
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural characterization of 13C-enriched humins and alkali-treated 13C humins by 2D solid-state NMR†
Ilona
van Zandvoort‡
a,
Eline J.
Koers‡
b,
Markus
Weingarth
b,
Pieter C. A.
Bruijnincx
*a,
Marc
Baldus
*b and
Bert M.
Weckhuysen
*a
aInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: b.m.weckhuysen@uu.nl; p.c.a.bruijnincx@uu.nl
bNMR Spectroscopy, Bijvoet Center for Biomolecular Research, Department of Chemistry, Faculty of Science, Utrecht University, Padualaan 8, 3584 CH, Utrecht, The Netherlands. E-mail: m.baldus@uu.nl
First published on 9th July 2015
Abstract
Humin by-products are formed during the acid-catalyzed dehydration of carbohydrates to bio-based platform molecules, such as hydroxymethylfurfural and levulinic acid. The molecular structure of these humins has not yet been unequivocally established. 1D 13C solid-state NMR data reported have, for example, provided considerable insight, but do not allow for the unambiguous assignment of key structural motifs. Complementary (2D) techniques are needed to gain additional insight into the molecular structure of humins. Here, the preparation of 13C-enriched humins is reported, together with the reactive solubilization of these labeled humins and their characterization with complementary 1D and 2D solid-state NMR techniques. 1D cross polarization (CP) and direct excitation (DE) 13C solid-state NMR spectra, 2D 13C-detected double-quantum single-quantum (DQSQ) as well as 2D 1H-detected heteronuclear correlation (HETCOR) were recorded with different excitation schemes. These experiments unambiguously established that the original humins have a furan-rich structure with aliphatic linkers and allowed for a refinement of the molecular structure proposed previously. Solid-state NMR data of alkali-treated 13C-labeled humins showed that an arene-rich structure is formed at the expense of the furanic network during alkaline pretreatment.
Introduction
Humins are carbonaceous, polymeric by-products that are almost inevitably formed during acid-catalyzed, hydrothermal processing of sugars to bio-based platform molecules, such as hydroxymethylfurfural (HMF) and levulinic acid (LA).1–3 Under such conditions, C6-sugars, such as glucose and fructose, are first dehydrated to form HMF, which in turn can be readily rehydrated to form LA and formic acid. The humins form as a result of uncontrolled cross-polymerization reactions of HMF and other reaction intermediates. Up to 30 wt% of the carbohydrate feedstock can end up in these humin by-products, which severely limits the efficiency and, as a result, economic viability of the biorefinery operation.3 To increase the economic potential of such biorefineries, the formation of humins should either be limited and/or value-added outlets should be found for any humins that are formed. In addition to serving as a source for heat and power or to direct application in functional materials,4 such valorization can be achieved by further conversion by gasification,3,5 pyrolysis,6 or chemo-catalytic routes to value-add chemicals or fuel components. Any attempt at humin valorization is hampered, however, by the fact that still little is known about the chemical structure and properties of these humins, as well as by the general insolubility of these materials.7 Indeed, more insight is needed in their molecular structure and mechanism of formation. The various suggestions made previously for the molecular structure of humins are discussed below.5,6,8–10Much of the current knowledge about the humin molecular structure is derived from studies of the molecular structure of the related hydrothermal carbons (HTC). The chemical and physical properties of these HTC are much better studied, but conflicting structures are still found in literature.11,12 HTC is also formed by hydrothermal treatment of sugars, but under non-acidic conditions, and are thought to share structural similarities to humins. For example, 13C solid-state NMR studies by Baccile et al.11 of HTC obtained from combinations of unlabeled glucose, 13C1- and 13C6-labeled glucose provided much insight in their molecular structure. A direct excitation (DE) 13C spectrum showed the HTC molecular structure to be complex with 13% of the carbon being part of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, 64% found in sp2 CC (of which 29% furan Cα, 29% furan Cβ and 6% arene sheets) and 23% in aliphatic groups. The 1
O groups, 64% found in sp2 CC (of which 29% furan Cα, 29% furan Cβ and 6% arene sheets) and 23% in aliphatic groups. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio observed for the signals assigned to the Cα and Cβ atoms of an oxygenated or oxygen-containing aromatic ring gave a first indication that the aromatic rings are furan-based rather than arene-based in nature. A refocused, INEPT experiment13 was used to study through-bond 1H–13C interactions, while Cross Polarization (CP) and Inversion Recovery Cross Polarization (IRCP) measurements provided information on 1H–13C through-space interactions of the aromatic and aliphatic carbons in the HTC, respectively. Insight in the 13C–13C connectivity of the various HTC constituents was obtained from a Double-Quantum Single-Quantum (DQSQ) experiment. Taken together, these NMR techniques revealed a furanic network in HTC, in which the furan rings are mainly linked through the α-position by a methylene group. Minor linkages included direct bonds between furan rings via the Cα or Cβ carbons. The DQSQ spectrum furthermore suggested LA molecules to be physically embedded in the polymer.11
:1 ratio observed for the signals assigned to the Cα and Cβ atoms of an oxygenated or oxygen-containing aromatic ring gave a first indication that the aromatic rings are furan-based rather than arene-based in nature. A refocused, INEPT experiment13 was used to study through-bond 1H–13C interactions, while Cross Polarization (CP) and Inversion Recovery Cross Polarization (IRCP) measurements provided information on 1H–13C through-space interactions of the aromatic and aliphatic carbons in the HTC, respectively. Insight in the 13C–13C connectivity of the various HTC constituents was obtained from a Double-Quantum Single-Quantum (DQSQ) experiment. Taken together, these NMR techniques revealed a furanic network in HTC, in which the furan rings are mainly linked through the α-position by a methylene group. Minor linkages included direct bonds between furan rings via the Cα or Cβ carbons. The DQSQ spectrum furthermore suggested LA molecules to be physically embedded in the polymer.11
In another example of the detailed information that advanced solid-state NMR studies can bring on complex organic structures, Schmidt-Rohr et al.14 reported on spectrally-edited 2D 13C solid-state NMR data for the characterization of glucose-derived and 13C-enriched carbon materials, obtained by pyrolysis and by carbonization with fuming sulfuric acid. Dipolar-dephased DQSQ measurements allowed to selectively study correlations between non-protonated carbon atoms. In addition, Exchange between protonated and non-protonated carbons via the so called EXPANSE spectral editing scheme14 allowed the authors to distinguish, for example, between signals from substituted furans and phenols. An advantage of both techniques is the absence of a diagonal ridge, which often obscures various expected cross-peaks in carbonaceous samples. It should be noted, that even though the materials reported by Baccile and Schmidt-Rohr are probably much more condensed than the humins studied here, the characteristic signals detected and identified for substituted furans, phenols, arenes, and polycyclic aromatic structures could be very relevant for humin structure elucidation.11,14,15
Only a few reports describe the structural characterization of actual humins rather than HTC materials. Zarubin et al., for instance, investigated humins formed during acid-catalyzed dehydration of different carbohydrates and HMF. Based on IR and pyrolysis-GC-MS analysis of the solids, it was concluded that the humins consist of about 60% furan rings and 20% aliphatic linkers. The authors suggested that humins are formed via a poly-condensation pathway leading to a network of furan rings linked by ether or acetal bonds.10 Lund et al. studied the molecular structure of HMF-derived humins by IR spectroscopy and proposed that the humins were formed by aldol condensations of HMF with 2,5-dioxo-6-hydroxy-hexanal (DHH), which is formed by rehydration of HMF. This leads to a conjugated network of CC and furanic rings with several aldehydes, ketones and furfuryl alcohols as functional groups.8 A comparison of the IR spectra of HMF- glucose-, fructose- and cellobiose-derived humins suggested that the humins could not be formed directly from the sugars.9 Recently, Seshan et al.16 presented a molecular structure of humins where furanic rings are connected via ethylene and methyl linkers with ketone functionalities. This structure was based on 1D 13C solid-state NMR, IR and pyrolysis data.16
Some of us previously reported on a multi-technique, multi-process parameter approach that provided new insights into the formation and molecular properties of humin by-products formed from various carbohydrate feeds. A combination of IR, 1D 13C solid-state NMR and pyrolysis-GC-MS measurements revealed a furan-rich structure containing several oxygen functionalities, in which the furan rings are connected directly or via aliphatic linkages. Based on this data a model for the molecular structure of glucose- and xylose-derived humins was proposed.17 Direct, conclusive evidence for the nature and connectivity of, in particular, the aromatic constituents of the humins was, however, difficult to obtain. Indeed, the 1D 13C solid-state NMR spectra did not allow unambiguous identification of the moieties giving rise to the broad signal intensity in the aromatic region. Furanic and phenolic aromatic groups could, for instance, both be formed under the conditions that lead to humin formation18 and would give very similar signal patterns around the same chemical shifts.14,19 In contrast, 2D solid-state NMR studies could provide the resolution and information needed to distinguish between such furanic or phenolic motifs and thus help to improve and strengthen the proposed model for the molecular structure of humins.11,14
Similar insight is needed into any changes that occur in the structure of humins upon further valorization. Such changes were previously noted in thermochemical conversion processes, such as gasification or pyrolysis. Seshan et al., for instance, showed using CP MAS 13C solid-state NMR studies that aromatic rings are formed in humins when gasified at 700 °C. A more detailed description of the remaining char could not be given, however.5 The hydrothermal alkaline treatment method for reactive solubilization of humins, which we recently developed, also changes the molecular structure of the humins considerably. IR, elemental analysis, 1D solid-state NMR and pyrolysis-GC-MS data indicate further aromatization of the structure, among other changes.20 More information is needed, nonetheless, to propose a model for the molecular structure of the alkali-treated humin. Also here, advanced 2D solid-state NMR studies could further advance our understanding of the reactions and structural changes that occur during the alkaline pretreatment.
Here, we describe the structural characterization of humin by-products obtained from 13C6-D-glucose by employing various 1D and 2D solid-state NMR techniques. Firstly, we compared one-dimensional DE and CP 13C NMR spectra that allowed us to observe 13C NMR signals in close proximity to protons (CP) in reference to the overall distribution of 13C signals that is detected with the DE method. Two-dimensional spectroscopy helped us to reduce spectral overlap by dispersing signals in two spectral dimensions. In the case of DQSQ (double quantum-single quantum) experiments, correlations between two dipolar coupled (i.e., in close spatial proximity) 13C spins are dispersed along the horizontal (single-quantum) axis with their individual 13C chemical shifts and resonate at the sum resonance frequency in the ω1 dimension.21 In addition, we employed proton-detected heteronuclear (13C,1H) HETCOR experiments to identify correlations of 1H–13C moieties that are in close spatial proximity. Please see the recent review by Baccile et al.22 for an introduction to the application of these solid-state NMR techniques in biomass characterization. Taken together, these experiments allowed us to refine the molecular structure of the humin by-products. The results provided new insight into the type and connectivity of the aromatic structures and improves our understanding of the chemical changes that occur during the alkaline pretreatment.
Results and discussion
13C-labeled humin synthesis and IR characterization
A 13C-labeled glucose-derived humin sample was prepared by heating 25 mL of an aqueous solution of 1 m 13C6-D-glucose and 0.01 m H2SO4 to 180 °C for 7 h. The humins were subjected to a Soxhlet extraction with water to remove any starting material, HMF or LA. The ATR-IR spectra of the labeled humins were found to be in good agreement with the humin samples reported in preceding work (Fig. 1),17 with vibrations being shifted to lower wavenumber as a result of the change in reduced mass caused by the 13C labels. These shifts actually help to improve the peak assignments made in literature and our preceding work.8,9,17 The vibration at 1700 cm−1 was originally assigned to the CO stretch from acids, aldehydes, and/or ketones. In the spectrum of 13C-labeled humin this band has shifted to 1640 cm−1. This shift of 60 cm−1 indicates that this vibration is strongly influenced by coupling with a labeled carbon–carbon bond and that the CO bond could be part of an enone functional group; the broadening of this band makes a firm attribution difficult, however, and points at multiple CO functional groups are present in different chemical environments. The peak ascribed to the CC stretch from a furanic ring shifted from 1605 cm−1 to 1546 cm−1, confirming its assignment. The peaks at 1292 and 1202 cm−1 shift to 1265 and 1179 cm−1, respectively. These smaller shifts of ∼20 cm−1 show that these peaks are, as expected, caused by C–O stretch vibrations. The C–H bending modes at 1020 cm−1 only shifted to 1010 cm−1, which indicates only a small contribution of the carbon atom to this vibration. The peaks below 1000 cm−1 have hardly shifted at all, suggesting they belong to C–H out of plane deformations of aromatic rings.23
 | ||
| Fig. 1 ATR-IR spectra of unlabeled humins (black) and 13C-labeled humins (blue). | ||
NMR analysis of 13C-labeled humins
1D 13C solid-state NMR spectra of the 13C-labeled humin sample were recorded by DE and by CP from 1H. The spectra are shown in Fig. 2 and peak assignments can be found in Table 1.11,14,24 The spectrum can be divided into three main regions, covering the aliphatic carbons between δ = 0–90 ppm, CC bonds and aromatic compounds from δ = 90 ppm to δ = 160 ppm, and CO signals between δ = 160 ppm and δ = 220 ppm.
 | ||
| Fig. 2 1D 13C solid-state NMR spectra of 13C-labeled humins obtained by DE (black) and CP via1H (grey). | ||
| δ (ppm) | Functional group | Chemical formula | Protonateda |
|---|---|---|---|
| a Based on comparison between DE and CP MAS NMR spectra shown in Fig. 2. | |||
| 207 | Ketone | CO |
No |
| 177 | Acid, ester | COOH/COOR | No |
| 151 | Cα phenol or linked furan | CC–OH or CC–O |
No |
| 142 | Cα free furan | CCH–O |
Yes |
| 129 | Conjugated CC |
C–CC–C |
No |
| 121 | Cβ phenol or furan linked |
CC–OH or C–CC–O |
No |
| 116 | Cβ phenol or furan protonated | HCC–OH or C–HCC–O |
Yes |
| 78 | Alcohol, ether, aliphatic | C–OH, C–O–C | Yes |
| 39 | Aliphatic | tert. C–H, quart. C | Yes |
| 31 | Aliphatic | sec. –CH2– | Yes |
| 15 | Aliphatic | prim. –CH3 | Yes |
In the DE spectrum, the highest signal intensity for the aliphatic carbons is observed at δ = 38 ppm, indicating that most aliphatic carbons are tertiary or quaternary, or could be located next to a CO group. From the limited signal intensity in the region δ = 60–90 ppm, considering the overall signal to noise in the spectrum and the low intensity in this region in the previously reported 2D PASS 13C solid-state NMR spectra,17 it can also be concluded that the humin structure contains only limited amounts of alcohols and ethers and that the amount of residual sugar is negligible. A typical pattern for oxygen-containing aromatics, such as (substituted) furanics and phenolics, is observed in the aromatic region. Protonated Cα and Cβ atoms of furanic rings are typically observed at δ = 140 ppm and δ = 110 ppm (see Fig. 3 for the labeling scheme), respectively, while (C-)substituted Cα and Cβ signals should give rise to signals around δ = 150 ppm and δ = 120 ppm, respectively. For phenol rings, Cα appears around δ = 150 ppm, while Cβ is observed at δ = 100–120 ppm depending on the substitution pattern. The similarity in signal position complicates distinguishing furanic from phenolic components in the humins based on 1D 13C NMR spectra alone. A first indication is nonetheless offered by the intensity ratio of the signals from Cα and Cβ being roughly 1:1, which points to the presence of furan rings rather than phenolics, for which a 1:2 ratio would be expected.14,19 The peak at δ = 177 ppm is assigned to acids or perhaps, esters. Finally, the signal at δ = 207 ppm can be ascribed to aldehydes and/or ketones, two functional groups that again cannot be readily distinguished in these DE 1D spectra.
 | ||
| Fig. 3 Labeling schemes for furan, phenol and benzofuran moieties. | ||
The 1D DE spectrum of the 13C-labeled humin is similar to the solid-state NMR spectra reported for HTC11 and the 2D PASS NMR spectra of unlabeled, glucose-derived humins shown in our previous study.17 Small differences can, nonetheless, be observed between the DE and 2D PASS spectra of labeled and unlabeled humins, respectively. The first shows a higher signal intensity in the aliphatic region and for the CO signal (δ = 207 ppm). Furthermore, a lower intensity in the C–O region was observed as well as a small downfield shift of the peaks at δ = 112 and 142 ppm in the aromatic region compared to the 2D PASS NMR spectrum. Another clear difference is the absence of conjugated aromatic carbons around δ = 130 ppm in the DE spectra. These differences could be caused by the 2D PASS spectrum being recorded after CP excitation, which is known to underestimate the amount of non-protonated carbons and methyl groups.25 Differences in sample preparation scale of the labeled and unlabeled humins, might also influence the structures somewhat. The heating and cooling rates of the small autoclave used to prepare the 13C-enriched humins, are faster than those of the 1 L autoclave that was used for the larger scale preparation of the unlabeled humins. This leads to longer residence times for the latter, which can result in further dehydration of the humin structure and a relatively more arene-rich structure.
The 1D CP MAS NMR spectrum was recorded with a contact time of 400 μs. Under such conditions, 13C magnetization is most efficiently generated by polarization transfer from directly bonded protons. Hence protonated carbons dominate the CP spectrum (Fig. 2, gray). As expected, the CP MAS 13C solid-state NMR spectrum had a much lower intensity than the one obtained by DE and showed strong peaks at δ = 30 and 38 ppm from aliphatic carbons. In the aromatic region, a pronounced peak at δ = 112 ppm from protonated Cβ is observed together with a much weaker signal at δ = 145 ppm, which is ascribed to Cα atoms. The fact that the signal at δ = 145 ppm is much weaker, indicates that a very limited, yet distinct amount of the Cα atoms are actually protonated, as was previously observed for HTC.11 As such unsubstituted, i.e. protonated, Cα atoms cannot be present in phenolic structures but can occur in furanic rings, these results already point towards the furanic nature of the humins (Fig. 3). Notable absences of intensity in the CP spectrum also provide additional information. The absence of a signal at δ = 207 ppm, for instance, shows that the carbonyl carbon is non-protonated and thus belongs to a ketone group.
A 1H-detected HETCOR (1H)–13C–(1H)–1H spectrum of 13C-labeled humins was recorded using a 1H detection-optimized probe with contact times of 900 μs for the transfer from H to C (optimized for maximum signal intensity) and 300 μs for the C to H transfer in order to study short-range 13C–1H correlations (Fig. 4a). Very few interactions between 13C and 1H nuclei were observed, again indicating that most carbon moieties are not protonated. The correlations seen between δ1H = 0–4 ppm/δ13C = 0–50 ppm and δ1H = 5–7.5 ppm/δ13C = 110–140 confirm that mainly the aliphatic carbons and furanic Cβ's are protonated. Furthermore, the absence of a C–H correlation peak for the carbonyl groups (at δ13C = 207 ppm) corroborates the assignment of this signal to ketones. The humins differ in this sense from the HTC samples studied by Baccile et al., who could identify aldehydes based on a HETCOR experiment with a longer contact time of 500 μs.11
 | ||
| Fig. 4 Top: Proton-detected HETCOR spectra of 13C-labeled humins recorded with the following CP contact times. A. 900 μs (1st CP) and 300 μs (2nd CP) and B. 2.5 ms for both CP steps. Cross correlations indicating transfer via more than one H–C bond are colored in black. Bottom: Molecular structures representing the C–H correlations observed in the HETCOR spectra of 13C-labeled, glucose-derived humins. | ||
Long-range 1H–13C correlations were studied with a second, longer contact time of 2500 μs (Fig. 4b). In addition to the strong signals from directly connected 1H–13C groups in the aliphatic and aromatic region, long-range interactions between Cα (δ13C = 150 ppm) and aliphatic protons were also detected. These interactions are also observed for Cβ, which shows that the aliphatic groups are located close to the aromatic rings. A cross peak between Cα and protons connected to Cβ was observed as well. At lower contour levels a weak cross peak indicates that the acids and ketone groups are located closer to the aliphatic groups than to the aromatic rings (see ESI, Fig. S1†).
Next, a CP-based 13C DQSQ of the 13C humins was recorded to get insight in carbon atom-connectivity in the different structural motifs (Fig. 5). A CP time of 600 μs was chosen to find a balance between the spectrum being dominated by signals from protonated carbons, and allowing sufficient sensitivity. It should thus be kept in mind that the spectrum does not exclusively consist of protonated carbon signals; indeed, the CP contact time used was longer than for the 1D CP MAS spectrum. In the aromatic region of the CP-DQSQ spectrum, correlation patterns typical for Cα and Cβ connectivities were observed. Such a pattern can, in principle, be assigned both to furanic and phenolic rings, as noted before for HTC11 and acid-treated glucose.14 Nonetheless, the clear correlation seen between the Cβ atoms (see Fig. 5 for the structures), is most probably the result of interactions in a furanic ring and is unlikely in phenolic structures.14 Furthermore we detect, weak, yet distinct correlations between Cα carbons and aliphatic carbons, which is again not possible in phenolic rings in which the Cα is by definition substituted with the OH-group only. In addition, the Cβ–Cγ connectivity pattern for phenolic rings, expected at Cβδ = 110–120 ppm and Cγδ = 130–140 ppm, is not observed. These observations provide further evidence for the furanic nature of the humins.
 | ||
| Fig. 5 2D CP-DQSQ 13C solid-state NMR spectrum of 13C-labeled humins obtained after CP via1H. Black regions are spinning side bands. | ||
The broad peak from the aliphatic carbons at δ = 0–60 ppm shows several 13C–13C correlations between the aliphatic carbon atoms. These peaks also show some correlation with the aliphatic carbons that are connected to Cα. Furthermore, the (weak) signals around δ = 175 ppm indicate the presence of aliphatic carboxylic acids (or their esters). Baccile et al.11 also observed these signals and ascribed them to physically embedded LA. Given that our samples are purified by extensive Soxhlet extraction, any residual, physically occluded LA should have been removed during purification, suggesting that the detected, probably LA-derived, carboxylic acids are actually chemically bound to the humin structure. The CP-DQSQ also shows a correlation between aliphatic carbons and a C–O group, which might be caused by alcohols but could also be from ether or acetal bonds. These two possibilities cannot be distinguished, however, based on the current data. The presence of ether or acetal bonds would partially explain the reactive solubilization and reduction in molecular weight of the humins after alkaline pretreatment, though, as suggested previously.20 Be it alcohols, ethers or acetals, these bonds are only present in small amounts given the (very weak) signal intensity in the δ = 60–90 ppm region in the DE 1D 13C solid-state NMR spectrum. A very weak correlation between Cβ (δ = 115 ppm) and aliphatic C (δ = 30 ppm) was finally observed indicating that linking via Cβ occurs to a very limited extent (not highlighted in Fig. 5). The correlations seen by Baccile et al.11 between CO and the furan rings, indicating aldehyde and acid functional groups on the furanic ring, were not observed in our spectrum.
It should be kept in mind that the mixing time chosen for the CP DQSQ measurement strongly influences to which extent protonated and non-protonated carbons are probed; indeed, the mixing time used for the spectrum depicted in Fig. 5 ensured that mainly protonated carbons were observed. Certain linkages that might be present in the molecular structure of the humins involving nonprotonated carbons, such as Cβ–Cβ linkages between furanic rings, other structural motifs, such as benzofuran and polycyclic aromatic sheets, and oxygen functionalities on aromatic rings are not expected to appear in the spectrum depicted in Fig. 5 or are at least underestimated. To study if such features are actually present, a second DQSQ was recorded with DE of the 13C atoms (Fig. 6). This DQSQ spectrum after DE of 13C is similar to the CP one, with the distinction that several interactions between non-protonated carbons are now indeed much more pronounced. In addition, signals from protonated carbons are weaker or not observed at all, indicating that these are present only to a minor extent in the molecular structure. Correlations between CC–O–Caliphatic and Cβ–Caliphatic were not observed, for example. Clear signals from Cα–Cβ and Cβ–Cβ correlations were seen. Taken together with the observed links between Cα and Caliphatic discussed previously, this proves that humins are indeed mainly composed of furanic rings and that aliphatic groups are the main linkers. Comparison with the 1D spectra shows that several Cβ's are substituted and therefore probably cross-linked as suggested by Baccile et al.11 To further confirm this, future efforts should include dipolar-dephased DQSQ measurements of the humins.14 In addition, Cα–Cα interactions are observed around δ = 140–145 ppm,14 indicating that direct, inter-furanic linkage between Cα's does occur, a possibility that furans offer but phenolics do not. Finally, strong signals from the functional groups acid and ketone functional groups are observed and both show a clear correlation with the aliphatic carbons. However, these aliphatic chains show little correlation with the aliphatic linkers that connect the furanic rings. This indicates that the acids and ketones are not located on these aliphatic linkers and could result from the chemical incorporation of LA or from sugar dehydration and HMF rehydration intermediates.
 | ||
| Fig. 6 2D DQSQ 13C solid-state NMR of 13C-labeled humins obtained after DE of 13C. Black regions are spinning side bands. | ||
The model for the molecular structure of humins proposed in a preceding paper, which was based on elemental analysis, IR, 1D and 2D (PASS) 13C solid-state NMR and pyrolysis-GC-MS data did already include short aliphatic linkers between the furanic rings.17 The Cα–Cα and C–O–C linkages that have now been identified in the 2D NMR spectra were not included, however, as the 1D (2D PASS) NMR spectra did not offer this insight. The CP-DQSQ spectrum furthermore suggests some linkages to involve Cβ as well, as does the peak at δ = 120 ppm in the 1D DE NMR spectrum, which indicates that some of the Cβ are not protonated. While clear signals from Cβ–Cβ correlations are also observed in the 2D spectra, it is still difficult to distinguish between correlations within one furan ring and between furan rings; direct linkages between the Cβ's of two furan rings could therefore not be excluded or proven. Based on this information, a refined model of the molecular structure of glucose-derived humins is proposed (Fig. 7), Note that given the random nature of the polymer and the difficulty in determining the relative abundance of the linkages, the fragment drawn should not be seen as ‘repeat unit’ but rather depicts the various different linkages thought to be present in the structure.17
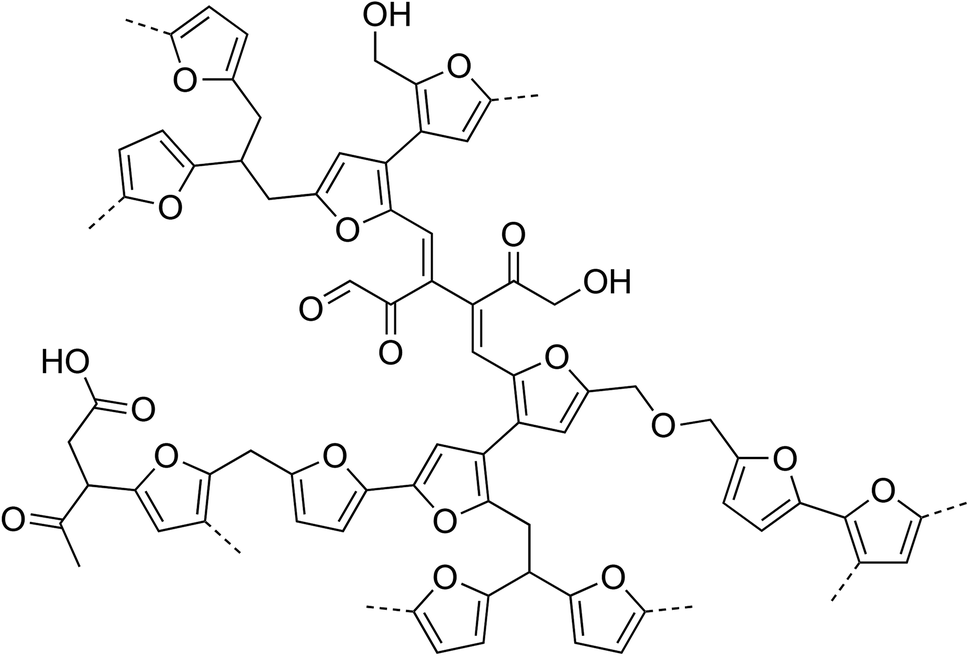 | ||
| Fig. 7 Revised model for the molecular structure of glucose-derived humin; linkages included in the fragment are based on literature precedence, our previously proposed model17 and have been refined based on the present NMR work. | ||
Some observations can be made with regards to previous proposals for the molecular structure of humins. Zarubin et al. proposed a furanic network where the furan rings were connected by C–O–C bonds.10 The C–O linkages that are observed in the CP-DQSQ spectrum suggests that some acetal or, alternatively, ether linkages are present, but in very limited amounts. These bonds were suggested to play a role in the solubilization by alkaline treatment of humins as described in earlier work and below.20 The alternative of solubilization by ester hydrolysis does not agree well with the IR spectra and solubilization behavior. An ether bond was therefore included as possible linker in the structure depicted in Fig. 7, but it should be stressed that the main linkages observed in our 2D 13C spectra are Cα–Cα and Caliphatic–Cα instead.
Lund et al.,8,9 proposed an aldol condensation mechanism for humin formation where HMF reacts with its rehydration product 2,5-dioxo-6-hydroxyhexanal; the correlations expected for the CC bonds of the conjugated enones formed are not observed in our DQSQ NMR spectra, however. Even though this would imply that inclusion of DHH does not happen to a large extent, we do think it plausible that some DHH is incorporated in the structure and one such linkage was therefore added to the proposed structure. It should finally be noted that both the Zarubin and Lund models do not account for the aliphatic C–C correlations that were observed in our DQSQ NMR spectra. In part, these differences might be due to the fact that the experimental conditions for the formation of these humins differ somewhat from our procedure.
The model proposed by Seshan et al.16 was based on humins that were prepared under the same conditions as described in this work. Their model, which was mainly based on 1D solid-state NMR, shows some important differences compared to the structure shown in Fig. 7. Our 2D NMR spectra, for instance, show that the ketone groups are not directly connected to the furanic rings and that most linkages are either direct Cα–Cα linkages or are longer than a single methylene group.16 The 2D NMR spectra thus provide valuable new information about the specific linkages in the molecular structure of humins.
The NMR data can also be compared to the two conflicting molecular structures that are found in literature for HTC. On the one hand, an arene-rich, polycyclic aromatic structure has been proposed for HTC from glucose and starch prepared at temperatures of 170–240 °C. This structure proposal was based on XPS, elemental analysis, IR and Raman data.12 No indications for the presence of polyaromatic sheets in the molecular structure of humins could be seen in our spectra, however. The other structure proposed for HTC from glucose involves a furan-rich network, in which the furan moieties are connected by methylene groups.11 While our spectra also strongly suggest a furanic motif, the proposed HTC structure would not give rise to the correlations seen between the aliphatic carbons that are present in our DQSQ spectra. Other sugar-derived materials, such as the acid-treated glucose-derived carbon reported by Schmidt-Rohr et al., are also furan-rich, yet much more acidic than our humins. In this case, the acid functional groups are directly connected to the aromatic rings instead of on the aliphatic chains as observed for our humins.14 This comparison shows that humins formed during the acid-catalyzed dehydration of glucose are similar to, yet distinct from HTC and acid-treated glucose, as different linkages and functional groups are observed.
NMR analysis of alkali-treated 13C-labeled humins
The 13C-labeled humins were treated in 2 M NaOH at 240 °C for 3 h, after which the humins were completely dissolved and a dark brown solution was obtained. The pH of the solution was decreased to 1 with 6 M H2SO4 to precipitate the humins from solution. Humins were isolated by filtration, washed extensively with water and dried under vacuum. The obtained alkali-treated, 13C-labeled humins were analyzed by 1D and 2D 13C (DQSQ) and 1H-detected HETCOR solid-state NMR.A comparison of the DE 1D 13C NMR spectra of alkali-treated humins (Fig. 8, Table 2) and the parent humin clearly shows that the molecular structure of humins significantly changed upon alkaline pretreatment. The largest differences are observed in the aromatic region where strong signals from conjugated systems can now be seen around δ = 130 ppm, indicating further aromatization by transformation of the furanic rings into arenes or polyaromatic structures. Less pronounced differences were observed in the aliphatic region (δ = 0–50 ppm) where the overall intensity decreased while the signal from methyl groups at δ = 19 ppm increased. All intensity between δ = 60–90 ppm is lost, indicating that all C–O bonds were hydrolyzed during alkaline treatment, as pointed out before.20 A signal for substituted Cα was still observed at δ = 151 ppm, but no signals for Cβ could be distinguished in the 1D spectrum. The latter might due to be overlap with the broad peak at δ = 130 ppm. The decrease in oxygen content of the material, as seen previously by elemental analysis,20 is reflected in the decrease of the signal from ketone groups, together with a slight shift downfield to δ = 210 ppm, with suggests that the carbonyls are on average less conjugated.14 The signal seen at 177 ppm is thought to arise from the overlap between a spinning side band and a signal from the carboxylic acids present in the alkali-treated humins, as also evidenced from the DQSQ spectrum detailed below. Given the low contribution such a spinning side band would make to the overall peak intensity, it is not labeled as such in Fig. 8. The aromatic region of the 1D spectrum of alkali-treated humins are most similar to the Total suppression Of Spinning Sidebands (CP TOSS) spectra reported by Schmidt-Rohr et al. of the sulfuric-acid treated carbon material that was formed by pyrolysis at 350 °C,14,15 with the exception that these materials show a much higher signal for carboxylic acid groups.
 | ||
| Fig. 8 1D 13C NMR spectra of 13C-labeled alkali-treated humins obtained by DE (black) and CP via1H (grey). | ||
| δ (ppm) | Functional group | Chemical formula | Protonateda |
|---|---|---|---|
| a Based on comparison between DE and CP MAS NMR spectra in Fig. 8. | |||
| 210 | Ketone | CO |
No |
| 177 | Acid | COOH | No |
| 153 | α carbon phenol or linked furan |
CC–OH or CC–O |
No |
| 130 | Conjugated CC |
CC–CC |
No |
| 125 | Conjugated CC |
CHC–CC |
Yes |
| 112 | β carbon phenol or furan protonated | HCC–OH or C–HCC–O |
Yes |
| 36 | Aliphatic | quart. C, tert. C–H, sec. –CH2–, | Yes |
| 18 | Aliphatic | prim. –CH3 | Yes |
The CP spectrum (400 μs contact time) again showed a much lower intensity, in particular in the aromatic region, than the DE spectrum (Fig. 8). This shows that that the aliphatic carbons are mainly protonated while the aromatic carbons are not. The shoulder peak at δ = 111 ppm suggests the presence of protonated Cβ, while the signals at δ = 125 and 153 ppm point at two more protonated aromatic carbons. The former can be attributed to protonated carbons in polycyclic aromatic systems, but assignment of the latter is more difficult. Furanic groups seem unlikely, as the chemical shift is too high for a protonated Cα; pyrone-like structures might give rise to such a signal, as suggested in our previous study.20,26
The CP-DQSQ (600 μs contact time, Fig. 9) spectrum of the alkali-treated humins also shows clear changes in the aromatic region compared to the parent humin. Some Cα–Cβ correlations were still present, but cross peaks from Cβ–Cβ correlations were not observed any more. While this could mean that phenolic structures have formed, Cβ–Cγ correlations expected for phenol were not found due to overlap with a new feature in the 2D spectrum; this new signal detected around 130 ppm can be attributed to the formation of aromatic conjugated structures.
 | ||
| Fig. 9 Aromatic region of the 2D CP-DQSQ spectra of 13C-labeled humins (blue) and alkali-treated humins (red). | ||
As the 1D CP 13C solid-state NMR showed that the alkali-treated humin does not contain many protonated carbon atoms, a DE DQSQ spectrum was recorded to study the structure in more detail (Fig. 10). Again, a correlation between Cα and Cβ was observed, but neither aliphatic linkages on the Cα nor Cβ–Cβ interactions, which would indicate the presence of furans, were present. If benzofuran would have formed upon alkaline treatment, as we previously suggested,20 a Cβ–Cβ correlation would be expected around δ = 110 ppm and δ = 130 ppm.27 This signal and Cβ–Cγ contacts from phenol, which is expected around the same chemical shift, were not observed in the DQSQ spectra. Formation of such species cannot be excluded, however, as such signals could be overlapped with the strong signals from arenes and fused rings. A strong peak at the diagonal around 130 ppm, reported to indicate the presence of graphene-like polycyclic aromatic sheets, was also absent.14,15 The spectrum thus suggests that the changes in molecular structure upon alkaline treatment involve further condensation of the humins to an arene-rich structure with some (benzo)furan or phenol rings (Fig. 11).
 | ||
| Fig. 10 2D DQSQ 13C solid-state NMR of alkali-treated, 13C-labeled humins obtained after DE of 13C. Please note, black regions are spinning side bands. | ||
 | ||
| Fig. 11 Proposed model for the molecular structure of alkali-treated, glucose-derived humins. | ||
Conclusions
The 1D and 2D solid-state NMR spectra of 13C-labeled humins provided further insight into the molecular structure of these highly complex and structurally heterogeneous materials and, most importantly, confirmed the furanic nature of these biorefinery by-products. Various different linkages could be identified in the 2D NMR spectra, ranging from the most abundant Cα–Caliphatic and Cα–Cα linkages to minor ones such as Cβ–Cβ and Cβ–Caliphatic cross-links. C–C correlations between the aliphatic carbons indicated that the linkers are not simply methylene groups, but rather comprise short aliphatic chains. The spectrum furthermore indicated that some LA, which is formed during acid-catalyzed conversion of sugars, is included in the molecular structure through covalent bonds. These results allowed us to refine the molecular structure previously proposed for humins (Fig. 7).Upon alkaline treatment of the humins, an arene-rich structure is formed at the expense of the furan content. A large increase in carboxylic acids was furthermore observed, which could in part explain the increased solubility of humins under alkaline conditions. It was also shown that the aliphatic and aromatic carbons are located far from each other, while CO functional groups are located on the aliphatic carbon atoms. Taken together, these observations allowed a molecular structure to be proposed also for the alkali-treated humins (Fig. 11).
Insight in the molecular structure of humins, with or without reactive solubilization, can contribute to the development of routes for their chemical valorization. Indeed, an analogy can be made here with the advanced NMR studies that have contributed much to the progress made in the structure elucidation of lignin, which is also a recalcitrant material from biomass with a complex, aromatic structure. These extensive lignin characterization efforts have led to increased understanding of the molecular structure including identification of the linkages within lignin, structural differences between lignins and changes in the structure upon (catalytic) conversion. For humins, comparable analytical strategies can be developed to further elucidate the molecular structure and its changes upon chemical treatment, which will aid the development of humin valorization processes.
Experimental section
Preparation of humins
Humins were prepared by heating 25 mL of an aqueous solution of 1 M D-glucose or 13C6D-glucose (99%, Buchem) and 0.01 M H2SO4 in an unstirred Teflon-lined autoclave vessel at 180 °C for 7 h in an oven. Humins were isolated by filtration, washed with water and dried at RT. The humins were submitted to a 24 h Soxhlet extraction with water and dried at 70 °C under vacuum.17 The alkaline pretreatment consisted of treating 0.5 g humin with 80 mL 0.5 M NaOH for 3 h in an unstirred Teflon-lined autoclave vessel in an oven at 240 °C. The humins were precipitated by decreasing the pH to 1 using 6 M H2SO4, isolated by filtration, washed with excess water and dried at 70 °C under vacuum.20 IR spectra were recorded on a Tensor 37 IR spectrometer using a diamond ATR crystal. For each spectrum 32 scans with a resolution of 4 cm−1 were averaged. The elemental composition of the humins was 59.6%C, 3.8%H and 36.6%O as determined with an automated Euro EA3000 CHNS. Oxygen content was calculated by difference.NMR analysis
DE spectra were recorded with a 5 μs 90° pulse and 14 kHz (alkali-treated humin) or 8 kHz (humin) MAS. The 13C CP MAS spectra were recorded with a CP contact time of 400 μs and 8 (humin) kHz or 14 kHz (alkali-treated humin) MAS. The processing was done using 100 Hz line broadening. Both type of experiments were performed on a 9.4 T NMR instrument (Bruker Biospin) using 3.2 mm MAS rotors.Proton-detected heteronuclear correlation (HETCOR) spectra were recorded at 60 kHz spinning using an 18.8 T Bruker spectrometer (Bruker Biospin) using 1.3 mm MAS rotors. Ramped CP contact times of 900 μs (1st CP) and 300 μs (2nd CP) (Fig. 4a) and 2.5 ms for both CP steps (Fig. 4b) were used. For both HETCOR experiments, the heteronuclear decoupling sequence PISSARRO28 was applied during t1 with 165 kHz and during t2 with 16 kHz. The number of TD points (and acquisition time) were 268 (9.0 ms), 250 (2.5 ms) for direct and indirect dimension respectively. For signal averaging, 16 scans were used. 4k and 1k zero filling were used for the direct and the indirect acquisition dimension, respectively. A sine-squared window function with a sine bell shift of 3 was applied to the spectrum.
Double quantum-single quantum experiments were done at 10 kHz MAS (3.2 mm rotor) at 9.4 T. The CP contact time was 600 μs and double-quantum excitation–reconversion was done using the SPC5 sequence29 for a total duration of 2 ms to compensate for residual offset effects and dipolar scaling due to motion.30
A recycle delay of 2 s was used. Heteronuclear decoupling during t1 (CW) and t2 (SPINAL64)31 were 83 kHz and during excitation–reconversion (CW) 120 kHz. The number of TD points (acquisition time) were 768 (7.7 ms), 120 (1.5 ms) for the direct and indirect dimension, respectively. For signal averaging, 128 scans were used. For both dimensions, 1k zero filling was applied. A sine-squared window function with a sine bell shift of 3 was applied to the spectrum.
Experimental conditions for the DE DQSQ were similar to the DQSQ (with CP). The recycle delay was 15 s. For signal averaging, 128 scans were used. For both dimensions, 1k zero filling was used and a sine-squared window function with a sine bell shift of 3 was applied to the spectrum.
Acknowledgements
This research has been performed within the framework of the CatchBio program. The authors gratefully acknowledge the support of the Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science. NMR studies were supported by NWO (grants 722.012.002 to M.W. and 700.26.121 and 700.10.443 to M.B.). Dr Peter de Peinder from VibSpec is thanked for help with the interpretation of the ATR-IR spectra.Notes and references
- J. Horvat, B. Klaić, B. Metelko and V. Šunjić, Tetrahedron Lett., 1985, 26, 2111–2114 CrossRef CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- D. J. Hayes, J. Ross, M. H. B. Hayes and S. W. Fitzpatrick, in Biorefineries - Industrial Processes and Products, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley Verlag, 2006, pp. 139–164 Search PubMed.
- J.-M. Pin, N. Guigo, A. Mija, L. Vincent, N. Sbirrazzuoli, J. C. van der Waal and E. de Jong, ACS Sustainable Chem. Eng., 2014, 2, 2182–2190 CrossRef CAS.
- T. M. C. Hoang, L. Lefferts and K. Seshan, ChemSusChem, 2013, 6, 1651–1658 CrossRef CAS PubMed.
- C. B. Rasrendra, M. Windt, Y. Wang, S. Adisasmito, I. G. B. N. Makertihartha, E. R. H. van Eck, D. Meier and H. J. Heeres, J. Anal. Appl. Pyrolysis, 2013, 104, 299–307 CrossRef CAS PubMed.
- E. Bartels, Über die Bildung von Huminstoffen aus Ketosen durch Einwirkung konzentrierter Halogenwasserstoffsäuren, Hamburg University, 1966 Search PubMed.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- S. K. R. Patil, J. Heltzel and C. R. F. Lund, Energy Fuels, 2012, 26, 5281–5293 CrossRef CAS.
- I. V. Sumerskii, S. M. Krutov and M. Y. Zarubin, Russ. J. Appl. Chem., 2010, 83, 320–327 CrossRef CAS.
- N. Baccile, G. Laurent, F. Babonneau, F. Fayon, M. M. Titirici and M. Antonietti, J. Phys. Chem. C, 2009, 113, 9644–9654 CAS.
- M. Sevilla and A. B. Fuertes, Chem. – Eur. J., 2009, 15, 4195–4203 CrossRef CAS PubMed.
- G. A. Morris and R. Freeman, J. Am. Chem. Soc., 1979, 101, 760–762 CrossRef CAS.
- R. L. Johnson, J. M. Anderson, B. H. Shanks, X. Fang, M. Hong and K. Schmidt-Rohr, J. Magn. Reson., 2013, 234, 112–124 CrossRef CAS PubMed.
- J. M. Anderson, R. L. Johnson, K. Schmidt-Rohr and B. H. Shanks, Carbon, 2014, 74, 333–345 CrossRef CAS PubMed.
- T. M. C. Hoang, E. R. H. van Eck, W. P. Bula, J. G. E. Gardeniers, L. Lefferts and K. Seshan, Green Chem., 2015, 17, 959–972 RSC.
- I. van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef CAS PubMed.
- G. C. A. Luijkx, F. van Rantwijk and H. van Bekkum, Carbohydr. Res., 1993, 242, 131–139 CrossRef CAS.
- M. Gomes and A. Gandini, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3759–3768 CrossRef CAS PubMed.
- I. van Zandvoort, E. R. H. van Eck, P. de Peinder, P. C. A. Bruijnincx and B. M. Weckhuysen, ACS Sustainable Chem. Eng., 2015, 3, 533–543 CrossRef CAS.
- M. Baldus, Prog. Nucl. Magn. Reson. Spectrosc., 2002, 41, 1–47 CrossRef CAS.
- N. Baccile, C. Falco and M. M. Titirici, Green Chem., 2014, 16, 4839–4869 RSC.
- W. Hübner and H. H. Mantsch, Biophys. J., 1991, 59, 1261–1272 CrossRef.
- C. Burket, R. Rajagopalan, a. Marencic, K. Dronvajjala and H. Foley, Carbon, 2006, 44, 2957–2963 CrossRef CAS PubMed.
- W. Kolodziejski and J. Klinowski, Chem. Rev., 2002, 102, 613–628 CrossRef CAS PubMed.
- D. Hobuß, S. Laschat and A. Baro, Synlett, 2005, 123–124 Search PubMed.
- T. Okuyama and T. Fueno, Bull. Chem. Soc. Jpn., 1974, 47, 1263–1266 CrossRef CAS.
- M. Weingarth, P. Tekely and G. Bodenhausen, Chem. Phys. Lett., 2008, 466, 247–251 CrossRef CAS PubMed.
- M. Hohwy, C. M. Rienstra, C. P. Jaroniec and R. G. Griffin, J. Chem. Phys., 1999, 110, 7983–7992 CrossRef CAS PubMed.
- R. Schneider, K. Seidel, M. Etzkorn, A. Lange, S. Becker and M. Baldus, J. Am. Chem. Soc., 2010, 132, 223–233 CrossRef CAS PubMed.
- B. Fung, A. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc00327j |
| ‡ These authors have equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2015 |