
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDownstream processing of Isochrysis galbana: a step towards microalgal biorefinery†
Bienvenida
Gilbert-López
a,
José A.
Mendiola
a,
Javier
Fontecha
b,
Lambertus A. M.
van den Broek
c,
Lolke
Sijtsma
c,
Alejandro
Cifuentes
a,
Miguel
Herrero
a and
Elena
Ibáñez
*a
aFoodomics Laboratory, Bioactivity and Food Analysis Department, Institute of Food Science Research (CIAL-CSIC), Campus de Cantoblanco, Calle Nicolás Cabrera 9, 28049 Madrid, Spain. E-mail: elena.ibanez@csic.es
bBioactivity and Food Analysis Department, Lipid Group, Institute of Food Science Research (CIAL-CSIC), Campus de Cantoblanco, Calle Nicolás Cabrera 9, 28049 Madrid, Spain
cWageningen UR Food & Biobased Research, P.O. Box 17, 6700 AA Wageningen, The Netherlands
First published on 16th July 2015
Abstract
An algae-based biorefinery relies on the efficient use of algae biomass through its fractionation of several valuable/bioactive compounds that can be used in industry. If this biorefinery includes green platforms as downstream processing technologies able to fulfill the requirements of green chemistry, it will end-up with sustainable processes. In the present study, a downstream processing platform has been developed to extract bioactive compounds from the microalga Isochrysis galbana using various pressurized green solvents. Extractions were performed in four sequential steps using (1) supercritical CO2 (ScCO2), (2) ScCO2/ethanol (Gas Expanded Liquid, GXL), (3) pure ethanol, and (4) pure water as solvents, respectively. The residue of the extraction step was used as the raw material for the next extraction. Optimization of the ScCO2 extraction was performed by factorial design in order to maximize carotenoid extraction. During the second step, different percentages of ethanol were evaluated (15%, 45% and 75%) in order to maximize the extraction yield of fucoxanthin, the main carotenoid present in this alga; the extraction of polar lipids was also an aim. The third and fourth steps were performed with the objective of recovering fractions with high antioxidant activity, eventually rich in carbohydrates and proteins. The green downstream platform developed in this study produced different extracts with potential for application in the food, pharmaceutical and cosmetic industries. Therefore, a good approach for complete revalorization of the microalgae biomass is proposed, by using processes complying with the green chemistry principles.
1. Introduction
A biorefinery involves biomass conversion processes and equipment to produce fuel, power, and added-value chemicals from organic materials1 such as renewable resources or microalgae. Microalgae are among the most promising raw materials for the sustainable supply of commodities and the use of algae.2,3 They use light energy, residual nutrients and carbon dioxide (that can be obtained from flue gas) with higher photosynthetic efficiency than plants for the production of biomass.4 Moreover, these organisms may be grown on non-arable land, thus, not competing with food needs for biofuel production. Microalgae biomass is an excellent source of oils (including high amounts of long chain polyunsaturated fatty acids (LC PUFAs)), proteins, polysaccharides (such as starch, xylans, pectins, glucans, extracellular polysaccharides (EPS)) and other high-added value compounds such as carotenoids, pigments, antioxidants, sterols and minerals. The potential for the production of these different components may even be tuned by setting particular growing conditions. Therefore, the microalgae-based biorefinery concept relies on the complete process chain ranging from optimization of biomass production to the development of a platform able to generate a wide range of products, from bulk chemicals, food supply (proteins, fibres), bioactive compounds, and oils with respect to its use as a biofuel.Isochrysis galbana is a small marine flagellate (Phylum: Haptophyta) widely used in aquaculture as a PUFA-rich microalga.5 It is commercially produced as feed for the early larval stages of mollusks, fish, and crustaceans. In fact, I. galbana cells produce antibacterial substances, which increase the toxicity of free fatty acids such as eicosapentanoic acid (EPA) to several pathogens, without the use of chemicals that might harm organisms under culture conditions or the environment.6 Besides polyunsaturated fatty acids, I. galbana is a valuable source of proteins, carbohydrates and photosynthetic pigments such as chlorophyll a and fucoxanthin.7
Fucoxanthin, a major carotenoid present in the chloroplasts of brown seaweeds, contributes to more than 10% of the estimated total production of carotenoids in nature. Although fucoxanthin is clearly a valuable pigment with various health benefits, its use has been limited due to the low extraction efficiency from marine materials and the difficulty to synthesize it. In this respect, algae, such as I. galbana, can be considered as a potential source of fucoxanthin.8
In order to fully develop the microalgae-based biorefinery concept, new aspects related to technologies for extraction, isolation and fractionation of the biomass into multiple products (lipids, proteins, polysaccharides, bioactives, etc.) should be studied. Also, steps into integrated approaches for multi-product biorefinery should be taken into account to improve the efficiency and minimize the energy and resource consumption,9 especially when green chemistry principles and sustainability issues are to be considered.
Traditionally, extraction of lipophilic compounds from algae, such as carotenoids and lipids, has been performed by means of toxic organic solvents like hexane. Nowadays there is a demand for fast, selective, efficient and greener processes able to provide extractions with high yields; besides, the costs associated have to be reduced, for instance, by minimizing the removal of solvent residues.
High-pressure extractions such as supercritical fluid extraction (SFE) and pressurized liquid extraction (PLE) using GRAS (generally recognized as safe) solvents such as CO2, ethanol or water, have emerged as promising alternatives to face these challenges.10 This was the subject of a specifically devoted workshop on Supercritical Fluids and Energy that was conducted in Brazil in December 2013,11 with the idea of assessing the potential of supercritical (pressurized fluids in general) technologies in the fields of energy, materials science, process technology, green chemistry and sustainable technologies.
SFE offers a fast extraction rate, high selectivity and is an ecofriendly technology with minimal or no use of organic solvents, although the low polarity of supercritical CO2 (ScCO2) limits its applications. ScCO2 has been reported as an interesting approach for the extraction of lipids with antimicrobial activity from the microalgae Chaetoceros muelleri,12 n-3 fatty acids from the seaweed Hypnea charoides,1 lutein and β-carotene from Scenedesmus almeriensis13 and fucoxanthin from the seaweed Undaria pinnatifida14 and Sargassum muticum,15 among others. In this latter application, the addition of ethanol as a co-solvent improved the yield of fucoxanthin in both algal species.15,16
Ethanol is often used as a modifier or a co-solvent of ScCO2 in order to overcome the CO2 limitations towards the extraction of medium polarity bioactive compounds. For instance, CO2 modified with ethanol has been applied for the extraction of astaxanthin from Haematococcus pluvialis17 and various pigments from Spirulina platensis.18 The use of a co-solvent at a higher concentration allows working in the region of gas-expanded liquids (GXLs),19 which is a promising intermediate between PLE and SFE for the extraction of medium or high-polarity compounds. Carbon dioxide expanded ethanol (CXE) has been recently used to obtain astaxanthin enriched extracts from H. pluvialis.20
Pressurized liquid extraction has demonstrated an interesting potential for extracting bioactive compounds from macro- and microalgae.10,20 This extraction technique allows obtaining higher yields than those achieved by conventional extraction techniques, in a shorter time and with less solvent consumption.10 PLE using ethanol has been reported for the extraction of carotenoids from Neochloris oleoabundans,22Dunaliella salina2 and Chlorella ellipsoidea.3 In addition, 90% ethanol was used for the extraction of fucoxanthin from Eisenia bicyclis23 and the mixture of ethanol/limonene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) has been proposed as a green approach for PLE extraction of lipids from microalgae.9
:1, v/v) has been proposed as a green approach for PLE extraction of lipids from microalgae.9
In the present study, we propose an integrated sequential extraction process based on the use of green compressed fluids, in increasing order of polarity, for the fractionation of bioactive compounds from the microalga I. galbana, as an approach to develop a microalgae biorefinery procedure.21 The developed process comprises the sequential extraction with ScCO2, CO2-expanded ethanol, PLE using ethanol and subcritical water extraction. Finally, different tools are employed for the chemical and functional characterization of the obtained fractions.
2. Materials and methods
2.1. Chemicals and samples
HPLC-grade methyl tert-butyl ether (MTBE), methanol, acetone, and ethanol were from VWR (Leuven, Belgium). Sea sand (0.25–0.30 mm diameter) and potassium persulfate were from Panreac. Butylated hydroxytoluene (BHT), formic acid (LC-MS grade), triethylamine (99.5%) and standards of β-carotene, fucoxanthin, chlorophyll a (from Anacystis nidulans algae), ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt), D-methionine and Trolox (6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid) were obtained from Sigma-Aldrich (St Louis, MO, USA). The water used was Milli-Q water (Millipore, Billerica, MA, USA). Dichloromethane, chloroform, hexane, methanol, isooctane, and isopropanol were HPLC-grade and purchased from LabScan (Gliwice, Poland).Freeze-dried samples of I. galbana (T-ISO) were obtained from Fitoplancton Marino S.A. (Cadiz, Spain), and stored under dry and dark conditions until further use. I. galbana was grown in outdoor vertical 400 L reactors. Air containing 2% CO2 is injected into the reactors, while natural light–dark cycles and ambient temperature are used (10–11 h of light, temperatures ranging from 10 to 22 °C). These reactors are inoculated with cultures grown in growth chambers under the standard conditions of Fitoplancton Marino S.A.
2.2. Extraction methods
Extractions were performed in four sequential steps using (1) supercritical CO2 (ScCO2), (2) ScCO2/ethanol (CXE), (3) pure ethanol (PLE), and (4) pure water (PLE) as solvents, respectively.
The different extraction steps were selected in increasing order of polarity (ScCO2 < CXE < ethanol < water), to exhaust the microalgae biomass of extractable compounds, fractionating its components in order to give valuable isolated fractions.
Step 1: ScCO2 extraction conditions were optimized using a response surface methodology (RSM) to reveal the functional relationship between the extraction responses (extract yield, total carotenoids and total chlorophylls of extracts) and independent variables (extraction pressure and extraction temperature). A three-level factorial design (32) was used. The studied factors were pressure (100–300 bar) and temperature (40–60 °C). To determine the extraction time of this step, a kinetic study was performed at the central point of the experimental design (200 bar, 50 °C), collecting the extract every 20 min and calculating the percentage of the extractable material. The parameters of the model were estimated by multiple linear regression using the Statgraphics Centurion XVI software (Statpoint Technologies, Warrenton, Virginia, USA), which allows both the creation and the analysis of experimental designs.
Step 2: The second step involved a carbon dioxide expanded ethanol (CXE) extraction in order to increase the polarity of the extracted fraction. This step was carried out in the residual biomass from the first step. The pressure was set at 70 bar, while the temperature was maintained at 50 °C to match the optimum temperature used in the first step in order to avoid unnecessary heating or cooling of the system and thus, minimizing operational costs. Three different percentages of ethanol were tested, 15%, 45% and 75%; the extraction time selected was 1 h. The extraction in the center point (45% EtOH) was performed in triplicate for the precision study.
Step 3: The residue from the previous extractions was extracted again using PLE at 100 bar and 80 °C for 30 min, using pure ethanol as an extracting solvent.
Step 4: In the fourth and last step, PLE was employed using water as a solvent under the same extraction conditions employed in step 3 (100 bar and 80 °C for 30 min).
All the collection recipients were protected from light and 0.1% (w/v) BHT was added to the extracts. Finally, the solvent (ethanolic extracts) was evaporated in a rotary evaporator (Buchi, Flawil, Switzerland) or the samples were freeze-dried (water extracts). The extracts were stored at −80 °C to prevent degradation until analysis.
952g at 4 °C for 10 min. The supernatant was collected, and the solvent was removed using a stream of N2. Dry acetone extracts were weighed and stored at −20 °C.
2.3. Total carotenoid and chlorophyll determination
A spectrophotometric method was used to determine the total carotenoid and total chlorophyll concentration, based on their characteristic absorbance. Extracts from steps 1 and 4 were dissolved in methanol at a concentration of 0.1 mg mL−1, while extracts of steps 2 and 3 were dissolved in methanol at a concentration of 0.05 mg mL−1. Absorbance of these solutions was recorded at two specific wavelengths, 470 and 665 nm, for carotenoids and chlorophylls, respectively. External standard calibration curves of fucoxanthin (0.5–10 μg mL−1) and chlorophyll a (0.5–7.5 μg mL−1) were used to calculate the total carotenoid and chlorophyll content. Total carotenoids were expressed as mg carotenoids per g extract, by interpolating the absorbance of the extract at 470 nm in the calibration curve of fucoxanthin. Total chlorophylls were expressed as mg chlorophyll per g extract, by interpolating the absorbance of the extract at 665 nm in the calibration curve of chlorophyll a.2.4. Analysis of carotenoids and chlorophylls by HPLC-DAD
The carotenoid and chlorophyll profile of I. galbana extracts was determined by HPLC-DAD (diode-array detector) according to a method previously described for N. oleoabundans by Castro-Puyana et al.22 HPLC analyses of the extracts were conducted using an Agilent 1100 series liquid chromatograph (Santa Clara, CA, USA) equipped with a diode-array detector, and using a YMC-C30 reversed-phase column (250 mm × 4.6 mm inner diameter, 5 μm particle size; YMC Europe, Schermbeck, Germany) and a pre-column YMC-C30 (10 mm × 4 mm i.d., 5 μm). The mobile phase was a mixture of methanol–MTBE–water (90:7:3 v/v/v) (solvent A) and methanol–MTBE (10:90 v/v) (solvent B) eluted according to the following gradient: 0 min, 0% B; 20 min, 30% B; 35 min, 50% B; 45 min, 80% B; 50 min, 100% B; 60 min, 100% B; 62 min, 0% B. The flow rate was 0.8 mL min−1 while the injection volume was 10 μL. The detection was performed at 280, 450 and 660 nm, although spectra from 240 to 770 nm were recorded using the DAD (peak width >0.1 min (2 s) and slit 4 nm). The instrument was controlled by LC ChemStation 3D Software Rev. B.04.03 (Agilent Technologies, Santa Clara, CA, USA). Extracts were dissolved in solvent A prior to HPLC analysis at a concentration of 1 mg mL−1 for the extract of steps 2 and 3; the extracts from the first (ScCO2) and fourth steps were analyzed at 10 mg mL−1 (and filtered through 0.45 μm nylon filters).
For the calibration curve, twelve different concentrations of fucoxanthin in ethanol, ranging from 0.97 × 10−4 to 0.2 mg mL−1, were analyzed using the LC-DAD instrument.
2.6. Identification of carotenoids by HPLC-APCI-MS/MS
LC-MS characterization of I. galbana extracts was performed according to the method previously described by Castro-Puyana et al.22 An Agilent (Santa Clara, CA, USA) 1200 liquid chromatograph equipped with a diode-array detector was directly coupled to an ion trap mass spectrometer (Agilent ion trap 6320) via an atmospheric pressure chemical ionization (APCI) interface. The HPLC conditions employed for performing the analysis were the same as those described in the previous section. MS analysis was conducted with APCI in positive ionization mode using the following parameters: capillary voltage, −3.5 kV; drying temperature, 350 °C; vaporizer temperature, 400 °C; drying gas flow rate, 5 L min−1; corona current (which sets the discharge amperage for the APCI source), 4000 nA; nebulizer gas pressure, 60 psi. Full scan was acquired in the range from m/z 150 to 1300. Automatic MS/MS analysis was also performed, fragmenting the two highest precursor ions (10000 counts threshold; 1 V Fragmentor amplitude).
2.7. Analysis of lipid class compositions by HPLC-evaporative light scattering detection
Separation of lipid classes was done using the method described by Castro-Gómez et al.24 The analysis was performed using an HPLC system (model 1260; Agilent Technologies Inc.) coupled with an evaporative light scattering detector (SEDEX 85 model; Sedere SAS, Alfortville Cedex, France) using prefiltered compressed air as the nebulizing gas at a pressure of 3.5 bar at 60 °C; the gain was set at 3. Two columns were used in series (250 × 4.5 mm Zorbax Rx-SIL column with 5 μm particle diameter; Agilent Technologies Inc.) and a precolumn with the same packing was used. Before analysis, samples were dissolved in CH2Cl2 (5 mg mL−1) and 50 μL was injected. The autosampler temperature was maintained at 4 °C, while the column temperature was set at 40 °C. Solvent mixtures and gradients are detailed in ref. 24.2.8. Antioxidant capacity assay
The TEAC (Trolox equivalent antioxidant capacity) value was determined using the method described by Re et al.25 with some modifications. The ABTS˙+ (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt) radical was produced by reacting 7 mM ABTS and 2.45 mM potassium persulfate in the dark at room temperature for 16 h. The aqueous ABTS˙+ solution was diluted with 5 mM sodium phosphate buffer pH 7.4 to an absorbance of 0.7 (±0.02) at 734 nm. Ten microliters of the sample (5 different concentrations) and 1 mL of the ABTS˙+ solution were mixed in an Eppendorf vial and 300 μL of the mixture was transferred into a 96-well microplate. The absorbance was measured at 734 nm every 5 min for 45 min in a microplate spectrophotometer reader (Synergy HT, BioTek). “Trolox” (6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid) was used as the reference standard and the results are expressed as TEAC values (mmol Trolox equivalents per g sample). These values are obtained from five different concentrations of each sample tested in the assay giving a linear response between 20 and 80% of the blank absorbance. All analyses were performed in triplicate.2.9. Protein analysis of PLE extracts
Protein analysis was performed according to the Dumas method26 by using a FlashEA 1112 nitrogen analyzer (Thermo Fisher Scientific, Waltham, MA, USA). Ten milligrams of the dry extract were weighed in a cup of tin and tightly pelleted and subsequently analyzed. A calibration curve of D-methionine was used within the range 1–20 mg. A N-to-protein conversion factor of 4.68 was used to calculate total protein from total nitrogen. The N-to-protein conversion factor was obtained by determination of the amino acid composition of I. galbana according to ref. 27. Analyses were performed in duplicate.2.10. Sugar composition analysis of PLE extracts
The hydrolysis of algae extracts was performed according to Saeman et al.28 75 mg of the extract was hydrolyzed for 1 h in 72% (w/w) H2SO4 at 30 °C and subsequently water was added giving 1 M H2SO4 and the mixture was incubated for 3 h at 100 °C. After hydrolysis the samples were cooled in ice and then centrifuged (3000g, 15 min, at room temperature). The supernatant of each sample was used for analysis of the sugar composition.The neutral sugar composition was determined according to de Keijzer et al.29 by high performance anion exchange chromatography (HPAEC) using an ICS-3000 ion chromatography HPLC system equipped with a CarboPac PA-1 column (2 × 250 mm) in combination with a CarboPac PA guard column (2 × 25 mm) and a pulsed electrochemical detector in pulsed amperometric detection mode (Dionex, Sunnyvale, USA). A flow rate of 0.3 mL min−1 was used and the column was equilibrated with 17 mM NaOH. Elution was performed in two steps: 0–0.5 min, 17–0 mM NaOH and 0.5–35 min, 0–35 mM NaOH in 0–350 mM sodium acetate. Detection of the monomers was possible after the post column addition of 0.5 M sodium hydroxide (0.2 mL min−1). Before analysis samples were diluted (1:3) in water and to a 1 mL sample, 2.5 μL 0.1% (w/v) bromophenol blue in ethanol was added. To adjust the pH, solid barium carbonate was added until a clear magenta color was obtained. Subsequently, the solution was filtered using a 0.45 μm PTFE filter. Fucose was used as an internal standard in the case where fucose was not present in the sample. Analysis was performed in duplicate.
3. Results and discussion
The strategy has been selected considering the compounds of interest that can be found in I. galbana (such as lipids, proteins, carbohydrates and carotenoids, mainly fucoxanthin and its isomers), the need for re-extracting the residual biomass from the previous extraction step, and the use of green solvents with increasing polarity.Experimental conditions of the different extraction steps were either optimized or selected according to the previous results obtained in our laboratory for the extraction of similar compounds in other microalgae samples. Moreover, minimization of operational and energy costs was also considered in the integrated process, thus minimizing heating/cooling operations and collection or treatment of the microalgae biomass.
3.1. Optimization of supercritical CO2 extraction of Isochrysis galbana (step 1)
As mentioned above, SFE using CO2 as a solvent is considered a green process for the extraction of non-polar compounds from natural sources.30,31 With the objective of maximizing the extraction of the less polar fraction of I. galbana biomass, supercritical CO2 extraction conditions were optimized using a three-level factorial design (32). Extraction time was selected after performing a kinetic study under the central conditions (200 bar, 50 °C) measuring the percentage of the extractable material vs. extraction time by collecting samples every 20 minutes (data not shown). An extraction time of 60 min was selected as the most appropriate since after that time the amount of extracted material did not increase. Table 1 shows the experimental design employed, together with the results of the different response variables measured, i.e. extraction yield and total carotenoids and total chlorophyll content. As shown in Table 1, extraction yields ranged from 0.31 to 5.00% while the carotenoid content can be as high as 16.15 mg per g extract at a pressure of 300 bar and medium temperature (50 °C). This is in agreement with the previous results obtained for the extraction of carotenoids from other microalgae such as D. salina.32,33| Extract (P.T) | P, bar | T, °C | Yield (%) | mg carotenoids per g ext. | mg chlorophylls per g ext. |
|---|---|---|---|---|---|
| 100.40 | 100 | 40 | 0.52 | 2.2 ± 0.2 | 3.1 ± 0.4 |
| 100.50 | 100 | 50 | 0.31 | 2.1 ± 0.2 | 3.3 ± 0.1 |
| 100.60 | 100 | 60 | 0.56 | 1.8 ± 0.1 | 2.7 ± 0.2 |
| 200.40 | 200 | 40 | 2.56 | 6.39 ± 0.09 | 4.6 ± 0.2 |
| 200.50 | 200 | 50 | 2.49 | 5.45 ± 0.08 | 1.9 ± 0.0 |
| 200.50 | 200 | 50 | 2.41 | 5.0 ± 0.2 | 1.22 ± 0.02 |
| 200.50 | 200 | 50 | 2.35 | 5.64 ± 0.07 | 1.24 ± 0.04 |
| 200.60 | 200 | 60 | 2.55 | 5.76 ± 0.06 | 0.8 ± 0.0 |
| 300.40 | 300 | 40 | 1.11 | 4.9 ± 0.2 | 1.09 ± 0.04 |
| 300.50 | 300 | 50 | 5.00 | 16.2 ± 0.3 | 4.5 ± 0.0 |
| 300.60 | 300 | 60 | 1.77 | 15.8 ± 0.8 | 2.70 ± 0.04 |
After performing the ANOVA (evaluation of the experimental design with Statgraphics Centurion XVI software) for each of the responses (data not shown), the statistical model was fitted and optimized. Considering that the goal of the first step was to maximize the yield and carotenoid content, while minimizing chlorophylls, a desirability function was selected for meeting these goals and giving to all responses the same weight. As shown in Fig. 1, this function provided an optimum of 299 bar and 51 °C to increase the extraction yield and carotenoid content while minimizing the chlorophyll content. The optimization desirability was equal to 0.66, while the values predicted by the model under the optimum extraction conditions were 4.41% for extraction yield, carotenoid content of 16.4 mg carotenoids per g extract and 4.3 mg chlorophylls per g extract for total chlorophylls. Experiments under the optimum conditions provided experimental values close to that predicted by the statistical model (Table 1, experiment 300.50).
 | ||
| Fig. 1 Surface of the desirability function in terms of pressure and temperature obtained to maximize the yield and carotenoid content, while minimizing the chlorophyll content. | ||
3.2. Design of the conditions of sequential extraction of Isochrysis galbana (steps 2–4)
Following this first step, three sequential extractions were studied in order to further fractionate the biomass achieving extracts with different compositions. The second step was selected to increase the polarity of the solvent mixture while taking advantage of the intermediate conditions, such as those provided by GXLs that allow working at lower pressures than those of SFE and using smaller volumes of solvents (compared to PLE). This approach has already been successfully applied to the extraction of astaxanthin from H. pluvialis microalgae.20 Thus, for the second step, a pressure of 70 bar was selected, which is lower than the CO2 critical pressure (73.8 bar). The temperature was fixed at the optimum value of the first step (50 °C) in order to minimize energy consumption due to heating or cooling of the system. Three different percentages of ethanol, corresponding to low (15%), medium (45%) and high (75%) levels were tested to fully study the possible advantages offered by this intermediate process.Steps 3 and 4 were performed under PLE conditions, using ethanol and water, respectively, which implies an increasing order of polarity. At this point, different bioactive compounds were sought such as polar lipids, proteins and carbohydrates. Moreover, the final objective was to extract all the valuable components contained in the microalgae biomass attaining different fractions and minimizing the leftovers. The extraction values selected included a pressure of 100 bar and a temperature of 80 °C. These values were maintained relatively low in order to avoid degradation of compounds.
The scheme of the overall extraction process, along with the target compounds expected in each step is depicted in Fig. 2.
 | ||
| Fig. 2 Scheme of the overall sequential extraction process. | ||
3.3. Chemical characterization of the extracts obtained in steps 1–4
| Peak # | t R (min) | Identification | UV-Vis max wavelength (nm) | Parent ion(s) | Main fragments |
|---|---|---|---|---|---|
| a Identification confirmed by comparison with commercial standards. | |||||
| 3 | 6.79 | Fucoxanthinol | 442 | — | |
| 4 | 7.63 | E-Fucoxanthina | 448 | 641.7 [M + H–H2O]+ | 641.6, 549.7 |
| 581.9 [M + H–H2O-60]+ | 563.5, 489.5 | ||||
| 5 | 8.13 | 13(′)Z-Fucoxanthin | 332, 442 | — | |
| 6 | 8.43 | 13(′)Z-Fucoxanthin | 332, 438 | — | |
| 7 | 10.61 | 9(′)Z-Fucoxanthin | 442 | — | |
| 9 | 13.82 | Carotenoid | 322, 422, 448 | — | |
| 10 | 15.07 | Carotenoid | 422, 446, 472 | — | |
| 11 | 15.64 | Diadinoxanthin | 428, 446, 474 | — | |
| 12 | 16.55 | Diadinoxanthin 5,8-epoxy derivative | 404, 428, 456 | 506.8 | 268.6 |
| 583.5 [M + H]+ | 565.6 | ||||
| 13 | 17.18 | Diadinoxanthin 5,8-epoxy derivative | 404, 428, 456 | 583.7 [M + H]+ | 565.6 |
| 14 | 18.85 | Carotenoid | 400, 424, 450 | — | |
| 16 | 19.35 | Carotenoid | 426, 450, 476 | — | |
| 17 | 22.90 | Carotenoid | 460 | — | |
| 18 | 25.92 | Carotenoid | 460 | — | |
| 19 | 26.70 | Carotenoid | 405, 426, 454 | — | |
| 20 | 28.31 | Carotenoid | 424, 450, 478 | — | |
| 21 | 29.14 | Carotenoid | 340, 420, 444, 470 | — | |
| 23 | 30.7 | Pheophytin a′ | 408, 666 | 871.9 [M]+ | 593.7 |
| 593.5 [M − C20H38]+ | 533.6 | ||||
| 25 | 32.99 | Carotenoid | 452, 476 | — | |
| 27 | 34.5 | Carotenoid | 446, 472 | — | |
| Peak # | t R (min) | Identification | UV-Vis max. wavelengths (nm) | Parent ion(s) | Main fragments |
|---|---|---|---|---|---|
| a Identification confirmed by comparison with commercial standards. | |||||
| 3 | 6.57 | Fucoxanthinol | 448 | 600.0 [M + H–H2O]+ | 581.6, 563.5 |
| 617.7 [M + H]+ | 599.5, 581.6 | ||||
| 4 | 7.63 | E-Fucoxanthina | 448 | 641.7 [M + H-18]+ | 641.6, 623.6, 581.6, 563.6, 549.6 |
| 581.6 [M + H–H2O-60]+ | 563.5, 489.5 | ||||
| 5 | 8.13 | 13(′)Z-Fucoxanthin | 332, 442 | 641.7 [M + H–H2O]+ | 641.6, 549.6 |
| 6 | 8.43 | 13(′)Z-Fucoxanthin | 581.9 [M − H2O-60 + H]+ | 563.5, 489.5 | |
| 8 | 13.36 | Carotenoid | 454 | — | |
| 11 | 15.57 | Diadinoxanthin | 420, 445, 475 | 583.6 [M + H]+ | 565.6, 547.6, 491.5 |
| 565.6 [M − H2O]+ | 547.6 | ||||
| 12 | 16.48 | Diadinoxanthin 5,8-epoxy derivative | 404, 428, 456 | 583.7 [M + H]+ | 565.6, 547.6, 491.5 |
| 13 | 17.10 | Diadinoxanthin 5,8-epoxy derivative | 565.7 [M − H2O]+ | 547.6 | |
| 15 | 18.88 | Chlorophyll aa | 432, 664 | 894.0 [M + H]+ | 615.7 |
| 567.8 | 549.6 | ||||
| 17 | 20.37 | Chlorophyll a′ | 432, 664 | 894.6 [M + H]+ | 615.5 |
| 22 | 29.72 | Pheophytin a | 408, 668 | 872.0 [M]+ | 593.6 |
| 593.6 [M − C20H38]+ | 533.6 | ||||
| 23 | 30.40 | Pheophytin a′ | 408, 666 | 871.9 [M]+ | 593.5, 533.5 |
| 593.5 [M − C20H38]+ | 533.6 | ||||
| 24 | 32.79 | Chlorophyll c1-like | 448, 582, 632 | — | |
| 26 | 33.39 | Chlorophyll c2-like | 456, 584, 634 | — | |
| 28 | 38.18 | Chlorophyll c2-like | 456, 584, 632 | — | |
Since the percentage of ethanol in the CXE step did not affect the chromatographic profile, the HPLC-DAD chromatogram obtained for 45% ethanol in CO2 has been used to illustrate the identification of carotenoids and chlorophylls in the second step of the sequential extraction (see Fig. S1, step 2, ESI†). Fucoxanthin was again the main compound present in the extracts, but several chlorophylls and chlorophyll derivatives were also detected (see Table 3). The protonated molecule [M + H]+ was not observed for any of the fucoxanthin isomers. Interestingly, E- and 13(′)Z-fucoxanthin isomers showed the same parent ions, corresponding to the dehydrated molecule ([M + H–H2O]+) and a fragment corresponding to a loss of 78 Da consistent with the sequential losses of the C-3 carbomethoxy group (acetic acid) and a water molecule. MS/MS analyses of these ions exhibited a loss of 92 Da that could be attributed to the loss of toluene from the polyene chain. Fucoxanthin metabolite fucoxanthinol (Table 3, peak 3) was tentatively identified by its protonated molecule. MS/MS analysis of fucoxanthinol led to dehydration of the molecule.
Diadinoxanthin (peak 11) was also identified in the extracts by the presence of its typical ions at m/z 583.6 ([M + H]+) and m/z 565.6 ([M + H–H2O]+). The same MS spectrum was obtained for peaks 12 and 13, but for these peaks, a hypsochromic shift of 15–20 nm was observed in all UV maxima. Therefore, these compounds can be tentatively identified as 5,8-epoxy derivatives of diadinoxanthin, according to Crupi et al.34
Chlorophyll a and its epimer chlorophyll a′ (peaks 15 and 17) lost the phytyl group (C20H39)35 and showed the same fragment, m/z 615.5, which corresponds to the chlorophyllides a and a′, respectively. Besides, the loss of the phythyl group (C20H39) can also be used for the identification of pheophytins a and a′ (peaks 22 and 23).36 The identification of chlorophyll a in the extract was confirmed by using a commercial standard, and thus peak 10 was assigned to chlorophyll a′. The same elution order was considered for pheophytins a and a′. Several chlorophyll c pigments were tentatively identified in the extracts, although no information could be obtained from the MS in this case. Nevertheless, they were grouped in chlorophyll c1-like (peak 24) and chlorophyll c2-like (peaks 26 and 28) compounds, on the basis of their UV-VIS spectra, since the band ratios (II/III) and the position of maxima are different. The ratios of band II (at ∼630 nm) to band III (at ∼580 nm) intensities are >1 for Chl c1-like chromophores, ≈1 for Chl c2-like chromophores and <1 for Chl c3-like chromophores.37
The vast majority of total fucoxanthin is formed by E-fucoxanthin, while the amount of the other isomers remains very low under the different extraction conditions, except for extractions at 300 bar and 60 °C (data not shown). Under these conditions, the sum of 13(′)Z isomer concentration is higher, although still extremely low compared to E-isomers, which could be due to an increase in their solubility under these extraction conditions. In general, the highest extraction of fucoxanthin occurred at 300 bar and 50 °C, over the experimental range that was explored.
Table 4 shows the quantification of fucoxanthin isomers, the total carotenoid amount and the total chlorophyll content of the extracts obtained after each step of the sequential integrated process. The highest total chlorophyll content (expressed as chlorophyll a) was found in the CXE extract obtained using 15% ethanol, while the highest content of total carotenoids (expressed as fucoxanthin) was obtained in the CXE extract containing 75% ethanol. In any case, total carotenoids and chlorophylls extracted with carbon dioxide expanded ethanol were higher than total carotenoids and chlorophylls extracted with acetone (146.58 vs. 57.19 mg per g extract and 96.56 vs. 44.48 mg per g extract, respectively, for carotenoids and chlorophylls). On the other hand, the highest content of E-fucoxanthin was found in the CXE extract containing 45% ethanol (40.69 ± 2.28 mg per g extract), and is comparable to the concentration of E-fucoxanthin obtained with acetone conventional solid-liquid extraction (44.60 ± 2.68 mg per g extract). Regarding Z isomers, the sum of 13Z + 13′Z isomers, as well as the amount of 9(′)Z isomers, is higher in acetone extracts, compared to CXE extracts. The content of fucoxanthinol, however, is comparable between acetone and CXE extracts. On the other hand, pooling both ethanol containing extracts (steps 2 and 3), the content of fucoxanthin isomers surpasses acetone extractions, thus validating the use of this new type of green technology for extraction of high value-added compounds.
| Samplea (%Step) | Yield (%) | Fucoxanthin isomers, mg per g extract | Total carotenoids, mg per g extract | Total chlorophylls, mg per g extract | TEAC (mmol g−1) | |||
|---|---|---|---|---|---|---|---|---|
| E-Fucox | 13Z + 13′Z-fucox | Fucoxanthinol | 9(′)Z-Fucox (2) | |||||
| a The name of the extracts corresponds to the %EtOH in the second step. Sequential steps have been named as 1 (ScCO2), 2 (CXE), 3 (PLE EtOH) and 4 (PLE water). b Average results from three independent extractions under the same conditions. | ||||||||
| 15.1 | 3.5 | 5.6 ± 0.4 | 0.22 ± 0.01 | 0.005 ± 0.001 | 0.026 ± 0.002 | 8.6 ± 0.2 | 3.0 ± 0.1 | |
| 15.2 | 6.3 | 36.8 ± 0.4 | 1.0 ± 0.1 | 0.26 ± 0.03 | — | 66.2 ± 0.7 | 65.4 ± 0.4 | |
| 15.3 | 15.0 | 21.8 ± 0.2 | 1.46 ± 0.03 | 0.20 ± 0.02 | <LOQ | 42.2 ± 1.2 | 39 ± 1 | 0.639 ± 0.006 |
| 15.4 | 5.8 | 0.870 ± 0.002 | 0.055 ± 0.003 | <LOQ | 0.0206 ± 0.0003 | 1.05 ± 0.07 | 1.10 ± 0.04 | 0.33 ± 0.01 |
| 45.1b | 4.4 ± 0.5 | 8 ± 2 | 0.23 ± 0.08 | 0.014 ± 0.006 | 0.033 ± 0.022 | 11 ± 1 | 5.6 ± 0.1 | |
| 45.2b | 11 ± 1 | 41 ± 2 | 1.2 ± 0.1 | 0.25 ± 0.04 | <LOQ | 62 ± 2 | 53 ± 11 | |
| 45.3b | 4.8 ± 0.4 | 21 ± 2 | 1.6 ± 0.3 | 0.22 ± 0.05 | 0.07 ± 0.01 | 49 ± 10 | 41 ± 5 | 0.54 ± 0.04 |
| 45.4b | 4.4 ± 0.5 | 0.7 ± 0.5 | 0.05 ± 0.03 | <LOQ | 0.011 ± 0.005 | 1.4 ± 0.9 | 1.6 ± 0.9 | 0.29 ± 0.05 |
| 75.1 | 3.9 | 4.61 ± 0.04 | 0.144 ± 0.003 | 0.004 ± 0.001 | 0.023 ± 0.0002 | 5.856 ± 0.2 | 3.8 ± 0.1 | |
| 75.2 | 14.7 | 38.2 ± 0.3 | 1.12 ± 0.07 | 0.216 ± 0.005 | — | 91.9 ± 0.8 | 58.1 ± 0.4 | |
| 75.3 | 2.0 | 9.00 ± 0.02 | 0.99 ± 0.01 | 0.041 ± 0.004 | 0.053 ± 0.002 | 45.2 ± 0.7 | 32.8 ± 0.2 | 0.482 ± 0.006 |
| 75.4 | 3.1 | 1.18 ± 0.02 | 0.093 ± 0.007 | 0.004 ± 0.0003 | 0.03 | 3.6 ± 0.2 | 1.918 ± 0.124 | 0.30 ± 0.02 |
| Acetone extractsb | 22.41 ± 0.09 | 44 ± 3 | 2.6 ± 0.4 | 0.23 ± 0.04 | 0.06 ± 0.04 | 57 ± 6 | 44 ± 4 | |
It is worth mentioning that the content of E-fucoxanthin in any of the CXE extracts (36–43 mg g−1) was higher than that previously reported for I. galbana using acetone extraction39 and for Isochrysis sp., using conventional extraction with methanol.34
 | ||
| Fig. 3 HPLC-ELSD chromatograms obtained from the analysis of the Isochrysis galbana sequential extraction using 45% EtOH in CO2 in the second step. The chromatograms have been divided into three segments to facilitate the discussion of the results. | ||
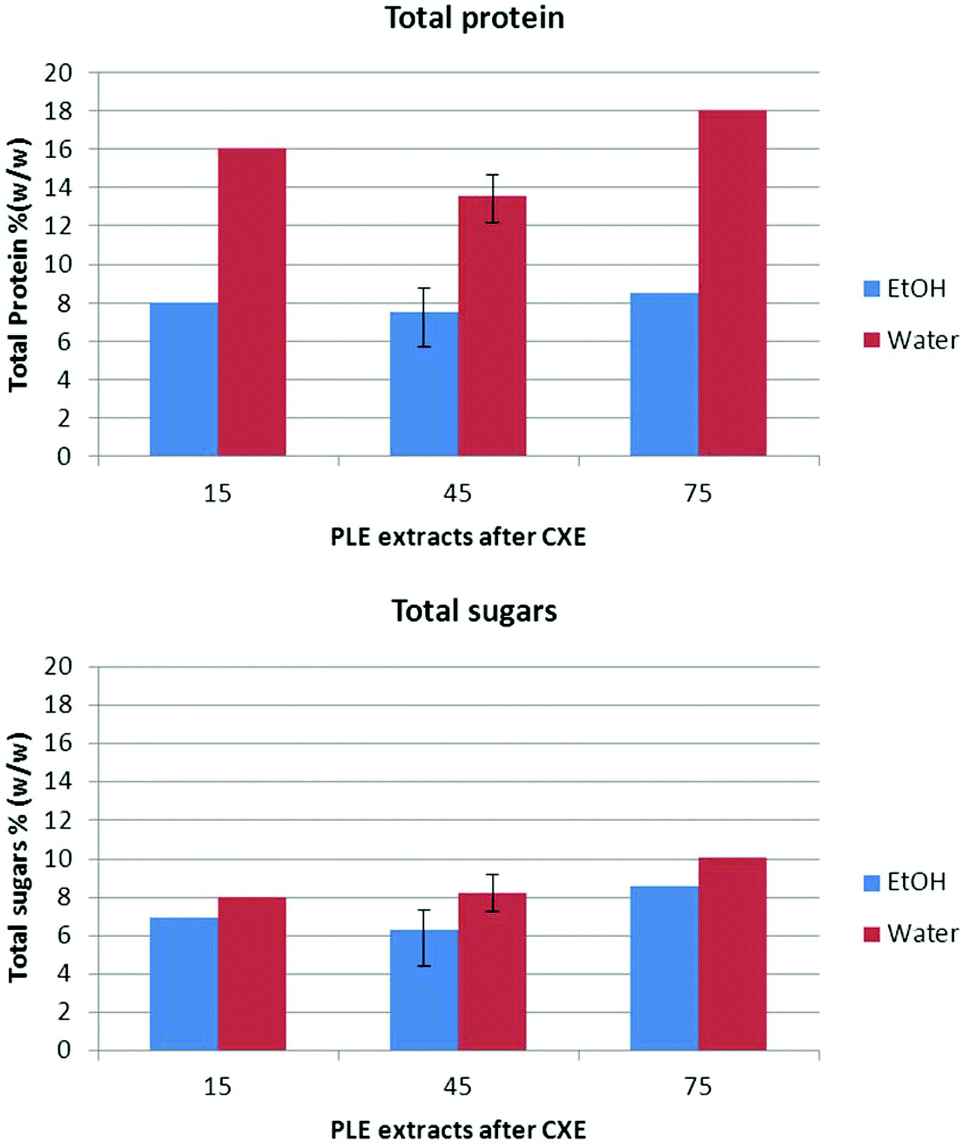 | ||
| Fig. 4 Total protein (%, w/w) and sugars (%, w/w) of ethanol and water PLE extracts. | ||
The sugar composition of ethanol and water PLE extracts was similar. Detailed results are shown in Table 5. Fucose, glucuronic acid, galacturonic acid, N-acetylglucosamine, N-acetylgalactosamine, glucosamine and galactosamine were not detected. Xylose (only present in CXE75-water) and mannose were found only in water extracts. A slightly higher amount of total sugars can be observed in the extracts obtained after CXE-75% ethanol compared to the extracts obtained after 15% and 45% ethanol. In any case, the total amount of sugars did not exceed 10% of the extract weight (see Fig. 4). Galactose is the main sugar in ethanol extracts, ranging from 5.69 to 6.68% of dry weight. In water extracts, galactose is present in a smaller amount (1.44–2.83% dry weight), while glucose is the main sugar found (3.91–4.11% dry weight).
| 15C | 15Da | 45Cb | 45Db | 75C | 75D | |
|---|---|---|---|---|---|---|
| a Duplicate sample was lost during analysis. b Average results from three independent extractions under the same conditions. c Ara is arabinose, Gal is galactose, Glc is glucose, Man is mannose, Rib is ribose and xyl is xylose. | ||||||
| Arac | 0.19 ± 0.01 | 1.1 | 0.18 ± 0.01 | 1.0 ± 0.2 | 0.21 ± 0.01 | 1.6 ± 0.1 |
| Galc | 5.7 ± 0.8 | 1.4 | 6.0 ± 0.1 | 2.1 ± 0.2 | 6.7 ± 0.1 | 2.8 ± 0.2 |
| Glcc | 0.56 ± 0.03 | 4.1 | 0.5 ± 0.1 | 4.1 ± 0.3 | 1.16 ± 0.04 | 3.9 ± 0.4 |
| Manc | 0 | 0.6 | 0 | 0.55 ± 0.07 | 0 | 0.59 ± 0.05 |
| Ribc | 0.47 ± 0.01 | 0.4 | 0.45 ± 0.04 | 0.46 ± 0.01 | 0.54 ± 0.03 | 0.53 ± 0.04 |
| Xylc | 0 | 0 | 0 | 0 | 0 | 0.69 ± 0.04 |
| Total sugars | 6.9 ± 0.9 | 8.0 | 7.2 ± 0.1 | 8 ± 1 | 8.6 ± 0.2 | 10.1 ± 0.7 |
The results corresponding to the antioxidant capacity assay (expressed as TEAC, mmol of Trolox per g sample), are shown in Table 4. As can be seen, ethanol extracts contained twice the activity as water extracts. This observation cannot be directly related to the total content of sugars, which was similar in both water and ethanol extracts. However, a different composition of sugars in ethanol and water extracts can be expected. Since ethanol is commonly used to precipitate polymeric sugars, monomers or oligomers may be preferably present in ethanol extracts, while oligomeric and polymeric sugars can be expected in water extracts. The total content of protein was lower in ethanol extracts, but proteins extracted in ethanol can be different from proteins present in water extracts, and therefore the activity can be different, too. On the other hand, the amount of fucoxanthin and total carotenoids in ethanol extracts is more than two times higher than the concentration of carotenoids in water extracts. Consequently, despite the fact that there is no linear relationship between the carotenoid content and antioxidant activity, data seem to indicate that higher antioxidant activity in ethanol extracts might be related to the fucoxanthin and fucoxanthin isomer content; Zhang et al.40 and Sachindra et al.41 previously confirmed the potent antioxidant activity of these compounds by using different methods.
4. Concluding remarks
A downstream processing platform is described for the first time to extract bioactive compounds from the microalga I. galbana using GRAS – generally recognized as safe – solvents and pressurized technologies. Extractions were performed in four sequential steps using (1) supercritical CO2 (ScCO2), (2) ScCO2/ethanol (Gas Expanded Liquids, GXLs), (3) PLE with pure ethanol, and (4) PLE with pure water as solvents, considering the residue of the previous extraction step as the raw material for extraction. The results obtained showed that the extraction process was partially selective according to the polarity of the solvent/mixture of solvents used. ScCO2 extracts were rich in triacylglycerides and showed less carotenoid and chlorophyll contents than ethanolic extracts. The main carotenoid identified was fucoxanthin which was found in highest amount in CXE extracts obtained with 45% ethanol. Steps 3 and 4 provide with extracts enriched in proteins and carbohydrates. Further studies should be carried out to determine more in depth the composition of the obtained extracts and their relationships with the antioxidant activity. Also, from our point of view, a scaling up to the industrial level of the process will be of interest.Acknowledgements
The authors acknowledge funding from the EU MIRACLES project (7th Framework Program - Grant Agreement No. 613588). B.G.L. thanks MINECO (Ministerio de Economía y Competitividad) for her Juan de la Cierva postdoctoral research contract. M.H. thanks MINECO for his Ramón y Cajal postdoctoral research contract.References
- P. C. K. Cheung, Food Chem., 1999, 65, 399–403 CrossRef CAS.
- M. Herrero, L. Jaime, P. J. Martín-Alvarez, A. Cifuentes and E. Ibáñez, J. Agric. Food Chem., 2006, 54, 5597–5603 CrossRef CAS PubMed.
- S. Y. Koo, K. H. Cha, D. G. Song, D. Chung and C. H. Pan, J. Appl. Phycol., 2011, 24, 725–730 CrossRef.
- R. Sayre, BioScience, 2010, 60(9), 722–727 CrossRef.
- E. Molina Grima, J. A. Sánchez Pérez, F. García Camacho, J. L. García Sanchez, F. G. Acién Fernández and D. López Alonso, J. Biotechnol., 1994, 37(2), 159–166 CrossRef CAS.
- C. A. Molina-Cárdenas, M. d. P. Sánchez-Saavedra and M. L. Lizárraga-Partida, J. Appl. Phycol., 2014, 26(6), 2347–2355 CrossRef.
- E. Valenzuela-Espinoza, R. Millán-Núñez and F. Núñez-Cebrero, Aquacult. Eng., 2002, 25(4), 207–216 CrossRef.
- S. M. Kim, S.-W. Kang, O.-N. Kwon, D. Chung and C.-H. Pan, J. Korean Soc. Appl. Biol. Chem., 2012, 55(4), 477–483 CrossRef CAS.
- M.-T. Golmakani, J. A. Mendiola, K. Rezaei and E. Ibañez, J. Supercrit. Fluids, 2014, 92, 1–7 CrossRef CAS.
- M. Herrero, A. P. Sánchez-Camargo, A. Cifuentes and E. Ibáñez, TrAC – Trends Anal. Chem., 2015, in press, DOI:10.1016/j.trac.2015.01.018.
- J. Supercr, Fluids, 2015, 96, 1–368 CrossRef.
- J. A. Mendiola, C. Torres, A. Tore, P. J. Martín-Alvarez, S. Santoyo, B. Arredondo, F. J. Señoráns, A. Cifuentes and E. Ibáñez, Eur. Food Res. Technol., 2007, 224, 505–510 CrossRef CAS.
- M. D. Macías-Sánchez, J. M. Fernandez-Sevilla, F. G. A. Fernández, M. C. C. García and E. M. Grima, Food Chem., 2010, 123, 928–935 CrossRef.
- A. T. Quitain, T. Kai, M. Sasaki and M. Goto, J. Agric. Food Chem., 2013, 61, 5792–5797 CrossRef CAS PubMed.
- E. Conde, A. Moure and H. Domínguez, J. Appl. Phycol., 2015, 27(2), 957–964 CrossRef CAS.
- M.-K. Roh, M. Uddin and B.-S. Chun, Biotechnol. Bioprocess Eng., 2008, 13, 724–729 CrossRef CAS.
- S. Machmudah, A. Shotipruk, M. Goto, M. Sasaki and T. Hirose, Ind. Eng. Chem. Res., 2006, 45(10), 3652–3657 CrossRef CAS.
- J. A. Mendiola, F. R. Marín, S. F. Hernández, B. O. Arredondo, F. J. Señorans, E. Ibáñez and G. Reglero, J. Sep. Sci., 2005, 28, 1031–1038 CrossRef CAS PubMed.
- G. R. Akien, G. R. Akien and M. Poliakoff, Green Chem., 2009, 11, 1083–1100 RSC.
- F. A. Reyes, J. A. Mendiola, E. Ibañez and J. M. del Valle, J. Supercrit. Fluids, 2014, 92, 75–83 CrossRef CAS.
- M. Herrero and E. Ibañez, J. Supercrit. Fluids, 2015, 96, 211–216 CrossRef CAS.
- M. Castro-Puyana, M. Herrero, I. Urreta, J. A. Mendiola, A. Cifuentes, E. Ibáñez and S. Suárez-Alvarez, Anal. Bioanal. Chem., 2013, 405, 4607–4616 CrossRef CAS PubMed.
- Y. F. Shang, S. M. Kim, W. J. Lee and B. H. Um, J. Biosci. Bioeng., 2011, 111, 237–241 CrossRef CAS PubMed.
- M. P. Castro-Gómez, L. M. Rodriguez-Alcalá, M. V. Calvo, J. Romero, J. A. Mendiola, E. Ibañez and J. Fontecha, J. Dairy Sci., 2014, 97, 6719–6728 CrossRef PubMed.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed.
- J. B. A. Dumas, Procedes de l'Analyse Organique, Ann. Chim. Phys., 1831, 247, 198–213 Search PubMed.
- B. J. Meussen, A. N. T. van Zeeland, M. E. Bruins and J. P. M. Sanders, Food Anal. Methods, 2014, 7, 1047–1055 CrossRef.
- J. F. Saeman, W. E. Moore, R. L. Mitchell and M. A. Millett, Tappi J., 1954, 37(8), 336–343 CAS.
- J. de Keijzer, L. A. M. van den Broek, T. Ketelaar and A. A. van Lammeren, PLoS One, 2012, 7(7), e39604 CAS.
- M. Herrero, M. Castro-Puyana, J. A. Mendiola and E. Ibáñez, TrAC, Trends Anal. Chem., 2013, 43, 67–83 CrossRef CAS.
- J. A. Mendiola, M. Herrero, M. Castro-Puyana and E. Ibáñez, in Natural Product Extraction: Principles and Applications, ed. M. A. Rostagno, RSC Publishing, 2013, ch. 6, pp. 196–230 Search PubMed.
- L. Jaime, J. Mendiola, E. Ibáñez, P. J. Martin-Álvarez, A. Cifuentes, G. Reglero and F. J. Señorans, J. Agric. Food Chem., 2007, 55, 10585–10590 CrossRef CAS PubMed.
- M. D. Macías-Sánchez, C. Mantell, M. Rodríguez, E. Martínez de la Ossa, L. M. Lubián and O. Montero, Talanta, 2009, 77(3), 948–952 CrossRef PubMed.
- P. Crupi, A. T. Toci, S. Mangini, F. Wrubl, L. Rodolfi, M. R. Tredici, A. Coletta and D. Antonacci, Rapid Commun. Mass Spectrom., 2013, 27, 1027–1035 CrossRef CAS PubMed.
- J. Wei, H. Li, M. P. Barrow and P. B. O'Connor, J. Am. Soc. Mass Spectrom., 2013, 24, 753–760 CrossRef CAS PubMed.
- S. M. Milenkovic, J. B. Zvezdanović, T. D. Anđelković and D. Z. Marković, Adv. Technol., 2012, 1, 16–24 Search PubMed.
- M. Zapata, J. L. Garrido and S. W. Jeffrey, in Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, 2006, ch. 3, pp. 39–53 Search PubMed.
- Y. Nakazawa, T. Sashima, M. Hosokawa and K. Miyashita, J. Funct Foods, 2009, 1, 88–97 CrossRef CAS.
- S. W. Jeffrey, Biochem. J., 1961, 80, 336–342 CrossRef CAS PubMed.
- Y. Zhang, H. Fang, Q. Xie, J. Sun, R. Liu, Z. Hong, R. Yi and H. Wu, Molecules, 2014, 19(2), 2100–2113 CrossRef PubMed.
- N. M. Sachindra, E. Sato, H. Maeda, M. Hosokawa, Y. Niwano, M. Kohno and K. Miyashita, J. Agric. Food Chem., 2007, 55(21), 8516–8522 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01256b |
| This journal is © The Royal Society of Chemistry 2015 |