
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient catalytic hydrotreatment of Kraft lignin to alkylphenolics using supported NiW and NiMo catalysts in supercritical methanol†
Anand
Narani
a,
Ramesh Kumar
Chowdari
b,
Catia
Cannilla
c,
Giuseppe
Bonura
c,
Francesco
Frusteri
c,
Hero Jan
Heeres
*b and
Katalin
Barta
*a
aStratingh Institute for Chemistry, Faculty of Mathematics and Natural Science, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
bChemical Engineering Department, Faculty of Mathematics and Natural Science, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
cCNR-ITAE, Istituto di Tecnologie Avanzate per l'Energia “Nicola Giordano”, Via S. Lucia sopra Contesse 5, 98126 Messina, Italy. E-mail: k.barta@rug.nl; h.j.heeres@rug.nl; Tel: +31 (0)503634174
First published on 28th August 2015
Abstract
Efficient catalytic hydrotreatment of Kraft lignin to yield aromatic monomers was demonstrated in supercritical methanol using a variety of NiW and NiMo catalysts on acidic, basic and neutral supports. It was found that NiW catalysts on neutral or basic supports are highly suitable for depolymerization of Kraft lignin to methanol soluble organics in high yields at 320 °C and 35 bar H2 pressure. An extensive analysis of the product mixtures was carried out using GC-MS-FID, GC × GC-FID, 2D HSQC NMR, GPC and elemental analysis, and several techniques were used for the characterization of the prepared catalysts in order to determine the acidity and basicity of the support and morphological changes after the catalytic reaction. The best results were obtained with sulphided NiW catalysts supported on activated carbon. Efficient depolymerization of Kraft lignin and a total 28 wt% monomer yield was obtained within 8 h and 76% of the products were alkylphenolics and guaiacolics. Over prolonged reaction times, the total monomer yield reached 35 wt%, containing up to 26 wt% alkylphenolics. During catalytic processing, deoxygenation was the most prevalent reaction and, importantly, no competing aromatic ring hydrogenation or undesired repolymerization to insoluble char was observed. The catalytic system described here represents a highly efficient and selective method for the production of alkylphenolics and guaiacolics from Kraft lignin.
1 Introduction
Kraft lignin comprises the majority of the total worldwide lignin production, which currently accounts for over 50 million tons per annum.1–3 However only a very few examples exist for the use of Kraft lignin as a raw material for the production of value added chemicals.4,5 More than 90% of lignin is treated as a low value fuel and burned to provide heat during paper processing, because there is lack of better methods for its utilization. Therefore the development of new catalytic technologies for more efficient conversion of Kraft lignin to aromatics is of crucial importance, as it would largely contribute to the sustainable use of the sizeable amount of waste streams.6–10Kraft lignins are obtained through the sulphide pulping process, and typically contain 1–3% sulphur in both inorganic as well as organically bound forms.11,12 Moreover, due to the extensive pulping, the structure of Kraft lignins is more condensed, and contains a higher amount of C–C bonds compared to other types of lignins.13–16 From this follows, that Kraft lignin is a particularly challenging substrate for catalytic processing, especially with noble metal catalysts which are frequently poisoned by sulphur.3,4,7,8,17
Over the past decade, significant advances have been made in the development of new processes for the depolymerization of lignin. Most of these methods typically targeted organosolv lignins and ranged from thermal or hydrothermal depolymerization18–21 to milder oxidative or reductive methods22–24 most of which have been extensively reviewed.4,7,8,17,18,25–28 Among the developed methods, reductive approaches have received particular attention, since they enable the direct hydrogenolysis of the most prevalent β-O-4 lignin linkage.29–33 Alternatively, protic solvents such as methanol,34 ethanol,35–37 isopropanol,38 formic acid,39–42 or water/alcohol mixtures43,44 could serve as hydrogen donors for the various depolymerization and deoxygenation processes. Furthermore, water has been proven as a suitable reaction medium.45,46
Methods that specifically target Kraft lignin have also been described. For instance, Miller et al. described the efficient depolymerization of Kraft and organosolv lignins in supercritical methanol and ethanol in the presence of KOH.47 In similar work, Shabtai described two step depolymerization of lignin in the presence of a base.48 Weckhuysen and co-workers reported a two-step process for the conversion of various lignins, including Kraft to aromatic compounds (<12 wt% yield) during which lignin was depolymerized using a Pt/γ-Al2O3 catalyst in alkaline ethanol/water medium and the obtained bio-oil was subsequently deoxygenated with CoMo/Al2O3 and Mo2C/CNF catalysts.49 In another process, Kraft lignin was depolymerised using formic acid as a hydrogen source in water/ethanol medium without any catalyst.50
Recent work by Ma et al. has reported that high yields (61.3 wt%) of aromatic compounds can be obtained from Kraft lignin in supercritical ethanol over MoC1−x/AC catalysts.51 In this system ethanol acted as a hydrogen source, but was also found to incorporate into the formed products by alkylation reactions. Side reactions of the solvent alone resulting in aliphatic products also occurred. Nonetheless this important work pointed at the suitability of molybdenum as an active metal in Kraft lignin depolymerization. More recently, a one-pot complete catalytic conversion of Kraft lignin into C6–C10 chemicals, has been reported in ethanol, using Mo-based catalysts.52
The depolymerization of Kraft lignin is a challenging task due to the highly condensed structure of the substrate as well as its relatively high sulphur content, which may lead to catalyst deactivation.4,12 Therefore the selection of the catalyst should take into account the high sulphur content of the lignin source. It is well known that NiMo and CoMo catalysts are tolerant towards sulphur and are widely used in the hydrodeoxygenation of aromatic compounds to simpler aromatics or hydrocarbons.53,54 They have recently been reported as suitable catalysts for the hydrodeoxygenation of lignin model compounds,55 and for lignin as discussed above.
Here we report the highly efficient catalytic hydrotreatment of Kraft lignin to yield value added low molecular weight aromatics, specifically alkylphenolics and guaiacolics in supercritical methanol in the presence of hydrogen. A variety of supported Mo and W catalysts promoted by Ni and Co on various supports have been tested and the best composition identified. Under optimal reaction conditions, the main products are alkylphenolics and gratifyingly no ring hydrogenation or char formation takes place. The catalyst is thoroughly characterized before and after the reaction.
2 Experimental section
2.1 Materials
Ni(NO3)2·6H2O (99%), Co(NO3)2·6H2O (>99%), Ce(NO3)3·6H2O (99.5%), and KOH (85%) were purchased from Acros Organics, Mg(NO3)2·6H2O (98%), La(NO3)3·6H2O (>99%), (NH4)6Mo7O24·4H2O, (NH4)6H2W12O40·H2O (>99%), ZrO(NO3)2·xH2O, dimethyldisulphide (DMDS), and K2CO3 (>99%), were purchased from Sigma-Aldrich. Indulin-AT (Kraft lignin) was from MWV Specialty Chemicals and was kindly provided by Dr R. Gosselink from the Wageningen University and Research Centre, The Netherlands. Indulin-AT is a purified form of Kraft pine lignin, which is free of the hemicellulosic material. The lignin content is 97 wt% on dry basis. Elemental analysis of this lignin is as follows: 61.1% of carbon, 5.6% of hydrogen, 1.6% sulphur and 30.6% of oxygen. Methanol (anhydrous) was purchased from Macron Fine Chemicals, Activated Charcoal (AC) was purchased from Merck Millipore, and ammonium-ZSM-5 (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Si/Al) was purchased from Alfa-Aesar.
:1 ratio of Si/Al) was purchased from Alfa-Aesar.
2.2 Preparation of the catalysts
Apart from the directly purchased supports (i.e., activated charcoal and ammonium-ZSM-5), three different mixed oxides were used as catalyst supports and prepared by co-precipitation methods in accordance with literature procedures.56 The support MgO–La2O3 (ML) with an atomic ratio of Mg/La of 3 was prepared by co-precipitation methods. In a typical procedure, required amounts of Mg(NO3)2·6H2O (0.39 mol) and La(NO3)3·6H2O (0.13 mol) were dissolved in 0.5 L of deionized water and precipitated with a mixture of K2CO3 (0.25 M) and KOH (1 M) in 0.52 L of deionized water at a constant pH of 10. After complete precipitation, the solid was filtered and thoroughly washed with water to reach neutral pH. The resultant solid was oven dried at 120 °C for 12 h and finally calcined at 650 °C for 5 h. The MgO–CeO2 (MC) and MgO–ZrO2 (MZ) supports were prepared by similar procedures and calcined at 500 °C for 5 h (MgO–CeO2) and 650 °C for 4 h (MgO–ZrO2).The catalysts NiO–MoO3/AC (NiMo/AC), CoO–MoO3/AC (CoMo/AC) and NiO–WO3/AC (NiW/AC) with 5 wt% of NiO or CoO and 15 wt% of WO3 or MoO3 were deposited on activated carbon by impregnation of about 3–4 mL of aqueous solutions of Ni(NO3)2·6H2O or Co(NO3)2·6H2O and (NH4)6Mo7O24·4H2O or (NH4)6H2W12O40·H2O followed by evaporation to dryness at 100 °C for 12 h and calcination under a N2 atmosphere at 450 °C for 5 h. In a similar method 5 wt% NiO–15 wt% MoO3, 5 wt% CoO–15 wt% MoO3 as well as 5 wt% NiO–15 wt% WO3 on different supports were prepared.
Sulphided catalysts were generated in situ by the addition of dimethyldisulphide (DMDS) to the reaction mixture at the start of the reaction.57 (For the detailed pathways related to in situ preparation of the catalysts see the ESI, Appendix A.†) The sulphided NiW catalysts supported on AC, ZSM-5, MgO–La2O3, MgO–CeO2, MgO–ZrO2 catalysts are designated as S-NiW/AC, S-NiW/ZSM-5, S-NiW/ML, S-NiW/MC, S-NiW/MZ respectively. Similarly, sulphided Ni, W, NiMo, and CoMo supported on AC catalysts are abbreviated as S-Ni/AC, S-W/AC, S-NiMo/AC and S-CoMo/AC. For more detailed information of catalyst characterization techniques, see the ESI, Appendix B.†
2.3 Catalytic hydrotreatment of Kraft lignin and product analysis
The catalytic hydrotreatment of Kraft lignin was carried out in a 100 ml high pressure Parr autoclave with an overhead stirrer. Typically, the autoclave was charged with the 0.25 g respective catalyst, 1 g of Kraft lignin, 0.1 g of dimethyl-disulphide and 30 ml of methanol. The reactor was sealed and purged with nitrogen several times and then pressurised with 35 bar H2 at room temperature. The reactor was heated to 320 °C for a predetermined amount of time, and stirred at 800 rpm. After reaction, the reactor was cooled to room temperature and the gas was collected in a Tedlar gas-bag. The reactor was washed with several portions of methanol to allow quantitative transfer of the reactor content, which was subjected to a general workup procedure (Scheme 1). In the first step, the solid residues were filtered and the solvent was removed by rotary evaporation. The resultant oily product (named methanol soluble oil) was further characterized. The remaining solids were washed with dichloromethane (DCM). The solids extracted in this way (DCM soluble products) were obtained after evaporation of the solvent. The remaining solid residue contained the catalyst, small amounts of unconverted lignin and char. These solids were further washed with dimethylsulfoxide (DMSO) to remove the unconverted lignin. | ||
| Scheme 1 Procedure developed for the fractionation of the crude product mixture obtained after catalytic treatment of Kraft lignin. | ||
Methanol soluble oils were analysed by GC-MS-FID analyses using a Quadruple Hewlett Packard 6890 MSD attached to a Hewlett Packard 5890 GC equipped with a 60 m × 0.25 mm i.d. and a 0.25 μm sol–gel capillary column. The injector temperature was set at 250 °C. The oven temperature was kept at 40 °C for 5 minutes then heated up to 250 °C at a rate of 3 °C min−1 and then held at 250 °C for 10 minutes.
GC × GC-FID analysis was performed on organic samples with a trace GC × GC from Interscience equipped with a cryogenic trap system and two columns: a 30 m × 0.25 mm i.d. and a 0.25 μm film of the RTX-1701 capillary column connected by using a meltfit to a 120 cm × 0.15 mm i.d. and a 0.15 μm film Rxi-5Sil MS column. An FID detector was used. A dual jet modulator was applied using carbon dioxide to trap the samples. Helium was used as the carrier gas (continuous flow 0.6 ml min−1). The injector temperature and FID temperature were set at 250 °C. The oven temperature was maintained at 40 °C for 5 minutes then heated up to 250 °C at a rate of 3 °C min−1. The pressure was set at 70 kPa at 40 °C. The modulation time was 6 s.
For GC × GC-FID and GC-MS-FID analyses, the samples were diluted with tetrahydrofuran (THF) and 500 ppm di-n-butyl ether (DBE) was added as an internal standard. For more detailed information see the ESI, Appendix C.†
Gel Permeation Chromatography (GPC) analyses of the samples were performed using a HP1100 equipped with three 300 × 7.5 mm PL gel 3 μm MIXED-E columns in series using a GBC LC 1240 RI detector. Average molecular weight calculations were performed using the PSS WinGPC Unity software from Polymer Standards Service. The following conditions were used: THF as the eluent at a flow rate of 1 ml min−1; 140 bar, a column temperature of 40 °C, 20 μl injection volume and a 10 mg ml−1 sample concentration. Toluene was used as a flow marker.
The gas phases were collected after the reaction and stored in a gas bag (SKC Tedlar 3 L sample bag [9.5′′ × 10′′]) with a polypropylene septum fitting. GC-TCD analyses were performed using a Hewlett Packard 5890 Series II GC equipped with a Poraplot Q Al2O3/Na2SO4 column and a molecular sieve (5 A) column. The injector temperature was set at 150 °C and the detector temperature at 90 °C. The oven temperature was maintained at 40 °C for 2 minutes then heated up to 90 °C at 20 °C min−1 and maintained at this temperature for 2 minutes. A reference gas was used to identify the peaks by retention time and to quantify the products (gas product mixture: 55.19% H2, 19.70% CH4, 3.00% CO, 18.10% CO2, 0.51% ethylene, 1.49% ethane, 0.51% propylene and 1.5% propane).
NMR spectra were acquired at 25 °C using an Agilent 400 MHz spectrometer. Approximately 50 mg of methanol soluble oil was dissolved in 0.7 ml dimethylsulfoxide-d6 (DMSO). For analysis of the Kraft lignin, approximately 100 mg was dissolved in 0.7 ml dimethylsulfoxide-d6 (DMSO). 1H–13C HSQC spectra were acquired using a standard pulse sequence HSQC programme with a spectral width of 160 ppm, 16 scans, 128 increments (256 increments for Kraft lignin), on F1 dimension and data were processed using the MestReNova software.
Elemental analyses (C, H, N and S) were performed using a Euro Vector 3400 CHN-S analyzer. The oxygen content was determined by difference.
3 Results and discussion
Inspired by previous reports36,37,49–52 as well as our own experiences regarding the catalytic conversion of various lignocellulose sources,58–60 we anticipated that supercritical methanol (at 300–320 °C) in the presence of hydrotreatment catalysts and hydrogen, would be a suitable reaction medium for the depolymerisation of Kraft lignin to aromatic monomers. These aromatic fragments would undergo further deoxygenation towards simpler aromatics, such as alkylphenolics, over the various hydrotreatment catalysts.53,55 Thus, a systematic study was conducted using various hydrodeoxygenation catalysts (Ni, Co promoted with Mo or W) on various supports.3.1 Catalyst screening and fractionation of products
A series of supported NiMo, CoMo and NiW catalysts on acidic (ZSM-5), neutral (activated carbon) or basic (various MgO/lanthanide oxide combinations) supports were prepared. The nature of the acidic and basic sites was confirmed by TPD measurements (discussed below). The corresponding BET areas as well as pore size parameters are shown in Table S1, Fig. S1 and S2.†These catalysts were tested in the depolymerization of Kraft lignin at 320 °C, 35 bar H2 for 8 h in supercritical methanol and dimethyldisulphide was used as a sulphur source. The reaction products were fractionated by the developed procedure (see Scheme 1 in the Experimental section). This consisted of a separate treatment of reaction solids followed by quantification of the amount of the methanol soluble material, a crucial parameter and indication for the activity of the respective catalysts (for calculation of the main parameters determined, see Appendix D in the ESI†). Gel permeation chromatography measurements confirmed that the methanol soluble oil mainly consists of aromatic monomers and oligomers of lower molecular weight ranges (see the ESI, Table S2 and Fig. S3-a†). The DCM soluble solids were identified as higher molecular weight materials (see the ESI, Table S2 and Fig. S3-b†). The brown DMSO soluble fraction was believed to be unreacted lignin, however due to its low solubility in THF, its molecular weight could not be determined accurately. The rest of the solids were visually assessed to be a mixture of char and catalysts.
The catalyst screening was conducted at 320 °C and 35 bar of H2 using the prepared catalysts in order to determine the most suitable composition and support for maximizing the yield of methanol soluble products. First, simply heating Kraft lignin without a catalyst and hydrogen resulted in only 15 wt% of methanol soluble oil and 30 wt% of insoluble char (Table 1, entry 1). When S-NiMo on activated carbon without external hydrogen was tested, the results were comparable to a blank experiment with only 22 wt% of methanol soluble oil yields and a more substantial (30 wt%) amount of char obtained (Table 1, entry 2). The same catalyst (S-NiMo/AC) with external hydrogen delivered a promising 57 wt% of methanol soluble oil yield and only traces of char (Table 1, entry 3). These findings indicate that the presence of external hydrogen is important, as it likely reduces the extent of recondensation reactions and/or enhances the rate of lignin depolymerisation reactions. In addition, unlike during our previous studies with CuPMO (Cu doped Mg–Al porous metal oxide) in supercritical methanol, where methanol reforming was the only source of hydrogen,60 the NiMo catalysts required external hydrogen that played a key role in affecting lignin depolymerization, as no significant methanol reforming was observed in the present case, as expected. For a related catalyst, comprising Co instead of Ni, (S-CoMo/AC) only 41 wt% of methanol soluble oil yield and 9 wt% of char were obtained (Table 1, entry 4). This is likely due to the increase in the acidity of the catalyst when Ni was replaced with Co (Table 2, entry 7). This indicates that Ni promoted Mo is more active than Co promoted Mo for lignin depolymerization. Subsequently, when Mo was replaced with W (i.e., S-NiW/AC), the acidity of the catalyst further decreased (Table 2, entry 1), causing the methanol soluble oil yield to increase to 82 wt% without any char formation (Table 1, entry 8), representing the best product yield among the screened catalysts. As a comparison, the methanol soluble oil yields dropped to 62 wt% and 8 wt% of char (Table 1, entry 5) obtained with the non sulphided NiW/AC catalyst. Thus, the sulphided catalyst was clearly more active. Further studies confirmed that the sulphided Ni promoted W catalysts are more active than the corresponding non-sulphided NiW/AC and S-NiMo/AC catalysts or Ni/AC and S-W/AC catalysts alone (Table 1, entries 5, 3, 7 and 6).
| Entry | Catalyst | Methanol soluble oil (wt%) | DCM soluble solids (wt%) | DMSO soluble solids (wt%) | Char (wt%) | Mass balance (wt%) |
|---|---|---|---|---|---|---|
| Reaction conditions: catalyst (0.25 g), Kraft lignin (1 g), methanol (30 ml), H2 (35 bar), temperature (320 °C), 8 h.a Without hydrogen added.b With 2 g of Kraft lignin. | ||||||
| 1 | Blank | 15 | 8 | 20 | 30 | 73 |
| 2 | S-NiMo/ACa | 22 | 10 | 8 | 30 | 70 |
| 3 | S-NiMo/AC | 57 | 10 | 8 | Trace | 75 |
| 4 | S-CoMo/AC | 41 | 6 | 10 | 9 | 66 |
| 5 | NiWOx/AC | 62 | 14 | 8 | 8 | 92 |
| 6 | S-W/AC | 45 | 2 | 30 | 5 | 82 |
| 7 | S-Ni/AC | 70 | 5 | 7 | 0 | 82 |
| 8 | S-NiW/AC | 82 | 10 | Trace | 0 | 92 |
| 9 | S-NiW/ZSM-5 | 40 | 3 | 13 | 30 | 86 |
| 10 | S-NiW/ML | 80 | 9 | Trace | 0 | 89 |
| 11 | S-NiW/MC | 75 | 11 | 9 | 0 | 95 |
| 12 | S-NiW/MZ | 68 | 6 | 12 | 4 | 90 |
| 13 | S-NiW/ACb | 60 | 20 | 15 | 0 | 95 |
| 14 | S-NiW/MLb | 60 | 22 | 16 | 0 | 98 |
| Entry | Catalyst | NH3 uptake (μmol gcat−1) | CO2 uptake (μmol gcat−1) |
|---|---|---|---|
| 1 | NiW/AC | 18.4 | 0.6 |
| 2 | NiW/ZSM-5 | 507.6 | — |
| 3 | NiW/MgO–La2O3 | — | 291.0 |
| 4 | NiW/MgO–CeO2 | — | 188.3 |
| 5 | NiW/MgO–ZrO2 | — | 104.7 |
| 6 | NiMo/AC | 44.5 | 0.6 |
| 7 | CoMo/AC | 77.0 | 0.7 |
Once established that a bimetallic Ni/W catalyst is the best regarding product yield, we have subsequently investigated whether the support also plays a significant role. To this end, NiW catalysts supported on basic MgO–La2O3 (ML), MgO–CeO2 (MC), MgO–ZrO2 (MZ) and acidic ZSM-5 catalysts (for acid and base properties, see Table 2, entries 2–5) were screened and indeed the variations in product yields were significant. Using the NiW catalyst on the acidic ZSM-5 support, 30 wt% of the insoluble residue and only 40 wt% of methanol soluble oil yields were observed (Table 1, entry 9), showing more pronounced recondensation processes likely due to acid catalysed side reactions leading to reactive unsaturated intermediates, such as dehydration products. The results using basic supports MgO–La2O3 were not significantly different from those obtained with the catalyst on neutral support (Table 1, entry 10). When MgO–CeO2 was used, the methanol soluble oil yields dropped to 75 wt% and 9 wt% DMSO soluble solids obtained (Table 1, entry 11). In the case of MgO–ZrO2, the methanol soluble oil yields further dropped to 68 wt%, increase in DMSO soluble solids (12 wt%) and 4 wt% of char was observed (Table 1, entry 12). This phenomenon is likely due to the decrease in basicity or the increase in acidity of the catalysts when La2O3 was replaced in MgO–La2O3 with CeO2 and ZrO2 (Table 2, entries 4 and 5). Lastly, with increased lignin loading to 2 g, both S-NiW/AC and S-NiW/ML catalysts gave 60 wt% of methanol soluble oil yields, whereby more DCM and DMSO soluble solids were obtained with both catalysts. Char formation was not observed with both catalysts even at increased substrate loading.
3.2 Surface properties of the catalysts
Surface concentrations of acidic and basic sites of NiMo, CoMo and NiW catalysts were determined by temperature programmed desorption of ammonia (NH3-TPD) and carbon dioxide (CO2-TPD), respectively (Table 2).It was found that NiW supported on ZSM-5 presents the highest number of acidic sites (507.6 μmol gcat−1), while the highest basic capacity was recorded on NiW supported on MgO–La2O3 (291 μmolCO2 gcat−1). Moreover, when La2O3 was exchanged with CeO2 (Table 2, entry 4) or ZrO2 (Table 2, entry 5), the basicity significantly decreased, resulting in CO2 uptake values of 188.3 and 104.7 μmolCO2 gcat−1 for NiW/MgO–CeO2 and NiW/MgO–ZrO2, respectively. Still, when NiW was supported on activated carbon (Table 2, entry 1), not only the NH3 adsorption capacity appeared quite low (18.4 μmol gcat−1), but also the basicity was practically negligible (0.6 μmolCO2 gcat−1), giving the sample an almost neutral character. A net increase of acidity was observed on the same support when W was exchanged with Mo (44.5 μmolNH3 gcat−1 for NiMo/AC), likely due to an increased presence of clusters, which are able to delocalize protons among the neighboring MoOx species more than in the presence of isolated groups. Lastly, the substitution of NiO with CoO on activated carbon (Table 2, entry 7) also promoted a further increase of acidity (77.0 μmolNH3 gcat−1 for CoMo/AC), while the basicity remained almost unchanged. In conclusion, it is clear that the supports play a key role in determining the acid–base behaviour, while the metal loading mainly affects the temperature of peak desorption, depending on a weak or strong metal–support interaction (see also Fig. S4 and S5 and related comments in the ESI†). So, from these results the following scale of acidity and basicity can be drawn:

In the attempt of confirming the sulphidation process, the spent catalyst NiW/AC, recovered after the reaction, was characterized by X-ray diffraction and in Fig. 1 the patterns of the fresh (a) and used (b) catalysts are compared. Before the reaction, typical crystalline peaks referred to the <100>, <−111> and <002> planes of a monoclinic phase of NiWO4 (JCPDS 150755) were evident, along with Ni metallic (JCPDS 040850) diffraction peaks at 44.55°, 51.91°, 76.34° and 92.89°, corresponding to <111>, <200>, <220> and <311> planes, respectively. The spent catalyst, indeed, shows the characteristic signals of tungsten sulphide (WS2) at 14°, 32.7°, 33.4°, 39.4°, 43.8°, 49.8° and 58.7° which are referred to the <002>, <100>, <101>, <006>, <105> and <110> planes (JCPDS 080237) and the peaks of the trigonal nickel sulphide Ni3S2 phase, observed at 21.65°, 30.9°, 37.6°, 44.4°, 20.1°, 54.9°, and 55.2°, corresponding to the <101>, <012>, <003>, <021>, <202>, <211>, <104>, <122> planes (JCPDS 020772).
 | ||
| Fig. 1 XRD patterns of NiW/AC catalysts: (a) sulphided (after reaction), (b) fresh (before reaction). | ||
The formation of sulphided NiW/AC was further confirmed by SEM-EDAX measurement, which showed the presence of about 2 wt% of sulphur in the used catalyst (see EDAX values reported in Table S3†). Fig. 2 exhibits the micrographs of fresh and used NiW/AC catalysts, showing that the spent catalyst maintains the original structure characterized by particles in the range comprised between 10 and 100 μm. The insert reported in Fig. 2-b is the mapping of the S-NiW/AC catalyst, which shows the almost homogeneous distribution of sulphur in correspondence to Ni and W.
 | ||
| Fig. 2 SEM micrographs of NiW/AC catalysts (a) fresh, and (b) used. In the insert, the mapping measurement is shown. | ||
3.3 In depth analysis of the methanol soluble products by GC-MS-FID measurements
The composition of the methanol soluble products obtained in catalytic tests described in Table 1 was investigated by GC-MS-FID chromatography. A representative chromatogram of the product mixture obtained using the S-NiW/AC catalyst is shown in Fig. 3. Though the GC-MS-FID trace is complex, only around 30 main components were found in significant quantities. All of these major products were identified as shown in Fig. 3. The main groups of compounds were: (a) mono oxygenated phenolics, such as phenol (1), anisole (4), o-, p-, m-cresols (3, 5, 6), dimethyl phenols (10, 13, 14, 19), ethyl-phenols (13, 15, 16), propyl phenols (25), ethyl-methyl phenols (21, 22), methylated propyl phenols (27, 28), tri- and tetra-methyl phenols (12, 17, 23, 24) and (b) guaiacolics (2, 7, 8, 18, 20, 26). Some extractives, not directly derived from lignin depolymerization were also seen (29, 30).61 Interestingly, tri-oxygenated compounds, mainly expected from syringyl lignin subunits were not found. Next, this product mixture was compared to that obtained with the same catalyst (NiMo/AC) under hydrogen free conditions. The major products in this case were tri- and tetra-methylated phenols as well as 2-isopropyl-1-methoxy-4-methylbenzene (Fig. S6†), a product mixture markedly different from that shown in Fig. 3, where phenolics and guaiacolics were the major products. Clearly, in the absence of hydrogen, the hydrodeoxygenation processes are limited and reactions involving the solvent are predominant. Similarly, in the case of non-sulphided NiW/AC catalysts, substituted guaiacolics were prevalent over phenolics (Fig. S7†) showing that the sulphided form of the NiW/AC catalyst enhances the deoxygenation activity of the catalyst, and further processing of the lignin derived monomers towards phenolics. All other sulphided NiMo/AC, CoMo/AC, W/AC and NiW supported on acidic ZSM-5 and basic MgO–La2O3 (ML), MgO–CeO2 (MC), and MgO–ZrO2 (MZ) catalysts gave similar product distributions.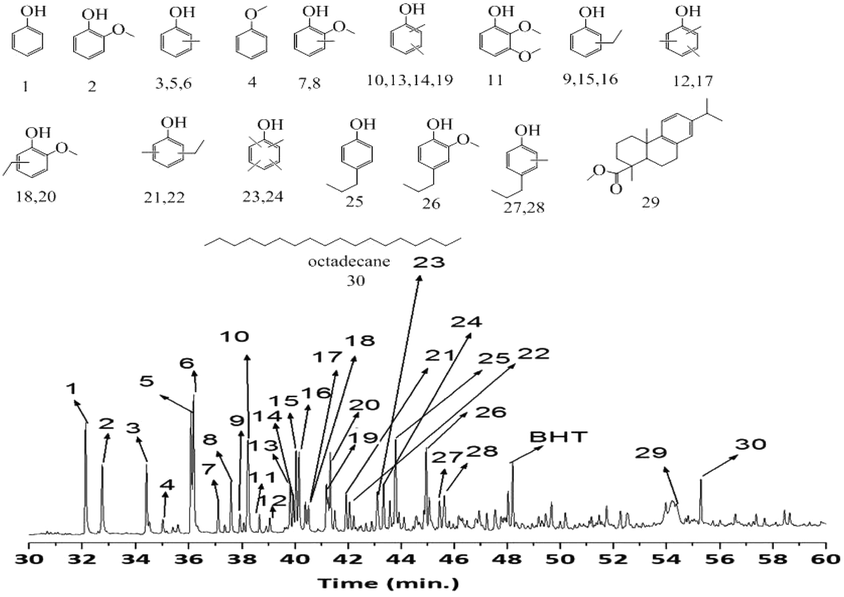 | ||
| Fig. 3 GC-MS-FID chromatogram of methanol soluble oil obtained at 320 °C for 8 h with 35 bar H2 using S-NiW/AC catalysts. | ||
3.4 Quantification of methanol soluble products by 2D-GC × GC measurements
In addition to identification of the components by GC-MS-FID measurements shown above, the product mixtures obtained in the catalytic runs shown in Table 1 were quantified using the GC × GC-FID technique. Quantification was based on calibration of the various types of compounds present in these product mixtures and the use of di n-butylether (DBE) as an external standard (Fig. 4). A representative GC × GC-FID chromatogram is given in Fig. 6b and the various areas corresponding to the different types of products in the mixture are displayed (for calibration details of GC × GC-FID, see Appendix C in the ESI†). Quantification based on such GC × GC-FID measurements, for all product mixtures obtained with each catalyst after 8 h reaction time, is displayed in Fig. 4. | ||
| Fig. 4 Effect of catalysts on alkylphenolics, guaiacolics and monomer yields. Reaction conditions: catalyst (0.25 g), Kraft lignin (1 g), methanol (30 ml), H2 (35 bar), temperature (320 °C), 8 h. | ||
The two most important groups of products (alkylphenolics and guaiacolics) were quantified and are displayed for each run in Fig. 4. The total number of monomers is also given, which include, besides the alkylphenolics and guaiacolics, other compounds such as naphthalenes, catechols, other aromatics and linear and branched alkanes (see the ESI, Table S4†). Similarly to the results obtained by gravimetric quantification of the various fractions (Table 1), there is a clear difference between the individual runs in terms of yields of total alkylphenolics and guaiacolics, which depend on the activity of the respective catalyst towards depolymerization. Also, the hydrodeoxygenation activity of the catalyst is correlated with the amount of alkylphenolics versus guaiacolics present.
Without hydrogen only 6.5 wt% of monomers were obtained with S-NiMo/AC and this amount increased to 14.5 wt% in the presence of hydrogen. Better results were obtained when Mo was replaced by W. With S-NiW/AC, a significantly higher (28.5 wt% monomer yield) was obtained; including 16 wt% of alkylphenolics and 5.5 wt% of guaiacolics (see also Table S4,† entry 7).
Good results were obtained with the other types of catalysts as well: S-NiMo/AC (14.5 wt%), S-CoMo/AC (9 wt%) and non-sulphided NiWOx/AC (16.5 wt%). Without the Ni promoter or with S-Ni/AC without W, the monomer yields decreased to 20.5 wt% and 19 wt% respectively. The acidic ZSM-5 support generally gave lower monomer yields (18%) than the basic supports. Especially MgO–La2O3 (ML) gave monomer yields comparable to the activated carbon support (26.5 wt%). Thus the monomer yields depend on reaction conditions, type of metal, promoter and type of support as expected. Sulphidation and combination of Ni with W on neutral support were ideal for obtaining higher monomer yields.
3.5 Further improvement of monomer yields and study of reaction parameters
Next, the effect of temperature on the product yield for the catalytic hydrotreatment of Kraft lignin in methanol using the S-NiW/AC catalyst (35 bar H2, 8 h) was investigated. Table 3 shows yields of methanol soluble oil as well as monomers (see also Table S5†) as a function of the temperature (entries 1–3). The runs differed in the extent of lignin conversion as well as the amount of the DCM soluble product, however good product yields could even be achieved at the lowest temperature tested (280 °C). The liquid phases were also analysed by GPC, which showed the occurrence of depolymerization of the lignin at all reaction temperatures with a slight variation in the average molecular weight (Fig. S8 and Table S2†).| Entry | Temp. (°C) | Methanol soluble oil (wt%) | DCM soluble products (wt%) | DMSO soluble solids (wt%) | Monomer yields (wt%) |
|---|---|---|---|---|---|
| Reaction conditions: catalyst (0.25 g), Kraft lignin (1 g), methanol (30 ml), H2 (35 bar), 8 h. | |||||
| 1 | 280 | 60 | 1 | 35 | 20 |
| 2 | 300 | 65 | 4 | 28 | 23 |
| 3 | 320 | 82 | 10 | Trace | 28.5 |
Next, we have carried out experiments under standard reaction conditions (320 °C, 35 bar H2 with the S-NiW/AC catalyst) with different reaction times to enhance the yield of aromatic monomers and to determine the extent of possible competing ring hydrogenation reactions that may lead to alkanes. The results summarized in Table 4 indicate that 4 h of reaction time was not sufficient for full lignin conversion. In this case, the yield of the methanol soluble product fraction was 40 wt% (14.5 wt% of monomer yield), which is substantially lower than that at standard reaction times (8 h, 82 wt% methanol soluble oil and 28.5 wt% of monomers). An increase in overall monomer yield to 32.5 wt% took place at 16 hours, as depolymerization of oligomeric components in the methanol soluble fraction progressed. No significant change was further observed after a prolonged reaction time (24 h) and no char was formed. The slight increase in monomer yields (35 wt%) was likely due to further methylation of the formed fragments as confirmed by various techniques (GC-MS-FID, GC × GC-FID, HSQC NMR).
| Entry | Time (h) | Methanol soluble oil (wt%) | DCM soluble products (wt%) | DMSO soluble solids (wt%) | Monomer yields (wt%) |
|---|---|---|---|---|---|
| Reaction conditions: catalyst (0.25 g), Kraft lignin (1 g), methanol (30 ml), H2 (35 bar), temperature (320 °C). | |||||
| 1 | 4 | 40 | 1 | 30 | 14.5 |
| 2 | 8 | 82 | 10 | Trace | 28.5 |
| 3 | 16 | 77 | 4 | Trace | 32.5 |
| 4 | 24 | 79 | 7 | Trace | 35.0 |
The GC-MS-FID and GC × GC-FID chromatograms (Fig. 5 and 6) of the methanol soluble oils versus time show a clear change in product distribution. Guaiacol (1) and substituted guaiacols (2, 3, 4) were the major products and small amounts of alkylphenolics including phenol were observed after 4 h. At prolonged reaction times, the amounts of guaiacolics are reduced and more alkylphenolics are formed. After 24 h of reaction time almost all guaiacolics were absent and only alkylphenolics were observed. In addition, methylation was more extensive after 24 h, as is evident by the formation of tetra alkylated phenolics (14, 15, 16). Notably, further hydrodeoxygenation or competing ring-hydrogenation did not occur even after 24 h of reaction time. Thus it can be concluded, that the catalyst (S-NiW/AC) is highly suitable, active and selective towards the formation of mono-oxygenated alkylphenolics from Kraft lignin.
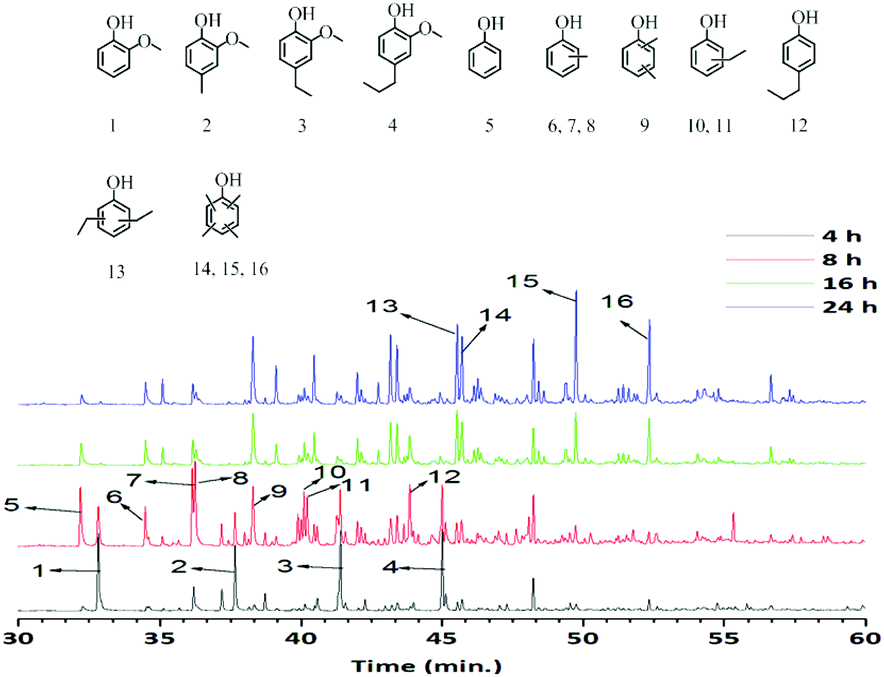 | ||
| Fig. 5 GC-MS-FID chromatograms of methanol soluble fractions obtained at different reaction times over the S-NiW/AC catalyst at 320 °C for 8 h with 35 bar H2. | ||
 | ||
| Fig. 6 GC × GC-FID chromatograms of methanol soluble fractions obtained after catalytic hydrotreatment of Kraft lignin over S-NiW/AC catalysts at 320 °C with 35 bar H2 (a) 4 h, (b) 8 h, (c) 16 h, and (d) 24 h. | ||
In addition, the composition of the obtained product mixtures at various time points was quantified by GC × GC-FID analysis, and the results are summarized in Fig. 7. The amount of the major components (alkylphenolics and guaiacolics) vs. time is displayed in Fig. 8. After 4 h of reaction time, 10 wt% of guaiacolics and only 2.5 wt% of alkylphenolics were present in the reaction mixture. At prolonged reaction times the amount of guaiacolics decreased to about 1.5 wt%, whereas the amount of alkylphenolics increased to a maximum of 26 wt% after 24 h. The graphs convincingly show that no substantial aromatic ring hydrogenation activity was observed, even after prolonged reaction times.
 | ||
| Fig. 7 GC × GC-FID results of product distribution with the time (a) 4 h, (b) 8 h, (c) 16 h, and (d) 24 h (reaction conditions: catalyst (0.25 g), Kraft lignin (1 g), methanol (30 ml), H2 (35 bar), temperature (320 °C)). | ||
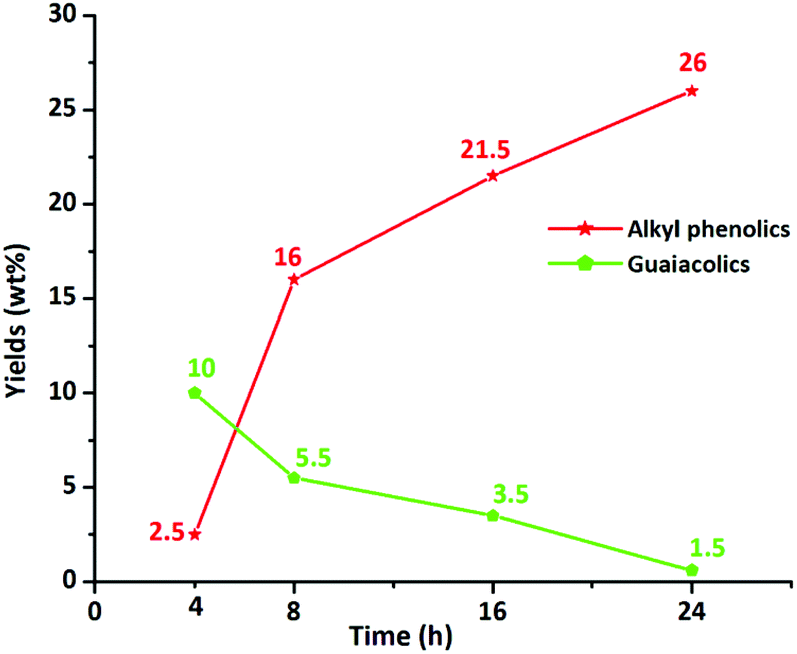 | ||
| Fig. 8 Yields of alkylphenolics and guaiacolics vs. batch time, in the product mixture obtained using the S-NiW/AC catalyst at 320 °C with 35 bar H2. | ||
HSQC NMR measurements confirmed the structural changes during the catalytic hydrotreatment. This NMR method was used before for the elucidation of the Kraft lignin structure.62,63 The HSQC NMR spectrum of the starting lignin in DMSO is shown in Fig. 9-a. The main inter-unit linkages: β-O-4, β–β, β-5, α-O-4 are clearly seen in the δ 3–5.5 ppm (1H) and δ 50–90 ppm (13C) regions. Signals corresponding to the aromatic region of the respective subunits are observed between δ 6–8 ppm (1H), δ 100–140 ppm (13C). The chemical shift regions corresponding to the various methoxy-groups belonging to guaiacyl or syringyl subunits are detected in the range of δ 3.2–4.2 ppm (1H), δ 56–58 ppm (13C), consistent with the literature. The 2D-HSQC NMR spectrum of the methanol soluble oil obtained after depolymerization of Kraft lignin after 8 h of reaction time with the S-NiW/AC catalyst at 320 °C is shown in Fig. 9-b. After depolymerisation, the signals belonging to the interunit lignin linkages are lacking and new peaks arise, that are related to the main types of aromatic products confirmed by GC-MS-FID or GC × GC-FID measurements. There is also a decrease of methoxy signals belonging to lignin in the region as well as a marked increase of signals in the aliphatic region, belonging to the formed alkyl-phenolics and guaiacolics. In addition, signals in Area 1 of the aromatic region correspond to aromatic protons of both guaiacolics and phenolics, these being practically indistinguishable due to extensive overlap. Area 2 of the aromatic region however, corresponds to only phenolics and alkylphenolics. These results confirm that under catalytic hydrotreatment conditions, the interunit linkages of lignin have been cleaved to produce guaiacolics, which are further demethoxylated to alkylphenolics.
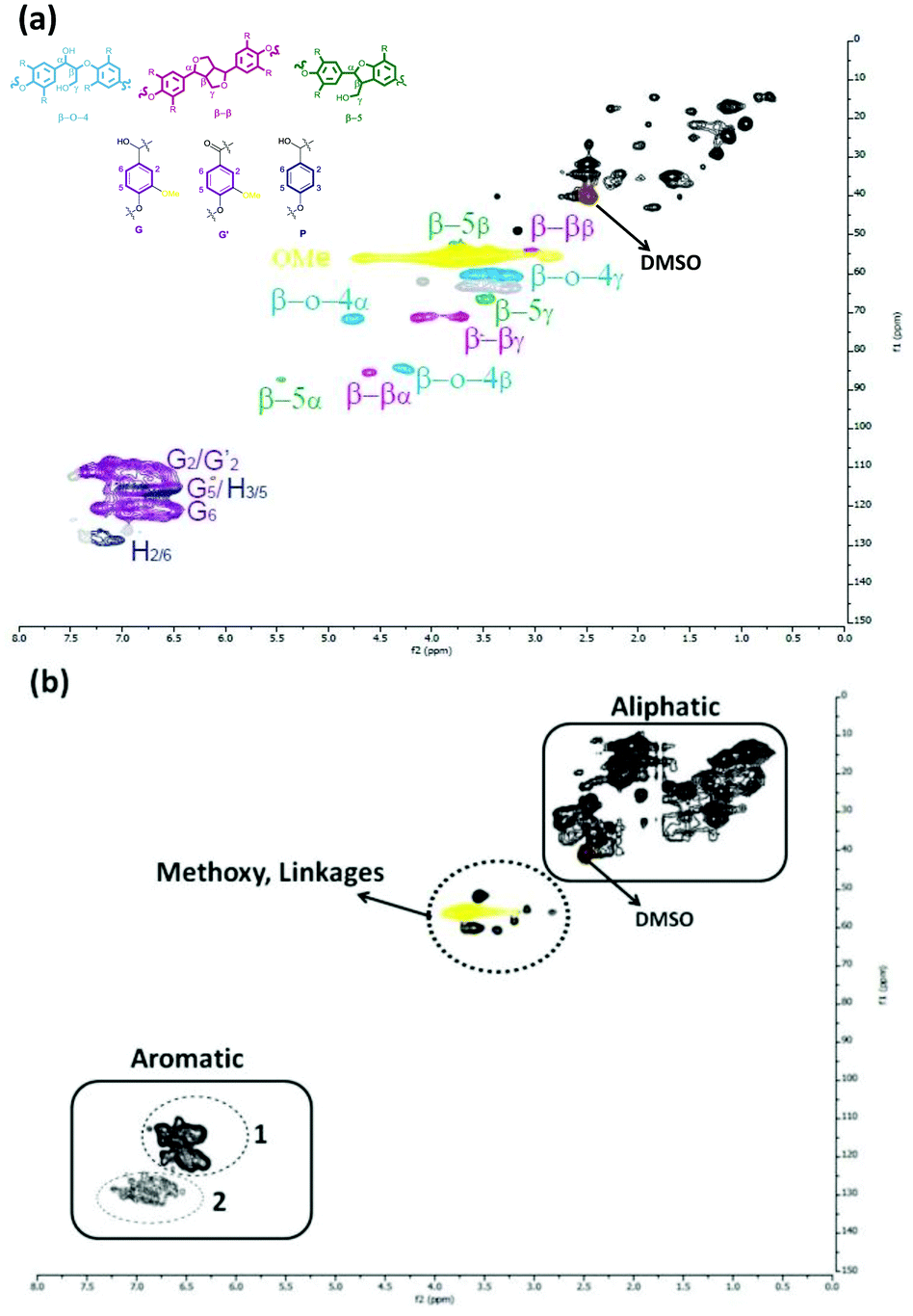 | ||
| Fig. 9 1H–13C HSQC NMR spectra of Kraft lignin and methanol soluble fractions obtained after catalytic treatment using S-NiW/AC catalysts at 320 °C with 35 bar H2 (a) Kraft lignin, (b) product mixture obtained after 8 h. | ||
3.6 Catalyst characterization after reaction
In Fig. 10, TEM images of the NiW/AC catalyst, recovered after the reaction, and the fresh catalyst are shown. In both these catalysts, the particles were in spherical shape; this confirmed that there is no change in the morphology of the particles after the reaction, as further confirmed by N2 adsorption–desorption measurements (see Table S1, Fig. S1 and S2†): the fresh catalyst exhibited a surface area of 857 m2 g−1 which decreased to 161 m2 g−1 after reaction, along with a corresponding increase in the average pore diameter from 21 Å to 39 Å. Nonetheless, despite particle agglomeration, the catalyst remained active even after 24 h of reaction time.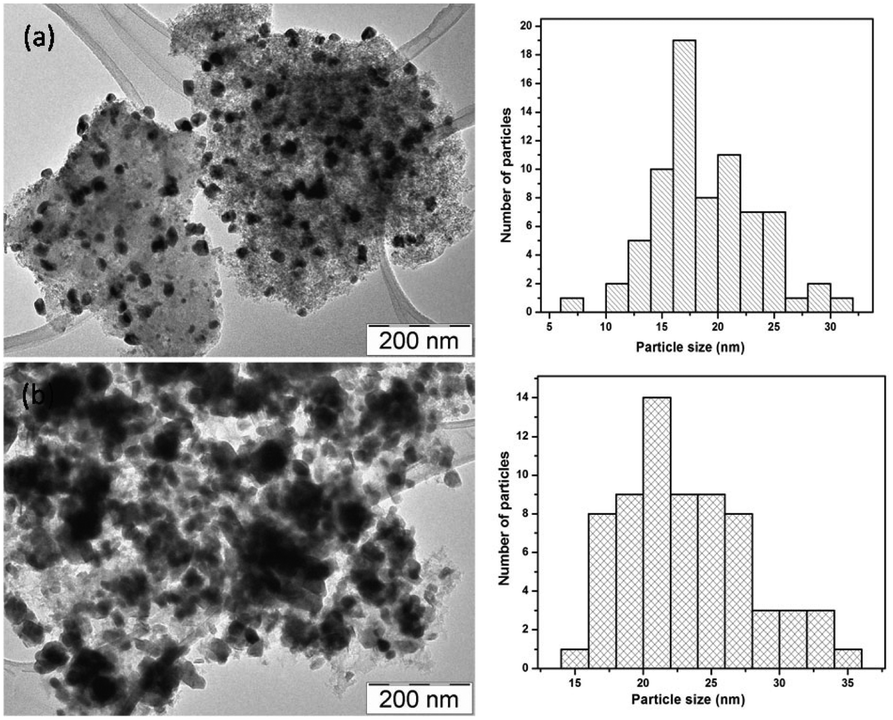 | ||
| Fig. 10 TEM images of NiW/AC catalysts (a) before (b) after the reaction. | ||
Finally, Fig. 11 shows the changes in the O/C and H/C atomic ratios determined by elemental analysis in the product oils obtained at 320 °C with S-NiW/AC catalysts at different reaction times. The Kraft lignin used in this study contains 61.1% of carbon, 5.6% of hydrogen, 1.6% sulphur and 30.6% of oxygen. The observed continuous decrease in the O/C value from 0.375 (Kraft lignin) to 0.112 and a marked increase in the H/C value of 1.102 (Kraft lignin) to 1.298 reflects the nature of transformations expected during catalytic hydrotreatment of Kraft lignin in supercritical methanol.
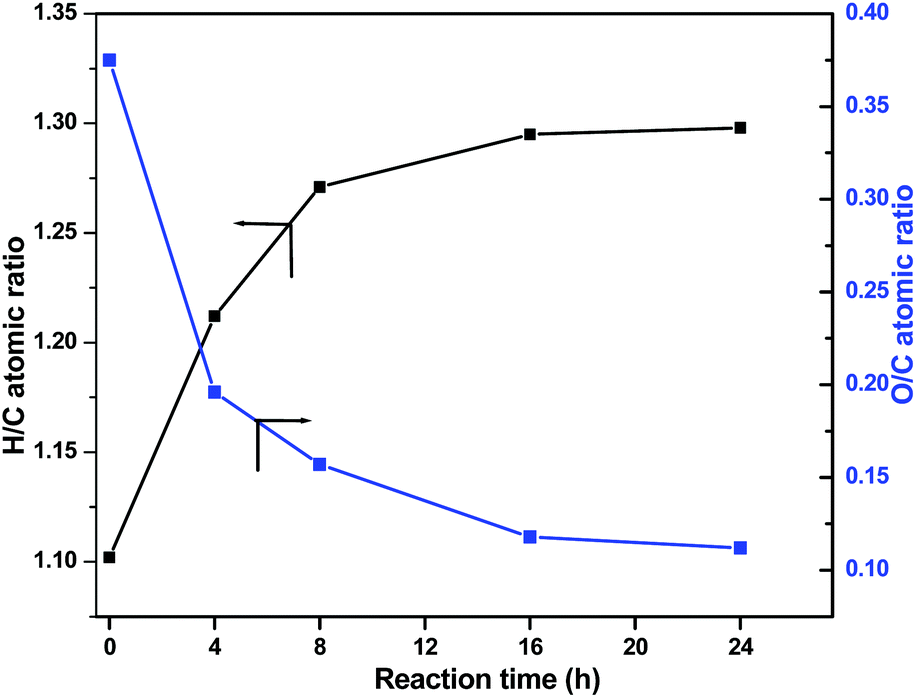 | ||
| Fig. 11 Elemental composition of Kraft lignin and derived methanol soluble fraction after reaction over S-NiW/AC catalysts at 320 °C with 35 bar H2 (blue line: O/C atomic ratio, black line: H/C atomic ratio). | ||
The decrease in the O/C atomic ratio value is due to the deoxygenation reactions, and the slight increase in the H/C atomic ratio is primarily due to methylation reactions and not because of ring hydrogenation, as confirmed by GC analysis.
4 Conclusions
This study presents a very efficient method for the catalytic conversion of Kraft lignin to aromatic monomers, in supercritical methanol. A variety of bimetallic Ni, Mo, Co and W catalysts on various supports were prepared, characterized and evaluated leading to several conclusions regarding catalyst structure–activity relationships. It was found that the activity towards lignin depolymerisation and further processing of the obtained aromatic fragments, depended on several factors: (i) sulphided catalysts were more active than oxide catalysts. (ii) W was a more suitable metal than Mo. (iii) Ni was a more ideal promoter than Co. (iiii) The role of the support was found to be significant and activated carbon or basic MgO–La2O3 gave highest product yields, whereas acidic supports promoted char formation.Excellent, 35 wt% monomer yields were achieved with a sulphided S-NiW/AC catalyst and the product mixture contained up to 26 wt% alkylphenolics, moreover, competing ring hydrogenation or severe repolymerisation to insoluble char was not observed. This stands among the best results in the conversion of Kraft lignin and will lead to the development of efficient methods for the valorisation of lignin waste streams in the future.
Acknowledgements
This research has been performed within the framework of the CatchBio program. The authors gratefully acknowledge the support of the Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science. The authors gratefully acknowledge Dr Arjan Kloekhorst for his help with the GC × GC-FID measurements.References
- A. Tejado, C. Pena, J. Labidi, J. Echeverria and I. Mondragon, Bioresour. Technol., 2007, 98, 1655 CrossRef CAS PubMed.
- A. J. Ragauskas, M. Nagy, D. H. Kim, C. A. Eckert, J. P. Hallett and C. L. Liotta, Ind. Biotechnol., 2006, 2, 55 CrossRef CAS.
- F. G. C. Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227 CrossRef PubMed.
- J. Zakzeski, P. C. A. Bruijninx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, in Top Value-Added Chemicalsfrom Biomass. Results of Screening for Potential Candidates from BiorefineryLignin, U.S. Department of Energy (DOE) by PNNL, Richland, WA, USA, 2007, vol. II, PNNL-16983 Search PubMed.
- Ch. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695 CrossRef CAS PubMed.
- S. Dutta, K. C. W. Wu and B. Saha, Catal. Sci. Technol., 2014, 4, 3785 CAS.
- C. Xu, R. Arneil, D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485 RSC.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. N. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- Z. Strassberger, S. Tanase and G. Rothenberg, RSC Adv., 2014, 4, 25310 RSC.
- J. Gierer, Wood Sci. Technol., 1980, 14, 241 CrossRef CAS.
- W. O. Doherty, P. Mousavioun and C. M. Fellows, Ind. Crops Prod., 2011, 33, 259 CrossRef CAS PubMed.
- J. Gierer, I. Noren and S. Wannstrom, Holzforschung, 1987, 41, 79 CrossRef CAS.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131 CrossRef CAS PubMed.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph and J. H. Christensen, Phytochem. Rev., 2004, 3, 29 CrossRef CAS.
- E. Adler, Wood Sci. Technol., 1977, 11, 169 CrossRef CAS.
- W. J. Liu, H. Jiang and H. Q. Yu, Green Chem., 2015 10.1039/C5GC01054C.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29 CrossRef CAS PubMed.
- D. Mohan, Ch. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249 CrossRef CAS PubMed.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2014, 16, 191 RSC.
- P. J. Deuss and K. Barta, Coord. Chem. Rev., 2015 DOI:10.1016/J.CCR.2015.02.004.
- G. Chatel and R. D. Rogers, ACS Sustainable Chem. Eng., 2014, 2, 322 CrossRef CAS.
- M. Zaheer and R. Kempe, ACS Catal., 2015, 5, 1675 CrossRef CAS.
- H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci. Technol., 2012, 2, 869 CAS.
- E. E. Harris, J. D'Ianni and H. Adkins, J. Am. Chem. Soc., 1938, 60, 1467 CrossRef CAS.
- K. M. Torr, D. J. van de Pas, E. Cazeils and I. D. Suckling, Bioresour. Technol., 2011, 102, 7608 CrossRef CAS PubMed.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. t. Liu and Y. Kou, ChemSusChem, 2008, 1, 626 CrossRef CAS PubMed.
- C. Li, M. Zheng, A. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383 CAS.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994 CAS.
- K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47, 1503 CrossRef CAS PubMed.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. Hensen, ChemSusChem, 2014, 7, 2276 CrossRef CAS PubMed.
- R. Ma, W. Hao, X. Ma, Y. Tian and Y. Li, Angew. Chem., Int. Ed., 2014, 126, 7438 CrossRef PubMed.
- X. Huang, T. I. Korányi, M. Boot and E. J. Hensen, Green Chem. 10.1039/C5GC01120E.
- X. Wang and R. Rinaldi, Angew. Chem., Int. Ed., 2013, 52, 11499 CrossRef CAS PubMed.
- W. Xu, S. J. Miller, P. K. Agrawal and C. W. Jones, ChemSusChem, 2012, 5, 667 CrossRef CAS PubMed.
- S. Huang, N. Mahmood, M. Tymchyshyn, Z. Yuan and C. C. Xu, Bioresour. Technol., 2014, 171, 95 CrossRef CAS PubMed.
- G. Gellerstedt, J. Li, I. Eide, M. Kleinert and T. Barth, Energy Fuels, 2008, 22, 4240 CrossRef CAS.
- A. Kloekhorst, Y. Shen, Y. Yie, M. Fang and H. J. Heeres, Biomass Bioenergy, 2015, 80, 147 CrossRef CAS PubMed.
- A. K. Deepa and P. L. Dhepe, ACS Catal., 2014, 5, 365 CrossRef.
- S. K. Singh and J. D. Ekhe, RSC Adv., 2014, 4, 53220 RSC.
- V. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. Li and J. A. Lercher, Chem. – Eur. J., 2011, 17, 5939 CrossRef CAS PubMed.
- J. A. Onwudili and P. T. Williams, Green Chem., 2014, 16, 4740 RSC.
- J. Miller, L. Evans, A. Littlewolf and D. Trudell, Fuel, 1999, 78, 1363 CrossRef CAS.
- J. S. Shabtai, US pat, US5959167A, 1999 Search PubMed.
- A. L. Jongerius, P. C. Bruijnincx and B. M. Weckhuysen, Green Chem., 2013, 15, 3049 RSC.
- S. Huang, N. Mahmood, M. Tymchyshyn, Z. Yuan and C. C. Xu, Bioresour. Technol., 2014, 171, 95 CrossRef CAS PubMed.
- R. Ma, W. Hao, X. Ma, Y. Tian and Y. Li, Angew. Chem., Int. Ed., 2014, 126, 7438 CrossRef PubMed.
- X. Ma, R. Ma, W. Hao, M. Chen, F. Yan, K. Cui, Y. Tian and Y. Li, ACS Catal., 2015, 5, 4803 CrossRef CAS.
- E. Furimsky, Catal. Rev., 1983, 25, 421 CAS.
- E. Furimsky and F. E. Massoth, Catal. Rev., 1999, 52, 381 CAS.
- (a) S. Mukundan, M. Konarova, L. Atanda, Q. Ma and J. Beltramini, Catal. Sci. Technol., 2015, 5, 4422 RSC; (b) V. N. Bui, D. Laurenti, P. Delichere and C. Geantet, Appl. Catal., B, 2011, 101, 246 CrossRef CAS PubMed.
- M. L. Kantam, R. Kishore, J. Yadav, M. Sudhakar and A. Venugopal, Adv. Synth. Catal., 2012, 354, 663 CrossRef CAS PubMed.
- (a) L. Ding and Y. Zheng, Catal. Commun., 2006, 7, 1035 CrossRef CAS PubMed; (b) F. Bataille, J. L. Lemberton, G. Perot, P. Leyrit, T. Cseri, N. Marchal and S. Kasztelan, Appl. Catal., A, 2001, 220, 191 CrossRef CAS.
- W. Yin, R. H. Venderbosch, G. Bottari, K. K. Krawzcyk, K. Barta and H. J. Heeres, Appl. Catal., B, 2015, 166–167, 56 CrossRef CAS PubMed.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090 CrossRef CAS PubMed.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640 RSC.
- M. Garcia-Perez, The Formation of polyaromatic hydrocarbons and dioxins during pyrolysis: a review of the literature with descriptions of biomass composition, fast pyrolysis technologies and thermochemical reactions, Washington State University, WA, USA, 2008, p. 5 Search PubMed.
- T. M. Liitiä, S. L. Maunu, B. Hortling, M. Toikka and I. Kilpeläinen, J. Agric. Food Chem., 2003, 51, 2136 CrossRef PubMed.
- C. Fernández-Costas, S. Gouveia, M. Sanromán and D. Moldes, Biomass Bioenergy, 2014, 63, 156 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01643f |
| This journal is © The Royal Society of Chemistry 2015 |