
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective terminal C–C scission of C5-carbohydrates†
Frits
van der Klis
a,
Linda
Gootjes
a,
Jacco
van Haveren
a,
Daan S.
van Es
*a and
Johannes H.
Bitter
*b
aWageningen UR Food & Biobased Research, Bornse Weilanden 9, 6708WG, Wageningen, The Netherlands. E-mail: daan.vanes@wur.nl; Tel: +31 317-481160
bWageningen University Biobased Chemistry and Technology, Bornse Weilanden 9, 6708WG, Wageningen, The Netherlands. E-mail: harry.bitter@wur.nl; Tel: +31 317-480303
First published on 2nd June 2015
Abstract
The selective catalytic production of C4-tetritols (erythritol and threitol) from C5-sugars is an attractive route for the conversion of non-digestible sugars to C4-building blocks from agro residues. Here we show that an unprecedented high selectivity of 20–25% C4-tertritols can be achieved under mild conditions (138 °C, 6 bar H2, and 24 h) in the aqueous conversion of xylose over a 5 wt% Ru/C catalyst. A mechanistic study revealed that the dominant reaction mechanism for C5-sugar conversion involves a formal decarbonylation step leading to the initial formation of the desired C4-tetritols. Subsequently the formed C4-tetritols undergo further terminal C–C scissions to glycerol and ethylene glycol. Remarkably, potentially competing reactions like internal C–C chain scission (fragmentation) or hydrodeoxygenation (HDO) do not occur to any significant extent under the applied conditions.
Introduction
Decreasing fossil feedstock reserves and political instability, combined with an increasing awareness on global climate change, have sparked a demand for more sustainable alternatives for both energy applications and materials.Carbohydrates are attractive feedstocks since they have numerous applications in food, feed and chemicals production. A prime example of versatile carbohydrates are the C4-polyols erythritol and threitol. They are currently commercially produced by glucose fermentation and are well known as low-calorie sweeteners.1–3
In addition, the C4-polyols can also serve as versatile chemical building blocks, for e.g. coating applications4,5 or the production of (bio)-butadiene.6–9
The production of the C4-polyols from non-edible sugars is however preferred over the use of edible sugars in order to prevent interference with food production.
Since C5-sugars (xylose, arabinose) are non-digestible, and hence do not compete with food, they are interesting alternative starting materials for producing C4-polyols. C5-sugar containing feedstocks are xylan-rich streams such as straw or wood residues, or arabinan-rich streams such as citrus- or sugar beet pulp10 (Scheme 1).
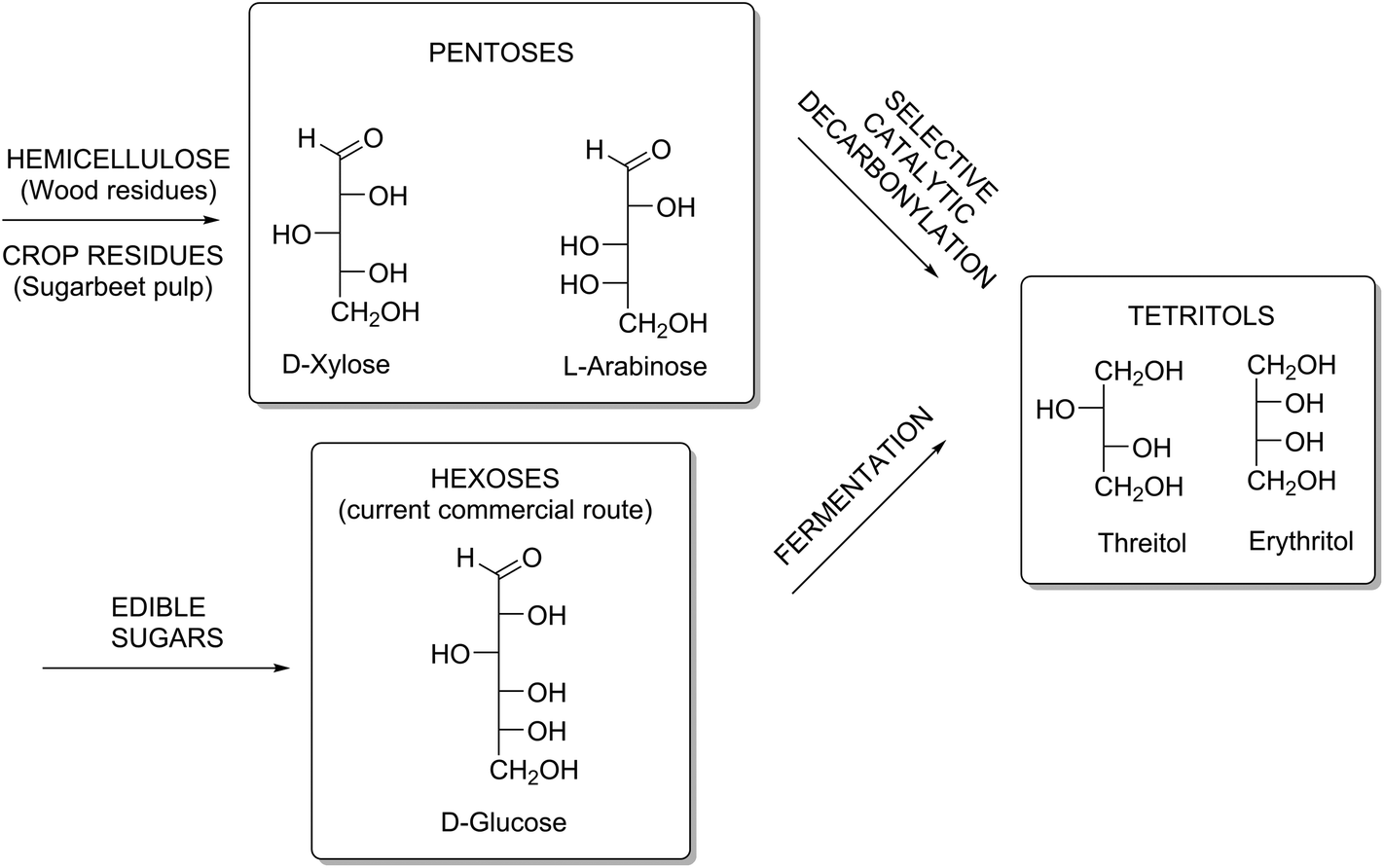 | ||
| Scheme 1 Desired catalytic route from non-edible agricultural residues towards C4-tetritols, compared to the current commercial route based on the fermentation of edible sugars. | ||
In order to achieve an efficient route from C5-sugars to tetritols, selective terminal C–C scission is required. In principle there are two ways to achieve this: a decarbonylation reaction (mainly reported for homogeneous catalysts), or terminal C–C scission in the presence of hydrogen. The latter is also called a hydrogenolysis reaction, and is mainly reported for heterogeneous catalysts. However, in general the latter reaction is not selective for the terminal position.
The only reported catalytic systems with a high selectivity (70–80% isolated yield) for sugar decarbonylation are based on homogeneous Ru- and Rh-catalysts.11,12 Unfortunately, these homogeneous catalysts require complex (high boiling) solvent mixtures in order to dissolve both the sugars and the catalysts. Air and moisture sensitivity are unfavourable when working with biomass. In addition, product separation from the catalyst is cumbersome and often energy intensive. In order to overcome these issues, the C5-sugar conversion should preferably be carried out under aqueous conditions (which is nature's carbohydrate solvent) using a heterogeneous catalyst.
The aqueous hydrogenolysis of carbohydrates using heterogeneous catalysts is a challenging topic. A wide variety of reactions are possible i.e., (de)hydrogenation, isomerisation, retro-aldol, retro-Claisen and decarbonylation reactions. In order to understand the potential influence of these reactions on the product distribution these reactions will be briefly discussed first.
As an example of these five different reaction pathways the conversion of xylose is shown in Scheme S1† ((de)hydrogenation and isomerization) and Scheme 2 (retro-aldol, retro-Claisen and decarbonylation). The reactions of Scheme S1† do not result in chain scission, but the reactions described in Scheme 2 all do.
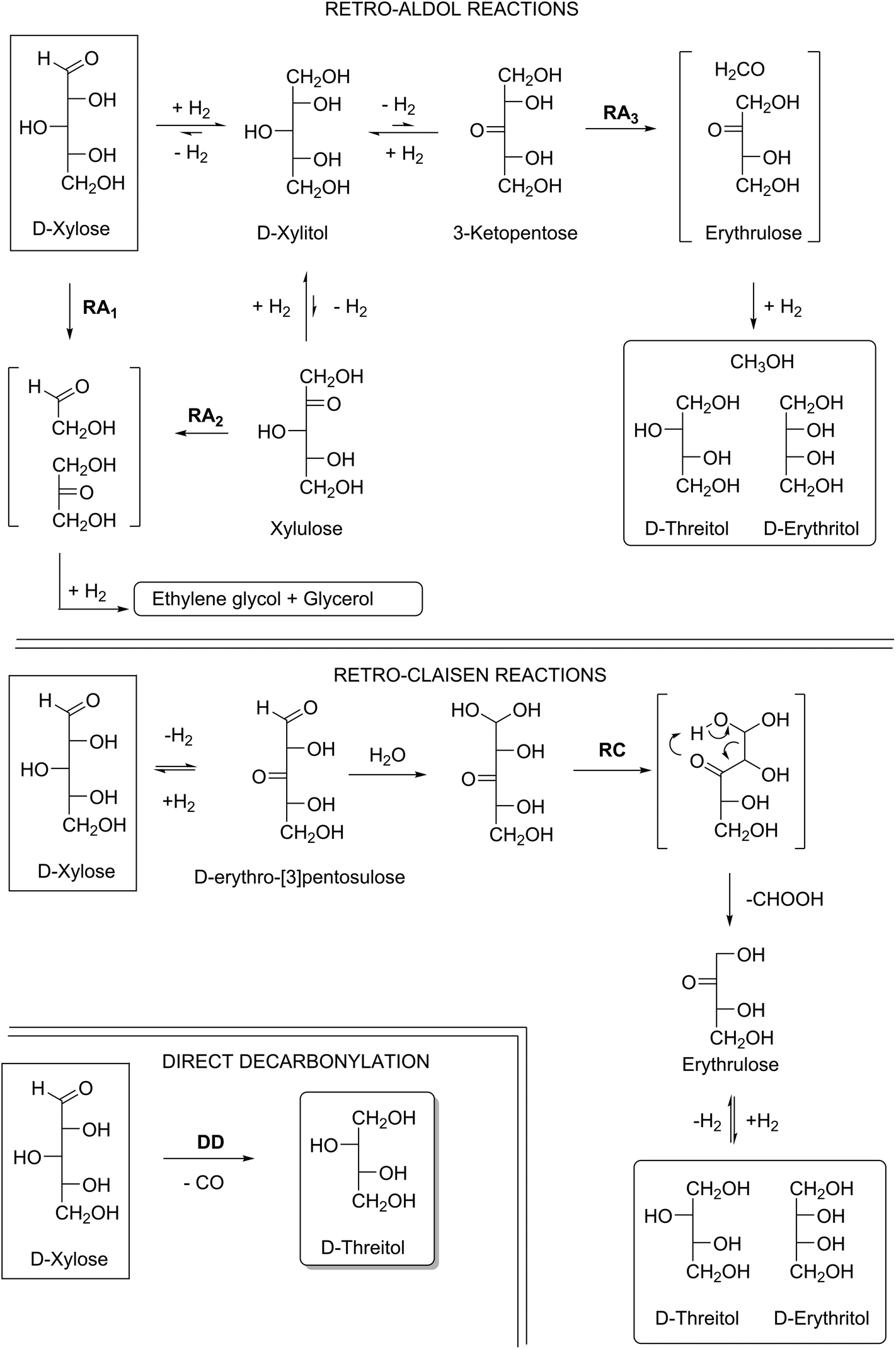 | ||
| Scheme 2 Possible chain scission mechanisms of xylose initiated by hydrogenation and dehydrogenation reactions: Retro-Aldol (RA) reactions, Retro-Claisen (RC) reactions and Direct Decarbonylation (DD). | ||
The retro-aldol reaction (RA) requires a carbonyl functionality on one of the carbons in the carbohydrate chain since the reaction proceeds via enolates. The use of base enhances the enolisation, and therefore bases like Ca(OH)2 are often applied in retro-aldol reactions. The other typical reaction parameters are temperatures between 200–240 °C and hydrogen pressures around 60–100 bar.13–16 Since the parent xylose already has an aldehyde functionality, it can give the retro-aldol products glycolaldehyde and dihydroxyacetone according to route RA1. These products, under the applied hydrogenation conditions, will give ethylene glycol (C2) and glycerol (C3) as the final products. Carbonyl groups can also be generated on all other carbons in the chain: dehydrogenation at carbons 1, 2, 4 and 5 of xylose can lead to the formation of C2 and C3 products via retro-aldol reactions RA1 and RA2 and (de)hydrogenation.
Only dehydrogenation at C3 leads to the desired tetritol products viaRA3.13 The RA-pathway to C4 involves erytrulose as an intermediate, a compound with a pro-chiral carbonyl on C2. Therefore two products can be formed after hydrogenation i.e., erythritol and threitol.
The retro-Claisen (RC) pathway involves the formation of a beta-keto aldehyde (i.e.D-erythro-[3]pentulose), via dehydrogenation of the aldose at position 3, followed by hydration of the aldehyde and subsequent loss of formic acid via a six membered ring transition state.17 This mechanism also leads to the desired tetritols and formic acid as a side product.
The last pathway described in Scheme 2 is the direct decarbonylation (DD) of xylose, leading to the formation of carbon monoxide and threitol. This mechanism is dominant in the previously mentioned homogeneous catalyst systems (reflux at 130–162 °C under inert atmosphere).11,12 However, this reaction is not well studied for heterogeneous catalysts, and conditions under which this reaction can occur are therefore the subject of this investigation. A summary of typical reaction conditions for the 3 different pathways is given in Table S1.†
For the selective production of C4-tetritols from C5-sugars, it is clear that of all these reactions, at least retro-aldol reactions should be avoided: they give rise to the formation of C2 and C3 fragments via four different pathways, while only one pathway leads to the desired C4-products. The formation C2 and C3 products is not only a matter of statistics: quantum chemical calculations clearly show a preference for other products than C4.18,19 Thus, conditions favouring retro-aldol reactions are applied when ethylene glycol and propylene glycol are the desired products. The formation of these products is non-selective and occurs at high temperatures (200–240 °C), H2-pressures (60–100 bar H2) and alkaline conditions.17,20,21 Similar conditions can also be applied to substrates like cellulose,22–24 glucose25 or sorbitol,13,14 which again mainly results in internal C–C scission.
Despite the large number of reports on carbohydrate C–C scission chemistry using heterogeneous catalysts for hydrogenolysis, little attention has been paid to selective terminal C–C scission. In one of the few reports, Deutsch et al.26 studied the conversion of various carbohydrates over (sulphur modified) ruthenium on carbon catalysts. By investigating the order of appearance of the products, these authors demonstrated that terminal C–C scission is dominated by a decarbonylation mechanism. They also found that retro-aldol reactions were not significantly contributing to terminal C–C scission. However, in their study typical retro-aldol conditions were applied: high temperatures (205–240 °C) and hydrogen pressure (100 bar), in combination with alkaline conditions. As a result, severe hydrogenolysis did occur all over the chain (instead of selectively at the terminal position) and the reported tetritol selectivities (at 20–60% conversion) were therefore rather low (1–4%).
Fabre et al.27,28 investigated a completely different reaction, namely the reduction of arabinonic acid to arabitol. For that purpose, they screened a set of carbon supported catalysts (Pt, Pd, Rh and Ru) and ruthenium on other supports (TiO2 and HY). Although production of tetritols was not the aim of the investigation, it was observed in small amounts i.e., 3% for Rh/C and over 4% for Ru/C. The authors showed that all ruthenium catalysts did form small amounts of tetritols (ca. 1%) while other metals did not. The experiments were conducted between 80–140 °C and 100 bar H2, with the highest tetritol formation at 140 °C. Since C2 and C3 products were not observed, it was concluded that retro-aldol reactions did not occur. This is probably due to the low reaction temperatures.
In a paper by Sun et al.,29 the hydrogenolysis of xylitol was optimized for the production of ethylene glycol and propylene glycol (conditions: 160–240 °C, 0–100 bar H2). Similar to the work of Fabre,27,28 they studied various carbon supported catalysts (Pt, Pd, Rh and Ru) and ruthenium on other supports (in this study TiO2, Al2O3, ZrO2 and Mg2AlOx). Out of a broad scope of catalysts, only the ruthenium catalysts showed tetritol formation (up to 7% at 200 °C and 40 bar H2 for Ru/Mg2AlOx).
Based on the results of Deutsch,26 Fabre27,28 and Sun,29 it was decided to use Ru/C (5 wt% Ru/C, Escat 4401) for our study. Here the aim is to investigate the conditions under which selective terminal C–C scission (decarbonylation) can occur and to maximize the formation of tetritols.
As described earlier it can be considered that selective C–C scission at the terminal position can only be achieved when retro-aldol reactions are suppressed. This requires low reaction temperatures (<180 °C), hydrogen pressures (<60 bar H2) and avoiding the use of a base.
Independent of the mechanism (either direct decarbonylation or retro-Claisen), the formal decarbonylation has to occur at the terminal aldehyde. It is therefore preferred to start directly from the aldose sugars (xylose and arabinose as commercial available feedstock), instead of using the corresponding alditols.
Results and discussion
Design of experiment (DOE)
A Design of Experiment (DOE) was performed in order to find the conditions at which terminal C–C scission occurs. As discussed earlier, temperature and hydrogen pressure are expected to be key for steering the selectivity, and therefore these variables were investigated.27–29 Details on the study can be found in the ESI.†It was found that the reaction mixtures contained only C5–C2 polyols and no other deoxygenated species were formed (see Fig. S1†). The DOE showed that, in agreement with our hypothesis, higher C4 yields can indeed be achieved when applying lower hydrogen pressures and temperatures (optimal conditions: 138 °C and 6 bar H2). The maximum calculated yield (24% C4) is, to the best of our knowledge, the highest reported tetritol yield using a heterogeneous catalyst system.
Product formation over time
The experiments in the DOE were performed for 24 h and full conversion of the starting xylose was observed. To gain more insight in the reaction path, product formation was followed over time.Fig. 1 shows the mole fraction of the identified compounds as a function of time during the conversion of xylose. The total mass balance is also shown in the graph. Table 1 gives an overview of all components, mass balance and C4 selectivity as function of time.
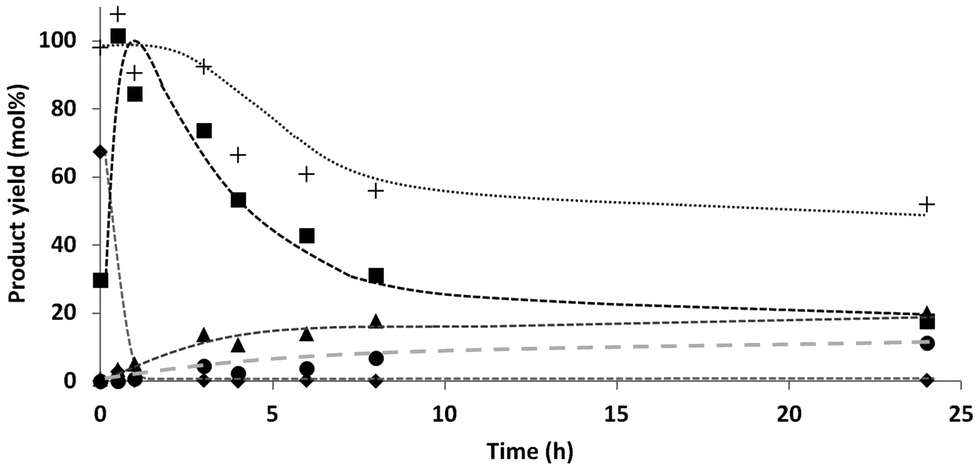 | ||
| Fig. 1 D-Xylose conversion over time; ◆ xylose, ■ C5 alditols, ▲ C4 alditols, ● C3 (glycerol), +total mass balance. Reaction conditions.: D-Xylose (1.00 g), 5 wt% Ru/C (2.2 mol% Ru relative to xylose), deoxygenated demineralized water (25 mL), 10 bar initial H2 pressure, 140 °C. | ||
| Entry | Time (h) | Conv.b (mol%) | C5c | C4c | C3c | C2c | MB.d (mol%) | C4 Sel.e (%) |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions (75 mL pressure reactor): D-Xylose (1.00 g, 6.7 mmol), 5% Ru/C (2.2 mol% metal relative to xylose), deoxygenated demineralized water (25 mL), 10 bar initial H2 pressure, 140 °C. b Conversion of starting material determined by GC-FID after acetylation. c Total amount of product fraction in mol%, C5 and C4 products are the sum of stereoisomers. d Mass balance, sum of starting material and C5–C2 polyols (mass balance exceeding 100% is due to experimental error). e C4 fraction/sum of C4–C2 polyols × mass balance (%) (selectivity exceeding 100% is due to experimental error). | ||||||||
| 1 | 0 | 33 | 30 | 1 | 0 | 0 | 98 | 98 |
| 2 | 0.5 | 97 | 102 | 3 | 0 | 0 | 108 | 108 |
| 3 | 1 | 100 | 84 | 5 | 1 | 0 | 90 | 79 |
| 4 | 3 | 100 | 74 | 14 | 5 | 0 | 92 | 70 |
| 5 | 4 | 100 | 53 | 11 | 2 | 0 | 66 | 54 |
| 6 | 6 | 100 | 43 | 14 | 4 | 0 | 61 | 48 |
| 7 | 8 | 100 | 31 | 18 | 7 | 0 | 56 | 40 |
| 8 | 24 | 100 | 18 | 20 | 11 | 3 | 52 | 31 |
Already at t = 0 part of the xylose was converted to C5-alditol. This indicates that hydrogenation already occurred during the initial heating of the reactor. Within 1 h all xylose was converted and the C5-alditols reached a maximum yield. Subsequently the C4 concentration increased while the C5 concentration decreased over time. However, from 8–24 h, no significant changes in total C4 yield were observed whereas the C5 selectivity steadily decreased. C3 formation was not observed at the start of the reaction (<1 h), but slowly increased from 1% after 1 h reaction time to 11% after 24 h. At this time also a small amount (2%) ethylene glycol was observed.
These results indicate a continuous selective chain scission process, going from pentitols, to tetritols, glycerol and ethylene glycol. Such a stepwise mechanism (sequential reaction) results in the slow conversion of all alditols into the next lower homologues. As long as C5 is present, the desired C4-products can be formed (rate determining step). However, since the initially formed C4 are further converted into C3 and C2, it is difficult to improve the C4 yield at high substrate conversion, hence the moderate “optimal” yields in our DOE.
From Table 1 is can be seen that high selectivities towards C4-products can be obtained at low conversion, while obtaining an almost complete mass balance. This offers opportunities to perform the reaction at low conversion and separate the C5 and C4 components by means of industrial chromatographic methods.30–32 The remaining C5-fraction can be re-used as a starting material for the production of C4.
These results support the hypothesis that very selective terminal C–C scission can indeed occur under mild conditions, when unselective retro-aldol reactions are minimized. Furthermore the absence of significant amounts of hydrodeoxygenation (HDO) products such as C5-tetra/tri/diols or C4-tri/diols is another reason for the high selectivity (see ESI†).
Influence of H2 pressure and gas phase analysis
A decrease in the H2 pressure over time was observed during the reaction, and especially in the beginning of the reaction. The activity measurements as function of time (Fig. 1) showed initially a fast conversion of xylose to xylitol, which explains the fast initial hydrogen consumption. However, also after this initial step, still some hydrogen consumption was observed, although to a much lesser extent (see discussion on gas phase products vide infra).From the results of the DOE it is clear that the hydrogen pressure is of great influence on the reaction steps. Based on the hydrogen consumption and subsequent pressure drop, a change in the kinetics of the reaction steps was expected. Therefore a reaction under constant pressure was performed in order to compare these results to the previous results under non-continuous pressure. The product distribution was monitored over time (Fig. 2), corresponding with the data shown in Table 2, entries 11–17.
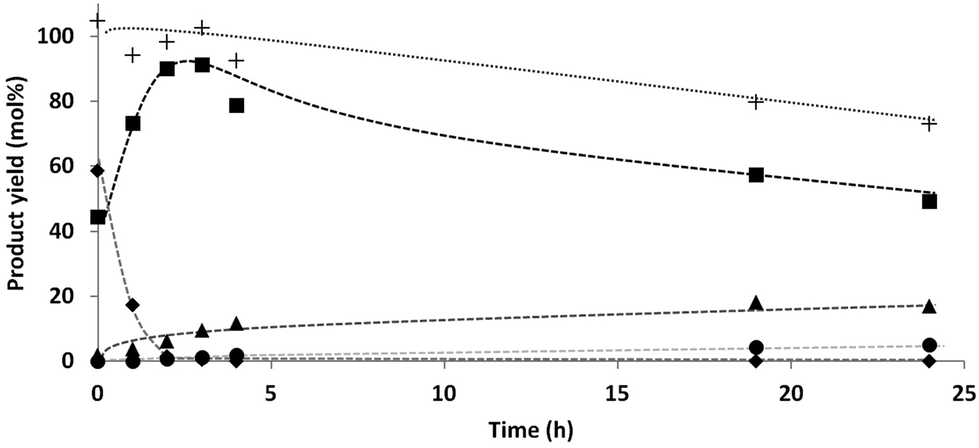 | ||
| Fig. 2 D-Xylose conversion over time: ◆ xylose, ■ C5 alditols, ▲ C4 alditols, ● C3 (glycerol), +total mass balance. Conditions: D-Xylose (16.0 g), 5% Ru/C (2.2 mol% Ru relative to xylose), deoxygenated demineralized water (400 mL), 10 bar continuous H2 pressure, 140 °C. | ||
| Entry | Substrate | Time (h) | Cat. (Mol%) | Conv.c (mol%) | C5 Adod (rel.)e | C5 Adod (abs.)f | C5 Arad (rel.)e | C5 Arad (abs.)f | C5 Xyld (rel.)e | C5 Xyld (abs.)f | C4 Eryd (rel.)e | C4 Eryd (abs.)f | C4 Thrd (rel.)e | C4 Thrd (abs.)f | C5 (total)g | C4 (total)h | C3 (total)h | C2 (total)h | Mass balancei (Mol%) | C4 Select.h (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions (75 mL pressure reactor): Substrate (1.00 g), 5% Ru/C (2.2 mol% Ru relative to substrate), 25 mL deoxygenated demineralized water, 10 bar H2 (initial pressure) 140 °C, 24 h. b Reaction conditions (600 mL pressure reactor): Substrate (16.0 g), 5% Ru/C (2.2 mol% Ru relative to xylose), deoxygenated demineralized water (400 mL), 10 bar continuous H2 pressure, 140 °C, 0–24 h. c Conversion of starting material determined by GC-FID after acetylation. d Ado = Adonitol; Ara = Arabitol; Xyl = Xylitol; Ery = Erythritol; Thr = Threitol. e Relative ratio of polyol (sugar alcohol) product compared to the other isomers. f Absolute amount of polyol (sugar alcohol) product in mol% relative to the starting material. g Total amount of product fraction in mol%, C5 and C4 products are the sum of stereoisomers. h Sum of starting material and C5–C2 polyols (mass balances exceeding 100% are due to experimental errors). i C4 fraction/sum of C4–C2 polyols × mass balance (%) (selectivity exceeding 100% is due to experimental errors). | ||||||||||||||||||||
| 1 | D-Xylosea | 24 | 2.3 | 100 | 17 | 3 | 53 | 9 | 30 | 5 | 52 | 10 | 49 | 10 | 18 | 20 | 11 | 3 | 52 | 31 |
| 2 | D-Xylitola | 24 | 2.3 | 81 | 13 | 6 | 48 | 21 | 39 | 17 | 40 | 9 | 60 | 13 | 44 | 21 | 9 | 3 | 77 | 49 |
| 3 | L-Arabinosea | 24 | 2.3 | 100 | 18 | 4 | 51 | 11 | 31 | 7 | 50 | 8 | 50 | 8 | 21 | 16 | 10 | 1 | 48 | 28 |
| 4 | L-Arabitola | 24 | 2.3 | 94 | 18 | 2 | 51 | 7 | 31 | 4 | 51 | 8 | 49 | 8 | 14 | 16 | 12 | 1 | 43 | 24 |
| 5 | D-Adonitola | 24 | 2.3 | 96 | 18 | 4 | 53 | 10 | 29 | 6 | 47 | 9 | 53 | 11 | 20 | 20 | 13 | 3 | 55 | 31 |
| 6 | Erythrulosea | 24 | 1.8 | 100 | — | — | — | — | — | — | 49 | 8 | 51 | 8 | — | 16 | 11 | 1 | 29 | — |
| 7 | Erythritola | 24 | 1.8 | 80 | — | — | — | — | — | — | 46 | 18 | 54 | 21 | — | 39 | 22 | 4 | 65 | — |
| 8 | D,L-Threitola | 24 | 1.8 | 0 | — | — | — | — | — | — | 8 | 8 | 92 | 98 | — | 107 | 1 | 0 | 107 | — |
| 9 | Glycerola | 24 | 1.4 | 74 | — | — | — | — | — | — | — | — | — | — | — | — | 26 | 6 | 32 | — |
| 10 | Ethylene glycola | 24 | 0.9 | 76 | — | — | — | — | — | — | — | — | — | — | — | — | — | 24 | 24 | — |
| 11 | D-Xyloseb | 0 | 2.3 | 42 | 0 | 0 | 1 | 0 | 99 | 44 | 0 | 0 | 100 | 2 | 44 | 2 | 0 | 0 | 105 | 105 |
| 12 | D-Xyloseb | 1 | 2.3 | 83 | 0 | 0 | 12 | 9 | 88 | 64 | 0 | 0 | 100 | 4 | 73 | 4 | 0 | 0 | 94 | 94 |
| 13 | D-Xyloseb | 2 | 2.3 | 99 | 1 | 1 | 9 | 8 | 91 | 82 | 8 | 1 | 93 | 6 | 90 | 6 | 1 | 0 | 98 | 84 |
| 14 | D-Xyloseb | 3 | 2.3 | 100 | 3 | 2 | 21 | 20 | 76 | 69 | 22 | 2 | 78 | 8 | 91 | 10 | 1 | 0 | 103 | 94 |
| 15 | D-Xyloseb | 4 | 2.3 | 100 | 7 | 5 | 32 | 25 | 62 | 49 | 26 | 3 | 74 | 9 | 79 | 12 | 2 | 0 | 93 | 80 |
| 16 | D-Xyloseb | 19 | 2.3 | 100 | 9 | 5 | 42 | 24 | 50 | 28 | 35 | 6 | 65 | 12 | 57 | 18 | 4 | 0 | 80 | 65 |
| 17 | D-Xyloseb | 24 | 2.3 | 100 | 9 | 4 | 43 | 21 | 48 | 24 | 35 | 6 | 65 | 11 | 49 | 17 | 5 | 2 | 73 | 52 |
| 18 | L-Arabinoseb | 0 | 2.3 | 44 | 0 | 0 | 100 | 34 | 0 | 0 | 99 | 1 | 1 | 0 | 34 | 1 | 1 | 0 | 91 | 46 |
| 19 | L-Arabinoseb | 1 | 2.3 | 81 | 1 | 1 | 99 | 49 | 0 | 0 | 97 | 2 | 4 | 0 | 49 | 3 | 1 | 0 | 72 | 54 |
| 20 | L-Arabinoseb | 2 | 2.3 | 99 | 9 | 7 | 91 | 65 | 0 | 0 | 97 | 4 | 3 | 0 | 72 | 4 | 1 | 1 | 79 | 53 |
| 21 | L-Arabinoseb | 3 | 2.3 | 99 | 17 | 17 | 78 | 80 | 5 | 5 | 69 | 7 | 31 | 3 | 102 | 10 | 2 | 1 | 115 | 88 |
| 22 | L-Arabinoseb | 5 | 2.3 | 100 | 16 | 11 | 70 | 49 | 14 | 9 | 54 | 7 | 46 | 6 | 69 | 13 | 4 | 1 | 86 | 62 |
| 23 | L-Arabinoseb | 24 | 2.3 | 100 | 15 | 7 | 66 | 30 | 19 | 9 | 53 | 8 | 47 | 8 | 45 | 16 | 6 | 2 | 68 | 45 |
Surprisingly, the C4 selectivities at short reaction times (0–3 h) were in accordance with the previous results. The C4 yield reached a maximum around 20%. The final mass balance was however better. A possible explanation might be that the higher H2 concentration leads to a lower concentration of aldehyde functionalities and therefore lower decarbonylation rates.
Fig. 2 shows that the mass balance steadily decreased over time. In an attempt to close the mass balance and to determine the final products formed from the C3 and C2 fraction, the water phase was analyzed by GC (before evaporation of the water) in order to check for the presence of small (volatile) compounds (e.g. formic acid, methanol, ethanol, acetic acid, 1-propanol and 2-propanol). This was done for the experiment shown in Table 2, entry 8; (xylose, 10 bar H2, 140 °C, 24 h). Although MeOH, EtOH, 1-PrOH and 2-PrOH were present, the amounts were too low (≪1%) to have a significant contribution on the mass balance.
Gas phase analysis showed the presence of the gasses CO2, CH4, C2H6 and C3H8 in a relative ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :79:14:6. Carbon monoxide was not detected. The pH of the reaction mixture dropped from pH 7 before reaction to pH 3 after reaction, which might be explained by dissolved CO2 in the aqueous phase. Furthermore, the relatively large concentration of methane is not uncommon for Ru-catalyzed hydrogenolysis reactions. Maris et al.33 showed that this can result from the hydrogenation of formaldehyde and methanol.
:79:14:6. Carbon monoxide was not detected. The pH of the reaction mixture dropped from pH 7 before reaction to pH 3 after reaction, which might be explained by dissolved CO2 in the aqueous phase. Furthermore, the relatively large concentration of methane is not uncommon for Ru-catalyzed hydrogenolysis reactions. Maris et al.33 showed that this can result from the hydrogenation of formaldehyde and methanol.
Although for the xylose conversion over Ru/C oxidized products (ketones) and HDO products were only observed in trace amounts, apparently the smaller C3 and C2 molecules undergo these types of transformations, resulting in the formation of short hydrocarbons.17
Reaction mechanism
The data shown above are a strong indication that, under these specific mild conditions (Ru/C, 140 °C, 10 bar H2), the reaction follows a terminal C–C scission pathway. Analogous to the work of Deutsch26 it was decided to further investigate the order of formation of the various epimers, since this will provide valuable information for possible reaction mechanisms.For this purpose, 10 different starting materials and intermediates were exposed to the standard reaction conditions (initial H2 pressure 10 bar, 140 °C), and the product distribution and mass balance after 24 h were analysed (Table 2). Furthermore, product formation over time for the conversion of both xylose (reaction from Fig. 2) and arabinose under constant pressure (10 bar H2) at 140 °C were compared.
Isomerisation reactions
A comparison of xylose (Table 2, entries 11–17) and arabinose (Table 2, entries 18–23) showed that in both cases initially the respective alditol was formed via hydrogenation of the aldose. Over the course of the reaction all other epimers were observed, indicating isomerisation reactions as discussed in Scheme S1 (ESI†).A difference in the order of appearance of the epimerisation products was observed for both pentoses. Xylitol was first mainly converted into arabitol (Table 2, entry 12) and subsequently into adonitol (Table 2, entries 14–17). This can on one hand be simply rationalised based on statistics; epimerisation at both C2 and C4 of xylitol yields arabitol, while only epimerisation at C3 can yield adonitol. However, hydroxyl group orientation also plays a role in the reaction rate, as is known from literature, e.g. in dehydrogenation reactions.34
The latter is apparent from the behaviour of arabitol, which was first converted into adonitol (C2 inversion, Table 2, entries 19–20) and subsequently into xylitol (C4 inversion, Table 2, entries 21–22). Independent of the stereochemistry of the starting C5-aldose or alditol, the final relative ratio of C5-pentitols and C4-tetritols remained the same: about 17% xylitol, 52% arabitol and 31% adonitol was found in all cases after 24 h (Table 2, entries 1–5) and the ratio of erythritol to threitol was always approx. 1:1.
The mixture of isomers indicates that the catalyst is highly active in the isomerisation of the sugar alcohols. This is also observed by the fact that C4 selectivities are highly comparable for arabinose and xylose (16–21 mol% yield) and therefore independent of the initial stereochemistry.
The equilibrium depends however on the applied conditions, as we have observed different ratio's at various temperatures and pressures (data of experimental design not presented, but also clear from the results of the xylose conversion, constant pressure vs. initial (variable) pressure: Table 2, entries 1 and 17, and for arabinose: entries 3 and 23).
Erythrulose and erythritol underwent isomerisation to threitol (Table 2, entries 6–8) while this was observed only to a small extent for (recrystallized) threitol (Table 2, entries 6–8). The lack of conversion of threitol was probably caused by deactivation of the catalyst due to impurities present in the starting material, since the commercial sample (before recrystallization) showed no conversion at all. However, Deutsch et al.26 also found that in their system, the reaction rate of threitol was lower compared to erythreitol. Since we applied much milder conditions in order to achieve selectivity for terminal C–C scission, our reaction rates in general are much lower, explaining the bigger difference in our system between erythritol and threitol.
Chain scission reactions
The initial selectivity towards the two C4-tetritols was different for xylose (∼100%, Table 2, entries 11 and 12) and arabinose (∼50%, Table 2, entries 18 and 19). In the case of xylose, first threitol was observed as secondary product (Table 2, entries 11 and 12), followed eventually by erythritol (Table 2, entries 16 and 17). Erythritol can however also be formed from arabitol and adonitol, which were formed via epimerisation of the initially formed xylitol over the course of the reaction, or via isomerisation of the initially formed threitol. In the case of arabinose the initial C4-product was erythritol (Table 2, entries 18 and 19), followed by threitol (Table 2, entries 21–23).Also the lower polyols undergo further conversion to the shorter chain products. Since all reactions in Table 2, entries 1–10 were performed with 1 g substrate, the catalyst to substrate ratio decreased with the molecular weight of the substrates, yet the conversion was much higher. The apparently higher reaction rate of the C2 and C3 components compared to the C4 and C5-polyols explains why the amount of C3 and C2 remained relatively low over the course of the reaction (Fig. 2 and Table 2, entries 11–23). Based on these observations the reaction pathway of Scheme 3 is proposed.
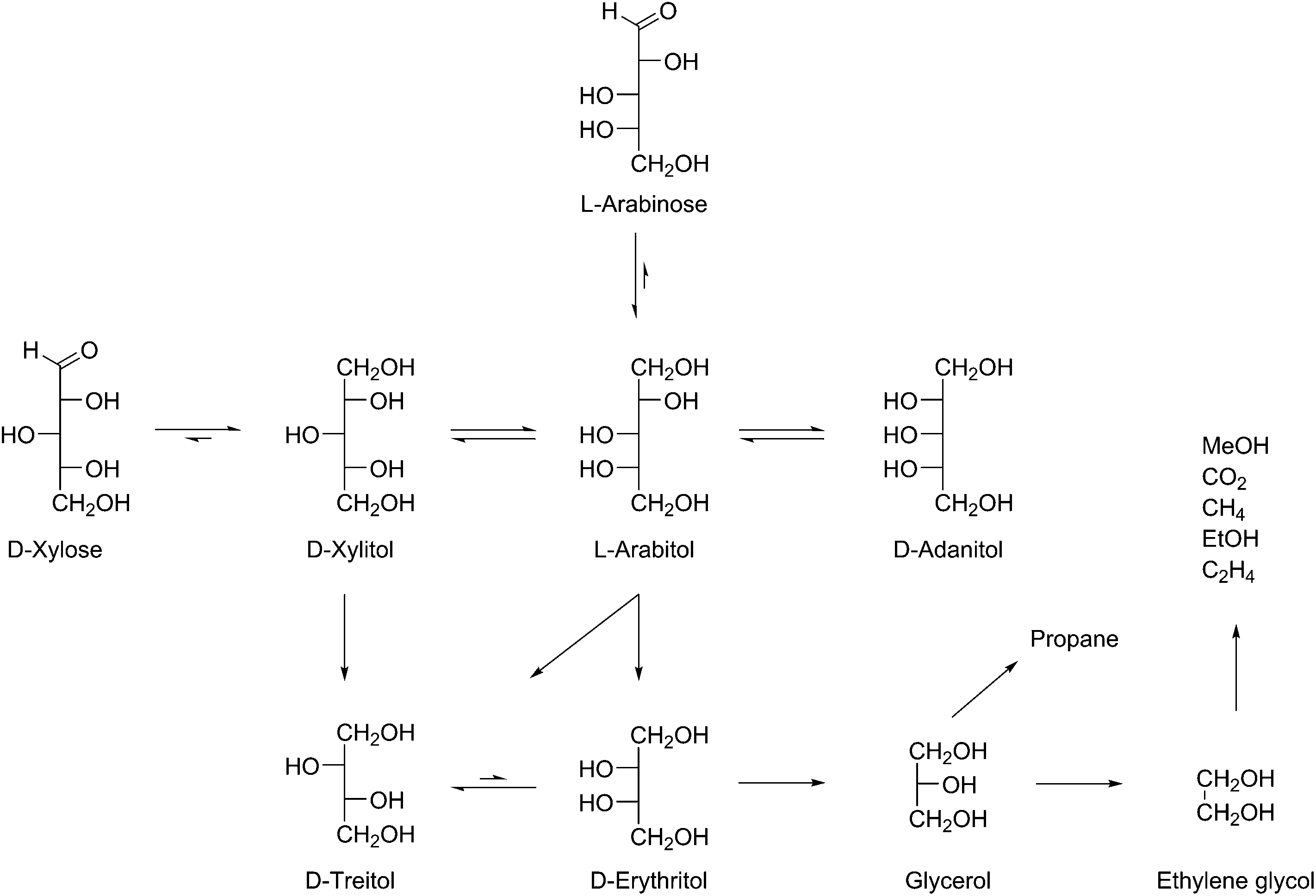 | ||
| Scheme 3 Proposed reaction pathways for the conversion of aldoses into alditols. | ||
From the proposed reaction pathway it is suggested that predominantly consecutive formal decarbonylation occurs. According to the previously discussed literature (at least) three fragmentation mechanisms are suggested to occur with carbohydrates; i.e. the retro-aldol mechanism, the retro-Claisen mechanism and a direct decarbonylation mechanism (Scheme 2).
Based on the initial exclusive formation of C4 products (and the absence of C3 and C2-products in the beginning of the reaction), we can in agreement with Deutsch26 exclude the retro-aldol reaction. As described earlier, this mechanism will not have a preference for the terminal positions.18,19 Most importantly, a retro-aldol mechanism fails to explain why starting from xylose, threitol is the first product to appear (Table 2, entries 11 and 12) and starting from arabinose, erythritol is the initial product (Table 2, entries 18 and 19). The intermediate tetrulose products have a carbonyl group at the C2 position, which after hydrogenation should give both erythritol and threitol independent of the starting aldose (see RA mechanism in Scheme 2).
The alternative retro-Claisen mechanism could however explain the selectivity for terminal C–C scission (see RC mechanism in Scheme 2). The retro-Claisen mechanism requires the presence of 2 carbonyl groups, and one would expect tetritol formation to occur immediately from t = 0, when the presence of aldoses is still high. Since a fast reduction of the aldoses to the corresponding alditols was observed before C4 formation started, this is in contrast to the experimental results. The observed order of formation of the C4-products (first threitol from xylose, Table 2, entries 11 and 12 and erythritol from arabinose, Table 2, entries 18 and 19) is also not in line with this mechanism: since in the retro-Claisen reaction tetrulose is formed as an intermediate, this does not provide an explanation for the initial retention of the aldose configuration.
A simpler explanation for the observed step-wise dehomologation mechanism is the direct decarbonylation of aldoses, comparable to routes known for homogeneous Ru- and Rh-catalysts.12,35–37 More detailed research is necessary to elucidate the exact reaction mechanism, which will be the subject of future work.
To stimulate this follow-up, we have added the results of a small screening of common hydrogenolysis catalysts (supported Ru-, Rh-, Ir-, Au-, Pt-, Pd- and Ni-catalysts) to the ESI.† This very limited test shows that various Ru-catalysts were active (10–20% tetritols), while the other metals were not (0–2% tetritols). It would be interesting to investigate what makes the Ru-catalysts special, and what is the role of their active sites. The long term stability of new developed catalysts should also be taken into account.
Conclusions
This work has shown that selective terminal C–C scission of C5-sugars (xylose and arabinose) to C4-tetritols (erythritol and threitol) can be achieved using a commercial 5 wt% Ru/C catalyst. High selectivity (>90%) for the terminal position was achieved at low conversions (<20%). The conversion of C5-sugars under the optimized conditions (138 °C, 6 bar H2) occurs via a direct decarbonylation mechanism. At higher conversions, the initially formed C4-tetritols undergo further decarbonylation to glycerol and eventually ethylene glycol. Due to this continuous chain scission mechanism, the selectivity for C4-products drops at higher conversion. As a result, the highest achieved yield of C4-tetritols in a batch system was 20–25% which is to the best of our knowledge the highest yield reported thus far.Experimental
Materials
The following chemicals and solvents were used as received: D-xylose (minimum 99%, Sigma Aldrich), L-arabinose (minimum 99%, Sigma Aldrich), D-xylitol (minimum 99%, Sigma Aldrich), L-arabitol (>99% Fluka), D-adonitol (99+%, Acros Organics), D,L-threitol (97%, Aldrich), Erythritol (Cerestar), L-erythrulose (>85% HPLC, Sigma Aldrich), glycerol (reagent plus, >99.0% Aldrich), ethylene glycol (anhydrous, 99.8%, Sigma Aldrich), pyridine (for analysis, Merck), ethanol (for analysis, Merck), n-hexane (99+%, for analysis, Merck), Sicapent (phosphorus pentoxide with indicator, Merck), activated charcoal (NORIT A SUPRA), acetic anhydride (>99%, Sigma Aldrich) and diphenylmethane (>99%, Fluka).Experiments with commercial D,L-threitol showed no conversion, which was expected to be caused by impurities in the starting material. The commercial product was therefore recrystallized via the following procedure: D,L-threitol (4.0 g, 33 mmol) was dissolved in 75 mL ethanol to give a clear yellow solution. Activated carbon (400 mg) was added and the suspension was stirred for 1 h. The activated carbon was removed by filtration to give a clear colourless solution. Hexane was added until the solution became slightly turbid. The solution was placed in the refrigerator for 19 h. The white crystals that formed over this period were collected by filtration and dried in a vacuum oven, 40 °C, over Sicapent to give 3.0 g (75%) of D,L-threitol as white crystals.
The industrial catalyst used in the investigation was 5% Ru/C (5% Ruthenium on activated carbon, reduced, 50% water wet paste, Escat 4401, %H2O 53.96, STREM). Analysis of this catalyst can be found in the ESI.†
Activated carbon (corresponding to the support used in commercially available 5% Pd/C catalyst) was kindly supplied by BASF.
General reaction procedure
The following procedure was used for the experiments shown in Table 1, Table S2,†Table 2 (entries 1–10) and Fig. 1: In a typical experiment, D-xylose (1.0 g), 5 wt% Ru/C (0.663 g) and deoxygenated demineralised water (25 mL) were charged into a 75 mL Hastelloy reactor (Parr MRS 5000 system). A magnetic stirring bar was added and the reactor was closed and flushed 3× with N2, followed by flushing 3× with H2. After applying the desired H2 pressure, stirring was started (600 rpm) and the reactor was heated to the desired temperature. The starting time of the reaction was determined as the point where the reactor reached the desired temperature (approx. 30 min.). To stop the reactions, the reactors were allowed to cool to room temperature (approx. 90 min.) after which they were depressurized, flushed 3× with N2 and opened. The catalyst was removed by filtration over filter paper and washed with distilled water. The combined aqueous phases were concentrated on a rotary evaporator at 40 °C under reduced pressure and the remaining product was further dried in a vacuum oven, 40 °C, over Sicapent. The obtained samples were weighed and analysed by GC-FID.Reactions under constant pressure
Reactions under constant H2 pressure (experiments of Fig. 2 and Table 2, entries 11–23) were performed in 600 mL Hastelloy reactor (Parr 4560). In a typical experiment, D-xylose (16.0 g), 5 wt% Ru/C (10.575 g) and deoxygenated demineralised water (400 mL) were charged to the reactor. The reactor was closed and flushed 3× with N2, followed by flushing 3× with H2. After applying the desired H2 pressure, stirring was started (600 rpm) and the reactor was heated to the desired temperature in approx. 30 min. The starting time of the reaction was determined as the point where the reactor reached the desired temperature. Over the course of the reaction, the H2 pressure was kept constant by manual re-pressurisation. At regular time intervals small samples (5 mL) were taken via the dip-tube, which were filtered, concentrated and dried as described above. The obtained samples were weighed and analysed by GC-FID.Analytical methods
GC-FID samples were prepared by dissolving the dried reaction mixtures in pyridine, followed by acetylation with acetic anhydride for 20 min. at 70 °C and the addition of a known concentration of internal standard (diphenylmethane). GC-FID analyses were performed on an Interscience Focus GC with a AS 3000 series auto sampler (He carrier gas, flow 50 mL min−1, split ratio 1:33; Restek GC column Rxi-5 ms 30 m × 0.25 mm × 0.25 μm; GC program hold 2 min at 70 °C, ramp 10 K min−1 to 300 °C, hold 2 min. Quantification of the products (expressed in mol%) was based on weight of the isolated product, and the peak areas of the GC samples (corrected for the response factors as obtained by comparison of commercial references to the internal standard).
Gas phase analysis was performed on a dual channel Intersience Compact GC with TCD detectors using He as the carrier gas. Carbon dioxide and hydrocarbons were separated on Poraplot Q, while CH4 and CO were separated on a Molsieve 5A. Samples for gas phase analysis were collected in a gas bag, by releasing the residual pressure of the reactors at room temperature. Results were compensated for relative response factors.
Acknowledgements
This research has been performed within the framework of the CatchBio program. The authors gratefully acknowledge the support of the Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science. The authors also want to thank Drs. Tomas van Haasterecht (Wageningen University) for gas-phase analysis and hydrogen chemisorption, Dr Jovana Zečević (Utrecht University), Drs. Wouter Lamme (Utrecht University) and Dr Rajeesh Pazhavelikkakath Purushothaman (WUR-FBR) for TEM/HAADF imaging, Dr Guus Frissen (WUR-FBR) for XRD measurements and Dr Eric Boer (WUR-FBR) for the DOE. The authors would also like to thank Dr Peter Witte (BASF) for supplying the activated carbon support.Notes and references
- J.-C. M.-P. G. de Troostembergh, I. A. Debonne and W. R. Obyn, US Pat, 0127667, 2002 Search PubMed.
- H.-J. Moon, M. Jeya, I.-W. Kim and J.-K. Lee, Appl. Microbiol. Biotechnol., 2010, 86, 1017 CrossRef CAS PubMed.
- M. Hara, T. Kawaguchi and M. Ueda, JP Pat, 11266887, 1999 Search PubMed.
- D. G. Barret, W. Luo and M. N. Yousaf, Polym. Chem., 2010, 1, 296 RSC.
- S. B. Kim, S. J. You, Y. T. Kim, S. M. Lee, H. Lee, K. Park and E. D. Park, Korean J. Chem. Eng., 2011, 28, 710 CrossRef CAS.
- S. Raju, J. T. B. H. Jastrzebski, M. Lutz and R. J. M. Klein Gebbink, ChemSusChem, 2013, 6, 1673 CrossRef CAS PubMed.
- I. Ahmad, G. Chapman and K. M. Nicholas, Organometallics, 2011, 30, 2810 CrossRef CAS.
- E. Arceo, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2010, 132, 11408 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Angew. Chem., 2012, 124, 8206 ( Angew. Chem., Int. Ed. , 2012 , 51 , 8082 ) CrossRef PubMed.
- A. Oosterveld, G. Beldman, M. J. F. Searle-van Leeuwen and A. G. J. Voragen, Carbohydr. Res., 1996, 288, 143 CrossRef CAS.
- M. A. Andrews, G. L. Gould and S. A. Claeren, J. Org. Chem., 1989, 54, 5257 CrossRef CAS.
- R. N. Monrad and R. Madsen, J. Org. Chem., 2007, 72, 9782 CrossRef PubMed.
- D. K. Sohounloue, C. Montassier and J. Barbier, React. Kinet. Catal. Lett., 1983, 22, 391 CrossRef CAS.
- L. Zhao, J. H. Zhou, Z. J. Sui and X. G. Zhou, Chem. Eng. Sci., 2010, 65, 30 CrossRef CAS PubMed.
- Z. Srokol, A.-G. Bouche, A. van Estrik, R. C. J. Strik, T. Maschmeyer and J. A. Peters, Carbohydr. Res., 2004, 339, 1717 CrossRef CAS PubMed.
- K. Y. Wang, M. C. Hawley and T. D. Furney, Ind. Eng. Chem. Res., 1995, 34, 3766 CrossRef CAS.
- C. Montassier, J. C. Ménézo, L. C. Hoang, C. Renaud and J. Barbier, J. Mol. Catal., 1991, 70, 99 CrossRef CAS.
- B. S. Kedel'baev, N. N. Imankulov, A. M. Kuatbekov and T. M. Ismailov, Chem. Nat. Compd., 2000, 36, 213 CrossRef.
- M. Vasiliu, K. Guynn and D. A. Dixon, J. Phys. Chem. C, 2011, 115, 15686 CAS.
- M. G. Musolino, L. A. Scarpino, F. Mauriello and R. Pietropaolo, ChemSusChem, 2011, 4, 1143 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827 CrossRef CAS PubMed.
- S. van de Vyver, J. Geboers, M. Dusselier, H. Schepers, T. Vosch, L. Zhang, G. van Tendeloo, P. A. Jacobs and B. F. Sels, ChemSusChem, 2010, 3, 698 CrossRef CAS PubMed.
- R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972 RSC.
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576 RSC.
- J. Liu, X. N. Tian and X. S. Zhao, Aust. J. Chem., 2009, 62, 1020 CrossRef CAS.
- K. L. Deutsch, D. G. Lahr and B. H. Shanks, Green Chem., 2012, 14, 1635 RSC.
- L. Fabre, P. Gallezot and A. Perrard, Catal. Commun., 2001, 2, 249 CrossRef CAS.
- L. Fabre, P. Gallezot and A. Perrard, J. Catal., 2002, 208, 247 CrossRef CAS.
- J. Sun and H. Liu, Green Chem., 2011, 13, 135 RSC.
- J. B. Park, B. C. Seo and J. R. Kim, US Pat, 6060291, 2000 Search PubMed.
- H. Paananen, J. Kuisma, V. Ravanko, N. Mayra, H. Heikkila and J. Lewandowski, Int. Pat, WO 0227037, 2002 Search PubMed.
- R. Mizanur, K. Takeshita, H. Moshino and G. Takada, J. Biosci. Bioeng., 2001, 237 CrossRef CAS.
- E. P. Maris and R. J. Davis, J. Catal., 2007, 249, 328 CrossRef CAS PubMed.
- G. de Wit, J. J. de Vlieger, A. C. Kock-van Dalen, R. Heus, R. Laroy, A. J. van Hengstum, A. P. G. Kieboom and H. van Bekkum, Carbohydr. Res., 1981, 91, 125 CrossRef CAS.
- S. Murahashi, T. Naota, K. Ito, Y. Maeda and H. Taki, J. Org. Chem., 1987, 52, 4319 CrossRef CAS.
- S. Galvagno, G. Capannelli, G. Neri, A. Donato and R. Pietropaolo, J. Mol. Catal., 1991, 64, 237 CrossRef CAS.
- S. Kolarić and V. Šunjić, J. Mol. Catal. A: Chem., 1996, 111, 23 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01012h |
| This journal is © The Royal Society of Chemistry 2015 |