
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct asymmetric reduction of levulinic acid to gamma-valerolactone: synthesis of a chiral platform molecule†
József M.
Tukacs
a,
Bálint
Fridrich
a,
Gábor
Dibó
b,
Edit
Székely
a and
László T.
Mika
*a
aDepartment of Chemical and Environmental Process Engineering, Budapest University of Technology and Economics, Budapest, Hungary. E-mail: laszlo.t.mika@mail.bme.hu; Fax: +36 1 463 3197; Tel: +36 1 463 1263
bJános Selye University, Faculty of Education, Department of Chemistry, Bratislavská str. 3322, Komárno SK-94501, Slovakia
First published on 28th July 2015
Abstract
Levulinic acid was directly converted to optically active (S)-gamma-valerolactone, a proposed biomass-based chiral platform molecule. By using a SEGPHOS ligand-modified ruthenium catalyst in methanol as a co-solvent, eventually, 100% chemoselectivity, and 82% enantioselectivity were achieved. The effect of the catalyst composition and reaction parameters on the activity and selectivity was investigated in detail. The conversion of a “real” biomass derived levulinic acid to optically active GVL without decreasing the enantioselectivity was also demonstrated.
Introduction
Currently, one of the most pressing challenges for the chemical industry is the gradual replacement of fossil resources with renewable ones. Since it is difficult to estimate the exact reserves of fossil resources, which provide more than 95% of our carbon based chemicals, the development of novel strategies to provide carbon-based building blocks should be accelerated. The selective biomass transformation, as one of the most preferred solutions, offers several alternative methods for the production of value-added chemicals and has led to the identification of key platform molecules, e.g. 5-hydroxymethyl furfural,1 levulinic acid (LA),2 and γ-valerolactone (GVL),3 which could replace the currently used fossil-based building blocks or serve as a “green” and renewable feedstock for their production. Due to its outstanding physical and chemical properties, Horváth et al. have first suggested GVL as a sustainable liquid.3 Later, it was shown that GVL can be used for the production of fuels,4 fuel additives,3 alkanes,5 and fine chemicals.6 In addition, GVL was also utilized as a green solvent for the conversion of carbohydrates to LA and subsequently to GVL.7Obviously, the most efficient protocol for the manufacture of GVL is the conversion of the carbohydrate content of biomass7b,8 including cellulose9 and/or biomass wastes10 to LA followed by the selective hydrogenation of LA to 4-hydroxyvaleric acid (4-HVA) by using either heterogeneous11 or homogeneous catalysis.6f,12,13 Recently, several catalytic systems have been reported for the conversion of LA to GVL. However, the asymmetric reduction of LA to optically active 4-HVA, which subsequently was converted to optically active GVL via ring closure dehydration (Scheme 1), has not been reported yet. The one-pot conversion of LA to optically active GVL could result in a green process representing the elegant approach of biomass waste valorization to a value-added chiral building block.
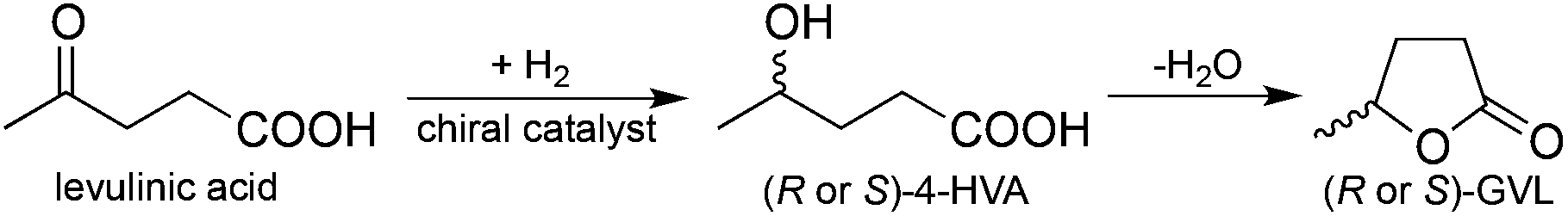 | ||
| Scheme 1 | ||
Optically active γ-lactones occur naturally14 and can be used as chiral building blocks for the synthesis of several biologically active compounds.15 The optically active GVL can be found in the synthetic schemes of several agricultural or pharmaceutical compounds16e.g. the aggregation pheromone Sulcatol,17 the antihypertensive WS75624B,18 the antileukemic Steganacin,19 and the insecticide Geodiamolide (Fig. 1).20 Accordingly, we propose here that the enantiopure GVL can be used as a promising chiral starting material for the synthesis of fine chemicals and other valuable intermediates i.e. chiral pentane-1,4-diol, 5-methyl-3-methylenedihydrofuran-2(3H)-one and its derivatives, unsaturated esters, ionic liquids etc. (Fig. 1). Since chiral solvents are of utmost importance in chiral recognition,21 due to its good solvating properties, the optically active GVL could serve as a renewable and non-toxic chiral reaction media in asymmetric synthesis, as well. It was demonstrated that chiral ionic liquids were successfully applied in asymmetric synthesis.22 GVL can easily be converted to ILs,6a,b and the application of chiral 4-hydroxyvalerate-based ionic liquids as chiral reaction media is also proposed in the same way.
 | ||
| Fig. 1 Selected applications of optically pure GVL. | ||
The enantioselective reduction of prochiral substrates including ketones is of utmost importance for the synthesis of optically pure substances both on laboratory and industrial scale.23 Although, several catalytic systems have been developed for the asymmetric reduction of the carbonyl group, most of them were tested on aromatic ketones e.g. acetophenone. It was revealed that the enantioselectivity could be dramatically influenced by the functional groups in the vicinity of the carbonyl group.23d Compared to the reduction of aromatic substrates, reduction of aliphatic ketones resulted in slightly lower yield and a small ee value.24,25 Several methods have been developed for the reduction of the carbonyl group of γ-oxo carboxylates; however, to the best of our knowledge, no direct asymmetric hydrogenation of the free carboxylic acid has been reported yet.
Karnik et al. reported the stoichiometric synthesis of optically active γ-lactones from (S)-menthyl or (S)-bornyl esters of 4-carboxylates by using NaBH4, however, the yield of (S)-GVL was moderate.26 Hilterhaus et al. suggested a chemoenzymatic reaction sequence to produce (S)-GVL via ethyl levulinate (EL). As expected, due to the enzymatic conversion, the ee values and overall yields were high (∼90%).27 So far, a few studies on the reduction of alkyl levulinates in the presence of Ru-based catalysts to optically active GVL have been published.28,29 Vinogradov et al. reported the Ru/BINAP-catalyzed asymmetric hydrogenation of LA by applying HCl in ethanol under 60 bar H2 at 60 °C.29a It was also shown that, instead of the reduction of LA, the in situ formed EL was reduced. Jacobs et al. demonstrated that the bakers’ yeast assisted reduction of alkyl levulinates and subsequent HCl-catalyzed hydrolysis of the corresponding hydroxy esters resulted in (S)-GVL with a yield of 73%.30 It is important to note that the ester hydrolysis step requires mineral acid. Noteworthily, the release of HCl from the reaction mixture into the atmosphere can result in serious environmental concerns, moreover, the aqueous HCl is extremely corrosive.
We report here the direct conversion of levulinic acid to optically active γ-valerolactone via asymmetric hydrogenation using various Ru-based catalyst systems.
Results and discussion
Firstly, concerning the ring opening/closing, the stability of the chiral center of GVL is a crucial point for further synthetic schemes. The ring opening of GVL under acidic conditions leads to the formation of 4-HVA. Subsequently, 4-HVA forms GVL via ring closure under neutral conditions. Accordingly, the stability of the chiral center of GVL is of utmost importance and was investigated by using the 18O-labelling technique as follows: 0.3 mmol of (S)-GVL having 98.5% ee was treated with 2.7 mmol of H218O (97 atom % 18O) in the presence of 1 mmol HCl at room temperature. The in situ NMR showed peaks at 176.6 ppm in 13C-NMR, and 1.2 ppm (3H, d, J = 5.9 Hz), 1.6 ppm (2H, m), 3.75 ppm (1H, s, J = 5.9 Hz) in 1H-NMR spectra. These data proved the equilibrium reaction between GVL and [4-HVA]. To neutralize the solution, an equimolar amount of sodium hydroxide was added to the reaction mixture after 1 h. The incorporation of an 18O-isotope into the (S)-GVL was verified by GC-MS. Chiral GC analysis of 18O-labelled GVL established that the ring opening and reclosing had no effect on the enantiopurity of (S)-GVL, as expected (Fig. 2).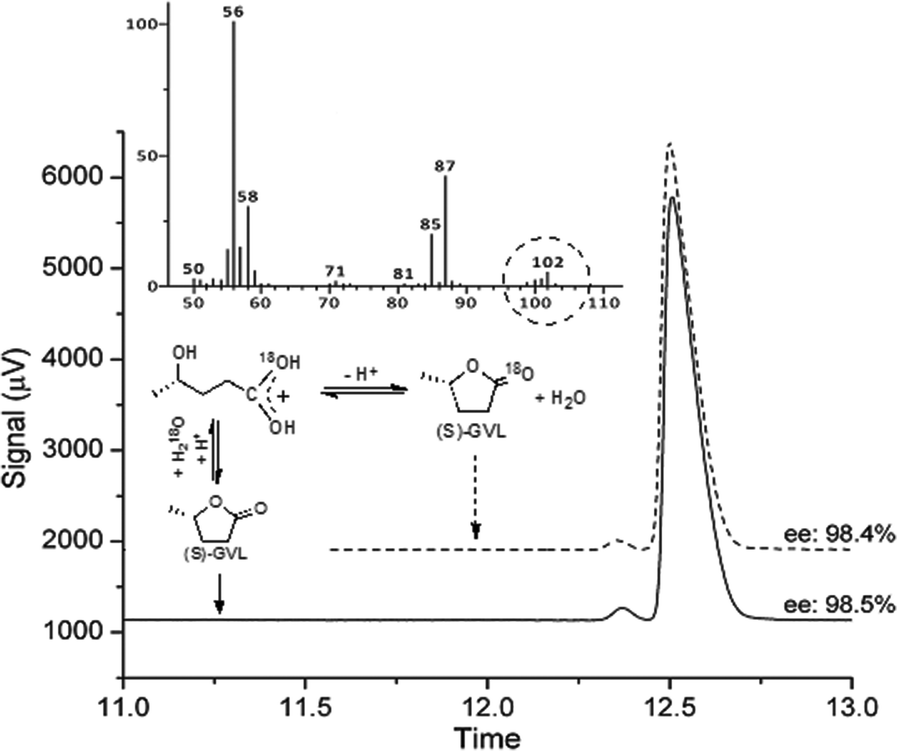 | ||
| Fig. 2 Chromatograms of (S)-GVL before and after ring opening, and MS spectrum of 18O-(S)-GVL. | ||
It was demonstrated, that LA was efficiently converted to GVL in the presence of bidentate phosphine-modified Ru catalysts without any added solvent and TOF = 100–21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 values were obtained. In the case of the BINAP ligand, 98.6% conversion was achieved with TOF = 6978 h−1.12c Firstly, reduction of LA (1 mL, 9.85 mmol) was attempted by using a catalyst formed in situ from Ru(acac)3 (1.56 μmol) and (R)-BINAP (15.6 μmol) under 60 bar H2, at 140 °C and 500 rpm. After 12 h, full conversion was obtained with ee = 26%. The reaction was repeated in a 120 mL Parr high-pressure reactor (equipped with a propeller stirrer) loaded with 30 mL (293.02 mmol) of LA, 0.023 mmol of Ru(acac)3 and 10-fold excess (0.233 mmol) of (R)-BINAP under identical conditions to monitor the possible change of ee with increasing conversion (Fig. 3). By using the propeller stirrer, the rate of hydrogen transfer from the gas to liquid phase was significantly higher. Thus, the reaction time required for full conversion of LA to GVL was decreased to 2.5 h, however, it was perceived that the ee value was almost the same (ee = 23%). To conclude, the ee values were not affected by the conversion rate. Moreover, addition of (R,R)-1,2-diphenyl-1,2-diaminoethane (R,R-DPEN) neither had a significant effect on the final ee values. When 9.85 mmol of LA was reduced, under identical conditions, in the presence of 0.001 mmol Ru(acac)3, 0.01 mmol (R)-BINAP, and 0.02 mmol of (R,R-DPEN), similarly 26.4% ee was obtained. Although the ee value was moderate, to the best of our knowledge, so far the solvent-free asymmetric reduction of levulinic acid to optically active GVL has not been reported yet.
000 h−1 values were obtained. In the case of the BINAP ligand, 98.6% conversion was achieved with TOF = 6978 h−1.12c Firstly, reduction of LA (1 mL, 9.85 mmol) was attempted by using a catalyst formed in situ from Ru(acac)3 (1.56 μmol) and (R)-BINAP (15.6 μmol) under 60 bar H2, at 140 °C and 500 rpm. After 12 h, full conversion was obtained with ee = 26%. The reaction was repeated in a 120 mL Parr high-pressure reactor (equipped with a propeller stirrer) loaded with 30 mL (293.02 mmol) of LA, 0.023 mmol of Ru(acac)3 and 10-fold excess (0.233 mmol) of (R)-BINAP under identical conditions to monitor the possible change of ee with increasing conversion (Fig. 3). By using the propeller stirrer, the rate of hydrogen transfer from the gas to liquid phase was significantly higher. Thus, the reaction time required for full conversion of LA to GVL was decreased to 2.5 h, however, it was perceived that the ee value was almost the same (ee = 23%). To conclude, the ee values were not affected by the conversion rate. Moreover, addition of (R,R)-1,2-diphenyl-1,2-diaminoethane (R,R-DPEN) neither had a significant effect on the final ee values. When 9.85 mmol of LA was reduced, under identical conditions, in the presence of 0.001 mmol Ru(acac)3, 0.01 mmol (R)-BINAP, and 0.02 mmol of (R,R-DPEN), similarly 26.4% ee was obtained. Although the ee value was moderate, to the best of our knowledge, so far the solvent-free asymmetric reduction of levulinic acid to optically active GVL has not been reported yet.
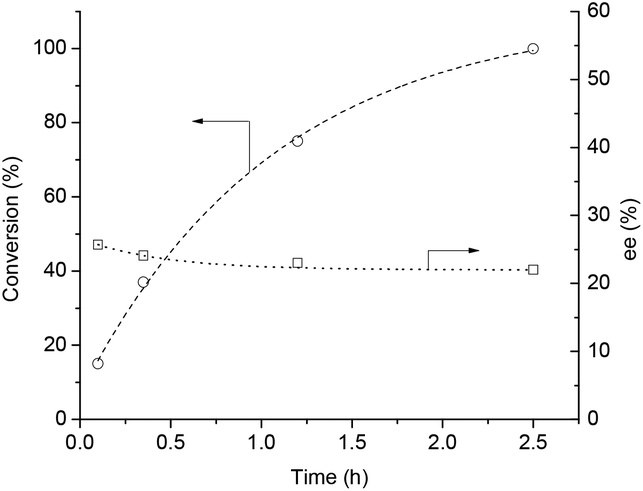 | ||
| Fig. 3 Solvent free asymmetric reduction of LA to optically active GVL. Conditions: [Ru] = 7.6 × 10−4 mol dm−3, [(R)-BINAP] = 7.6 × 10−3 mol dm−3, T = 140 °C, p = 60 bar, RPM = 600. | ||
It was established that in situ generated and preformed complexes can be used for the reduction, however, the latter showed higher activity and selectivity.23d Noyori et al. demonstrated that dialkyl ketones, e.g. 4-phenylbutan-2-one, were reduced with ee = 51%23d by applying an XylBINAP/(S,S)-DPEN-modified Ru catalyst (1) (ESI Fig. S1†). Significantly higher selectivity with the opposite sense of asymmetric induction was achieved for the RuCl2[(S)-XylBINAP][(R,R)-DAIPEN] (2) (ESI Fig. S1†) catalyzed reduction of cyclopropyl ketones.31 Accordingly, we performed the reduction of LA in the presence of 1 and 2. Firstly, in the absence of a catalyst no conversion was detected demonstrating the ineffectiveness of the high-pressure Hastelloy-C reactor. When LA was used as a substrate and a solvent without any additive, all substrates were converted to GVL using 1. However, the enantioselectivity was modest (ee = 11%). Similarly, ee = 13% was achieved when methanol was used as a co-solvent (ESI Table S1,† entries 1 and 2). Importantly, the composition of the reaction mixture was continuously changing from LA to LA/GVL/H2O and finally a GVL/H2O mixture was obtained. When different alcohols were used as co-solvents, either with or without a base, the ee was not affected significantly (ESI Table S1,† entries 4–10). Although, by replacing DPEN with DAIPEN, higher enantioselectivity was proposed,23d however, no further increase could be achieved by us (ESI Table S1,† entry 4).
By using a bidentate phophine-modified Ru catalyst, both the electronic and steric effects of the bis(diarylphosphine) backbone significantly enhanced the reactivity, and improved the stereorecognition.32 Recently, Saito et al. introduced a new ligand family (SEGPHOS) with a smaller dihedral angle representing an outstanding catalytic performance for the reduction of carbonyl compounds.33 By using a substrate/catalyst ratio of 1000, ethyl levulinate was hydrogenated at 50 °C for 20 h, and ethyl 4(R)-hydroxypentanoate was obtained with a yield of 99%.33b The conversion of LA to GVL in methanol, as the preferred solvent,33b was screened and further improved by using various SEGPHOS-based Ru catalysts (Table 1). When a mononuclear (R)-RuCl2[(p-cymene)(SEGPHOS)] precursor was applied, full conversion was achieved with ee = 18% (similar yields were obtained under solvent-free conditions where BINAP-based catalysts were used). When various substituents on the SEGPHOS ligand were used, a small decrease of the ee values was observed (Table 1, entries 1–3). Comparing DPEN and DAIPEN analogues of Noyori's catalyst, no significant change in the ee was detected (Table 1, entries 4 and 5); similar results were observed when an (S)-Ru(OAc)2(SEGPHOS) precursor was applied. However, the use of the (S)-[(RuCl(SEGPHOS))2(μ-Cl)3][NH2(CH3)2] (3) (Fig. 4) precursor resulted in a dramatic increase in the enantioselectivity. The reduction of 9.8 mmol of LA in the presence of 0.004 mmol 3 leads to the complete formation of (S)-GVL with ee = 56% (Table 1, entry 7), that was 4.5 and 2.5 times higher than that obtained by the use of Noyori's catalyst in methanol or a Ru/BINAP catalyst under solvent-free conditions.
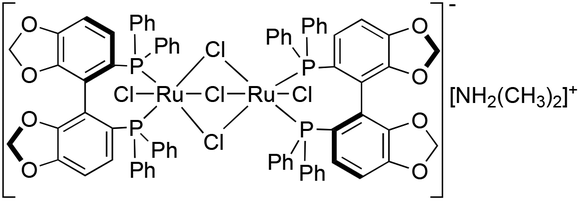 | ||
| Fig. 4 (S)-[(RuCl(SEGPHOS))2(μ-Cl)3][NH2Me2] catalyst (3). | ||
| Entry | Catalyst | Conv. (%) | ee |
|---|---|---|---|
| Reaction conditions: 1 mL (9.8 mmol) LA in 1.4 mL MeOH, T = 140 °C, t = 20 h, p = 60 bar, catalyst: 0.004 mmol, S/C = 2400. | |||
| 1 | (R)-RuCl2[(p-cymene)(SEGPHOS)] | 100 | 18 |
| 2 | (R)-RuCl2[(p-cymene)(DM-SEGPHOS)] | 100 | 9 |
| 3 | (R)-RuCl2[(p-cymene)(DTMB-SEGPHOS)] | 95 | 13 |
| 4 | RuCl2[(S)-(DM-SEGPHOS)][(S,S)-DPEN] | 100 | 16 |
| 5 | RuCl2[(S)-(DM-SEGPHOS)][(S)-DAIPEN] | 100 | 13 |
| 6 | Ru(OAc)2((S)-SEGPHOS) | 100 | 12 |
| 7 | (S)-[(RuCl(SEGPHOS))2(μ-Cl)3][NH2Me2] | 100 | 56 |
Since the asymmetric induction could be strongly affected by the solvent, we screened the conversion of LA (9.8 mmol) to GVL by using various alcohols, methylene chloride and supercritical CO2 at 60 bar H2 and 140 °C. Although, full conversion was obtained in all cases, the enantiomeric excess varied in a wide range depending on the solvent (Fig. 5). Without any added solvent, no chiral induction occurred, and negligible values were obtained when methylene chloride or supercritical carbon-dioxide was used. However, significantly higher enantioselectivities were obtained in alcohols, a protic reaction media, in accord with literature data concerning the reduction of non-aromatic ketones (Saito et al.).33b Hydrogenation of 9.8 mmol LA in 1.4 mL methanol resulted in ee = 56% (Fig. 5). When 9.8 mmol of LA was reduced in 0.7 mL methanol in the presence of 0.004 mmol of 3, the ee decreased to 43%. By using 3 mL methanol, the ee was 53%. Supposedly, methanol acts as a co-solvent and may have an effect on the formation and stability of the catalytically active species. Importantly, the ring closure of 4-hydroxypentanoic acid resulted in the formation of an equimolar amount of water and GVL. Accordingly, the application of a water-free solvent is inefficient; however, the initial water content may have an effect on the ee. When, the reaction was performed in 96% ethanol, a slight decrease of ee (37%) was found.
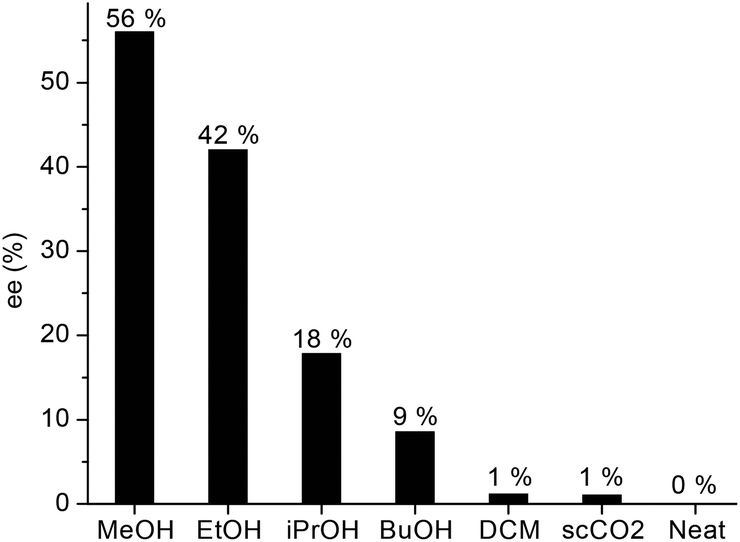 | ||
| Fig. 5 Asymmetric hydrogenation of LA to GVL in different solvents. Conditions: [Ru] = 0.004 mol dm−3, T = 140 °C, p = 60 bar, RPM = 400. | ||
Subsequently, the effect of the reaction temperature on the activity and selectivity was investigated (Fig. 6). The Ru-catalyzed asymmetric reductions are usually performed in a range of 20–40 °C, although, the SEGPHOS-based catalysts operate at a higher (65 °C) temperature. Noteworthily, hydrogenation of LA by a homogeneous Ru catalyst was unsuccessful below 80 °C and minimum 140 °C was necessary to obtain the reduction product.12a Interestingly, ethyl and methyl levulinate were converted to the corresponding alcohols at 30 °C28 and 60 °C,29 respectively. When, 9.8 mmol LA in 1.4 mL MeOH was reduced, negligible conversion (8%) and ee (3%) was obtained at 80 °C and 60 bar H2 for 20 h; and only a slight increase was observed at 120 °C. Although, full conversion was obtained over 130 °C, the maximum ee value was 56% at 140 °C (Scheme 2(a)).
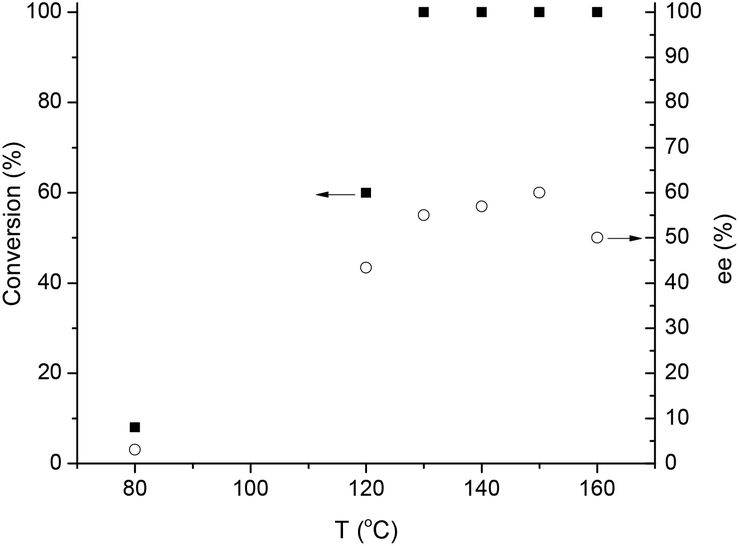 | ||
| Fig. 6 Asymmetric hydrogenation of LA to GVL at different temperatures. Conditions: 9.8 mmol LA in 1.4 mL MeOH, [Ru] = 0.004 mol dm−3, pH2 = 60 bar, RPM = 400. | ||
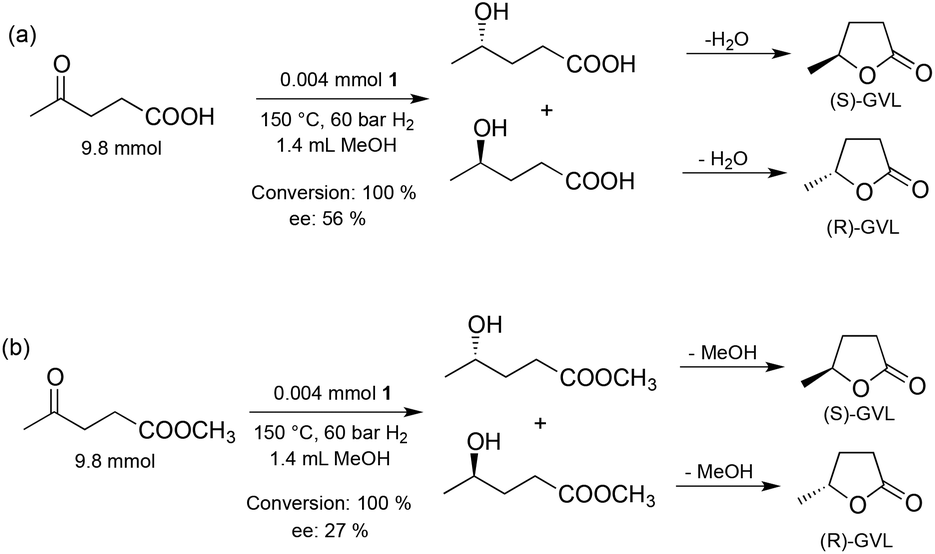 | ||
| Scheme 2 Reduction of LA and methyl levulinate. | ||
Supposedly, the reduction of LA to GVL in the presence of HCl with a Ru-BINAP-HCl system occurs via in situ formation of ethyl levulinate in ethanol. Although no in situ spectroscopic data were included, ethyl levulinate, ethyl 4-hydroxyvalerate and GVL were detected in the product mixture.29a When LA was reduced by catalyst 1, the concentration of methyl levulinate in the final reaction mixture was below the detection limit. For comparison, methyl levulinate (9.8 mmol) in methanol (1.4 mL) was reduced under identical conditions resulting in full conversion and with ee = 27% (cf.Scheme 2(b)) which is significantly lower than the value obtained for LA (56%). In addition, methyl levulinate was reduced at a lower temperature. Although, the in situ equilibrium formation of methyl levulinate from methanol and LA cannot be completely excluded, if LA is hydrogenated to (S)-4-hydroxyvaleric acid it will spontaneously dehydrate to (S)-GVL. Unexpectedly, for the reduction of LA, significant improvement in enantioselectivity was detected by varying the Ru concentration between 0.002–0.016 mol dm−3 (Fig. 7). For example, 9.8 mmol LA was hydrogenated in 1.4 mL of methanol as a co-solvent at [Ru] = 0.016 mol dm−3 at 60 bar and 150 °C, full conversion was obtained with ee = 82% for (S)-GVL. This selectivity fits the ee values obtained for dialkyl ketones, however further increase of the amount of catalyst had no effect on the selectivity.
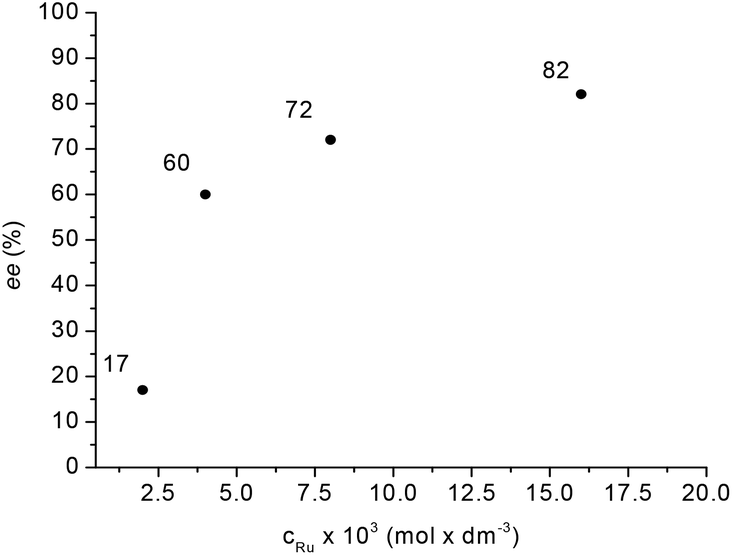 | ||
| Fig. 7 Influence of catalyst concentration on ee. Reaction conditions: 9.8 mmol LA in 1.4 mL MeOH, pH2 = 60 bar, T = 150 °C, RPM = 400. | ||
The use of “real” biomass-based levulinic acid to chiral γ-valerolactone is fundamentally important to show and establish that no “chemical” (product or side product) formed, which could interfere with chiral induction of the catalyst. In order to investigate the catalyst's applicability, firstly D-fructose (4 g, 22.2 mmol) was converted to LA under optimized conditions.8b As a result: 1.65 g of LA was isolated as a light brown, viscous liquid. 1 mL of this product was mixed with 1.4 mL of methanol and subsequently hydrogenated by using 3 at 60 bar H2 and 150 °C. Quantitative formation of GVL was achieved with ee = 78% after 20 h (Scheme 3A). Finally, to demonstrate the conversion of a “real” biomass waste containing wheat straw, rice husk, corn straw, nut and pea-pod was treated as follows: 3 g of dried biomass waste was heated in 2 M H2SO4 at 170 °C for 8 h. After our published workup procedure was performed,8bca. 1 mL of a deep dark brown solution was obtained. After vacuum distillation 560 mg of LA was isolated as a colorless liquid (yield: 18%). It was subsequently reduced under optimized conditions (0.75 mL MeOH, 0.009 mmol 3, 60 bar H2, 150 °C) resulting in 100% conversion of LA with 80% enantioselectivity for (S)-GVL (Scheme 3B). Indeed, GVL with high enantiomeric excess can be produced from biomass-based levulinic acid under optimized conditions.
 | ||
| Scheme 3 Conversion of D-fructose (A) and mixed biomass waste (B) to (S)-GVL. | ||
The reproducibility of the experiments were confirmed by repeating the reduction of LA (9.8 mmol) at 60 bar H2 and 140 °C (Table 1, entry 7). Complete conversion was achieved after 20 h with ee = 55.2%. When LA (9.8 mmol) in 1.4 mL of methanol was reduced by using 3 ([Ru] = 0.016 mol dm−3) under 60 bar of H2 at 150 °C, full conversion with ee = 83% was achieved. After the removal of methanol and H2O by vacuum distillation 712 mg of colorless (S)-GVL was obtained. Isolated yield: 62.4% (ee = 83%).
Experimental
Levulinic acid, catalyst precursors, (S)-GVL, were purchased from Sigma-Aldrich Kft, Budapest, Hungary and used as received. Solvents were obtained from Molar Chemicals Ltd, Budapest, Hungary and used without further purification (ESI†).Hydrogenation reactions were performed in a 25 mL high-pressure Hastelloy-C reactor with magnetic stirring (Parr Inst, IL, USA) equipped with a manometer, a safety relief and a magnetic stirring bar using external heating. In hydrogenation experiments using in situ generated Ru-based catalysts, the high-pressure reactor was charged with 1.14 g (1 mL, 9.8 mmol) levulinic acid, 1.4 mL methanol, 0.62 mg (0.00156 mmol) ruthenium(III)-acetylacetonate and 10 eq. (0.0156 mmol) of the corresponding chiral phosphine ligand. In the case of a pre-prepared Ru-based catalyst, the reactor was charged with 1.14 g (1 mL, 9.8 mmol) levulinic acid, 1.46 mL methanol, and 0.0048 mmol ruthenium-based chiral complex resulting in a colorful solution. In both cases, the reaction mixture was pressurized up to the desired pressure and heated to a given temperature. After completing the reaction, the reactor was cooled down to ambient temperature, and stirring was stopped. The conversion was determined by 1H-NMR spectroscopy by the signals of methyl protons of levulinic acid (δ: 2.11 ppm, s, 3H) and GVL (δ: 1.41 ppm, d, 3H). The NMR measurements were performed on a Bruker-Avance 250 MHz instrument. The enantiomeric excess (ee) was determined on an HP-CHIRAL-20B capillary column (30 m × 0.25 mm × 0.25 μm) with a Finnigan Trace GC Ultra (Thermo Electron Corporation) using H2 as a carrier gas. For the analysis, 10 μL of the reaction mixture was dissolved in 1 mL methanol.
Conclusions
We demonstrated that levulinic acid could be directly converted to optically active (S)-GVL, a proposed chiral platform molecule. In contrast to previously published procedures, no alkyl levulinate was necessary to synthesize optically active GVL. It was revealed that in the presence of a catalyst in situ generated from Ru(III)-acetylacetonate and (S)-BINAP, levulinic acid was converted to (S)-GVL with ee = 26% without adding any solvent and/or additive. By applying an (S)-{[RuCl(SEGPHOS)]2(μ-Cl)3}−[NH2(CH3)2]+ catalyst precursor in methanol, the enantiomeric excess was increased resulting in enantioselectivity of 82%. The conversion of “real” biomass waste to optically active GVL was also demonstrated.Acknowledgements
This work was funded by the Budapest University of Technology and Economics under project number KMR_12-1-2012-0066 and Hungarian National Scientific Research Fund (OTKA–PD 116559). E. Székely and L. T. Mika are grateful for the support of the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. The authors would like to thank Prof. István T. Horváth (City University of Hong Kong) and Prof. József Bakos (University of Pannonia) for helpful discussions.Notes and references
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538 RSC.
- (a) B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339 CrossRef CAS; (b) J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenschwander, W. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227 CrossRef.
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238 RSC.
- J. Q. Bond, J. A. Dumesic, D. M. Alonso, D. Wang and R. M. West, Science, 2010, 327, 1110 CrossRef CAS PubMed.
- H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49 CrossRef CAS.
- (a) D. Fegyverneki, L. Orha, G. Láng and I. T. Horváth, Tetrahedron, 2010, 66, 1078 CrossRef CAS PubMed; (b) A. Strádi, M. Molnár, M. Óvári, G. Dibó, F. U. Richter and L. T. Mika, Green Chem., 2013, 15, 1857 RSC; (c) P. K. Wong, C. Li and L. Stubbs, Patent Appl, WO2012/134397A1, 2012 Search PubMed; (d) M. Chalid, H. J. Heeres and A. A. Broekhuis, J. Appl. Polym. Sci., 2011, 123, 3556 CrossRef PubMed; (e) F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510 CrossRef CAS PubMed; (f) J.-P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479 CrossRef CAS PubMed.
- (a) L. Qui and I. T. Horváth, ACS Catal., 2012, 2, 2247 CrossRef; (b) L. Qi, Y. F. Mui, S. W. Lo, M. Y. Lui, G. R. Akien and I. T. Horváth, ACS Catal., 2014, 4, 1470 CrossRef CAS.
- (a) G. Novodárszki, N. Rétfalvi, G. Dibó, P. Mizsey, E. Cséfalvay and L. T. Mika, RSC Adv., 2014, 4, 2081 RSC; (b) Á. Szabolcs, M. Molnár, G. Dibó and L. T. Mika, Green Chem., 2013, 15, 439 RSC; (c) M. Kang, S. W. Kim, J.-W. Kim, T. H. Kim and J. S. Kim, Renewable Energy, 2013, 54, 173 CrossRef CAS PubMed; (d) K. W. Omari, J. E. Besaw and F. M. Kerton, Green Chem., 2012, 14, 1480 RSC; (e) A. M. R. Galletti, C. Antonetti, E. Ribechini, M. P. Colombini, N. N. o. Di Nasso and E. Bonari, Appl. Energy, 2012, 102, 157 CrossRef PubMed; (f) D. M. Rackemann and W. O. S. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198 CrossRef CAS PubMed and reference therein; (g) G.-T. Jeong and D.-H. Park, Appl. Biochem. Biotechnol., 2010, 161, 41 CrossRef CAS PubMed; (h) L. Deng, J. Li, D.-M. Lai, Y. Fu and Q.-X. Guo, Angew. Chem., Int. Ed., 2009, 48, 6529 CrossRef CAS PubMed; (i) B. Girisuta, L. P. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339 CrossRef CAS.
- (a) J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184 CrossRef CAS PubMed; (b) S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Energy Environ. Sci., 2012, 5, 8199 RSC; (c) D. M. Alonso, J. M. R. Gallo, M. A. Mellmer, S. G. Wettstein and J. A. Dumesic, Catal. Sci. Technol., 2013, 3, 927 RSC.
- (a) S. Tabasso, E. Montoneri, D. Carnaroglio, M. Caporasob and G. Cravotto, Green Chem., 2014, 16, 73 RSC; (b) J. Li, Z. Jiang, L. Hu and C. Hu, ChemSusChem, 2014, 7, 2482 CrossRef CAS PubMed.
- (a) W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657 CrossRef CAS PubMed; (b) J. M. Tukacs, R. V. Jones, F. Darvas, G. Dibó, G. Lezsák and L. T. Mika, RSC Adv., 2013, 3, 16283 RSC.
- (a) J. M. Tukacs, M. Novák, G. Dibó and L. T. Mika, Catal. Sci. Technol., 2014, 4, 2908 RSC; (b) J. M. Tukacs, D. Király, A. Strádi, G. Novodárszki, Z. Eke, G. Dibó, T. Kégl and L. T. Mika, Green Chem., 2012, 14, 2057 RSC; (c) W. Li, J.-H. Xie, H. Lin and Q.-L. Zhou, Green Chem., 2012, 14, 2388 RSC; (d) J. Deng, Y. Wang, T. Pan, Q. Xu, Q.-X. Guo and Y. Fu, ChemSusChem, 2013, 6, 1163 CrossRef CAS PubMed.
- V. Fábos, L. T. Mika and I. T. Horváth, Organometallics, 2014, 33, 181 CrossRef.
- (a) W. G. Jennings and M. R. Sevenants, J. Food Sci., 1964, 16, 252 Search PubMed; (b) M. R. Sevenants and W. G. Jennings, J. Food Sci., 1966, 31, 81 CrossRef CAS PubMed; (c) C. S. Tang and W. G. Jennings, J. Agric. Food Chem., 1968, 16, 252 CrossRef CAS; (d) B. Willhalm, E. Palluy and M. Winter, Helv. Chim. Acta, 1966, 49, 65 CrossRef CAS PubMed; (e) T. Fujimori, R. Kasuga, H. Matsushita, H. Kaneko and M. Noguchi, Agric. Biol. Chem., 1976, 40, 303 CrossRef CAS; (f) U. Ravid, R. M. Silverstein and L. R. Smith, Tetrahedron, 1978, 34, 1449 CrossRef CAS.
- (a) L. Hilterhaus and A. Liese, Building Blocks, in White Biotechnology, Advances in Biochemical Engineering/Biotechnology, ed. R. Ulber and D. Sell, Springer-Verlag, Berlin, Heidelberg, 2007, vol. 105, pp. 133–173 Search PubMed; (b) S. K. Taylor, R. F. Atkinson, E. P. Almli, M. D. Carr, T. J. van Huis and M. R. Whittaker, Tetrahedron: Asymmetry, 1995, 6, 157 CrossRef CAS; (c) T. Kiyoshi, I. Tsuneo and K. Koga, J. Chem. Soc., Chem. Commun., 1979, 652 Search PubMed; (d) A. B. Smith, T. A. Rano, N. Chida, G. A. Sulikowski and J. L. Wood, J. Am. Chem. Soc., 1992, 114, 8008 CrossRef CAS; (e) C. M. Blackwell, A. H. Davidson, S. B. Launchbury, C. N. Lewis, E. M. Morrice, M. M. Reeve, J. A. R. Roffey, A. S. Tipping and R. S. Todd, J. Org. Chem., 1992, 57, 5596 CrossRef CAS; (f) U. David, R. M. Silverstein and L. R. Smith, Tetrahedron, 1978, 34, 1449 CrossRef.
- (a) P. J. Fagan and C. J. Brandenburg (Du Pont), Patent Appl, US20060030719, 2004 Search PubMed; (b) H. G. Gorissen, J.-P. van Hoeck, A. M. Mockel, G. H. Journée, C. Delatour and V. R. Libert, Chirality, 1992, 4, 286 CrossRef CAS PubMed; (c) J. E. Baldwin, R. M. Adlington and S. H. Ramcharitar, Synlett, 1992, 875 CrossRef CAS.
- K. Mori, Tetrahedron, 1975, 31, 3011 CrossRef CAS.
- E. L. Stangeland and T. Sammakia, J. Org. Chem., 2004, 69, 3281 CrossRef PubMed.
- K. Tomioka, T. Ishiguro and K. Koga, Tetrahedron Lett., 1980, 21, 2973 CrossRef CAS.
- J. D. White and J. C. Amedio, J. Org. Chem., 1989, 54, 738 CrossRef.
- (a) C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley VCH, Weinheim, Germany, 3rd edn, 2003 Search PubMed; (b) S. K. Tulashie, H. Lorenz, L. Hilfert, F. T. Edelmann and A. Seidel-Morgenstern, Cryst. Growth Des., 2008, 8, 3408 CrossRef CAS; (c) S. K. Tulashie, H. Lorenz and A. Seidel-Morgenstern, Cryst. Growth Des., 2009, 9, 2387 CrossRef CAS.
- C. Baudequin, D. Bregeon, J. Levillain, F. Guillen, J.-C. Plaquevent and A.-C. Gaumont, Tetrahedron: Asymmetry, 2005, 16, 3921 CrossRef CAS PubMed.
- (a) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS; (b) N. B. Johnson, I. C. Lennon, P. H. Moran and J. A. Ramsden, Acc. Chem. Res., 2007, 40, 1291 CrossRef CAS PubMed; (c) W. Zhang, Y. Chi and X. Zhang, Acc. Chem. Res., 2007, 40, 1278 CrossRef CAS PubMed; (d) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS; (e) H. U. Blaser, Adv. Synth. Catal., 2002, 344, 17 CrossRef CAS.
- Q. Jiang, Y. Jiang, D. Xiao, P. Cao and X. Zhang, Angew. Chem., Int. Ed., 1998, 37, 1100 CrossRef CAS.
- R. Patchett, I. Magpantay, L. Saudan, C. Schotes, A. Mezzetti and F. Santoro, Angew. Chem., Int. Ed., 2013, 52, 10352 CrossRef CAS PubMed.
- A. V. Karnik, S. T. Patil, S. S. Patnekar and A. Semwal, New J. Chem., 2004, 28, 1420 RSC.
- K. Götz, A. Liese, M. Ansorge-Schumacher and L. Hilterhaus, Appl. Microbiol. Biotechnol., 2013, 97, 3865 CrossRef PubMed.
- T. Ohkuma, M. Kitamura and R. Noyori, Tetrahedron Lett., 1990, 31, 5509 CrossRef CAS.
- (a) E. V. Starodubtseva, O. V. Turova, M. G. Vinogradov, L. S. Gorshkova and V. A. Ferapontov, Russ. Chem. Bull., 2005, 54, 2374 CrossRef CAS; (b) O. V. Turova, E. V. Starodubtseva, M. G. Vinogradov and V. A. Ferapontov, J. Mol. Catal. A: Chem., 2009, 311, 61 CrossRef CAS PubMed.
- H. Jacobs, K. Berryman, J. Jones and A. Gopalan, Synth. Commun., 1990, 20, 999 CrossRef CAS PubMed.
- T. Ohkuma, M. Koizumi, H. Doucet, T. Pham, M. Kozawa, K. Murata, E. Katayama, T. Yokozawa, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1998, 120, 13529 CrossRef CAS.
- H. Shimizu, I. Nagasaki and T. Saito, Tetrahedron, 2005, 61, 5407 CrossRef PubMed.
- (a) H. Shimizu, I. Nagasaki, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385 CrossRef CAS PubMed; (b) T. Saito, T. Yokozawa, T. Ishizaki, T. Moroi, N. Sayo, T. Muira and H. Kumobayashi, Adv. Synth. Catal., 2001, 343, 264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01099c |
| This journal is © The Royal Society of Chemistry 2015 |