||
3.6. Synthetic wood and synthetic wood composites
The main rationale for using ILs in the field of polysaccharide based composites is the excellent dissolving power of some ILs for most of the biopolymers, which can be exploited for the production of highly useful hybrid composites consisting of both biodegradable and non-biodegradable components. In the work of Simmons et al.,194 synthetic wood was fabricated from the three major components of lignocellulosic biomass i.e., cellulose, hemicellulose and lignin, as shown in Fig. 17c. The artificial wood films were obtained by dissolution of individual biopolymer components (cellulose/xylan/lignin, 5/3/2, wt%) in [C2C1im][OAc] at 90 °C for 3 h and reconstituting these using water as anti-solvent. As compared to the individual components, synthetic wood showed remarkably improved physicochemical properties. In addition, a wide variety of synthetic wood based composite materials could be fabricated having special properties like mechanical strength, color, conductivity or anti-microbial properties etc. Simmons and co-workers194 also prepared synthetic wood composites by dissolution of different polymers such as chitosan or nanoparticle like multi-wall carbon nanotubes (MWNT) in [C2C1im][OAc] along with wood components followed by regeneration with water. This excellent property of synthetic wood MWNTs composites makes them a suitable candidates as flexible high dielectric insulator materials in applications such as batteries and supercapacitors.1953.7. Chemical and surface modification of lignocellulosic materials with ionic liquids
The properties of synthetic fibers can be engineered during their manufacturing process to make them suitable for different end uses.196 In the case of natural fibers, various pretreatment steps are highly desirable in order to impart a range of suitable characteristics for different applications as engineered materials.197 For example, in order to improve the interfacial compatibility between individual components in composites containing natural fibers, the modification of the natural fiber is an extremely important step prior to hybrid injection molding. Thus, the chemical or surface modification of lignocellulose and wood has been extensively studied with the objective of modifying or improving its compatibility with thermoplastics or thermosets, as well as for improving dimensional stability.84,198 However, to obtain a hydrophobic wood matrix by heterogeneous chemical modification so that it could be molded into any desired shape is a very difficult task, due to the poor solubility of its lignocellulosic components in conventional solvents.199 Recently, the efficient lignoellulose dissolution capabilities of ILs have opened a new platform for novel homogeneous modification of wood and lignocellulose for derivative biopolymers.In the study of Wen et al.,200 complete dissolution and homogeneous lauroylation of abundantly available hydroxyl groups in ball-milled lignocellulose bamboo meal was carried out in [C4C1im]Cl at 130 °C for 6 h. The thermal stability of the esterified derivatives was found to be lower than that of the unmodified bamboo meal and the rough appearance of the bamboo meal changed into a relatively homogeneous and smooth surface morphology after lauroylation. Another work that describes the development of an IL-assisted pretreatment technology for efficient conversion of chemically modified wood material into thermoplastic material was reported by King et al.201 Benzoylation and lauroylation of spruce wood thermomechanical pulp was carried out in the IL solution 4% w/w spruce in [C4C1im]Cl followed by the incremental addition of benzoyl chloride and lauroyl chloride, respectively. Subsequently, these highly substituted benzoylated and laurolyated lignocellulosic spruce fibers were used as reinforcement for thermoplastic composites of polypropylene and polystyrene. Excellent dispersion of the IL-modified wood fibers material into the synthetic polymers was achieved throughout the composite material, as highlighted by SEM images. Utilization of [C4C1im]Cl for homogeneous benzolyation of wood meal was also reported by Yuan et al.202 The focus of this study was to enhance the compatibility as well as photostability of wood with hydrophobic synthetic polymers for the production of green composites that could be used for outdoor applications. The article describes a highly promising IL based modification of wood fiber with improved properties for photostable green composites applications.
For the application of ILs in wood based composite materials, most of the studies have been focused on the effect of ILs on the bulk properties of lignocellulosic biomass and wood, and there are only a few reports related to the IL based modification of wood surface properties. In the study by Croitoru et al.,203 the effect of four types of imidazolium-based ILs, namely 1-butyl-3-methylimidazolium tetrafluoroborate ([C4C1im][BF4]), 1-butyl-3-methylimidazolium hexafluorophosphate ([C4C1im][PF6]), [C4C1im]Cl and 1-butyl-3-methylimidazolium tetrachloroferrate ([C4C1im][FeCl4]), on the various surface properties of poplar wood veneers was studied in terms of contact angle, electrical conductivity measurement and photographic image analysis and compared with those of untreated wood. It was found that IL treatment improved the flexibility of the cellulose matrix and decreased its crystallinity. Indeed, the workability of wood could be remarkably enhanced by IL surface pretreatment, which decreased its rigidity as well as preventing the accumulation of static electric charges on the surface of the wood during finishing. The changes that occurred in the structure and surface properties of maple wood veneers treated with three different types of IL, [C4C1im][BF4], [C4C1im][PF6] and 1-hexyl-3-methylimidazolium chloride ([C6C1im]Cl) were studied after being subjected to an electron beam irradiation with a 50 kGy dose.204 Also, an interesting observation was noted that a higher resistance to water penetration and spreading on the wood surface resulted after the electron beam irradiation, due to covalent bonding of the imidazolium moiety to wood structure (Table 3).
| Entry | Ionic liquid | Raw material | Conditions | Modification reaction | Ref. |
|---|---|---|---|---|---|
| 1 | [C4C1im]Cl | Bamboo meal | 130 °C | Lauroylation | 200 |
| 6 h | |||||
| 2 | [C4C1im]Cl | Southern pine Thermomechanical pulp | 70 °C | Lauroylation | 201 |
| 2 h | |||||
| 3 | [C4C1im]Cl | Wood meal | 130 °C | Benzoylation | 200 |
| 5 h | |||||
| 4 | [C4C1im]Cl | Poplar veneers | 22 °C | Surface treatment | 203 |
| [C4C1im][PF6] | 15 min | ||||
| [C4C1im][BF4] | |||||
| 5 | [C4C1im]Cl | Poplar veneers | 22 °C | Electron beam irradiation | 204 |
| [C4C1im][PF6] | 15 min | ||||
| [C4C1im][BF4] | |||||
| 6 | [C4C1im]Cl | Spruce thermomechanical pulp | 130 °C | Acylation | 205 |
| [(C1C2)C1im]Cl |
4–6 h | Carbanilation | |||
| 7 | [C4C1im]Cl | Poplar wood | 125 °C | Acylation | 206 |
| 1 h | |||||
| 8 | [(C1C2)C1im]Cl |
Cellulose | 80 °C | Acetylation | 183 |
| 1 h | |||||
| 9 | [C4C1im]Cl | Poplar wood | 130 °C | Benzoylation | 202 |
| 5 h | |||||
| 10 | [C4C1im]Cl | sspen veneer | 22 °C | Surface treatment | 207 |
| [C4C1im][PF6] | 15 min | ||||
| [C4C1im][BF4] |
Whether wood is used for interior applications or under exterior conditions, the important properties that could be significantly affected are its texture, color, wettability and roughness. Patachia et al.207 studied the effect of four types of imidazolium-based IL on the chemical modification and photo-degradation of the surface of wood veneers. The ILs treated wood showed some useful properties including enhanced wettability that could increase the compatibility of the wood with adhesives in fabrication of wood based composite materials.
3.8. Ionic liquids as plasticizers for biodegradable thermoplastic polymers
In nature, most vegetal structures are based on matrices of various biopolymers. In terms of the vocabulary of polymer material scientists, one could state that nature has developed extremely efficient composites of biopolymers.208 The glass transition temperature (Tg) of a polymeric material is an important parameter in determining its mechanical performance during composite manufacturing and service.209 Unfortunately, Tg of most of the biopolymers is experimentally inaccessible due to thermal degradation of these polymers at elevated temperatures.210 Consequently, the plasticization of different biopolymers using various plastisizing agents and employing different mechanical techniques, such as shear mixing, melt-processing, extrusion etc., has been demonstrated through the decrease in the Tg of the native biopolymer.211,212 Recently, ILs have been shown to be efficient thermoplasticizers for different bipolymers leading to significantly different properties, such as water sensitivity and thermomechanical properties, from those of classical plasticizers such as glycerol, glycerine, glycol, urea etc.213,214 ILs are able to disrupt the semicrystalline structure of native biopolymer granules and destroy the intermolecular hydrogen bonding among hydroxyl groups of polysaccharides.215 Among various biopolymers, starch has been recognized as a promising substrate due to its wide availability, low cost and renewable character.Sankri et al.79 reported modification of starch with [C4C1im]Cl in a microcompounder and a twin screw extruder. To the best of the author's knowledge, this is currently the only article describing a continuous method of starch plasticization using ILs. The remarkable reduction in molecular weight of starch extruded with [C4C1im]Cl compared to glycerol-plasticized polysaccharide was assigned to thermo-mechanical treatment rather than interaction with IL. The use of [(C1C2)C1im]Cl as a corn starch plasticizer to fabricate solid biopolymer electrolytes has been described.215 The films were obtained by solution casting starch plasticized with 10 and 30 wt% IL respectively. Another study216 showed the plasticization of starch by mixing with same IL in the presence of lithium chloride. It was noted that thermal decomposition of starch was stimulated by high Cl− ion concentration.
Cellulose acetate is one of the most valuable cellulose derivatives, with a wide range of applications including films, coatings, membrane separation, textile and cigarette filters. Availability from renewable sources, biodegradability, non-toxicity and low cost are the combination of characteristics that make the cellulose acetate a highly promising biopolymer matrix. However, its functionalization and processing for different applications is limited by its properties such as high crystallinity and thermal decomposition temperature, which is very close to its melt processing temperature. Bendaoud and Chalamet217 utilized a micro-compounding technique to convert cellulose acetate into a thermoplastic polymer using [C4C1im]Cl. The biocomposites were thermo-molded into dumbbells and disk-shaped plates of dimensions of 30 mm × 10 mm × 2 mm and 25 mm × 2 mm (Fig. 18), respectively. It was noted that the presence of the IL strongly reduced the interactions between cellulose acetate chains as evidenced from mechanical tensile tests and rheology analyses of plasticized cellulose acetate (Table 4).
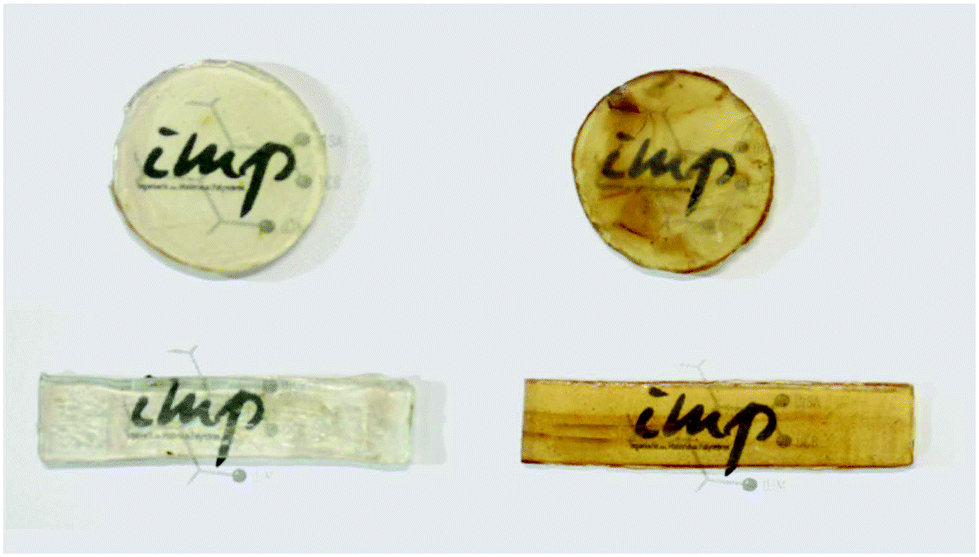 | ||
| Fig. 18 Visual observations of pressed plasticized cellulose acetate samples.217 | ||
| Entry | Ionic liquid | Raw material | Compounding method | Ionic liquid proportion (%) | Conditions | Ref. |
|---|---|---|---|---|---|---|
| 1 | [C4C1im]Cl | Maize starch | Melt-processing | 15–30 | 160 °C, 2 min | 79 |
| 2 | [(C1C2)C1im]Cl |
Corn starch | Mixing | 23 | 75 °C, 20 min | 215 |
| 3 | [C4C1im]Cl | Cellulose acetate | Micro-compounder | 20–40 | 150 °C, 150 rpm | 217 |
| 4 | [C4C1im]Cl | Starch, zein | Micro-compounder | 23 | 130 °C, 3 min | 80 |
| 5 | [C2C1im]Cl | Wood flour | Twin-screw extruder | 1–5 | 150–175 °C | 218 |
3.9. Role of ILs in fiber-reinforced polymeric composite (FRC)
Recently, wood plastic composites (WPCs) have attracted significant attention from both industrial and scientific communities due to their increasing applications as engineered materials in automobile components, building materials and infrastructure etc. and have achieved commercial success.219,220 Natural lignocellulosic resources can efficiently be utilized in WPC manufacturing and thus have high potential to reduce the environmental hazard mainly created by waste from non-degradable plastic products.174 However, in general increasing the wood contents resulted in higher viscosity of the WPC melts. During the processing of WPC, higher viscosity undoubtedly requires high torque for efficient mixing of the blends which will increase the energy consumption. Moreover, this can stimulate high internal friction and can generate local overheated spots, which will ultimately cause the thermal degradation of the wood flour.221,222 Hence, the overall performance and surface quality of WPC will be remarkably reduced. Also, at high wood contents, the rigid cell wall architecture of wood lacks sufficient plastic deformability during extrusion processing, which under extreme conditions may induce structural damage of lignocellulose particles and consequently poor reinforcing effects.223,224 Therefore, current technologies such as controlling die stresses or altering the flow of plastics do not work efficiently for high levels of wood flour. Recently, ILs have emerged as alternative and benign processing media for the manufacture of natural fiber reinforced polymeric composites.Ou et al.218 proposed that improving the thermoplasticity of rigid cell walls of lignocellulose using ILs may be a viable strategy to enhance the processability of wood based thermoplastic composites. They pretreated the wood flour with [(HO)2C2C1im]Cl to different weight percent gains and the pretreatment effect on the rheological properties of the resulting wood flour/high density polyethylene (HDPE) composites was investigated. It was observed that [(HO)2C2C1im]Cl treatment significantly reduced the crystallinity and improved the thermoplasticity of wood fibers and thus broadened the processing window of wood fiber/HDPE composites with stable flow and remarkably smooth product surfaces. It was further concluded that filler–filler interaction had a dominant effect on the extrusion/injection processing of WPCs and that IL pretreatment of wood fiber laid the groundwork for future research in WPCs. A recent report193 also describes the novel use of ILs in production of WPCs. Ball-milled mulberry wood (BMMW) was firstly kneaded in [C4C1im]Cl or [C4C1im][OAc] with the help of the cosolvent DMSO. Plasticization of BMMW for injection-moulding was achieved after the structural destruction of the wood cell walls in the presence of ILs (Fig. 19). The thermal flow and mechanical results revealed that the WPC obtained with DMSO/[C4C1im][OAc] exhibited better thermal and mechanical properties than those obtained with DMSO/[C4C1im]Cl, indicating that the thermoplasticity of AWP was related to the ILs’ anionic ability to disrupt the hydrogen bond networks in the rigid crystalline region of cellulose.
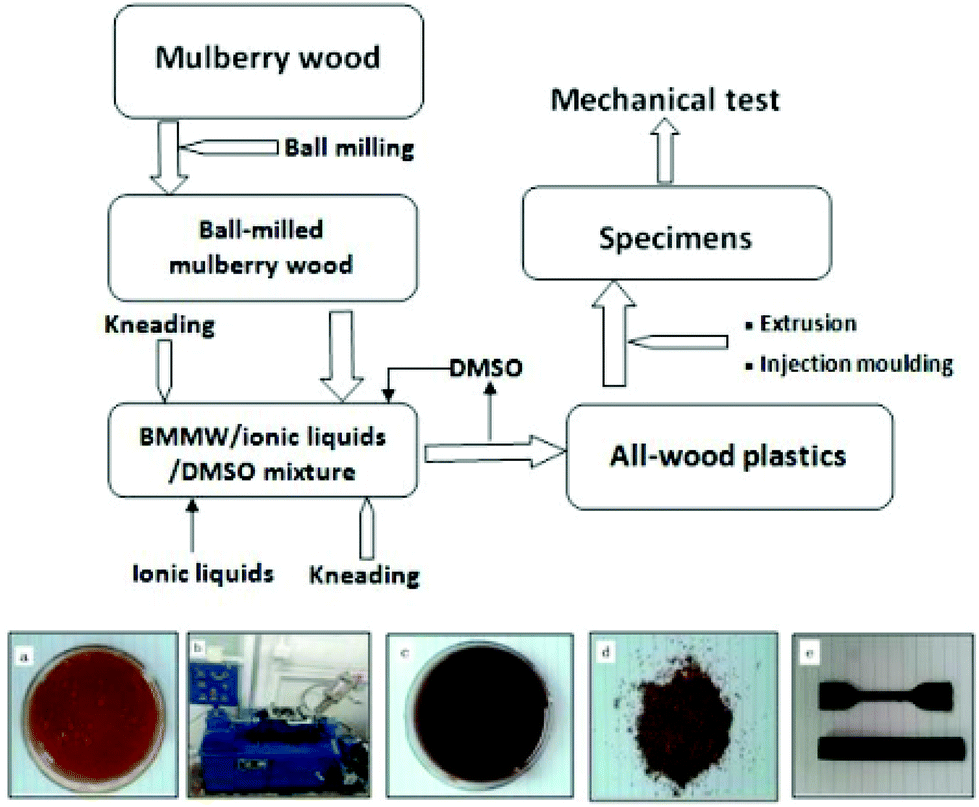 | ||
| Fig. 19 Process flow diagram of the preparation AWP and appearance of mulberry wood before kneading (a), the kneader (b), mulberry wood after kneading (c), final AWP (d) and injection moulded AWP specimens (e).193 | ||
4. Could ionic liquids be helpful to provide alternative raw materials for the wood composite industry?
Among the various lignocellulosic resources, woody biomass has been the most promising raw material for widely known and used natural organic fibers for the production of composite panels.225 However, the massive deforestation, forest degradation and simultaneously increasing demand for wood based composite panels has created a critical raw material issue in the wood composite industry for a long time. Considering these factors, recently research has been focused on the utilization of a wide variety of agro-based renewable lignocellulosic residues as a supplement to, or as a direct substitute for, wood fiber in the production of industrial products such as particleboard, fiberboard, plywood etc.226,227 The availability of these inexpensive lignocellulosic materials in every part of the world together with exclusive advantages in terms of low density, CO2 neutrality (when burned), non-abrasive nature and safe working environment have fueled their use for composite panels manufacturing in the past few years. Malaysia is one of the most important producers of palm oil in the world. During oil palm fruit harvesting, the pruning of oil palm frond (OPF) generates approximately 44 million tonnes dry weight in the oil palm fields annually. Conversely, considering the international scenario, each year about 184.6 million tonnes of lignocellulosic residue is available worldwide from the palm oil industry alone.228 The exploration of this lignocellulosic residue as a biobased renewable resource for manufacturing of industrial products will open a new avenue for the efficient utilization of agricultural waste by reducing the need for disposal and environmental degradation and simultaneously add value to the development of agricultural based economies in rural areas.227Recently, we have reported the pretreatment of lignocellulosic oil palm biomass with imidazolium based ILs.84 The oil palm biomass was ground to a particle size range of 250–300 μm and dissolved in [C2C1im][Et2PO4] or [C4C1im]Cl at 80 °C for 3 h and then regenerated using an acetone–water mixture as an anti-solvent. Afterward, the precipitated solid residue was separated by decantation and washed with distilled water 4–5 times to ensure complete removal of any residual IL. The anti-solvent acetone/water was removed from the supernatant using a rotary evaporator to leave an IL/lignin rich phase. The dried regenerated cellulose-rich fiber was compounded with a thermoplastic starch polymer and was thermally pressed at 160 °C for 10 min in a Carver Laboratory compression molding machine (CARVER, INC. USA) to fabricate a ‘green’ composite board (Fig. 20). The cellulose contents in the regenerated fibers after IL pretreatment were significantly increased, which resulted in a more accessible surface area for binder interaction and improvement in the thermal stability of the fibers. Due to removal of non-cellulosic impurities from the fiber surface as a result of the IL pretreatment, the composite panels fabricated from IL pretreated fiber exhibited superior mechanical and thermal properties. It was clearly indicated that IL-assisted pretreatment could be a new, highly promising and green technology for effective utilization of lignocellulosic biomass in the wood composites and related industries.
 | ||
| Fig. 20 Manufacturing steps to fabricate composite board from ILs treated oil palm fiber.84 | ||
5. Parameters affecting the practical applications of ILs for green composites processing
Several criteria must be considered for the large scale use of ILs for pretreatment and dissolution of lignocellulose and other biobased polymers for the manufacture of sustainable biocomposites. The most significant ones include price, toxicity, corrosivity, physical properties, availability, regeneration requirement, biodegradability and water tolerance. Unfortunately, until now these issues have been rarely discussed. Utilization of ILs in manufacturing engineered green composites based on renewable and biodegradable resources is possible only if both ILs and dissolution conditions are properly optimized.14,24,26,2295.1 Toxicity and corrosivity
Toxicity of ILs is one of the most important parameters when considering their practical applications on industrial scale. The toxicological effect on human health must be further evaluated for the implementation of ILs as solvents in the biocomposite industry. The ease of modification and tunability of IL compositions holds great potential to design targeted properties of ILs which may be useful in this regard.230–232 In addition, IL corrosivity is a very important economic issue while selecting the material of construction for equipment to be used in large scale applications for lignocellulose pretreatment. Halogen-free ILs may be suitable candidates as non-corrosive IL solvents.233,2345.2 Viscosity and water content
Viscosity is another important parameter from the practical point of view, because the higher viscosity of an IL will require a higher dissolution temperature and also more energy for mixing and fluid transfer. Furthermore, this could potentially give rise to other side-effects, such as the generation of unwanted side-reactions.235–237 One promising approach to reduce the viscosity of ILs is to use a co-solvent such as DMSO, polyethylene glycol etc.67,84 Similarly, the water content of an IL also has an impact on its dissolution efficiency. It has been observed that the presence of water in ILs significantly hampers their cellulose dissolution ability.8 Therefore, drying of woody material at 90 °C prior to the dissolution experiments in ILs is strongly recommended to reduce its water content.74 In addition, the use of water as precipitating solvent for dissolved lignocellulose has also been demonstrated.84 The dissolved lignocellulosic material can be easily regenerated for subsequent biorefinery applications by precipitating it from an IL through the addition of water as an anti-solvent.49,74 When water is added as an anti-solvent into the IL and lignocellulose mixture, the IL ions form hydrogen bonds with the water molecules and they become highly hydrated. The interactions between the IL and lignocellulosic particles are hence shielded by the hydration shells formed by the water molecules around the ions of IL, which ultimately provokes the precipitation of the dissolved material.265.3 Recycling and re-use of ILs
The efficient recycling and reuse of ILs is indeed critical to reduce the cost of pretreatment processes as well as to realize the environmental and economic benefits of ILs. This is an inherent advantage in the IL based processing of biomass that biomass dissolved in IL can be regenerated rapidly using precipitation from solution by addition of various anti-solvents, such as water, acetone etc.49,238,239 The IL used can then be easily recovered upon removal of the non-solvent through evaporation. The selection of suitable anti-solvent is considered to be one of the most important factors that has had a significant impact on the yields of the regenerated material and the recycled IL, which ultimately affects the overall cost of the pretreatment process.5.4 Unwanted reactions of solvated biopolymers with IL
The side reaction of IL with biopolymer species during dissolution processes to form intermediate products could not only cause IL recycling and product separation issues but also deteriorate the quality of regenerated material for downstream applications. The reactivity of carboxylate ILs towards carbohydrates was first demonstrated by Ebner et al.184 and later thoroughly explored by Welton and co-workers.240 Mixtures of [C2C1im][OAc] with carbohydrates were found to undergo reaction of the C2 carbon of the imidazolium ring yielding a terminal adduct species 1-ethyl-2-(hydroxymethyl)-3-methylimidazolium acetate by elimination of successive formaldehyde units from the sugar adduct. Such unwanted side reactions of solvated biopolymers with IL presents potential problems for both laboratory and industrial processes, due to the influence of new chemical species on both the physical and rheological properties of the mixture. It is crucial, therefore, to understand factors contributing towards unwanted side reactions, and how they might be prevented or controlled.2405.5 Batch vs. continuous pretreatment
Disregarding its various advantages, currently IL-assisted pretreatment is not feasible for industrial-scale operations due to many reasons, including the large initial biomass loading requirements. The considerable number of studies that investigated IL-mediated pretreatment operations with low biomass loadings (5–10%) could be because of the effective mixing obstructions of conventional stirred reactors for ‘semi-solid’ high biomass loads systems (>20 wt%).241,242 Recently, continuous pretreatment of lignocellulosic material with [C2C1im][OAc] mediated by a twin-screw extruder was reported at biomass loadings as high as 50 wt%. The extrusion process can be carried out continuously and is suitable for industrial-scale lignocellulose processing allowing higher throughputs and reactant concentrations, improved mixing rates as well as more uniform products as compared to batch operations.836. Future prospects
While there is a growing literature of the use of ionic liquids in the processing of renewable resources for the fabrication of biodegradable composite materials, this remains an underdeveloped field of research. The literature is characterized by a large number of initial proof of concept studies. For the future success of ionic liquids in this endeavor, deeper mechanistic understanding of the processes involved is required to enable the optimization of these processes, since their application will only be achieved if the advantages of ILs outweigh their drawbacks, most prominently the prices of ILs relative to the value of the material processed by these.243 Generally the development of pretreatment processes and selection of suitable conditions for efficient processing requires a compromise between conflicting objectives which can be overcome by applying multi-objective optimization techniques in pretreatment process design and selection.Modeling of the process is essential to estimate the energy requirement and cost for dissolution processes, so that the comparison with other pretreatment methods will be possible. The first techno-economic analysis of IL based processing system was carried out and the following order of importance/sensitivity among the investigated variables was reported: IL price > biomass loading > recycling rate.243 Furthermore, molecular level simulation should be employed for better understanding the interaction mechanism of ILs with biobased molecules.244 Additionally, an optimized selection of both cations and anions as well as processing conditions may allow the design of IL systems with unique physicochemical characteristics that could lead to more effective utilization of ILs in lignocellulosic and biopolymer based composite industries.53
Despite all of these advantages and potential applications, ILs currently suffer from clear and significant disadvantages of their high cost that stand in the way of many commercial applications. It is believed that ILs normally fall in the range of 5–20 times more expensive than molecular solvents.245 The most common criticism of ILs that the authors encounter is that of the ‘severe’ limitations placed upon their large-scale deployment by their high cost. But are ILs inherently expensive, or is this opinion a consequence of the specific ILs that are historically prominent (dialkylimidazolium cations with polyfluorinated anions)? To answer this question, Welton and co-workers evaluated the economic feasibility and determined the cost price of two ILs synthesized from cheap raw materials.246 The cost prices of triethylammonium hydrogen sulfate and 1-methylimidazolium hydrogen sulfate were estimated as $1.24 kg−1 and $2.96–5.88 kg−1, respectively, which compares favourably with organic solvents such as acetone or ethyl acetate, which sell for $1.30–$1.40 kg−1. These results indicate that ionic liquids are not necessarily expensive, and therefore large-scale IL-based processes can become a commercial reality. In addition, organic electrolyte solutions containing a small fraction of ILs with potential dissolution capabilities for large amounts of cellulose can lead in the future to very interesting developments.247 Dissolution and depolymerization of cellulose in organic acid-catalyzed 30 wt% aqueous solution of NaCl under mild reaction conditions (100–125 °C) and some pressure (10–30 bar) has also been observed.248 It was proposed that such salt solutions disrupt the hydrogen-bond matrix among cellulose fibrils in a mechanism similar to that of ILs. Development of such ILs based solvent systems, in the future, might open new pathways in the field of biopolymer processing in which environmental concerns can be tackled, while simultaneously keeping process costs (energy, substrates, catalysts, etc.) at an acceptable level.
It must also be emphasized that conventional dissolution processes employ various solvent systems under extreme conditions and also are limited due to their dissolving capability, solvent recovery, toxicity, uncontrollable side reactions and high cost during biomass processing and derivatisation, the following list summarizes the major aspects for IL based processing of natural fibers and polymers that should receive attention for future research.24
■ Low cost of ILs and co-solvents (typically less than $2.50 kg−1)
■ Decreased IL loading
■ Short dissolution time
■ Applicability to a wide range of raw materials
■ End of use IL recovery
■ Optimized recycling to reduce ILs losses
■ Reduced influence of residual IL on downstream processing
■ ‘Greenness’ of ILs (environmental and health impact).
Acknowledgements
The authors would like to acknowledge Universiti Teknologi PETRONAS for University Research Internal Fund (URIF) under grant 0153AA-B80 which enabled this research work to be accomplished. TW would like to thank the EPSRC for funding (EP/K014676/1 and EP/L017679/1). We also extend apologies to those whose biocomposite research using ILs was not cited due to space limitations.References
- P. Wasserscheid and T. Welton, Ionic liquids in Synthesis, John Wiley & Sons, New York, 2008 Search PubMed.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790–2794 RSC.
- M. J. Earle, J. M. Esperança, M. A. Gilea, J. N. C. Lopes, L. P. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS PubMed.
- O. Aschenbrenner, S. Supasitmongkol, M. Taylor and P. Styring, Green Chem., 2009, 11, 1217–1221 RSC.
- J. L. Anderson, J. Ding, T. Welton and D. W. Armstrong, J. Am. Chem. Soc., 2002, 124, 14247–14254 CrossRef CAS PubMed.
- T. Welton, Green Chem., 2011, 13, 225–225 RSC.
- M. Kosmulski, J. Gustafsson and J. B. Rosenholm, Thermochim. Acta, 2004, 412, 47–53 CrossRef CAS.
- P. Verdía, A. Brandt, J. P. Hallett, M. J. Ray and T. Welton, Green Chem., 2014, 16, 1617–1627 RSC.
- M. Freemantle, Chem. Eng. News, 1998, 76, 32–37 Search PubMed.
- V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459–2477 CrossRef CAS.
- A. E. Visser, R. P. Swatloski, S. T. Griffin, D. H. Hartman and R. D. Rogers, Sep. Sci. Technol., 2001, 36, 785–804 CrossRef CAS.
- M. S. Rajabi, M. Moniruzzaman, H. Mahmood, M. Sivapragasam and M. A. Bustam, J. Mol. Liq., 2017, 227, 15–20 CrossRef CAS.
- Y. Zhou, Curr. Nanosci., 2005, 1, 35–42 CrossRef CAS.
- M. Antonietti, D. Kuang, B. Smarsly and Y. Zhou, Angew. Chem., Int. Ed., 2004, 43, 4988–4992 CrossRef CAS PubMed.
- J. L. Anderson and D. W. Armstrong, Anal. Chem., 2005, 77, 6453–6462 CrossRef CAS PubMed.
- F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef CAS PubMed.
- M. Moniruzzaman, K. Ino, N. Kamiya and M. Goto, Org. Biomol. Chem., 2012, 10, 7707–7713 CAS.
- W. L. Hough and R. D. Rogers, Bull. Chem. Soc. Jpn., 2007, 80, 2262–2269 CrossRef CAS.
- M. Moniruzzaman and M. Goto, J. Chem. Eng. Jpn., 2011, 44, 370–381 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- P. Mäki-Arvela, I. Anugwom, P. Virtanen, R. Sjöholm and J.-P. Mikkola, Ind. Crops Prod., 2010, 32, 175–201 CrossRef.
- S. Lems, H. Van der Kooi and J. de Swaan Arons, Green Chem., 2002, 4, 308–313 RSC.
- K. Protocol, Kyoto Protocol, Kyoto, 1997.
- C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 17, 40–71 RSC.
- H. Khoo, W. Ee and V. Isoni, Green Chem., 2016, 18, 1912–1922 RSC.
- H. Mahmood, M. Moniruzzaman, S. Yusup and H. M. Akil, Chem. Eng. Trans., 2015, 45, 709–714 Search PubMed.
- C. Vila, A. Campos, C. Cristovao, A. Cunha, V. Santos and J. Parajó, Compos. Sci. Technol., 2008, 68, 944–952 CrossRef CAS.
- M. Abe, K. Kuroda and H. Ohno, ACS Sustainable Chem. Eng., 2015, 3, 1771–1776 CrossRef CAS.
- M. H. Zamri, H. M. Akil, A. A. Bakar, Z. A. M. Ishak and L. W. Cheng, J. Compos. Mater., 2011, 46, 51–61 CrossRef.
- A. Engström, M. Ek and G. Henriksson, Biomacromolecules, 2006, 7, 2027–2031 CrossRef PubMed.
- S. Righi, A. Morfino, P. Galletti, C. Samorì, A. Tugnoli and C. Stramigioli, Green Chem., 2011, 13, 367–375 RSC.
- T. Rosenau, A. Potthast, H. Sixta and P. Kosma, Prog. Polym. Sci., 2001, 26, 1763–1837 CrossRef CAS.
- F. Hermanutz, F. Gähr, E. Uerdingen, F. Meister and B. Kosan, Macromol. Symp., 2008, 262, 23–27 CrossRef CAS.
- P. Langan, L. Petridis, H. M. O'Neill, S. V. Pingali, M. Foston, Y. Nishiyama, R. Schulz, B. Lindner, B. L. Hanson and S. Harton, Green Chem., 2014, 16, 63–68 RSC.
- M. Arshadi, T. M. Attard, R. M. Lukasik, M. Brncic, A. M. da Costa Lopes, M. Finell, P. Geladi, L. N. Gerschenson, F. Gogus and M. Herrero, Green Chem., 2016, 18, 6160–6204 RSC.
- A. Tilahun and B. S. Chun, J. Cleaner Prod., 2016, 120, 3719–3727 Search PubMed.
- X. Meng, T. Wells, Q. Sun, F. Huang and A. Ragauskas, Green Chem., 2015, 17, 4239–4246 RSC.
- Z. Zhang, M. D. Harrison, D. W. Rackemann, W. O. Doherty and I. M. O'Hara, Green Chem., 2016, 18, 360–381 RSC.
- F. Teymouri, L. Laureano-Perez, H. Alizadeh and B. E. Dale, Bioresour. Technol., 2005, 96, 2014–2018 CrossRef CAS PubMed.
- H. Mahmood, M. Moniruzzaman, S. Yusup and H. M. Akil, Clean Technol. Environ. Policy, 2016, 18, 2217–2226 CrossRef CAS.
- R. Xie, Y. Yuan and J.-j. Huang, Ecol. Econ., 2017, 132, 104–112 CrossRef.
- G. F. De Gregorio, C. C. Weber, J. Gräsvik, T. Welton, A. Brandt and J. P. Hallett, Green Chem., 2016, 18, 5456–5465 RSC.
- A. George, A. Brandt, K. Tran, S. M. N. S. Zahari, D. Klein-Marcuschamer, N. Sun, N. Sathitsuksanoh, J. Shi, V. Stavila and R. Parthasarathi, Green Chem., 2015, 17, 1728–1734 RSC.
- M. Moniruzzaman and T. Ono, Bioresour. Technol., 2013, 127, 132–137 CrossRef CAS PubMed.
- S. H. Lee, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2009, 102, 1368–1376 CrossRef CAS PubMed.
- H. Mahmood, M. Moniruzzaman, S. Yusup and H. M. Akil, Polym. Compos., 2016 DOI:10.1002/pc.24159.
- A. Idris, R. Vijayaraghavan, U. A. Rana, D. Fredericks, A. Patti and D. MacFarlane, Green Chem., 2013, 15, 525–534 RSC.
- B. Lu, A. Xu and J. Wang, Green Chem., 2014, 16, 1326–1335 RSC.
- S. Zhang, N. Sun, X. He, X. Lu and X. Zhang, J. Phys. Chem. Ref. Data, 2006, 35, 1475–1517 CrossRef CAS.
- Q. Dong, C. D. Muzny, A. Kazakov, V. Diky, J. W. Magee, J. A. Widegren, R. D. Chirico, K. N. Marsh and M. Frenkel, J. Chem. Eng. Data, 2007, 52, 1151–1159 CrossRef CAS.
- S. Aparicio, M. Atilhan and F. Karadas, Ind. Eng. Chem. Res., 2010, 49, 9580–9595 CrossRef CAS.
- P. A. Hunt, I. R. Gould and B. Kirchner, Aust. J. Chem., 2007, 60, 9–14 CrossRef CAS.
- I. Skarmoutsos, D. Dellis, R. P. Matthews, T. Welton and P. A. Hunt, J. Phys. Chem. B, 2012, 116, 4921–4933 CrossRef CAS PubMed.
- H. Passos, M. G. Freire and J. A. Coutinho, Green Chem., 2014, 16, 4786–4815 RSC.
- M. Moniruzzaman and T. Ono, Biochem. Eng. J., 2012, 60, 156–160 CrossRef CAS.
- A. Chapeaux, L. D. Simoni, M. A. Stadtherr and J. F. Brennecke, J. Chem. Eng. Data, 2007, 52, 2462–2467 CrossRef CAS.
- K. A. Fletcher, I. A. Storey, A. E. Hendricks, S. Pandey and S. Pandey, Green Chem., 2001, 3, 210–215 RSC.
- Y. Liu, K. Thomsen, Y. Nie, S. Zhang and A. S. Meyer, Green Chem., 2016, 18, 6246–6254 RSC.
- C. M. Hansen, Hansen solubility parameters: a user's handbook, CRC Press, 2007 Search PubMed.
- M. Ab Rani, A. Brant, L. Crowhurst, A. Dolan, M. Lui, N. Hassan, J. Hallett, P. Hunt, H. Niedermeyer and J. Perez-Arlandis, Phys. Chem. Chem. Phys., 2011, 13, 16831–16840 RSC.
- L. Cammarata, S. Kazarian, P. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192–5200 RSC.
- L. Wu, S.-H. Lee and T. Endo, Bioresour. Technol., 2013, 140, 90–96 CrossRef CAS PubMed.
- M. Sivapragasam, M. Moniruzzaman and M. Goto, Biotechnol. J., 2016, 11, 1000–1013 CrossRef CAS PubMed.
- R. Wu, X. Wang, F. Li, H.-Z. Li and Y. Wang, Bioresour. Technol., 2009, 100, 2569–2574 CrossRef CAS PubMed.
- H. Xie, S. Zhang and S. Li, Green Chem., 2006, 8, 630–633 RSC.
- Q. Chen, A. Xu, Z. Li, J. Wang and S. Zhang, Green Chem., 2011, 13, 3446–3452 RSC.
- K. Park, J. U. Ha and M. Xanthos, Polym. Eng. Sci., 2010, 50, 1105 CAS.
- Y. Pu, N. Jiang and A. J. Ragauskas, J. Wood Chem. Technol., 2007, 27, 23–33 CrossRef CAS.
- N. Sun, M. Rahman, Y. Qin, M. L. Maxim, H. Rodríguez and R. D. Rogers, Green Chem., 2009, 11, 646–655 RSC.
- W. Lan, C.-F. Liu and R.-C. Sun, J. Agric. Food Chem., 2011, 59, 8691–8701 CrossRef CAS PubMed.
- Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2008, 10, 44–46 RSC.
- M. Guo, Q. Gu, P. Chen, L. Zhao and H. Liang, J. Polym. Mater., 2015, 32, 31 CAS.
- M. Moniruzzaman, M. R. Talukder, Y. Hayashi and T. Kawanishi, Appl. Biochem. Biotechnol., 2007, 142, 253–262 CrossRef CAS PubMed.
- A. Sankri, A. Arhaliass, I. Dez, A. C. Gaumont, Y. Grohens, D. Lourdin, I. Pillin, A. Rolland-Sabaté and E. Leroy, Carbohydr. Polym., 2010, 82, 256–263 CrossRef CAS.
- E. Leroy, P. Jacquet, G. Coativy, A. laure Reguerre and D. Lourdin, Carbohydr. Polym., 2012, 89, 955–963 CrossRef CAS PubMed.
- A. A. Modenbach and S. E. Nokes, Biotechnol. Bioeng., 2012, 109, 1430–1442 CrossRef CAS PubMed.
- L.-P. Zhang, J. Zhang, C.-H. Li and J. Bao, Biochem. Eng. J., 2014, 90, 324–332 CrossRef CAS.
- A. S. A. da Silva, R. S. S. Teixeira, T. Endo, E. P. Bon and S. Lee, Green Chem., 2013, 15, 1991–2001 RSC.
- H. Mahmood, M. Moniruzzaman, S. Yusup and H. M. Akil, J. Cleaner Prod., 2016, 126, 677–685 CrossRef CAS.
- R. Sescousse, R. Gavillon and T. Budtova, Carbohydr. Polym., 2011, 83, 1766–1774 CrossRef CAS.
- R. Gavillon and T. Budtova, Biomacromolecules, 2007, 8, 424–432 CrossRef CAS PubMed.
- M. Ghasemi, M. Tsianou and P. Alexandridis, Bioresour. Technol., 2017, 228, 330–338 CrossRef CAS PubMed.
- R. Sescousse and T. Budtova, Cellulose, 2009, 16, 417–426 CrossRef CAS.
- F. Hermanutz, F. Gähr, E. Uerdingen, F. Meister and B. Kosan, Macromol. Symp., 2008, 262, 23–27 CrossRef CAS.
- F. Wendler, L. Todi and F. Meister, Thermochim. Acta, 2012, 528, 76–84 CrossRef CAS.
- L. Alves, B. Medronho, F. E. Antunes, D. Topgaard and B. Lindman, Carbohydr. Polym., 2016, 151, 707–715 CrossRef CAS PubMed.
- D. Liu, K. Xia, W. Cai, R. Yang, L. Wang and B. Wang, Carbohydr. Polym., 2012, 87, 1058–1064 CrossRef CAS.
- M. E. Zakrzewska, E. Łukasik and R. Łukasik, Energy Fuels, 2010, 24, 737–745 CrossRef CAS.
- S. Zhang, F. Li, J. Yu and Y. Hsieh, Carbohydr. Polym., 2010, 81, 668–674 CrossRef CAS.
- J. Cai and L. Zhang, Biomacromolecules, 2006, 7, 183–189 CrossRef CAS PubMed.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- S. M. Hudson and J. A. Cuculo, J. Macromol. Sci., Rev. Macromol. Chem., 1980, 18, 1–82 CrossRef.
- S. Sen, J. D. Martin and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2013, 1, 858–870 CrossRef CAS.
- A. Ostlund, D. Lundberg, L. Nordstierna, K. Holmberg and M. Nydén, Biomacromolecules, 2009, 10, 2401–2407 CrossRef PubMed.
- R. C. Remsing, G. Hernandez, R. P. Swatloski, W. W. Massefski, R. D. Rogers and G. Moyna, J. Phys. Chem. B, 2008, 112, 11071–11078 CrossRef CAS PubMed.
- J. Zhang, H. Zhang, J. Wu, J. Zhang, J. He and J. Xiang, Phys. Chem. Chem. Phys., 2010, 12, 1941–1947 RSC.
- R. L. Quirino, T. F. Garrison and M. R. Kessler, Green Chem., 2014, 16, 1700–1715 RSC.
- I. Šimkovic, Carbohydr. Polym., 2008, 74, 759–762 CrossRef.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- B. Li, J. Asikkala, I. Filpponen and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2010, 49, 2477–2484 CrossRef CAS.
- H. Zhang, J. Wu, J. Zhang and J. He, Macromolecules, 2005, 38, 8272–8277 CrossRef CAS.
- Z. Liu, H. Wang, Z. Li, X. Lu, X. Zhang, S. Zhang and K. Zhou, Mater. Chem. Phys., 2011, 128, 220–227 CrossRef CAS.
- A. Takegawa, M. Murakami, Y. Kaneko and J. Kadokawa, Carbohydr. Polym., 2010, 79, 85–90 CrossRef CAS.
- Z. Meng, X. Zheng, K. Tang, J. Liu, Z. Ma and Q. Zhao, Int. J. Biol. Macromol., 2012, 51, 440–448 CrossRef CAS PubMed.
- R. Li and D. Wang, J. Appl. Polym. Sci., 2013, 127, 2648–2653 CrossRef CAS.
- R. De Silva, X. Wang and N. Byrne, Cellulose, 2013, 20, 2461–2468 CrossRef.
- N. Singh, K. K. Koziol, J. Chen, A. J. Patil, J. W. Gilman, P. C. Trulove, W. Kafienah and S. S. Rahatekar, Green Chem., 2013, 15, 1192–1202 RSC.
- R. De Silva, K. Vongsanga, X. Wang and N. Byrne, Carbohydr. Polym., 2015, 121, 382–387 CrossRef CAS PubMed.
- S. Agarwal, A. Greiner and J. H. Wendorff, Prog. Polym. Sci., 2013, 38, 963–991 CrossRef CAS.
- Z.-M. Huang, Y.-Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS.
- L. Meli, J. Miao, J. S. Dordick and R. J. Linhardt, Green Chem., 2010, 12, 1883–1892 RSC.
- M. Polaskova, R. Cermak, V. Verney, P. Ponizil, S. Commereuc, M. F. C. Gomes, A. A. Padua, P. Mokrejs and M. Machovsky, Carbohydr. Polym., 2013, 92, 214–217 CrossRef CAS PubMed.
- Y. Ahn, S. H. Lee, H. J. Kim, Y.-H. Yang, J. H. Hong, Y.-H. Kim and H. Kim, Carbohydr. Polym., 2012, 88, 395–398 CrossRef CAS.
- G. Jiang, W. Huang, L. Li, X. Wang, F. Pang, Y. Zhang and H. Wang, Carbohydr. Polym., 2012, 87, 2012–2018 CrossRef CAS.
- S.-L. Quan, S.-G. Kang and I.-J. Chin, Cellulose, 2010, 17, 223–230 CrossRef CAS.
- Y. Qin, X. Lu, N. Sun and R. D. Rogers, Green Chem., 2010, 12, 968–971 RSC.
- B. Ma, A. Qin, X. Li and C. He, Carbohydr. Polym., 2013, 97, 300–305 CrossRef CAS PubMed.
- B. Ma, M. Zhang, C. He and J. Sun, Carbohydr. Polym., 2012, 88, 347–351 CrossRef CAS.
- G. Bentivoglio, T. Röder, M. Fasching, M. Buchberger, H. Schottenberger and H. Sixta, Lenzinger Ber., 2006, 86, 154–161 CAS.
- L. K. Hauru, M. Hummel, A. Michud and H. Sixta, Cellulose, 2014, 21, 4471–4481 CrossRef CAS.
- S. S. Rahatekar, A. Rasheed, R. Jain, M. Zammarano, K. K. Koziol, A. H. Windle, J. W. Gilman and S. Kumar, Polymer, 2009, 50, 4577–4583 CrossRef CAS.
- C. Zhu, J. Chen, K. Koziol, J. Gilman, P. Trulove and S. Rahatekar, eXPRESS Polym. Lett., 2014, 8, 154–163 CrossRef CAS.
- C. Zhu, R. M. Richardson, K. D. Potter, A. F. Koutsomitopoulou, J. S. van Duijneveldt, S. R. Vincent, N. D. Wanasekara, S. J. Eichhorn and S. S. Rahatekar, ACS Sustainable Chem. Eng., 2016, 4, 4545–4553 CrossRef CAS.
- R. De Silva, X. Wang and N. Byrne, Carbohydr. Polym., 2016, 153, 115–123 CrossRef CAS PubMed.
- N. Sun, W. Li, B. Stoner, X. Jiang, X. Lu and R. D. Rogers, Green Chem., 2011, 13, 1158–1161 RSC.
- J.-i. Kadokawa, M.-a. Murakami, A. Takegawa and Y. Kaneko, Carbohydr. Polym., 2009, 75, 180–183 CrossRef CAS.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243–4266 CrossRef CAS PubMed.
- J. A. Kenar, F. J. Eller, F. C. Felker, M. A. Jackson and G. F. Fanta, Green Chem., 2014, 16, 1921–1930 RSC.
- X. Shen, J. L. Shamshina, P. Berton, G. Gurau and R. D. Rogers, Green Chem., 2016, 18, 53–75 RSC.
- B. Balakrishnan and R. Banerjee, Chem. Rev., 2011, 111, 4453–4474 CrossRef CAS PubMed.
- C. García-González, M. Alnaief and I. Smirnova, Carbohydr. Polym., 2011, 86, 1425–1438 CrossRef.
- S. V. Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef PubMed.
- J. Li, Y. Lu, D. Yang, Q. Sun, Y. Liu and H. Zhao, Biomacromolecules, 2011, 12, 1860–1867 CrossRef CAS PubMed.
- R. Sescousse, R. Gavillon and T. Budtova, Carbohydr. Polym., 2011, 83, 1766–1774 CrossRef CAS.
- J. Kadokawa, M. Murakami and Y. Kaneko, Carbohydr. Res., 2008, 343, 769–772 CrossRef CAS PubMed.
- S. S. Silva, E. G. Popa, M. E. Gomes, M. B. Oliveira, S. Nayak, B. Subia, J. F. Mano, S. C. Kundu and R. L. Reis, Acta Biomater., 2013, 9, 8972–8982 CrossRef CAS PubMed.
- M. Xu, Q. Huang, X. Wang and R. Sun, Ind. Crops Prod., 2015, 70, 56–63 CrossRef CAS.
- N. Hameed and Q. Guo, Cellulose, 2010, 17, 803–813 CrossRef CAS.
- Y. Cao, H. Li, Y. Zhang, J. Zhang and J. He, J. Appl. Polym. Sci., 2010, 116, 547–554 CrossRef CAS.
- S. Shang, L. Zhu and J. Fan, Carbohydr. Polym., 2011, 86, 462–468 CrossRef CAS.
- S. Mahmoudian, M. U. Wahit, A. Ismail and A. Yussuf, Carbohydr. Polym., 2012, 88, 1251–1257 CrossRef CAS.
- R. Wu, X. Wang, Y. Wang, X. Bian and F. Li, Ind. Eng. Chem. Res., 2009, 48, 7132–7136 CrossRef CAS.
- L. Haverhals, T. A. Isaacs, E. Page, W. M. Reichert, H. De Long and P. C. Trulove, ECS Trans., 2009, 16, 129–139 CAS.
- C. Stefanescu, W. H. Daly and I. I. Negulescu, Carbohydr. Polym., 2012, 87, 435–443 CrossRef CAS.
- O. Kuzmina, T. Heinze and D. Wawro, ISRN Polym. Sci., 2012, 2012, 251950 Search PubMed.
- Y. Kang, Y. Ahn, S. H. Lee, J. H. Hong, M. K. Ku and H. Kim, Fibers Polym., 2013, 14, 530–536 CrossRef CAS.
- H. B. Kocer, I. Cerkez, S. Worley, R. Broughton and T. Huang, Carbohydr. Polym., 2011, 86, 922–927 CrossRef CAS.
- D. M. Phillips, L. F. Drummy, R. R. Naik, C. Hugh, D. M. Fox, P. C. Trulove and R. A. Mantz, J. Mater. Chem., 2005, 15, 4206–4208 RSC.
- J. Zhu, S. Wei, R. Patil, D. Rutman, A. S. Kucknoor, A. Wang and Z. Guo, Polymer, 2011, 52, 1954–1962 CrossRef CAS.
- G. Viswanathan, S. Murugesan, V. Pushparaj, O. Nalamasu, P. M. Ajayan and R. J. Linhardt, Biomacromolecules, 2006, 7, 415–418 CrossRef CAS PubMed.
- S. Xu, J. Zhang, A. He, J. Li, H. Zhang and C. C. Han, Polymer, 2008, 49, 2911–2917 CrossRef CAS.
- M. G. Freire, A. R. R. Teles, R. A. Ferreira, L. D. Carlos, J. A. Lopes-da-Silva and J. A. Coutinho, Green Chem., 2011, 13, 3173–3180 RSC.
- H. Zhang, Z. Wang, Z. Zhang, J. Wu, J. Zhang and J. He, Adv. Mater., 2007, 19, 698–704 CrossRef CAS.
- P. S. Barber, C. S. Griggs, J. R. Bonner and R. D. Rogers, Green Chem., 2013, 15, 601–607 RSC.
- N. D. Wanasekara, A. Michud, C. Zhu, S. Rahatekar, H. Sixta and S. J. Eichhorn, Polymer, 2016, 99, 222–230 CrossRef CAS.
- T. Park, Y. J. Jung, H. Park, S.-W. Choi, E. Kim, S. H. Lee and J. H. Kim, Macromol. Res., 2011, 19, 317–321 CrossRef CAS.
- X. Hu, K. Hu, L. Zeng, M. Zhao and H. Huang, Carbohydr. Polym., 2010, 82, 62–68 CrossRef CAS.
- X. Liang, B. Qu, J. Li, H. Xiao, B. He and L. Qian, React. Funct. Polym., 2015, 86, 1–6 CrossRef CAS.
- J. Wang, L. Wei, Y. Ma, K. Li, M. Li, Y. Yu, L. Wang and H. Qiu, Carbohydr. Polym., 2013, 98, 736–743 CrossRef CAS PubMed.
- L. Li, Z. Lin, X. Yang, Z. Wan and S. Cui, Chin. Sci. Bull., 2009, 54, 1622–1625 CAS.
- T. Oshima, T. Sakamoto, K. Ohe and Y. Baba, Carbohydr. Polym., 2014, 103, 62–69 CrossRef CAS PubMed.
- A. Demilecamps, C. Beauger, C. Hildenbrand, A. Rigacci and T. Budtova, Carbohydr. Polym., 2015, 122, 293–300 CrossRef CAS PubMed.
- K. Prasad, M.-a. Murakami, Y. Kaneko, A. Takada, Y. Nakamura and J.-i. Kadokawa, Int. J. Biol. Macromol., 2009, 45, 221–225 CrossRef CAS PubMed.
- O. Aaltonen and O. Jauhiainen, Carbohydr. Polym., 2009, 75, 125–129 CrossRef CAS.
- A. A. Shamsuri, D. K. Abdullah and R. Daik, Cellul. Chem. Technol., 2012, 46, 45–52 CAS.
- T. J. Trivedi, K. S. Rao and A. Kumar, Green Chem., 2014, 16, 320–330 RSC.
- K. G. Satyanarayana, G. G. Arizaga and F. Wypych, Prog. Polym. Sci., 2009, 34, 982–1021 CrossRef CAS.
- L. Wei, N. M. Stark and A. G. McDonald, Green Chem., 2015, 17, 4800–4814 RSC.
- T. Huber, S. Pang and M. P. Staiger, Composites, Part A, 2012, 43, 1738–1745 CrossRef CAS.
- T. Nishino, I. Matsuda and K. Hirao, Macromol., 2004, 37, 7683–7687 CrossRef CAS.
- H. Ma, B. Zhou, H.-S. Li, Y.-Q. Li and S.-Y. Ou, Carbohydr. Polym., 2011, 84, 383–389 CrossRef CAS.
- K. O. Reddy, J. Zhang, J. Zhang and A. V. Rajulu, Carbohydr. Polym., 2014, 114, 537–545 CrossRef CAS PubMed.
- W. Altmutter, J. Keckes, J. Plackner, F. Liebner, K. Englund and M. Laborie, Compos. Sci. Technol., 2012, 72, 1304–1309 CrossRef.
- L. M. Haverhals, W. M. Reichert, H. C. De Long and P. C. Trulove, Macromol. Mater. Eng., 2010, 295, 425–430 CrossRef CAS.
- L. M. Haverhals, H. M. Sulpizio, Z. A. Fayos, M. A. Trulove, W. M. Reichert, M. P. Foley, C. Hugh and P. C. Trulove, Cellulose, 2012, 19, 13–22 CrossRef CAS.
- J. Wu, J. Zhang, H. Zhang, J. He, Q. Ren and M. Guo, Biomacromolecules, 2004, 5, 266–268 CrossRef CAS PubMed.
- G. Ebner, S. Schiehser, A. Potthast and T. Rosenau, Tetrahedron Lett., 2008, 49, 7322–7324 CrossRef CAS.
- J. Reina, E. Velo and L. Puigjaner, Ind. Eng. Chem. Res., 1998, 37, 4290–4295 CrossRef CAS.
- J. A. Cooper, J. Air Pollut. Control Assoc., 1980, 30, 855–861 CrossRef CAS.
- A. Cobut, P. Blanchet and R. Beauregard, J. Cleaner Prod., 2015, 109, 232–246 CrossRef.
- D. Jochem, N. Janzen and H. Weimar, J. Cleaner Prod., 2016, 137, 1216–1227 CrossRef.
- M. Shibata, K. Yamazoe, M. Kuribayashi and Y. Okuyama, J. Appl. Polym. Sci., 2013, 127, 4802–4808 CrossRef CAS.
- M. Shibata, N. Teramoto, T. Nakamura and Y. Saitoh, Carbohydr. Polym., 2013, 98, 1532–1539 CrossRef CAS PubMed.
- B. J. Duchemin, A. P. Mathew and K. Oksman, Composites, Part A, 2009, 40, 2031–2037 CrossRef.
- Q. Zhao, R. C. Yam, B. Zhang, Y. Yang, X. Cheng and R. K. Li, Cellulose, 2009, 16, 217–226 CrossRef CAS.
- J. Chen, X. Chen, M. Su, J. Ye, J. Hong and Z. Yang, Chem. Eng. J., 2015, 279, 136–142 CrossRef CAS.
- T. J. Simmons, S. H. Lee, J. Miao, M. Miyauchi, T. Park, S. S. Bale, R. Pangule, J. Bult, J. G. Martin and J. S. Dordick, Wood Sci. Technol., 2011, 45, 719–733 CrossRef CAS.
- V. L. Pushparaj, M. M. Shaijumon, A. Kumar, S. Murugesan, L. Ci, R. Vajtai, R. J. Linhardt, O. Nalamasu and P. M. Ajayan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13574–13577 CrossRef CAS PubMed.
- M. Ho, H. Wang, J. Lee, C. Ho, K. Lau, J. Leng and D. Hui, Composites, Part B, 2012, 43, 3549–3562 CrossRef CAS.
- S. Kalia, B. Kaith and I. Kaur, Polym. Eng. Sci., 2009, 49, 1253–1272 CAS.
- S. Behera, R. Arora, N. Nandhagopal and S. Kumar, Renewable Sustainable Energy Rev., 2014, 36, 91–106 CrossRef CAS.
- M. Fasching, P. Schröder, R. P. Wollboldt, H. K. Weber and H. Sixta, Holzforschung, 2008, 62, 15–23 CrossRef CAS.
- J. Wen, Y. Sun, L. Meng, T. Yuan, F. Xu and R. Sun, Ind. Crops Prod., 2011, 34, 1491–1501 CrossRef CAS.
- H. Xie, P. Jarvi, M. Karesoja, A. King, I. Kilpelainen and D. S. Argyropoulos, J. Appl. Polym. Sci., 2009, 111, 2468–2476 CrossRef CAS.
- T. Yuan, L. Zhang, F. Xu and R. Sun, Ind. Crops Prod., 2013, 45, 36–43 CrossRef CAS.
- C. Croitoru, S. Patachia, N. Cretu, A. Boer and C. Friedrich, Appl. Surf. Sci., 2011, 257, 6220–6225 CrossRef CAS.
- C. Croitoru, S. Patachia, F. Doroftei, E. Parparita and C. Vasile, Appl. Surf. Sci., 2014, 314, 956–966 CrossRef CAS.
- H. Xie, A. King, I. Kilpelainen, M. Granstrom and D. S. Argyropoulos, Biomacromolecules, 2007, 8, 3740–3748 CrossRef CAS PubMed.
- X. Shen, Y. Xie and Q. Wang, BioResources, 2016, 12, 684–695 CrossRef.
- S. Patachia, C. Croitoru and C. Friedrich, Appl. Surf. Sci., 2012, 258, 6723–6729 CrossRef CAS.
- B. Boury and S. Plumejeau, Green Chem., 2015, 17, 72–88 RSC.
- R. J. Andrews and E. A. Grulke, Wiley Database Polym. Prop, 2003 Search PubMed.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC.
- K. Van de Velde and P. Kiekens, Polym. Test., 2002, 21, 433–442 CrossRef CAS.
- H. Liu, F. Xie, L. Yu, L. Chen and L. Li, Prog. Polym. Sci., 2009, 34, 1348–1368 CrossRef CAS.
- N. V. Shvedene, D. V. Chernyshov, M. G. Khrenova, A. A. Formanovsky, V. E. Baulin and I. V. Pletnev, Electroanalysis, 2006, 18, 1416–1421 CrossRef CAS.
- K. Wilpiszewska and T. Spychaj, Carbohydr. Polym., 2011, 86, 424–428 CrossRef CAS.
- W. Ning, Z. Xingxiang, L. Haihui and H. Benqiao, Carbohydr. Polym., 2009, 76, 482–484 CrossRef.
- W. Ning, Z. Xingxiang, L. Haihui and H. Na, Polym. Polym. Compos., 2010, 18, 53 Search PubMed.
- A. Bendaoud and Y. Chalamet, Carbohydr. Polym., 2014, 108, 75–82 CrossRef CAS PubMed.
- R. Ou, Y. Xie, Q. Wang, S. Sui and M. P. Wolcott, Composites, Part A, 2014, 61, 134–140 CrossRef CAS.
- P. F. Sommerhuber, T. Wang and A. Krause, J. Cleaner Prod., 2016, 121, 176–185 CrossRef CAS.
- S. K. Najafi, Waste Manage., 2013, 33, 1898–1905 CrossRef PubMed.
- L. Sobczak, R. W. Lang and A. Haider, Compos. Sci. Technol., 2012, 72, 550–557 CrossRef CAS.
- G. Koronis, A. Silva and M. Fontul, Composites, Part B, 2013, 44, 120–127 CrossRef CAS.
- N. Ayrilmis, S. Jarusombuti, V. Fueangvivat and P. Bauchongkol, Polym. Degrad. Stab., 2011, 96, 818–822 CrossRef CAS.
- C. Schuerch, M. P. Burdick and M. Mahdalik, Ind. Eng. Chem. Prod. Res. Dev., 1966, 5, 101–105 CrossRef CAS.
- Y. Kojima, A. Kawabata, H. Kobori, S. Suzuki, H. Ito, R. Makise and M. Okamoto, J. Wood Sci., 2016, 62, 518–525 CrossRef CAS.
- P. Klímek, R. Wimmer, P. K. Mishra and J. Kúdela, J. Cleaner Prod., 2017, 141, 812–817 CrossRef.
- M. F. N. dos Santos, B. Rosane Ap G, B. S. Bezerra and H. S. Varum, J. Cleaner Prod., 2014, 64, 345–355 CrossRef CAS.
- M. F. Awalludin, O. Sulaiman, R. Hashim and W. N. A. W. Nadhari, Renewable Sustainable Energy Rev., 2015, 50, 1469–1484 CrossRef CAS.
- F. Ibrahim, M. Moniruzzaman, S. Yusup and Y. Uemura, J. Mol. Liq., 2015, 211, 370–372 CrossRef CAS.
- H. Tadesse and R. Luque, Energy Environ. Sci., 2011, 4, 3913–3929 CAS.
- Y. Zhao, J. Zhao, Y. Huang, Q. Zhou, X. Zhang and S. Zhang, J. Hazard. Mater., 2014, 278, 320–329 CrossRef CAS PubMed.
- S. Stolte, M. Matzke, J. Arning, A. Böschen, W. Pitner, U. Welz-Biermann, B. Jastorff and J. Ranke, Green Chem., 2007, 9, 1170–1179 RSC.
- M. Uerdingen, C. Treber, M. Balser, G. Schmitt and C. Werner, Green Chem., 2005, 7, 321–325 RSC.
- R. G. Reddy, Z. Zhang, M. F. Arenas and D. M. Blake, High Temp. Mater. Processes, 2003, 22, 87–94 CAS.
- X. Wang, Y. Chi and T. Mu, J. Mol. Liq., 2014, 193, 262–266 CrossRef CAS.
- K. Ghandi, Green Sustainable Chem., 2014, 4, 43349 Search PubMed.
- N. Adawiyah, M. Moniruzzaman, S. Hawatulaila and M. Goto, MedChemComm, 2016, 7, 1881–1897 RSC.
- N. L. Mai, K. Ahn and Y. Koo, Process Biochem., 2014, 49, 872–881 CrossRef CAS.
- K. Haerens, S. Van Deuren, E. Matthijs and B. Van der Bruggen, Green Chem., 2010, 12, 2182–2188 RSC.
- M. T. Clough, K. Geyer, P. A. Hunt, S. Son, U. Vagt and T. Welton, Green Chem., 2015, 17, 231–243 RSC.
- Y. He, J. Zhang and J. Bao, Bioresour. Technol., 2014, 158, 360–364 CrossRef CAS PubMed.
- A. G. Cruz, C. Scullin, C. Mu, G. Cheng, V. Stavila, P. Varanasi, D. Xu, J. Mentel, Y.-D. Chuang and B. A. Simmons, Biotechnol. Biofuels, 2013, 6, 1–11 CrossRef PubMed.
- D. Klein-Marcuschamer, B. A. Simmons and H. W. Blanch, Biofuels, Bioprod. Biorefin., 2011, 5, 562–569 CrossRef CAS.
- M. Diedenhofen, F. Eckert and A. Klamt, J. Chem. Eng. Data, 2003, 48, 475–479 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC.
- R. Rinaldi, Chem. Commun., 2011, 47, 511–513 RSC.
- T. vom Stein, P. Grande, F. Sibilla, U. Commandeur, R. Fischer, W. Leitner and P. D. de María, Green Chem., 2010, 12, 1844–1849 RSC.
| This journal is © The Royal Society of Chemistry 2017 |