
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural changes in lignins isolated using an acidic ionic liquid water mixture†
Agnieszka
Brandt‡
a,
Long
Chen‡
bc,
Bart E.
van Dongen
d,
Tom
Welton
b and
Jason P.
Hallett
*a
aDepartment of Chemical Engineering, Imperial College London, London, SW7 2AZ, UK. E-mail: j.hallett@imperial.ac.uk
bDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK
cState Key Laboratory of Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
dSchool of Earth, Atmospheric, and Environmental Sciences and the Williamson Research Centre for Molecular Environmental Science, University of Manchester, Manchester M13 9PL, UK
First published on 2nd September 2015
Abstract
Recently, acidic ionic liquid water mixtures based on the hydrogen sulfate anion have been shown to effectively extract lignin from lignocellulosic biomass. This study analyses Miscanthus giganteus lignin isolated after extraction with the protic ionic liquid 1-butylimidazolium hydrogen sulfate ([HC4im][HSO4]) followed by precipitation with the antisolvent water. Several analytical techniques were employed, such as quantitative 13C-NMR, 1H–13C HSQC NMR, 31P-NMR, Py-GC-MS, GPC and elemental analysis. The analysis shows that the ionic liquid pretreatment breaks lignin-hemicellulose linkages and depolymerizes the lignin through the cleavage of glycosidic, ester and β-O-4 ether bonds. This is accompanied by solubilization of the newly generated lignin fragments. At longer pretreatment times, repolymerization of lignin fragments through condensation reactions occurs. The isolated lignins were carbohydrate-free, had low sulfur contents, low molecular weights. Early stage lignins were structurally similar to ball-milled lignin, while more treated lignins were enriched in p-hydroxyphenyl and guaiacyl units and had a high phenolic hydroxyl group content. We conclude that, depending on the treatment conditions, lignins with a variety of characteristics can be isolated using this type of ionic liquid solution.
Introduction
It is imperative to develop cost-competitive processes for the production of sustainable liquid fuels and chemicals. This is due to the significant contribution of petroleum use to climate change, compounded by the increasing demand for liquid fuels.Lignocellulosic biomass is a sustainable source of organic carbon that is a suitable feedstock for large-scale production of renewable fuels and materials.1,2 It is mainly composed of the polymers cellulose, hemicellulose and lignin and is naturally resistant to deconstruction by most microbes, enzymes and mechanical stress. This is collectively defined as recalcitrance.3 The recalcitrance makes fuel and chemical production from lignocellulose more complex, energy-intensive and currently also more expensive than fuel production from starch or sucrose.
To date, an array of pretreatment technologies has been explored to enable utilisation of the carbohydrates contained in the lignocellulose.4–6 Many attempts to enhance or simplify the pretreatment have been made, as this step represents a large capital investment (19–22% of total capital)7 for lignocellulosic biofuel production. However, inefficient deconstruction or the production of downstream inhibitors highlights the need for the development of improved methods.8
Recently, ionic liquid (IL)-based biomass pretreatments have generated increasing interest due to the unique behavior of ILs toward the component biomass polymers. The most widely investigated IL-based pretreatment is the dissolution of the entire biomass composite with dialkylimidazolium acetate ILs.9 Unfortunately, this approach suffers significant disadvantages, including limited IL thermal stability10 and high solvent cost.11 The “Ionosolv deconstruction” is an alternative IL-based pretreatment that demonstrates high delignification efficiency and high lignin yields.12,13 The lignin is recovered from the IL solution by reversible addition of water as an antisolvent. A recent techno-economic analysis of the ILs used in Ionosolv deconstruction indicated that these will have manufacturing costs similar to conventional organic solvents, such as toluene or acetone.14 Because of these cost advantages, we believe that Ionosolv deconstruction holds significant promise for industrial application.
A number of studies have demonstrated that the conversion of cellulose to biofuels benefits from reduced lignin content and alterations in lignin structure.15,16 Early separation of lignin from the carbohydrates is also desirable for obtaining high quality lignins, which is thought to greatly enhance the economic returns of a biorefinery.17 Separation of lignin and cellulose can only be achieved by a limited number of pretreatment technologies. The majority of these dissolve the lignin while they leave cellulose, the main carbohydrate component, as a solid. This separation requires a solvent with three capabilities: the ability to break the linkages between carbohydrates and lignin, a high solubility of the lignin fragments in the solvent and an effective way to recover the lignin from the solution.
Lignin is an irregular polyphenolic biopolymer in plants, synthesized from up to three phenylalanine-derived monomers that differ in their ring methoxylation: coniferyl, sinapyl and p-coumaryl alcohol. These monomers assemble into a racemic macromolecule via free radical polymerization, giving rise to guaiacyl (G), syringyl (S) and p-hydroxyphenyl (H) subunits in the lignin polymer. In grasses such as Miscanthus, p-coumaric acid (PCA) is also present in significant amounts. PCA is attached to the lignin via ester bonds with the γ-hydroxyl group of the lignin side chain.18 The core lignin polymer contains a wide range of linkages, such as β-O-4, β-5, β–β, 5–5, 4-O-5, and β-1, with no regular inter-unit repeating structure observed.19–21 In addition, lignin carbohydrate complex (LCC) linkages occur in grasses. These form during lignification between ferulic acid (FA) containing hemicellulose (feruloyted arabinoxylan) and the nascent lignin. The extent of this cross-linking has been correlated with increased plant cell wall recalcitrance.22
Miscanthus is a tall perennial grass with high lignin content (typically around 25%) and can be grown as a dedicated energy crop with high yields and low maintenance. Without pretreatment, enzymatic saccharification yields are very low. Miscanthus giganteus lignin has previously been characterized as a G/S/H type lignin (52%, 44% and 4%, respectively) with approximately 0.4 β-O-4 linkages per aromatic ring, and ca. 0.1 p-coumaric acid ester linkages per aromatic ring.23
Currently, detailed characterisation of the lignin precipitate recovered after Ionosolv pretreatment is not available. There is also a lack of understanding of the chemical transformations leading to the lignin extraction. Understanding these will provide important insights for controlling the chemical characteristics of the precipitated lignin and for achieving optimal carbohydrate-lignin separations.
Lignin's complexity as well as the difficulty to isolate native lignin from cell walls in an unaltered state, provides significant challenges to analytical techniques.24 Nevertheless, technological advances have enabled researchers to collect valuable information about the composition and chemical linkages in isolated lignins. In the present study, we have examined these transformations through the characterization of lignins isolated from Miscanthus at different time points. We report structural characteristics of Ionosolv lignins using several complementary analytical techniques. Quantitative 13C-NMR and two-dimensional 1H–13C heteronuclear single-quantum coherence NMR spectroscopy (HSQC NMR) provided information on changes in sub-unit composition, and inter-unit linkages. 31P-NMR spectroscopy was employed to determine the concentrations of hydroxyl functionalities and how these change throughout the course of the pretreatment. Pyrolysis-gas chromatography-mass spectrometry (Py-GC-MS) was applied to examine changes in the lignin subunit composition. The molecular weights of the lignins were measured by gel permeation chromatography (GPC) to elucidate the interplay between depolymerization and repolymerization reactions occurring during the IL pretreatment. Elemental analysis (EA) was used to assess the purity of the lignins and draw conclusions about its fuel value. We chose the representative ionic liquid 1-butylimidazolium hydrogen sulfate ([HC4im][HSO4]) as pretreatment solvent, an Ionosolv IL previously shown to have efficacy in biomass deconstruction.25
Materials and methods
Materials
The lignocellulosic feedstock used throughout this study was Miscanthus giganteus harvested from Silwood Park campus, Imperial College London, UK. It was air-dried, ground and sieved (0.18–0.85 mm, −20 to +80 of US mesh scale) before use. The biomass was then stored in plastic bags at room temperature. The synthesis of the IL 1-butylimidazolium hydrogen sulfate ([HC4im][HSO4]) is described in the ESI.†Ionosolv deconstruction and lignin isolation
Ionosolv lignins were isolated according to the method of Brandt et al.13 with slight modifications. 1 g biomass (on oven-dry weight basis) was placed in a wide mouthed Pyrex culture tube with a screw cap. 8 g dried [HC4im][HSO4] and 2 g water were then added. The tubes were sealed and samples incubated without stirring in an oven at 120 °C for a pre-determined time. After pretreatment, the samples were cooled to room temperature and washed with acetone (20 mL) followed by filtration, giving carbohydrate rich material (CRM) and a liquor. The liquor was concentrated by evaporating the acetone, then water was added as an anti-solvent to precipitate the lignin, and the recovered lignin was dried under vacuum at 40 °C overnight.Py-GC-MS experiments
The Miscanthus giganteus and Ionosolv lignin samples (approximately 1.1 to 1.4 mg) were pyrolyzed using a chemical data system (CDS) 5200 series pyroprobe pyrolysis unit by heating at 600 °C for 20 seconds to fragment macromolecular components. Fragments were analyzed using an Agilent 7890A gas chromatograph, fitted with a HP-5 fused capillary column (J+W Scientific; 5% diphenyl-dimethylpolysiloxane; 30 m length, 0.32 μm internal diameter, 0.25 μm film thickness), coupled to an Agilent 5975 MSD single quadrupole mass spectrometer operating in electron ionization (EI) mode (scanning a range of m/z 50 to 700 at 2.7 scans per second; ionization energy 70 eV). The pyrolysis transfer line and injector temperatures were set at 350 °C, the heated interface at 300 °C, the EI source at 230 °C and the MS quadrupole at 150 °C. The GC oven was programmed from 40 °C (held for 3 min) to 100 °C at 300 °C min−1 and then to 300 °C at 5 °C min−1 and held at this temperature for 15 min. Helium was used as the carrier gas (1 mL min−1) and the compounds were introduced in split mode (split ratio 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Prior to analyses, 1 to 3 μl of an internal standard (5α-androstane; 100 μl of a 0.256 mg mL−1 solution in dichloromethane) and in the case of GC-MS with tetramethylammonium hydroxide (TMAH) thermochemolysis (Py-TMAH-GC-MS), 10 μl of a TMAH solution (25%, w/w, in methanol) was added to each sample. The relative peak areas were obtained by normalization to the total integral of the areas.
:1). Prior to analyses, 1 to 3 μl of an internal standard (5α-androstane; 100 μl of a 0.256 mg mL−1 solution in dichloromethane) and in the case of GC-MS with tetramethylammonium hydroxide (TMAH) thermochemolysis (Py-TMAH-GC-MS), 10 μl of a TMAH solution (25%, w/w, in methanol) was added to each sample. The relative peak areas were obtained by normalization to the total integral of the areas.
NMR spectroscopy
:1 (v/v) of pyridine and deuterated chloroform was prepared. The solvent solution was used to prepare a mixture solution containing 20.5 mg ml−1 of cyclohexanol (as internal standard) and a second 5.6 mg ml−1 solution of chromium(III) acetylacetonate solution (relaxation reagent). Previously dried Ionosolv lignin (ca. 10 mg) was accurately weighed and dissolved in 100 μl of anhydrous pyridine/deuterated chloroform solvent solution (1.6:1, v/v). 50 μL of the cyclohexanol solution, 50 μL of the chromium(III) 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) were added to the lignin and the sealed vial was vortex mixed intensely until it was completely dissolved. The samples were transferred into Shigemi NMR tubes for subsequent NMR analysis. The NMR experiments were carried out at 298 K on a Bruker Avance 500 MHz NMR spectrometer. To obtain quantitative spectra a relaxation delay of 25 s was used between 30° pulses, the number of scans was 127 and an inverse gated decoupling pulse sequence was used. The acquisitions were performed at room temperature. Chemical shifts were calibrated relative to the internal standard, i.e. the cyclohexanol peak signal centred at δ 144.2 ppm. Integration regions that were used to assign the signals and the relative signal intensities used to calculate the concentration of hydroxyl groups are tabulated in Table S4 (ESI†).
GPC measurements
A JASCO instrument equipped with an LC-NetII/ADC interface, an RI-2031Plus refractive index detector, two PolarGel-M columns (300 × 7.5 mm) and two PolarGel-M guard columns (50 × 7.5 mm) was employed. Dimethylformamide (DMF) with 0.1% lithium bromide was used as the eluent. Samples were prepared at 1 mg ml−1 concentration in DMF with 0.1% LiBr. The flow rate was 0.7 mL min−1 and the analyses were carried out at 40 °C. Polystyrene standards (Sigma-Aldrich) ranging from 266 to 70000 g mol−1 were used for calibration.
Treatment of p-coumaric acid in [HC4im][HSO4] water mixtures
75 mg (0.46 mmol) of p-coumaric acid (Sigma-Aldrich) were dissolved in 1.315 g (4.74 mmol) of an 80 wt% solution of [HC4im][HSO4] with 20 wt% water in a small round bottomed flask with stir bar. The flask was heated at 120 °C for 1 h, 5 h, 8 h and 12 h. Samples for 1H-NMR analysis were dissolved in DMSO-d6 and analysed on Bruker Avance 400 MHz NMR spectrometer. At the end of the experiment, the IL solution was diluted with water and the resulting precipitate washed 3 times with water, followed by drying under vacuum at 40 °C. The yellowish powder was submitted to electrospray mass spectrometry analysis (Micromass LCT Premier, Waters) using methanol as a solvent.Elemental analysis
Elemental analysis was performed in duplicate by Medac Ltd (Chobham, UK). Raw data were processed by calculating the cation and anion contributions, which were subsequently subtracted to obtain the elemental composition of the IL free lignins. The molar carbon hydrogen ratio for the IL free lignin was calculated, as was the molar cation to anion ratio of the ionic liquid portion.Results and discussion
The Miscanthus gigantheus biomass utilised in this study contained 22.4% acid-insoluble and 4.0% acid-soluble lignin (exact composition shown in ESI, Table S1†). The lignin was extracted using a solution of 80 wt% 1-butylimidazolium hydrogen sulfate and 20 wt% water at 120 °C for between 1 h and 24 h. The lignin containing ionic liquid solutions were separated from the cellulose pulp by washing with acetone. The acetone was removed and the lignins precipitated by adding water. The washed and dried lignins were subjected to the analyses detailed below.Quantitative 13C-NMR
The composition of lignins isolated after 1 h and 12 h was probed using quantitative 13C-NMR spectroscopy. Due to the low sensitivity, the acquisition time was long and the signal-to-noise ratio poor (Fig. 1). Some of the signal groups overlap. Nevertheless the spectra provide useful clues about the composition of lignin linkages. We assigned areas of the spectrum to functional groups and functional group clusters and quantified by integration, similar to El Hage et al. (Table 1).23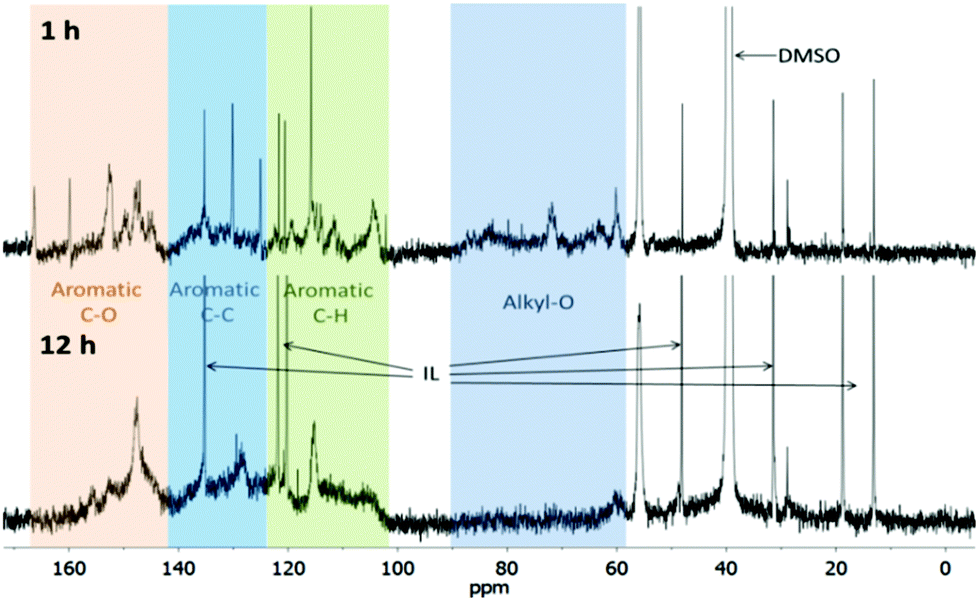 | ||
| Fig. 1 Quantitative 13C NMR spectra of Ionosolv lignin isolated after pretreatment for 1 h (top) and 12 h (bottom). Peaks caused by the NMR solvent (DMSO) and the ionic liquid (IL) cation were also detected. | ||
| δ (ppm) | Assignment | 1 h lignin | 12 h lignin |
|---|---|---|---|
| 1–6: positions on aromatic ring, α,β,γ: positions on side chain. | |||
| Clusters | |||
| 166–142 | Aromatic C–O | 1.91 | 1.81 |
| 142–124 | Aromatic C–C | 1.87 | 2.06 |
| 124–102 | Aromatic C–H | 2.11 | 2.03 |
| 90–58 | Alkyl-O | 2.65 | 0.30 |
| Individual signals | |||
| 166 | PCAγ | 0.17 | 0 |
| 160 | PCA4 | 0.11 | 0.04 |
| 156 | H4 | 0.08 | 0.06 |
| 151–154 | S3/5 (ether & condensed) | 0.68 | 0.30 |
| 148–151 | G3 (ether & condensed) | 0.31 | 0.44 |
| 147–148 | G4 (ether) & S3,5 (Non-ether) | 0.6 | 0.63 |
| 145 | G3,4 (non-ether) | 0.28 | 0.26 |
| 135 | S1/G1 | 0.33 | 0.34 |
| 130 | PCA2/6 | 0.35 | 0 |
| 127–130 | H2,6 | 0.24 | 0.53 |
| 125 | PCA1 | 0.16 | 0 |
| 119 | G6 | 0.21 | 0.14 |
| 116 | PCA3,5 & H3,5 | 0.32 | 0.17 |
| 115.5–114.5 | G5 | 0.25 | 0.25 |
| 113–114.5 | PCAβ | 0.14 | 0.17 |
| 110–113 | G2 | 0.26 | 0.30 |
| 103–106 | S2,6 | 0.46 | 0.19 |
| 88–77 | Cβ in β-O-4, Cα in β-5&β–β | 0.82 | 0.02 |
| 71–73 | Cα in β-O-4, Cγ in β–β | 0.35 | 0.02 |
| 65 | Cγ PCA ester | 0.24 | 0.01 |
| 63 | Cγ in β-5 | 0.27 | 0.04 |
| 58–61 | Cγ (in β-O-4 and primary OH) | 0.29 | 0.15 |
| 55 | MeO | 1.30 | 0.73 |
| 29 | α-β methylene groups | 0.09 | 0.08 |
| 22 | Acetic acid (CH3) | 0.02 | 0 |
The NMR spectrum of the early stage lignin (1 h) and the lignin isolated after extensive treatment (12 h) differed significantly. The 1 h lignin had significant intensity in the aliphatic region, while in the 12 h lignin almost all carbon atoms were vinylic or aromatic. The 1 h lignin was characterised by alternating high and low intensities in the aromatic region (102–165 ppm), while the 12 h lignin aromatic intensity was more evenly distributed, with only 3 distinct spikes. This suggests that there was a greater heterogeneity in the aromatic structure of the 12 h lignin. A general commonality was that both lignins appeared to be contaminated with a minor amount of butylimidazole. We have performed additional purification experiments that show a substantial reduction in imidazolium content, although it is noteworthy that it was never removed completely from the lignin.
The 13C-NMR spectrum of the 1 h lignin was similar to the spectrum reported for ball milled lignin,23 suggesting that the early Ionosolv lignin was similar in composition and linkages. Integration of functional group clusters revealed that the alkyl-O content in the 1 h Ionosolv lignin was high with, around 2.7 alkoxy groups per aryl ring. Strikingly, the alkoxy group content was only 0.3 per aryl ring in the 12 h lignin. This implies that the majority of alkoxy side chains had been modified during extended Ionosolv treatment, most likely by reactions such as dehydration, shortening of the side chain and transformations of alkoxy groups into carbonyl groups such as ketones, aldehydes or carboxylic acid groups (some intensity for these was found at 180–220 ppm).
In the 12 h lignin, we observed an increased abundance of aromatic C–C bonds, which we attribute to condensation reactions, leading to replacement of C–H bonds with C–C bonds. Lignin condensation is the formation of non-native bonds between lignin polymer chains and compounds cleaved from the native lignin. The condensation reaction most commonly postulated is in competition with β-O-4 ether hydrolysis. It forms a diphenylmethane structure between electron-rich C–H positions on subunit rings and α carbons on side chains with a non-hydrolyzed β-O-4 linkage (Fig. 2).28,29
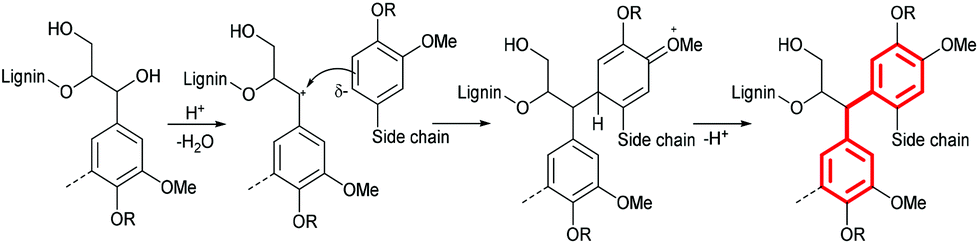 | ||
| Fig. 2 Formation of condensed diphenylmethane structures at the G6 position during pretreatment in acidic solvents (assumed mechanism). | ||
We also observed a slight decrease in C–O bond abundance. This could be due to a reduction in the content of S units, which has three C–O bonds, compared to H and G units which have only one or two C–O bonds. Further evidence for low S unit content in highly treated lignins is provided by the methoxy group content, which decreased from 1.30 to 0.73 methoxy groups per aromatic ring. The integrals of S peaks (151–154, 103–106 ppm) decreased in size, while the integrals of G peaks (119, 115, 145, 148–151 ppm) remained constant. The S/G unit ratio was calculated using the S2,6 and G2 peak integrals. It was 0.88 for the 1 h lignin, which agrees well with the literature value of 0.85 for ball milled Miscanthus lignin,23 but was only 0.32 for the 12 h lignin, again suggesting that the 12 h lignin may have a low S unit content. However, condensation reactions may create a false impression, as they will shift the S resonances to higher frequencies. The degree of the shifts will depend on the exact nature and number of substitutions that have taken place. It is conceivable that the electron-rich S units are more affected by such condensation than are the G units, resulting in an apparent reduction in the S/G ratio.
A very notable difference between early stage and late stage Ionosolv lignin was the disappearance of the PCA peaks. The 1 h lignin had a similar or slightly higher PCA content than ball milled Miscanthus lignin,23 while PCA signals were absent in the 12 h lignin. Conversely, signals for H units (visible in the 127–130 ppm region) were not detected for the 1 h lignin but were noticeable in the 12 h lignin spectrum. The acetyl group content was low in both early and late stage lignins, suggesting that acetyl groups are removed early in the pretreatment.
Overall, the quantitative 13C NMR spectra show that Ionosolv lignins can be structurally very different, depending on the length (severity) of the treatment.
HSQC NMR analysis
1H–13C HSQC NMR is a two-dimensional NMR technique resolving resonances that overlap in one-dimensional 13C NMR and 1H NMR spectra. Fewer signals are seen, as only carbon atoms with one or more protons provide a signal. It is a powerful technique giving equivalent or even better information compared to traditional wet chemistry methods.22A number of structural units can be detected by HSQC. They have been assigned by comparison with cell wall model compounds in an NMR database.30–35 Examples of substructures that are resolved by HSQC NMR are β-ether (β-O-4, A), resinol (β–β, B), phenylcoumaran (β-5, C) and Hibbert's ketone (H) side chains. Carbohydrates can also be detected. The aromatic rings of the G, S and H units are resolved, as are the rings and unsaturated side chains of p-coumaric acid (PCA) and ferulic acid (FA). The chemical structures of these substructures are shown in Fig. 3.
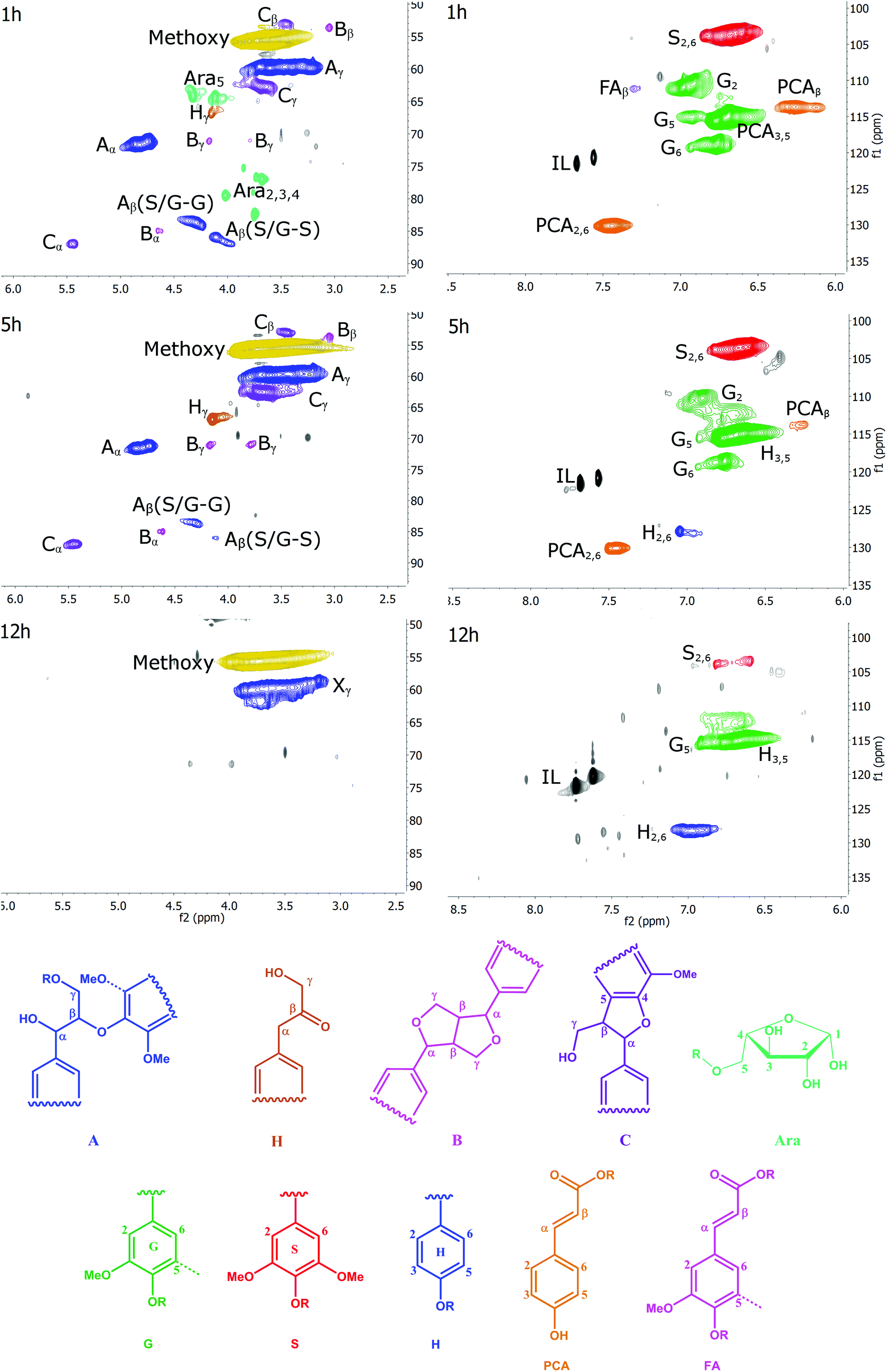 | ||
| Fig. 3 The HSQC spectra of the side chain (left) and aromatic region (right) of Ionosolv lignins after different pretreatment times and the substructures detected in it. Grey peaks: unassigned; Xy primary hydroxyl group on condensed lignin. H = Hibbert's ketone side chain. | ||
HSQC spectra of cell walls and lignins can be divided into three regions: the aliphatic region, the side chain region and the aromatic region. The aliphatic region does not provide useful structural information, with the exception of the presence of signals at δC/δH 20.7/1.7–1.9 ppm, corresponding to acetyl groups attached to the lignin polymer, as well as a peak at 20.6/2.0 ppm corresponding to acetyl groups attached to hemicellulosic components. Acetyl groups were not detected in the isolated lignins. Therefore, the aliphatic region will not be discussed.
We recorded HSQC spectra of ball milled whole cell walls of Miscanthus giganteus and the Ionosolv lignins obtained after 1 h, 5 h, 8 h, 12 h and 24 h of treatment. Full spectra are shown in the ESI (Fig. S7–S12†) and a list of assigned peaks is shown in Table S2.† The annotated side chain regions (δC/δH 50–90/2.5–5.8 ppm) and aromatic regions (δC/δH 110–130/6.0–9.0 ppm) of the 1 h, 5 h and 12 h spectra are shown in Fig. 3 and discussed below. The annotated HSQC spectra of the 8 h lignin is shown in Fig. S6 of the ESI.†
Side chain region
The side chain region of the HSQC provides valuable information about linkages in the lignin structure (Fig. 3, left column). In addition, the methoxyl substituents on the aromatic ring contribute to the most prominent peak at δC/δH 55.6/3.73 ppm.The most common linkage in the 1 h lignin was, as expected, the β-O-4 linkages (structure A). This lignin also contained carbohydrates. The signals correspond to a furanose saccharide, which strongly suggests that the 1 h lignin contained arabinose. Lignin with a significant arabinose content has been previously observed after mild Organosolv extraction.36 The arabinose correlations disappeared after 1 h of pretreatment. This suggests that the lignin-carbohydrate-complex (LCC) linkages, which are contributing to cell wall recalcitrance, are hydrolyzed rapidly during Ionosolv lignin extraction, and cleavage of the glycosidic bond between the arabinosyl substituent and the xylan core chain is fast.
For lignin isolated after short pretreatment times (1 and 5 h), a strong signal for the α methylene group of the β-O-4 linkage was observed, a signal for the Cγ–Hγ correlation and two Cβ–Hβ correlations, one for the side chain connected to a G unit and one for the side chain connected to an S unit. When examining the intensity of the β-O-4 ether peaks over time, it becomes evident that cleavage of this linkage is significant during Ionosolv deconstruction. The disappearance of the β-O-4 linkage signals after 12 hours of pretreatment was nearly quantitative. It is well known that cleavage of β-O-4 linkages is the most important depolymerization reaction in lignin in any pretreatment, although it rarely is as pronounced as seen here.
Interestingly, we were able to observe a signal for Hibbert's ketone (Hγ), the hydrolysis product of β-O-4 ether cleavage. We detected the γ signal at 67.0/4.2 ppm and also observed a peak for the corresponding α methylene group (at 44.3/3.6 ppm, just outside the range of the side chain region). The Hibbert's ketone resonances were observed at 1, 5 and 8 h, but disappeared at long treatment times (12 h).
The side chain region also yields information on resinol (β–β linkage, structure B) and phenylcoumaran (β-5 linkage, structure C) structures. The four correlations of the β–β linkage were observed and the three signals for the β-5 linkages (structure C) were readily identified. In contrast to the β-O-4 linkage, significant changes were not detected for the β–β and β-5 linkages before 8 h. However, their signals reduced significantly between 8 and 12 h of pretreatment. This suggests that the β–β and β-5 linkages were also chemically altered at longer treatment times. It is unclear whether the alterations led to actual cleavage, as it is unlikely that the C–C bonds in these linkages were broken.37
The most prominent correlation in the side chain region of the 12 h lignin was the methoxyl signal with a peak caused by primary aliphatic hydroxyl groups (Xγ). We called this signal Xγ because it occurs in the same location as the signal for the γ-methylol group of the β-O-4 linkage. However, the absence of the corresponding α and β signals means that Xγ signal must be caused by other aliphatic structures. At the moment we are not sure what these structures are.
Aromatic region
The aromatic region of the HSQC spectra contains correlations of the H, S and G aromatic rings and of ferulic and p-coumaric acid (Fig. 3, right column). Residual ionic liquid was detected in this region as C4-H and C5-H correlations of the imidazolium ring.The S units were represented by one prominent signal for the C2 and C6 correlations, whereas G units were represented by three correlations for their C2, C5, and C6 positions, respectively. The C5-H correlation overlaps with the PCA3,5-H and the H3,5-H correlation.
Ferulic acid (FA) peaks were small. The signals of p-coumaric acid (PCA) were prominent in the early stage lignins and decreased over time. A peak for the C2,6-H correlation of the H unit was prominent, in increasing amounts, in late-stage lignins.
Estimation of subunit composition using the HSQC aromatic region
The HSQC pulse sequence employed in this study is optimised for resolution and signal strength. Quantification is not completely reliable, as signal relaxation following each pulse will not be complete for some correlations, especially for end groups such as H38 and PCA, which relax more slowly than the bulk.22 Another limitation of integrating HSQC spectra is the potential for the earlier mentioned condensation reactions which replace aromatic C–H bonds with C–C bonds. These C–C bonded aromatic positions do not produce cross correlations, hence the calculated subunit composition may be skewed when examining condensed lignins.With these restrictions in mind, volume integration was attempted for estimating the subunit composition (Table 2). It should be noted that the G5 peak cannot be relied on for quantification of the guaiacyl content because of overlap with the PCA3,5 and H3,5 correlations. G6 is para to the methoxy group and hence has an increased potential to participate in condensation reactions (compare with Fig. 2). The G2 peak was therefore selected for estimating the S/G ratio to minimise these disturbances. HSQC spectra were recorded using the internal standard p-xylene (not shown). They showed that the G2 correlation was indeed the most stable signal per mg of lignin, with only a 7% drop in intensity; hence G2 was used as a reference peak for volume integration (integral set to 1.0).
The apparent subunit composition calculated this way is shown in Table 2. It can be seen that the S/G2 ratio for untreated Miscanthus is 0.69, which is lower than the ratio determined for ball-milled Miscanthus lignin by quantitative 13C-NMR (0.85 (ref. 23) and our value −0.88). The data in Table 2 further show that the Ionosolv lignin isolated after 1 h had a similar composition compared to ball-milled lignin in untreated Miscanthus. The S/G ratio appeared to drop with increasing pretreatment time. This trend agrees with our quantitative 13C NMR data, which provided an S/G ratio of 0.32 for the 12 h lignin.
Evidence of p-coumaric acid conversion and integration into treated lignins
Table 2 also shows that the H content increased by more than 5 times during treatment. Since the H content in ball-milled Miscanthus is generally low (ca. 3%, see Table 2), we speculate that this was not solely because of selective enrichment but because of p-coumaric acid conversion into H type structures. Initial evidence was provided by the interdependence of PCA removal and H accumulation observed in the HSQC, illustrated in Fig. 4.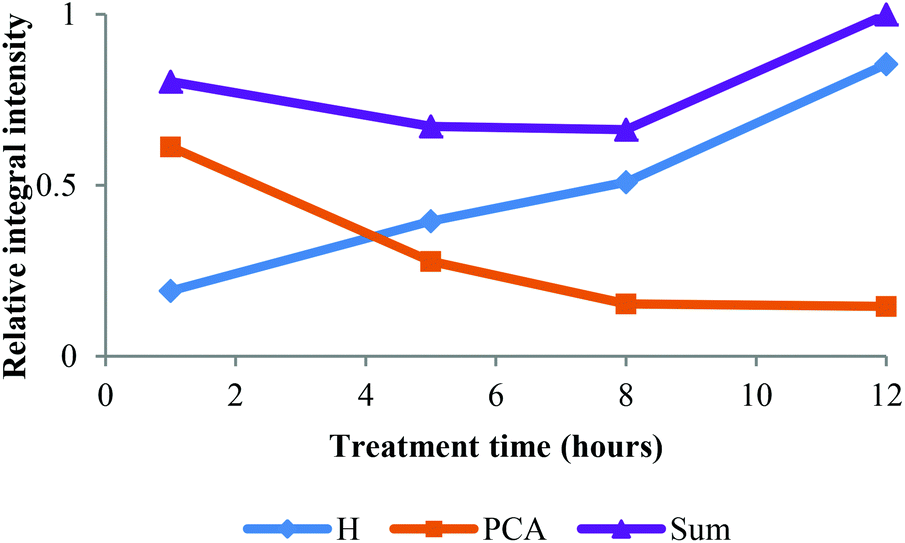 | ||
| Fig. 4 Time course for HSQC NMR volume integrals for H2,6 and PCA2,6 correlations over time. Values are relative to the sum at 12 h. | ||
To further verify the chemical incorporation of the hydroxycinnamic acid into isolated lignin as H type units, pure PCA was subjected to Ionosolv pretreatment. Samples were taken at intervals and submitted for 1H NMR analysis. The NMR spectra (see ESI†) indicate that PCA was consumed and new products were formed. To identify products, water was added, simulating the lignin recovery procedure. The precipitate was washed, dried and submitted for mass spectrometry. We were able to identify a polymeric product with a repeating unit of 120 g mol−1 (Fig. 5). Such a fingerprint agrees with the loss of the carboxylic acid group of PCA resulting in p-hydroxystyrene, followed by polymerisation to poly(p-hydroxystyrene). The presumed mechanism is shown in Fig. 6.
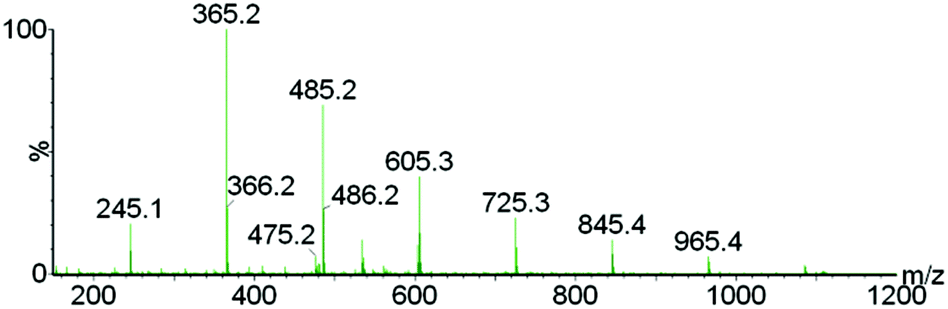 | ||
| Fig. 5 Electrospray mass spectrum of the product isolated after p-coumaric acid was treated in [C4Him][HSO4], showing the poly(p-hydroxystyrene) formed. | ||
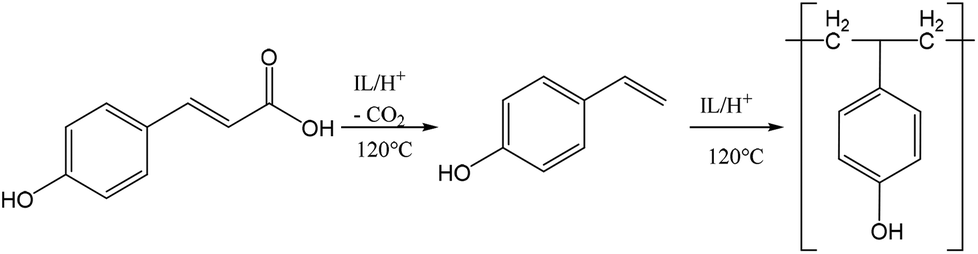 | ||
| Fig. 6 Suggested mechanism for the conversion of p-coumaric acid to poly(p-hydroxystyrene) in acidic Ionosolv solutions. | ||
These decarboxylation and polymerization reactions are likely catalyzed by the acidic protons in the IL–water mixture. Hence we conclude that the apparent enrichment of H units in the extensively treated lignin is due to the conversion of PCA into IL soluble oligomers that precipitate during lignin isolation. It is also possible that PCA co-polymerizes with lignin, but at present we do not have evidence for this. The oligomers appear to be short, as the most abundant oligomer was the trimer. MALDI ToF analysis (not shown) using a variety of matrices did not detect polymers with a molecular weight above 1000 g mol−1.
31P-NMR analysis
A quantitative 31P-NMR method was applied to study the major types of hydroxyl groups in the Ionosolv lignin and how their abundance changes over time. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) was employed in order to link hydroxyl groups in lignin arising from aliphatic, phenolic and carboxylic acids groups with phosphorous atoms. The phosphitylated lignin was then quantitatively assessed in a 31P NMR spectrum against an internal standard.39The original 31P-NMR spectra of the lignin samples are shown in the ESI,† as well as the numerical values of the concentrations derived from the integrals. Fig. 7 depicts the concentration changes of the lignin hydroxyl groups during Ionosolv pretreatment. The syringyl signal overlaps with signals of condensed G and S structures (β-5, β–β and others).39 It was observed that the concentration of phenolic hydroxyl groups (syringyl/condensed, guaiacyl and p-hydroxyphenyl) increased over time, while the aliphatic hydroxyl group content decreased.
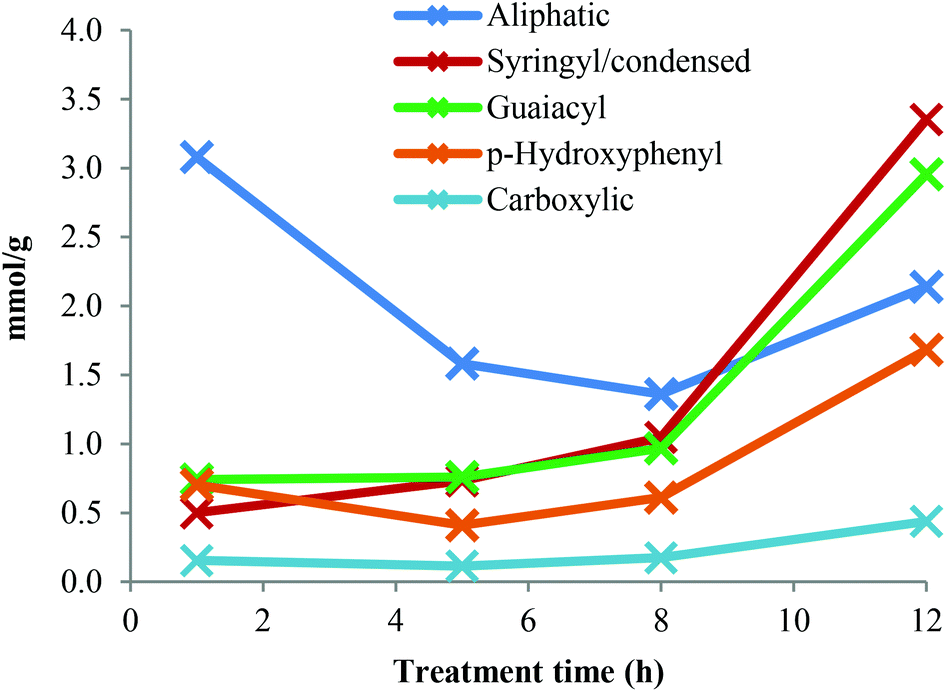 | ||
| Fig. 7 Hydroxyl group concentrations in Ionosolv lignins after different pretreatment times. | ||
For the lignin isolated after 1 h, the concentration of phenolic hydroxyl groups was low and the concentrations of G, S and H hydroxyl groups were similar. This suggests that most phenolic oxygen atoms were part of ether linkages. The lower content of S hydroxyl groups relative to G hydroxyls agrees with the observation that S units are preferably internal in native lignin and the relatively high content of p-hydroxyphenyl OH groups confirms that PCA and H units are terminal.38 The slow increase of phenolic hydroxyl content between 1 h and 5 h pretreatment time suggests that many of the phenolic ends produced by β-O-4 ether cleavage were not present in the isolated lignin. They are presumed to be part of small lignin fragments that were hydrophilic enough to remain in solution upon antisolvent (water) addition. A minimum of PCA and H content at 5 h pretreatment was observed, which also agrees with our HSQC data (Fig. 4). The drop in p-hydroxyphenyl hydroxyl concentration is attributed to the release of PCA into the IL solution.
The phenolic hydroxyl content increased after 5 h and particularly so after 8 h. This increase occurred when HSQC NMR indicate that condensation reactions become dominant. Condensation reactions are thought to involve the 2 and 6 positions of aromatic rings and the side chains but not the phenolic OH groups,40,41 hence phenolic hydroxyl groups become enriched when fragments are added to the lignin polymers via condensation. The hypothesis that soluble fragments rejoin the isolated lignin is in agreement with results previously published by our group, which show a continuing increase in lignin yield after 5 h of pretreatment.13
The abundance of aromatic OH groups in the 12 h lignin was the following: 42% syringyl/condensed, 37% guaiacyl and 21% PCA/p-hydroxyphenyl. The syringyl/condensed hydroxyl content increased steeply over time and was higher than the syringyl content estimated by quantitative 13C-NMR (24%) and HSQC NMR (33%), indicating that condensed units become abundant in highly treated Ionosolv lignins.
The aliphatic OH content decreased to a lower level with advancing pretreatment time. This is likely due to the loss of the Cα hydroxyl groups during β-O-4 ether breakage and during condensation. It is established knowledge that the first step of the β-O-4 ether hydrolysis in acidic media is the formation of a double bond between Cα and Cβ.42 The reduction in aliphatic hydroxyl content between 1 h and 5 h pretreatments is probably also caused by the removal of arabinose. The loss of γ-methylol groups during β-O-4 ether hydrolysis by splitting off as formaldehyde may also contribute to a reduction in aliphatic hydroxyl content. The carboxylic acid content increased slowly over time. It was low compared to the content of p-coumaric acid, confirming that PCA is esterified to the lignin polymer and that repolymerized PCA products do not contain carboxylic groups. The total hydroxyl content decreased after 1 h pretreatment and increased from 5 mmol g−1 at 1 h to 11 mmol g−1 at 12 h. This shows that the condensed lignin has more hydroxyl groups than the early-stage lignin. Increasing amounts of G and S phenolic hydroxyl groups have been observed before for Organosolv pulping under high severity (harsh) conditions,23 but still to a lesser degree than observed for the 12 h Ionosolv lignin.
In summary, our hydroxyl group analysis confirms that smaller lignin fragments are generated in the beginning of the Ionosolv treatment, some of them water-soluble. The solubilised fragments are not inert but rejoin the lignin precipitate, at least partially, through condensation reactions, increasing the over-all content of phenolic hydroxyl groups.
Py-GC-MS analysis
Pyrolysis-GC-MS was employed to characterize the compositions of untreated Miscanthus giganteus and Ionosolv lignin after 5 h and 12 h pretreatment (Fig. 8 and Table 3). The pyrolysis step cleaved the lignin polymeric framework into a multitude of fragments, which are separated by gas chromatography and identified by mass spectrometry. It should be noted that some of the lignin may remain as a non-volatile component (char). Hence caution should be used when interpreting the Py-GC-MS data.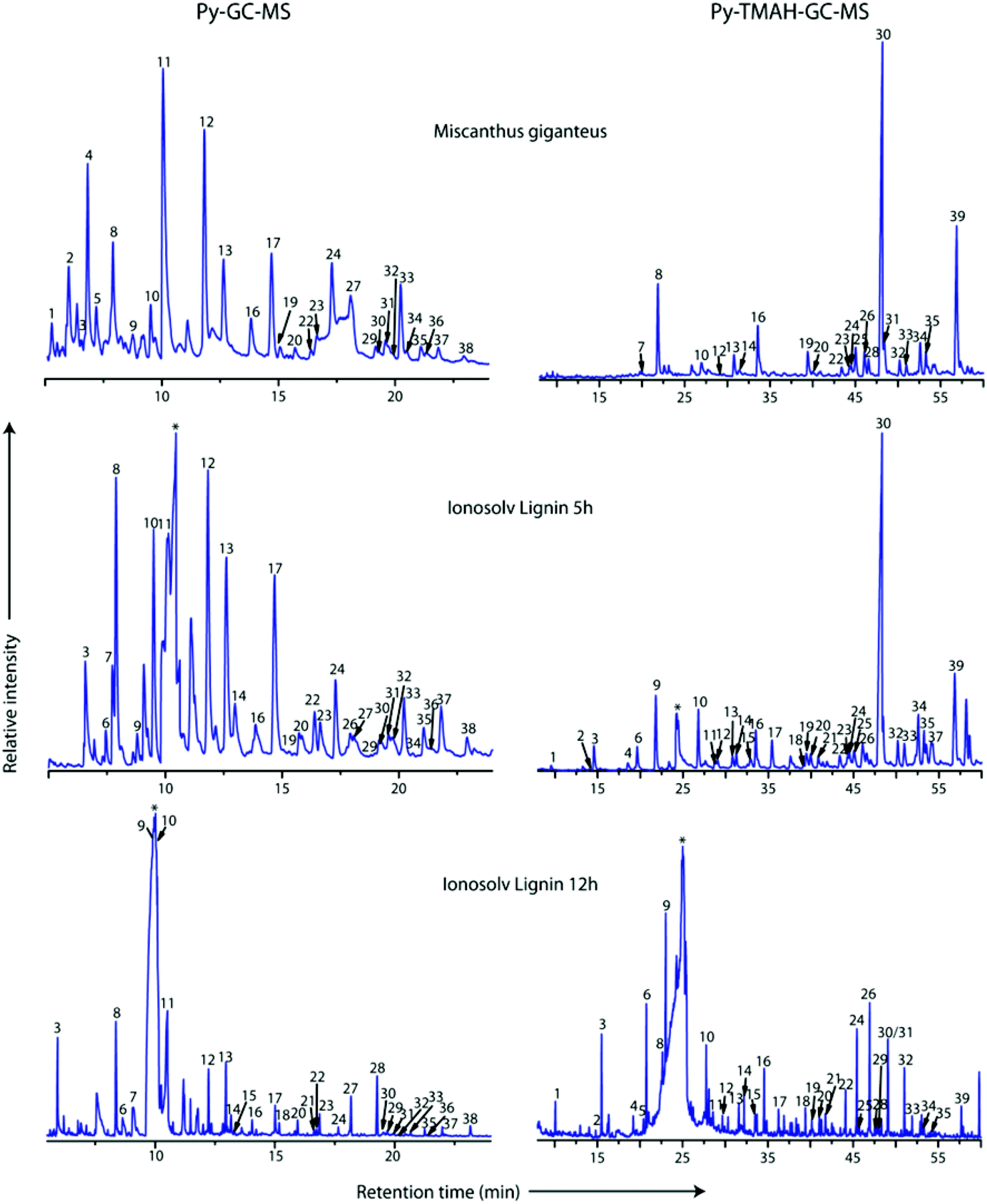 | ||
| Fig. 8 Py-GC-MS (left) and Py-TMAH-GC-MS (right) pyrograms of Miscanthus giganteus, and two Ionosolv lignins (5 h and 12 h). Numbers correspond to compounds listed in Tables 1 and 2 for Py-TMAH-GC-MS and Py-TMAH-GC-MS analyses, respectively. * indicates butylimidazole pyrolysis moieties. | ||
| Label | Compound | Origin | Relative peak area (%) | ||
|---|---|---|---|---|---|
| Miscanthus giganteus | Ionosolv lignin (5 h) | Ionosolv lignin (12 h) | |||
| PS: polysaccharide; H: Hydroxyphenyl unit; G: Guaiacyl unit; S: Syringyl unit; PCA: p-coumaric acid; FA: ferulic acid; BDL: Below detection limit. | |||||
| 1 | 2-Hydroxymethylfuran | PS | 1.64 | BDL | BDL |
| 2 | 2,3-Dihydro-5-methylfuran-2-one | PS | 3.63 | BDL | BDL |
| 3 | Phenol | H | 0.10 | 3.87 | 9.45 |
| 4 | 4-Hydroxy-5,6-dihydro-(2H)-pyran-2-one | PS | 7.03 | BDL | BDL |
| 5 | 3-Hydroxy-2-methyl-2-cyclopenten-1-one | PS | 2.44 | BDL | BDL |
| 6 | 4-Methylphenol | H | BDL | 1.05 | 3.56 |
| 7 | 2/3-Methylphenol | H | BDL | 3.34 | 9.52 |
| 8 | Guaiacol | G | 4.72 | 9.06 | 14.78 |
| 9 | 2,4-Dimethylphenol | H | 1.53 | 0.10 | 1.41 |
| 10 | 4-Methylguaiacol | G | 2.07 | 7.84 | 4.49 |
| 11 | 4-Vinylphenol | H/PCA | 18.60 | 21.22 | 13.99 |
| 12 | 4-Vinylguaiacol | G/FA | 9.88 | 12.12 | 6.80 |
| 13 | Syringol | S | 4.13 | 8.94 | 8.28 |
| 14 | 5-Ethylpyrogallol | S | BDL | 0.71 | 2.41 |
| 15 | 4-Propylguaiacol | G | BDL | BDL | 0.58 |
| 16 | Vanillin | G | 2.02 | 2.29 | 1.69 |
| 17 | 2,6-Dimethoxy-4-methylphenol | S | BDL | 6.99 | 2.91 |
| 18 | cis-Isoeugenol | G | 0.84 | 2.29 | 1.23 |
| 19 | Homovanillin | G | 0.57 | 0.07 | BDL |
| 20 | Acetovanillone | G | 0.60 | 0.85 | 1.69 |
| 21 | Vanillic acid methyl ester | G | BDL | BDL | 0.52 |
| 22 | 4-Ethylsyringol | S | 0.29 | 1.73 | 0.64 |
| 23 | Guaiacyl acetone | G | 0.71 | 1.88 | 2.39 |
| 24 | 4-Vinylsyringol | S | 3.34 | 3.63 | 0.75 |
| 25 | 1,6-Anhydro-β-D-glucopyranose | PS | 22.06 | BDL | BDL |
| 26 | Propiovanillone | G | BDL | 0.53 | 0.39 |
| 27 | 4-Allylsyringol | S | 1.09 | 0.51 | 0.22 |
| 28 | Benzophenone | Unknown | BDL | BDL | 7.05 |
| 29 | cis-4-Propenylsyringol | S | 0.56 | 0.38 | 0.33 |
| 30 | Dihydroconiferyl alcohol | G | 0.15 | 0.63 | 0.14 |
| 31 | Syringaldehyde | S | 0.75 | 0.59 | 0.47 |
| 32 | 1-(3,5-Dimethoxy-4-hydroxyphenyl)propyne | S | 0.22 | 1.17 | 0.14 |
| 33 | trans-4-Propenylsyringol | S | 3.55 | 2.69 | 0.41 |
| 34 | Homosyringe aldehyde | S | 0.48 | BDL | BDL |
| 35 | Acetosyringone | S | 0.75 | 1.42 | 0.86 |
| 36 | trans-Coniferyl aldehyde | G | 0.34 | 0.11 | 0.12 |
| 37 | Syringyl acetone | S | 0.73 | 2.23 | 1.12 |
| 38 | Propiosyringone | S | 0.34 | 0.87 | 1.59 |
The Py-GC-MS procedure can distinguish between carbohydrate derived products and the different subunits. However, chemical processes occurring during the pyrolysis can result in misleading results, as PCA and H, and FA and G units cannot be distinguished. Examples are 4-vinylphenol (peak 11) which may originate from PCA or the H unit, and 4-vinylguaiacol (peak 12), which may be derived from FA or the G unit.43,44
Since we detected 38 different pyrolysis moieties, we grouped the compounds according to subunits to facilitate data interpretation (Fig. 9). The aromatic compounds were assigned to three subunit groups using the substitution pattern on the aromatic ring. If the compound had no methoxy substituents ortho to the phenolic hydroxyl group, they were assigned to H or PCA; one methoxy substituent, assigned to G or FA and two methoxy substituents, assigned to S.
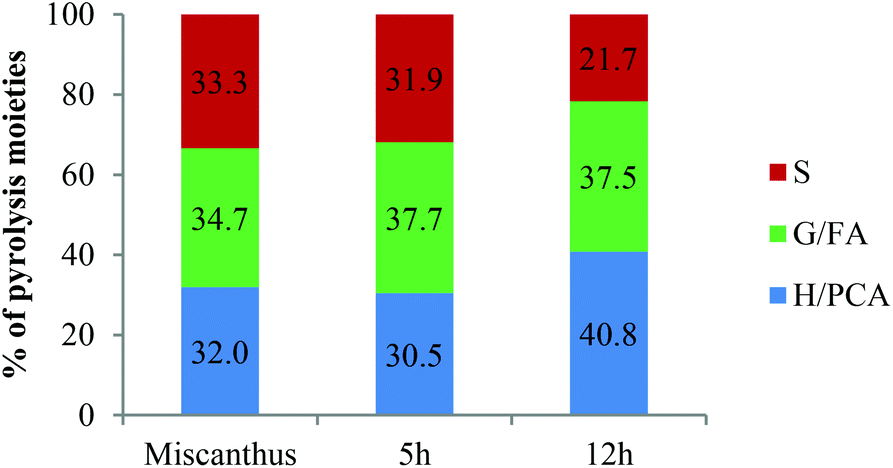 | ||
| Fig. 9 Relative abundances of Py-GC-MS moieties (excluding polysaccharides and benzophenone) of Miscanthus giganteus and isolated Ionosolv lignin. H: p-hydroxyphenyl unit; G: guaiacyl unit; S: syringyl unit; PCA: p-coumaric acid; FA: ferulic acid. | ||
In the pyrogram obtained for untreated Miscanthus, carbohydrate-derived moieties were detected (peaks 1, 2, 4 and 5). Interestingly, none of these were present in the 5 h and 12 h lignin pyrograms, indicating that the polysaccharides had been separated from the lignin during Ionosolv deconstruction. The ionic liquid solution is known to solubilize lignin and a large part of the hemicellulose.25 The Py-GC-MS data show that intact hemicelluloses do not precipitate with the lignin after 5 h. The lack of carbohydrate correlations in Ionosolv lignins isolated after longer pretreatment times agrees with the HSQC NMR analysis of the 5 h and 12 h lignins, which didn't detect carbohydrates either.
Fragments released from untreated Miscanthus originated from H/PCA, G/FA and S in roughly equal amounts (32%, 35% and 33%, respectively). In the 12 h lignin, the amounts of released G/FA fragments and H/PCA fragments increased to 38% and 41%, respectively, while the relative quantity of S derived compounds decreased to 22%. The H/PCA group of peaks was more abundant than expected for a typical Miscanthus lignin. The results suggest that the pyrolysis MS procedure may over-represent the H/PCA content.
An increased release of simple monomers such as phenol (peak 3), guaiacol (peak 8) and syringol (peak 13) was observed. These simple aromatic compounds comprised 14% of the moieties released from untreated Miscanthus, 22% of the 5 h moieties and 33% of the 12 h moieties. This indicates that aromatic rings in the depolymerised and/or condensed lignin have connectivities that facilitate release of simple monomers and suggests that pyrolytic valorisation of Ionosolv lignin may be more promising than whole untreated lignocellulose.
Py-TMAH-GC-MS analysis
Py-GC-MS in the presence of tetramethylammonium hydroxide (TMAH) is a method for determining the composition of lignin with fewer restrictions than conventional Py-GC-MS, since the compounds derived from the H and G units in lignin and the p-hydroxycinnamic acid derivatives of PCA and FA can be clearly differentiated. The resulting derivatives are also more robust, protecting thermolabile compounds and facilitating chromatographic separation.45The compound table for the Py-TMAH-GC-MS experiment is shown in Table 4, while Fig. 10 displays the abundance of the released compounds grouped according to source subunits. Table 4 shows that negligible amounts of carbohydrate derived products were detected among the pyrolysis products, even for untreated Miscanthus. This demonstrates that the TMAH procedure is more selective for lignin fragments. In addition, fewer S derived compounds were released with Py-TMAH-GC-MS than with Py-GC-MS, while nearly half of the moieties were derived from either PCA or H (untreated Miscanthus and 5 h Ionosolv lignin). Oxidized compounds, such as 3,4-dimethoxybenzoic acid methyl ester (peak 24), 3,4-dimethoxybenzene acetic acid methyl ester (peak 27) and 3,4,5-trimethoxybenzoic acid methyl ester (peak 32) were also observed (Table 4). Their abundance increased remarkably with treatment time. This agrees with the 31P NMR data (hydroxyl group analysis), which saw the abundance of carboxylic acid functionalities increase over time.
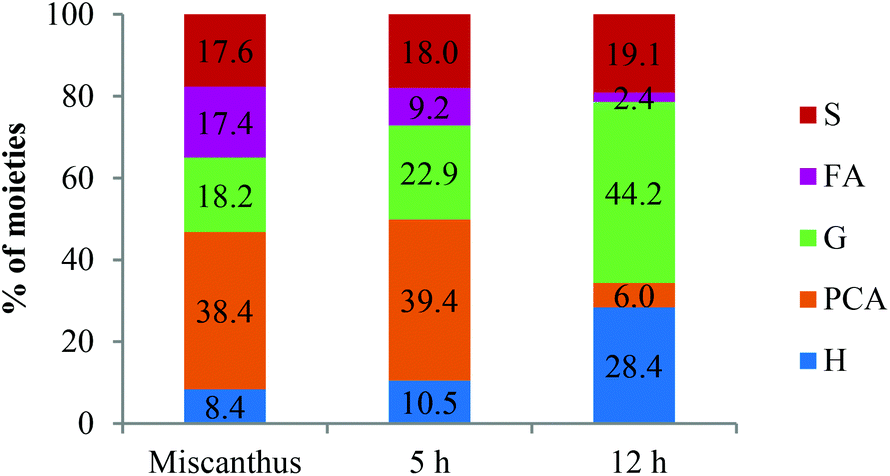 | ||
| Fig. 10 Relative abundances of Py-TMAH-GC-MS moieties (excluding polysaccharides) of Miscanthus giganteus and Ionosolv lignin (5 and 12 hour) in %. H: p-hydroxyphenyl unit; G: guaiacyl unit; S: syringyl unit; PCA: p-coumaric acid; FA: ferulic acid. | ||
| Label | Compound | Origin | Relative peak area (%) | ||
|---|---|---|---|---|---|
| Miscanthus giganteus | Ionosolv lignin (5 h) | Ionosolv lignin (12 h) | |||
| PS: polysaccharide; H: p-hydroxyphenyl subunit; G: guaiacyl subunit; S: syringyl subunit; PCA: p-coumaric acid; FA: ferulic acid; BDL: below detection limit; TR: traces. | |||||
| 1 | Methoxybenzene | H | BDL | 0.31 | 2.72 |
| 2 | 1-Methoxy-2-methyl benzene | H | BDL | 0.10 | 0.46 |
| 3 | 1-Methoxy-4-methyl benzene | H | BDL | 1.78 | 7.78 |
| 4 | 2-Methoxyphenol | H | BDL | 0.76 | 1.68 |
| 5 | 2-Methoxy-1,4-dimethyl benzene | H | BDL | 0.16 | 1.45 |
| 6 | 1-Ethyl-4-methoxy benzene | H | BDL | 1.85 | 0.88 |
| 7 | 3-Ethyl-2,4,-furandione | PS | 0.17 | BDL | BDL |
| 8 | 1,2-Dimethylbenzene | Unknown | BDL | 0.90 | 2.09 |
| 9 | 1-Ethenyl-4-methoxy benzene | H | 8.38 | 4.86 | 11.82 |
| 10 | 3,4-Dimethoxy-1-methyl benzene | G | TR | 4.21 | 5.41 |
| 11 | 3,4-Dimethoxy-1-ethyl benzene | G | BDL | TR | BDL |
| 12 | 1-Methoxy-4-(1-propenyl) benzene | H | BDL | 0.58 | 1.01 |
| 13 | 1,2,3-Trimethoxy benzene | S | 2.49 | 0.91 | 2.34 |
| 14 | 4-Ethyl-1,2-dimethoxy benzene | G | 0.20 | 1.02 | 2.47 |
| 15 | 2,6-Dimethoxyphenol | S | BDL | BDL | 1.40 |
| 16 | 4-Ethenyl-1,2-dimethoxy benzene | G | 5.23 | 2.67 | 4.93 |
| 17 | 1,2,3-Trimethoxy-5-methyl benzene | S | TR | 2.15 | 1.59 |
| 18 | 1-(4-Hydroxy-3,5-dimethoxyphenyl)ethanone | S | BDL | 0.25 | 0.25 |
| 19 | 3,4-Dimethoxybenzaldehyde | G | 2.60 | 1.46 | 1.24 |
| 20 | 1,2-Dimethoxy-4-(1-propenyl)benzene | G | 0.33 | 1.32 | 1.52 |
| 21 | 1,2-Dimethoxy-4-(2-methoxyethenyl) benzene | G | 0.45 | 0.98 | 2.37 |
| 22 | 1-(3,4-Dimethoxyphenyl)ethanone | G | 1.19 | 1.14 | 3.21 |
| 23 | cis-3-(4-Methoxyphenyl)-2-propenoic acid methyl ester | PCA | 0.73 | 0.74 | 0.40 |
| 24 | 3,4-dimethoxybenzoic acid methyl ester | G | 0.89 | 1.18 | 8.30 |
| 25 | 3,4,5-Trimethoxybenzaldehyde | S | 3.22 | 1.70 | 0.74 |
| 26 | cis-1,2-Dimethoxy-4-(2-methoxyethenyl) benzene | G | 2.81 | 2.16 | 0.92 |
| 27 | 3,4-Dimethoxybenzene acetic acid methyl ester | G | TR | 0.38 | 9.90 |
| 28 | trans-1,2-Dimethoxy-4-(2-methoxyethenyl) benzene | G | 1.93 | 1.38 | 0.48 |
| 29 | 1,2,3-Trimethoxy-5-(2-propenyl) benzene | S | 0.35 | 0.64 | 0.51 |
| 30 | trans-3-(4-Methoxyphenyl)-2-propenoic acid methyl ester | PCA | 37.62 | 38.35 | 5.44 |
| 31 | 3,4,5-Trimethoxyacetophenone | S | 3.65 | 2.42 | 4.54 |
| 32 | 3,4,5-Trimethoxybenzoic acid methyl ester | S | 1.68 | 2.12 | 6.01 |
| 33 | trans-1-(3,4-Dimethoxyphenyl)-3-methoxy-1-propene | G | 1.19 | 2.02 | 1.49 |
| 34 | cis-1-(3,4,5-Trimethoxyphenyl)-2-methoxy ethylene | S | 3.27 | 4.04 | 0.77 |
| 35 | trans-1-(3,4,5-Trimethoxyphenyl)-2-methoxy ethylene | S | 2.15 | 2.35 | 0.23 |
| 36 | threo/erythro-1-(3,4-Timethoxyphenyl)-1,2,3-trimethoxypropane | G | 0.57 | 1.19 | 0.50 |
| 37 | threo/erythro-1-(3,4-Dimethoxyphenyl)-1,2,3-trimethoxypropane | G | 0.75 | 1.60 | 0.56 |
| 38 | cis-1-(3,4,5-Trimethoxyphenyl)-methoxyprop-1-ene | S | 0.81 | 1.21 | 0.27 |
| 39 | 3-(3,4-Dimethoxyphenyl)-2-propenoic acids methyl ester | FA | 17.35 | 9.12 | 2.31 |
Using the peaks assigned to S and G in Table 4, the S/G ratio was determined for Miscanthus giganteus, Ionosolv lignin (5 h) and Ionosolv lignin (12 h) as 0.97, 0.78 and 0.43, respectively. While the first two ratios are reasonably close to the S/G ratio of native Miscanthus lignin as determined elsewhere (0.85),46 the 12 h lignin released high quantities of guaiacyl derived compounds, confirming that G units are enriched in the Ionosolv lignin.
The Py-TMAH-GC-MS further shows that the p-hydroxycinammic acids, p-coumaric acid and ferulic acid, contribute substantially to the range of pyrolysis products. Miscanthus giganteus and Ionosolv lignin isolated after 5 h released exceptionally high amounts of methylated derivatives of PCA (peaks 23 and 30) and of the methylated derivative of FA (peak 39). In untreated Miscanthus, PCA accounted for >80% of the pyrolysis fragments with a p-hydroxyphenyl ring, while ferulic acid accounted for ca. 50% of the fragments with a guaiacyl ring. These proportions are higher than the abundance of PCA and FA predicted by other techniques, such as 13C-NMR or HSQC-NMR. For example, volume integration of the HSQC estimated that PCA is ca 14% of the 1 h lignin and 7% in 5 h lignin.
This indicates that the Py-GC-MS analyses employed here do not represent the true composition of lignin, at least when it comes to ester bound components.
Release of the PCA and FA derivatives in the pyrograms decreased dramatically after prolonged Ionosolv extraction (6% and 2%, respectively, in the 12 h lignin pyrogram), while fragments assigned to H (28%) and G subunits (44%) increased. The reduction of PCA and FA content agrees with the 13C and HSQC NMR data. The increased abundance of H and G units is due to enrichment, at least partly caused by the chemical transformation of PCA and possibly also of ferulic acid into new polymers that create H and G type pyrolysis fragments. This hypothesis is supported by the observation that some moieties with H or G type substitution patterns were abundant in the Ionosolv lignin pyrograms (compounds 3 and 10 for example) while they were negligible in the untreated Miscanthus pyrogram.
In summary, the Py-(TMAH-)GC-MS results confirm the relative changes in Ionosolv lignin composition observed by NMR analyses, such as removal of carbohydrates, decrease in S unit, PCA and FA content and increase in H and G unit content. The technique appears to overestimate the content of p-coumaric acid and ferulic acid and to underestimate the amount of syringyl and guaiacyl subunits. This is likely due to the way the subunits are linked into the polymer.
Molecular weight determination
GPC analysis was performed to investigate the effect of Ionosolv deconstruction on the molecular weight of the resulting lignin. Fig. 11 and Table 5 summarize the drastic changes in molecular weight of the Ionosolv lignins with increasing pretreatment time.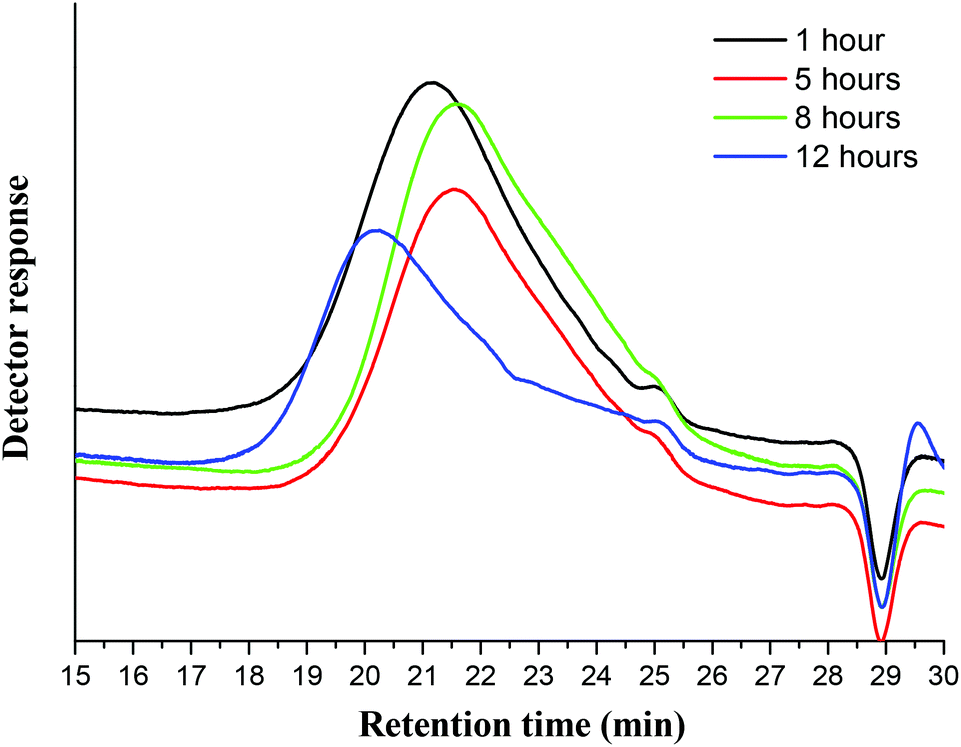 | ||
| Fig. 11 Gel permeation chromatography traces of Ionosolv lignin after different pretreatment times. | ||
The recovered lignin exhibited a substantial decrease in molecular weight, confirming that the lignin macromolecules undergo depolymerization through linkage cleavage. For example, Ionosolv lignin after 1 h pretreatment had a molecular weight of Mn = 2500 g mol−1. The Mn had decreased further by ∼36% to 1580 g mol−1 after 8 h of pretreatment. This represents a >80% reduction in molecular weight relative to milled Miscanthus lignin (8300)23 and is substantially lower than the reported Organosolv Miscanthus lignin value of 4690,23 though it is important to note that measured lignin Mn can vary from system to system. The results suggest that lignin solubilization takes place after substantial depolymerization inside the biomass. We conclude this because the 1 h lignin has a similar ether content and S/G ratio as native ball milled lignin, yet has much lower molecular weight. This suggests that the 1 h lignin has been depolymerized before extraction; otherwise, we would expect a higher molecular weight at shorter times (prior to solution-phase depolymerization).
Despite the initial hydrolysis in the biomass, further hydrolysis occurred in solution. The estimated average degree of polymerization (DP, obtained by assuming MW = 200 g mol−1 as the average weight of a lignin unit) decreased to ca. 8 by 8 hours.
Interestingly, the molecular weight of lignin increased noticeably after 12 hours to Mn = 2530. We infer that the condensation reactions (C–C bond formation) overtake the depolymerization reactions (ether cleavage) when the Ionosolv treatment is performed for exceedingly long times. This is supported by the 31P NMR results which show a rapid rise of the phenolic hydroxyl content between 8 and 12 h. The increased polydispersity index (PDI) at 8 h and 12 h (DP = 4.28 and 7.26, compared with 3.71 at 5 h) is also evidence of repolymerization, as repolymerization has been shown to increase the heterogeneity of resulting lignins.47
Elemental composition of Miscanthus Ionosolv lignin
The lignin was further analysed for elemental composition, looking at the carbon (C), hydrogen (H), nitrogen (N) and sulfur (S) content. The N and S contents indicate residual IL contamination; nitrogen is representative of the imidazolium cation which contains two nitrogen atoms and sulfur is representative of the hydrogen sulfate anion, which contains one sulfur atom. We noticed residual cation in some of the NMR experiments; here we report that anion was also present. The untreated Miscanthus sample was nitrogen and sulfur free (data not shown). Table 6 shows the content of the four elements in the isolated lignins. Carbon was the largest contributor, making up ca. 60 wt% of each sample, followed by H (5 wt%), N and S.| Pretreatment time | C % | H % | N % | S % |
|---|---|---|---|---|
| 1 h | 59.9 | 5.5 | 0.7 | 1.0 |
| 5 h | 61.5 | 5.6 | 1.2 | 1.3 |
| 8 h | 62.6 | 5.5 | 1.0 | 1.1 |
| 12 h | 58.5 | 5.3 | 3.4 | 3.7 |
For the 1 h, 5 h and 8 h lignin samples, the N and S content was roughly the same (around 1 wt% each), suggesting that the lignin contained 6–9 wt% of residual ionic liquid. At 10 wt% biomass loading and 25% lignin content in the biomass, this equates to a solvent loss of 0.2% or less into the lignin. In contrast, the 12 h lignin sample contained more than three times as much nitrogen and sulfur, pointing to substantial IL contamination, which we believe to be due to incomplete washing. This high IL content is reflected by a large peak for butylimidazole seen in the Py-GC-MS pyrograms of the 12 h lignin (Fig. 8) and particularly prominent signals for butylimidazole in the 13C-NMR and HSQC NMR spectra. The contamination of this sample highlights the need for thorough washing. A 24 h lignin sample had an IL content similar to the 1–8 h lignins (refer to HSQC NMR spectrum in Fig. S12 of the ESI†), confirming that the 12 h sample was unusual and that the IL content does not rise with increasing treatment time.
We used the N and S contents to calculate the molar cation/anion ratio for the IL portion, which is shown in Fig. 12 (raw EA data and the derived values are shown in Tables S4–6 in the ESI†). It can be seen that the cation-to-anion ratio was typically around 1:1. Only the 1 h lignin had an excess of 20% hydrogen sulfate anion, which we cannot explain at present. This, and the presence of intact butylimidazole in the pyrograms, indicates that the residual IL is simply adsorbed. Otherwise we would expect the ion ratio to be imbalanced, with either the cation or the anion being more abundant, depending on which component was more reactive towards the lignin.
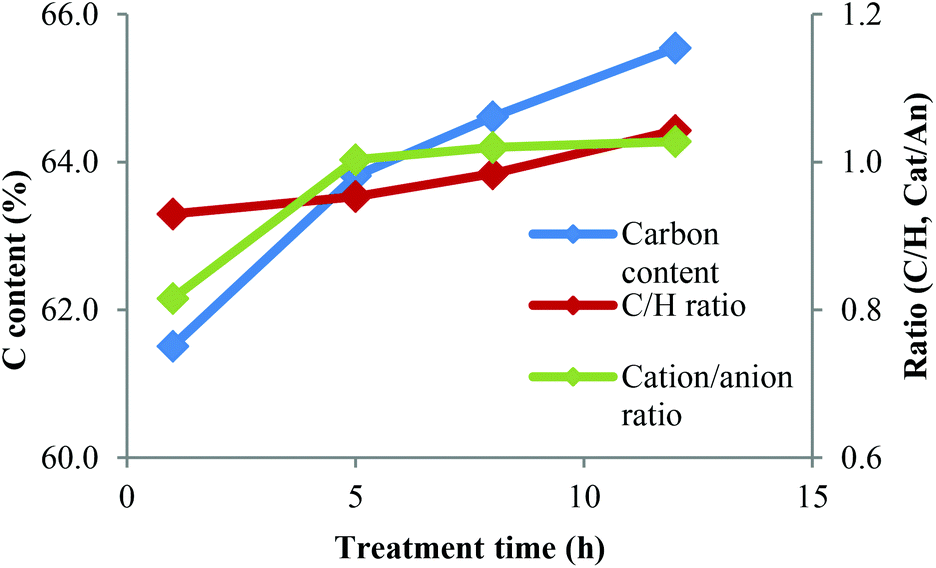 | ||
| Fig. 12 Carbon content and molar C–H ratio for IL ‘free’ portion of the lignin and the cation-to-anion ratio in the IL portion. | ||
We note that the amount of IL as found in the 12 h lignin would be considerable; hence this content needs to be minimized on economic grounds. An improved lignin washing protocol or alternative lignin recovery technique may achieve this. The interactions of residual IL with the lignin need to be explored in more detail in the future in order to identify an appropriate solution. Nevertheless, three of the Ionosolv lignins contained substantially lower amounts of sulfur than Kraft or sulfite lignins.48
The elemental analysis data also suggest that there may be a trend for increasing carbon content, with exception of the 12 h lignin. In order to focus on the lignin portion of the elemental analysis, we calculated the carbon content and the molar ratio of carbon and hydrogen for the ‘IL-free’ portion of the lignin samples.
The carbon content of the IL-free lignin increased substantially with treatment time (Fig. 12), as did the C/H ratio. This indicates that lignins treated for longer contained more C–C bonds and fewer C–O bonds. The overall amount of carbon plus hydrogen increased from 67% at 1 h to 71% at 12 h, indicating a reduction of oxygen content. High C and H contents are beneficial for energy applications where fuel value matters.
Model of Ionosolv delignification and isolation
Based on the preceding results, a summary of changes in the lignin properties is presented in Table 7. We believe that the mechanism for Ionosolv delignification is initiated by hydrolysis of the glycosidic bonds between xylose and arabinose in lignin carbohydrate complexes and the shortening of lignin polymers inside the biomass pulp. This is followed by solubilization of the shortened lignin polymers. The solubilized chains continue to fragment inside the IL liquor, but also begin to condense.| Impact | Experimental evidence | Technique |
|---|---|---|
| Carbohydrate free | No carbohydrates detected, apart from arabinose at very short treatment times | HSQC NMR, Py-GC-MS, HSQC NMR |
| Hydrolysis of ether and ester linkages | Disappearance of β-O-4 ether linkages | HSQC NMR, Py-TMAH-GC-MS, GPC |
| Disappearance of p-coumaric and ferulic acid from lignin | ||
| Decreased molecular weight | ||
| Changes in subunit composition over time | More H and G units | Py-TMAH-GC-MS, 13C-NMR, HSQC NMR |
| Fewer PCA, FA and S units | ||
| Condensation | Increased C–C aromatic bond content | 13C-NMR, GPC, HSQC NMR, 31P-NMR, EA |
| Increase in phenolic hydroxyl content | ||
| Molecular weight increased after 8 h | ||
| Increased carbon content |
We hence recovered lignin that is chemically similar to native Miscanthus lignin in the initial stages of the treatment, while at longer times, we recovered a condensed lignin that is rich in G units, H units and phenolic hydroxyl groups.
S/G ratios obtained with various techniques (Py-GC-MS, quantitative 13C-NMR, HSQC NMR) showed that S and G units were present in roughly equal proportions in ball milled Miscanthus and in early stage lignins, while the S/G ratio in highly treated Ionosolv lignins was around 1:3–1:4 (Fig. 13). We postulate that the enrichment of guaiacyl units is due to preferred C–C bond formation (condensation) at the G6 ring position, resulting in precipitation of G-rich lignin polymers. However, it is possible that the measured S/G ratios have been obscured by condensation reactions. Further investigations into the fate of the S units and the chemistry of lignin condensation in acidic solvents are urgently required.
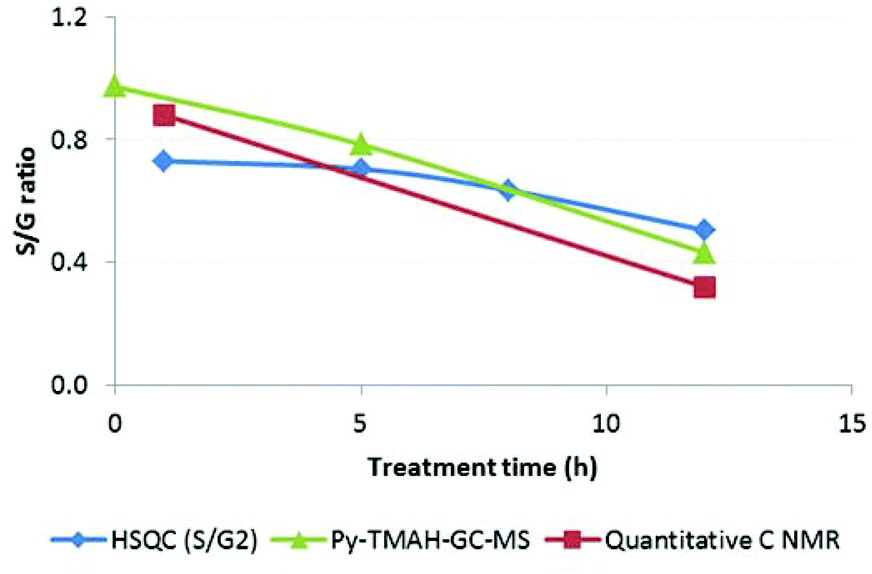 | ||
| Fig. 13 Changes in the of S/G ratio in Miscanthus derived Ionosolv lignins. | ||
The more treated lignins obtained from Miscanthus were high in H content, due to p-coumaric acid decarboxylation to p-hydroxystyrene followed by polymerisation with itself and potentially other fragments in solution. 15–20% of the Ionosolv lignin derived from Miscanthus is p-coumaric acid or a PCA derived H-like polymer. A similar conversion may be happening with the less abundant ferulic acid, contributing to the increased G unit content in highly treated lignins.
A preliminary analysis of the IL liquor (LC-MS and GC-MS, data not shown) revealed predominantly monomeric syringols, guaiacols and phenols (plus sugar degradation products), which are water-soluble or liquid and hence do not precipitate. The fate of these solubilized fragments will be explored in a future ionic liquid solvent recycling study.
Conclusions
In summary, this study illustrates the changes occurring during the Ionosolv delignification through comparison of lignin structures in the plant cell walls of Miscanthus giganteus to the isolated lignins.We have shown that lignin undergoes more than 80% depolymerization during the initial stage of the pretreatment through the cleavage of β-O-4 aryl ether linkages and ester linkages, confirmed by reduction in molecular weight and the β-O-4 ether bond signal in the HSQC NMR spectra.
At long pretreatment times, repolymerization (condensation) overtakes the depolymerization, as evidenced by increased lignin molecular weight (GPC), increased content of phenolic hydroxyl groups and increased C/H ratio in the later stage lignins. The composition of the later stage Miscanthus lignin was also modified, as p-hydroxyphenyl and guaiacyl subunits were enriched and p-coumaric acid and ferulic acid were removed.
These results suggest that the Ionosolv deconstruction can be tuned to selectively depolymerize lignin during pretreatment by eliminating ether and ester linkages, providing an opportunity to produce small aromatic molecules during the pretreatment itself. Alternatively, the pretreatment can be tuned to favour condensation reactions, increasing lignin molecular weight and phenolic hydroxyl content for the production of additives or resins and to maximize fuel value.
The Ionosolv deconstruction can therefore produce lignin that appears to be more fragmented or more condensed than Organosolv23 or other ionic liquid lignins.37,49 At very short times it can produce lignins that retain most of their native subunit composition and linkages. We hope that this information opens new avenues of scientific investigation in the development of the Ionosolv process as an alternative pretreatment technology and encourages the subsequent valorization of Ionosolv lignins.
Acknowledgements
Long Chen gratefully acknowledges the China Scholarship Council for providing a scholarship. Long Chen is also grateful to Jan Marchant for help with HSQC, and Dr Raquel Prado for help with GPC measurements. Additional support was obtained from the UK Engineering and Physical Sciences Research Council (EPSRC grant EP/K014676/1) and Shell Global Solutions.Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak and C. L. Liotta, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- Y. Pu, F. Hu, F. Huang, B. H. Davison and A. J. Ragauskas, Biotechnol. Biofuels, 2013, 6, 1–13 CrossRef PubMed.
- P. Kumar, D. M. Barrett, M. J. Delwiche and P. Stroeve, Ind. Eng. Chem. Res., 2009, 48, 3713–3729 CrossRef CAS.
- J. Zhu and X. Pan, Bioresour. Technol., 2010, 101, 4992–5002 CrossRef CAS PubMed.
- V. B. Agbor, N. Cicek, R. Sparling, A. Berlin and D. B. Levin, Biotechnol. Adv., 2011, 29, 675–685 CrossRef CAS PubMed.
- B. Yang and C. E. Wyman, Biofuels, Bioprod. Biorefin., 2008, 2, 26–40 CrossRef CAS PubMed.
- L. J. Jönsson, B. Alriksson and N.-O. Nilvebrant, Biotechnol. Biofuels, 2013, 6, 16 CrossRef PubMed.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- M. T. Clough, K. Geyer, P. A. Hunt, J. Mertes and T. Welton, Phys. Chem. Chem. Phys., 2013, 15, 20480–20495 RSC.
- D. Klein-Marcuschamer, B. A. Simmons and H. W. Blanch, Biofuels, Bioprod. Biorefin., 2011, 5, 562–569 CrossRef CAS PubMed.
- A. Brandt, J. P. Hallett, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2010, 12, 672–679 RSC.
- A. Brandt, M. J. Ray, T. Q. To, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2011, 13, 2489–2499 RSC.
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC.
- C. Chapple, M. Ladisch and R. Meilan, Nat. Biotechnol., 2007, 25, 746–749 CrossRef CAS PubMed.
- D. Haverty, K. Dussan, A. V. Piterina, J. Leahy and M. Hayes, Bioresour. Technol., 2012, 109, 173–177 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna and M. Keller, Science, 2014, 344, 1246843 CrossRef PubMed.
- J. Ralph, R. D. Hatfield, S. Quideau, R. F. Helm, J. H. Grabber and H.-J. G. Jung, J. Am. Chem. Soc., 1994, 116, 9448–9456 CrossRef CAS.
- F. Masarin, D. B. Gurpilhares, D. C. Baffa, M. H. Barbosa, W. Carvalho, A. Ferraz and A. M. Milagres, Biotechnol. Biofuels, 2011, 4, 55 CrossRef CAS PubMed.
- J. J. Stewart, T. Akiyama, C. Chapple, J. Ralph and S. D. Mansfield, Plant Physiol., 2009, 150, 621–635 CrossRef CAS PubMed.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protocols, 2012, 7, 1579–1589 CAS.
- R. El Hage, N. Brosse, L. Chrusciel, C. Sanchez, P. Sannigrahi and A. Ragauskas, Polym. Degrad. Stab., 2009, 94, 1632–1638 CrossRef CAS PubMed.
- A. Tolbert, H. Akinosho, R. Khunsupat, A. K. Naskar and A. J. Ragauskas, Biofuels, Bioprod. Biorefin., 2014, 8, 836–856 CrossRef CAS PubMed.
- P. Verdía, A. Brandt, J. P. Hallett, M. J. Ray and T. Welton, Green Chem., 2014, 16, 1617–1627 RSC.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- C. Crestini and D. S. Argyropoulos, J. Agric. Food Chem., 1997, 45, 1212–1219 CrossRef CAS.
- M. Funaoka, T. Kako and I. Abe, Wood Sci. Technol., 1990, 24, 277–288 CrossRef CAS.
- H. Nimz, Holzforschung: Int. J. Biol. Chem. Phys. Technol. Wood, 1969, 23, 84–88 CAS.
- S. Ralph, J. Ralph, L. Landucci and L. Landucci, NMR database of lignin and cell wall model compounds, US Forest Prod. Lab., Madison, WI, http://ars.usda.gov/Services/docs.htm, 2004 Search PubMed.
- E. A. Capanema, M. Y. Balakshin and J. F. Kadla, J. Agric. Food Chem., 2005, 53, 9639–9649 CrossRef CAS PubMed.
- T.-T. You, J.-Z. Mao, T.-Q. Yuan, J.-L. Wen and F. Xu, J. Agric. Food Chem., 2013, 61, 5361–5370 CrossRef CAS PubMed.
- M. Sette, R. Wechselberger and C. Crestini, Chem. – Eur. J., 2011, 17, 9529–9535 CrossRef CAS PubMed.
- T.-Q. Yuan, S.-N. Sun, F. Xu and R.-C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS PubMed.
- K. Cheng, H. Sorek, H. Zimmermann, D. E. Wemmer and M. Pauly, Anal. Chem., 2013, 85, 3213–3221 CrossRef CAS PubMed.
- S. Bauer, H. Sorek, V. D. Mitchell, A. B. Ibáñez and D. E. Wemmer, J. Agric. Food Chem., 2012, 60, 8203–8212 CrossRef CAS PubMed.
- J.-L. Wen, T.-Q. Yuan, S.-L. Sun, F. Xu and R.-C. Sun, Green Chem., 2014, 16, 181–190 RSC.
- C. Lapierre and C. Rolando, Holzforschung: Int. J. Biol. Chem. Phys. Technol. Wood, 1988, 42, 1–4 CAS.
- Y. Pu, S. Cao and A. J. Ragauskas, Energy Environ. Sci., 2011, 4, 3154–3166 CAS.
- R. Samuel, S. Cao, B. K. Das, F. Hu, Y. Pu and A. J. Ragauskas, RSC Adv., 2013, 3, 5305–5309 RSC.
- J. Li and G. Gellerstedt, Ind. Crops Prod., 2008, 27, 175–181 CrossRef CAS PubMed.
- M. R. Sturgeon, S. Kim, K. Lawrence, R. S. Paton, S. C. Chmely, M. Nimlos, T. D. Foust and G. T. Beckham, ACS Sustainable Chem. Eng., 2013, 2, 472–485 CrossRef.
- F. J. F. Lopes, F. O. Silvério, D. C. F. Baffa, M. E. Loureiro and M. H. P. Barbosa, J. Wood Chem. Technol., 2011, 31, 309–323 CrossRef CAS PubMed.
- K. Ross and G. Mazza, World J. Agric. Sci., 2011, 7, 763–776 CAS.
- J. C. Del Río, J. Rencoret, P. Prinsen, A. n. T. Martínez, J. Ralph and A. Gutiérrez, J. Agric. Food Chem., 2012, 60, 5922–5935 CrossRef PubMed.
- Y. P. Timilsena, C. J. Abeywickrama, S. K. Rakshit and N. Brosse, Bioresour. Technol., 2013, 135, 82–88 CrossRef CAS PubMed.
- J. Li, G. Henriksson and G. Gellerstedt, Bioresour. Technol., 2007, 98, 3061–3068 CrossRef CAS PubMed.
- N.-E. El Mansouri and J. Salvadó, Ind. Crops Prod., 2006, 24, 8–16 CrossRef CAS PubMed.
- N. Sathitsuksanoh, K. M. Holtman, D. J. Yelle, T. Morgan, V. Stavila, J. Pelton, H. Blanch, B. A. Simmons and A. George, Green Chem., 2014, 16, 1236–1247 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: IL synthesis and characterisation, the original HSQC spectra including volume integrals, the assigned 8 h HSQC spectra, the original 31P-NMR spectra and calculated values, the elemental analysis raw data. See DOI: 10.1039/c5gc01314c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |