
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn integrated biorefinery concept for olive mill waste management: supercritical CO2 extraction and energy recovery†
Andrea
Schievano
*a,
Fabrizio
Adani
*a,
Li
Buessing
b,
Alfonso
Botto
c,
Esteve N.
Casoliba
d,
Mara
Rossoni
a and
Jillian L.
Goldfarb
e
aDipartimento di Scienze Agrarie ed Ambientali (DISAA), Università degli Studi di Milano, via Celoria 2, 20133 Milano, Italia. E-mail: andrea.schievano@unimi.it; fabrizio.adani@unimi.it
bDepartment of Chemical Engineering, University of New Hampshire, 33 Academic Way, Durham, NH 03824, USA
cSepareco S.r.l., via Rivarossa 15, 10060 Piscina (TO), Italy
dVeolia Water Solutions & Technologies, 4760 World Houston Parkway, Ste. 140, Houston, TX 77032, USA
eDepartment of Mechanical Engineering and Division of Materials Science & Engineering, Boston University, 110 Cummington Mall, Boston, MA 02215, USA
First published on 3rd March 2015
Abstract
Commercial production of olive oil generates four times the amount of waste as it does oil, along with a number of environmental issues. We propose an integrated biorefinery concept for the management of pomace, i.e. solid Olive Mill Waste (OMW), that utilizes supercritical carbon dioxide (SCO2), coupled with a polar co-solvent (Ethanol), for extracting value-added polyphenols and mono/poly-unsaturated fatty acids (MUFA/PUFA), followed by thermochemical (oxidation or pyrolysis) recovery of energy, biofuels and materials. The SCO2 + EtOH extraction recovered 77.6 g of freeze-dried extract per kg of raw OMW, with relatively high concentrations in polyphenols (10.9 g kg−1 of which 60.1% of di-hydroxytyrosol), PUFA (20 g kg−1), MUFA (601 g kg−1) and other valuable compounds, such as squalene (10 g kg−1). All these substances are of extreme interest in pharmaceutical and nutraceutical market, for their antioxidant, anti-cancer, functional, anti-bacterial and nutritional properties. The SCO2 + EtOH flux acted as physical/chemical carrier for over 85% of humidity, leaving the exhaust OMW almost dry, with evident advantages for downstream. Using nonisothermal thermogravimetric analysis, the apparent activation energies required to pyrolyze OMW to produce fuel and biochar ranged from 20 to 140 kJ mol−1 depending on heating ramp rate and temperature regime. BET analysis of unactivated biochars show increased (+25%) mesopore surface areas after SCO2 extractions (up to 500 m2 g−1). A more in-depth view on the proposed biorefinery is needed, to consider the overall energy balance, as well as possible market values of the obtained extract, and evaluate the real feasibility of the proposed concept.
Introduction
According to the International Olive Council (IOC), the global production of olive oil reached 2.9 million metric tons in the 2012–2013 harvest.1 The Mediterranean Basin and the Middle East accounted for 95.9% of the total world olive oil production in this harvest (latest available data), with Spain alone accounting for nearly 34% of the total global production.1 The commercial production of olive oil generates upwards of four times the amount of waste as it does oil, representing a heavy burden on the olive oil industry, a threat to the environment, and the potential waste of a useable series of byproducts. The majority of olive mills utilize a three-phase centrifugation system, introduced in the 1970s, that requires large amounts of water and produces two types of waste: one in the form of a wastewater, known as olive mill wastewater, black water, or alpechin, and the other in the form of a solid waste, known as pomace, or sansa,2 hereafter called olive mill waste (OMW). These systems and types of waste are one of the most diffuse in the Mediterranean area.3A variety of sources demonstrate the high and variable – from 0.02 g kg−1 up to 10 g kg−1 – amounts of biophenols (e.g. hydroxytyrosol, tyrosol, caffeic acid, rutin, luteolin, flavonoids) present in OMW that vary seasonally and geographically, and depend on the type of milling.4,5 The polyphenols present in olives and in olive oil are known to have anti-oxidant, anti-inflammatory and anti-microbial properties.6 Most of them are insoluble in oil and thereby remain in OMW and in wastewaters.6 It is these biophenols that may hamper efforts to dispose of OMW and treat wastewater; high concentrations of phenolic compounds can be phytotoxic and bacteriostatic.7 In many regions of Europe, both OMW and wastewaters are often spread directly on land as fertilizers. Low concentrations of OMW in soils have been observed in some cases to increase the organic carbon, aggregate stability, available potassium, and cation-exchange capacity of soil, all of which aid crop production.8 On the other hand, especially when large amounts of such material are used in soil, the net effect on crops and on soil micro flora is questionable, given the toxicity at high doses.9
At the same time, one of the primary biophenols present, hydroxytyrosol, retails for upwards of $500 (U.S.) for 100 mg at 98% purity.10 Nutraceutical products like capsules with extracted concentrates from olives and/or olive-tree fractions may reach market prices of around 100–200 € for 100 mg of hydroxytyrosol.11 Removing these biophenols from OMW solves the phytotoxic disposal issue as well as provides a revenue stream for the use of polyphenols in the health food, cosmetic, and pharmaceutical industries.12
Moreover, other interesting and valuable compounds can be extracted from OMW, especially regarding the fat fractions, rich unsaturated fatty acids (UFA) and squalene.12 Squalene, in particular, is an intermediate metabolite in the synthesis of cholesterol and phytosterols. In humans, about 60% of dietary squalene is absorbed and is distributed ubiquitously in tissues, being one of the major components of the epidermal lipids. Supplementation of the diet with squalene can reduce cholesterol and triglyceride levels. Acting as a quencher of singlet oxygen, squalene functions in protecting skin surface from lipid peroxidation.13
There are several methods under consideration for the extraction of biophenols, UFA and squalene from olive byproducts. From olive mill wastewaters, the main separation strategy involves the use of successive micro- and nano-membrane filtrations.14 The concentrated sludge must undergo further extraction and refinement processes, similarly to those used for OMW. These methods include ultrasound-assisted extraction;15 solvent extraction;16 superheated liquid extraction17 and supercritical fluid extraction.18
The difficulty in separating phenolic compounds from the waste comes from the hydrophilic and amphiphilic natures of the phenolic compounds. Supercritical fluid extraction is suitable for extracting molecules for human consumption because it eliminates the harsh solvents used in conventional extractions. CO2 is the most common supercritical fluid used because it is a nontoxic, nonflammable, widely available and inexpensive at high purity solvent, exhibiting moderate critical conditions (31.1 °C and 73.8 MPa) and can be easily separated because of its high volatility at normal conditions.19 There are several advantages to Supercritical Carbon Dioxide (hereafter SCO2) over organic solvent extraction. The first is solvency power, which can be changed easily by adjusting operating conditions (temperature and pressure), which in turn change the extraction capacity and selectivity to extract the desired compounds. The near ambient temperatures at which SCO2 proceeds gives it the advantage over conventional solvent methods run at higher temperatures, as there is less thermal stress on the desired extract.20 The main issue with SCO2 is that it is usually limited to low or medium polarity compounds due to the low polarity of CO2. A co-solvent (modifier) can increase extraction efficiency immensely and can be used to reduce operating pressure, extraction time, and also for extraction of polar compounds.21 The most common co-solvents are ethanol and methanol. Le Floch18 found methanol to be a better co-solvent than ethanol for extracting polyphenols from olive leaves, but due to the toxicity of methanol, ethanol is preferred for downstream human consumption. The extracted compound yield increases with constant temperature and increasing pressure, but decreases with increasing temperature at constant pressure due to the solvent density reduction.22
While mitigating the phytotoxicity of OMW by polyphenols extraction would enable more widespread use as a soil amendment, the vast quantities of OMW produced over a short time (3–4 month harvest) suggest that using extracted OMW as a fertilizer cannot be the sole method of disposal unless the waste is transported long distances to agricultural sites, thus increasing both costs and the carbon footprint of the waste.23,24 There are several viable bioenergy conversion pathways to consider with the OMW following SCO2, as seen in Fig. 1.
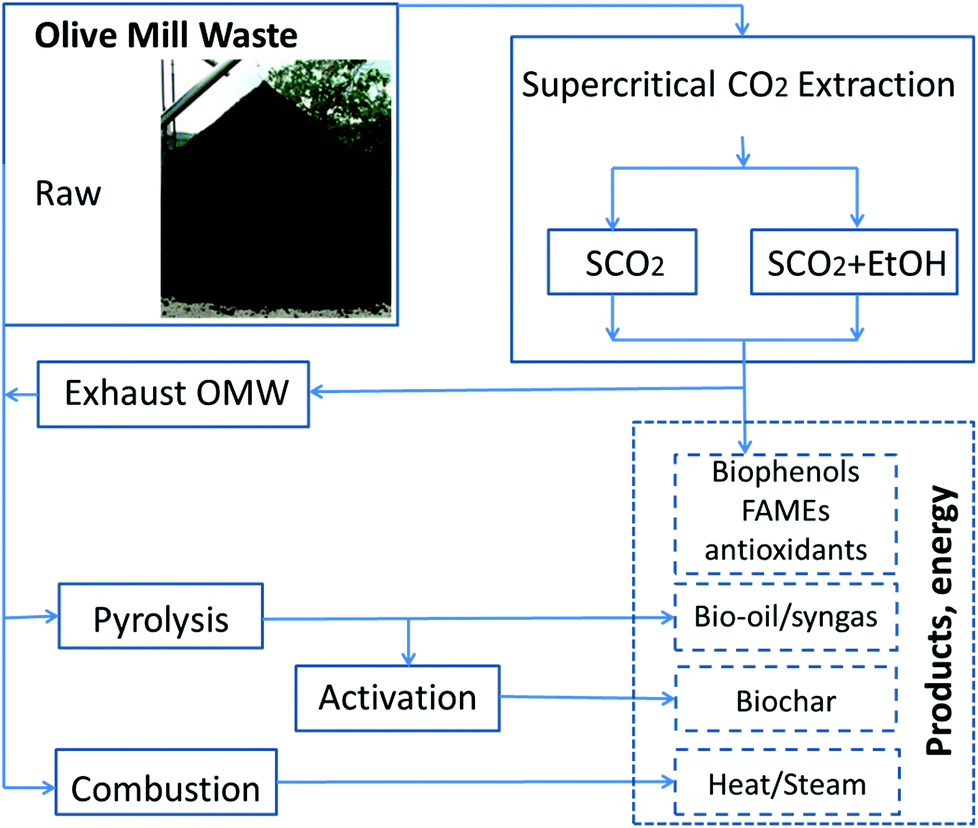 | ||
| Fig. 1 Potential biorefinery pathways for olive mill waste valorization; solid lines indicate OMW treatment paths, dashed lines indicate products recovered from treatment. | ||
One disposal method that is of interest in some locations is combustion of OMW. For centralized olive oil production facilities, where vast quantities of OMW are produced and land application is not an option, this process reduces the amount of waste disposed via oxidation, using the heat from combustion for evaporation of the humidity of the incoming waste streams and for other purposes.25 Combustion reduces the quantity of waste, but the extent of combustion, the profile of volatilized compounds, and the disposal of ash must all be addressed to insure that this process is industrially and environmentally feasible.
Another pathway that we consider here is the pyrolysis of OMW. Pyrolysis (heating in the absence of oxygen) can be used to produce a bio-oil or syngas that mimics petroleum-derived fuels, and a carbonaceous char with high specific surface areas.26 The temperature and heating rate of pyrolysis strongly affects the quantities of each product (bio-oil, syngas, char) recovered,27 and the development of industrial devolatilization units requires a complete understanding of pollutant evolution and kinetics modeling.28 There has been a significant amount of work done on the pyrolysis of raw OMW and OMW mixed with various other waste products.27,29–34 The calorific value of bio-oil extracted from raw OMW was found to be as high as 29 MJ kg−1 with a molecular formula of CH1.54N0.02O0.29, with maximum oil yield from fast pyrolysis at approximately 550 °C (E.U. 2011). Syngas produced from OMW pyrolyzed at 550 °C by Uzun35 was shown to contain approximately 50% CO2, 14% CO, 21% H2, balance roughly split between CH4, C2H4 and C2H6. However, overall little research has been done on such an integrated pathway, considering the changes in thermal decomposition of OMW following SCO2.
In this work, we explore an integrated biorefinery concept (Fig. 1) that aims to produce value-added products (high value antioxidants, biofuels, energy, sustainable source of carbon for soil) and at the same time solve concerns over the proper disposal of OMW. This new approach would further “green” the ancient practice of olive oil extraction.
Results and discussion
The total content of dry matter (DM) in fresh matter (FM), organic matter and ash in the raw OMW were 660, 587 and 63 g kg−1 FM; total polyphenols (TP) content was 1.809 g kg−1 FM (Table 1).| FM balance | DM concentration | DM balance | TP concentration | TP balance | |||||
|---|---|---|---|---|---|---|---|---|---|
| kg FM | % | gDM kg−1 FM | kg DM | % | gGAE kg−1 FM | g GAE | % | ||
| SCO2 | Raw pomace | 7.300 | 100 | 660 | 4.818 | 100 | 1.809 | 13.21 | 100 |
| Exaust pomace | 6.110 | 83.7 | 721 | 4.405 | 91.4 | 2.097 | 12.81 | 97.0 | |
| Extracts | |||||||||
| Acqueous extract | 1.008 | 13.8 | 240 | 0.242 | 5.0 | 0.252 | 0.25 | 1.9 | |
| Fat suspension | 0.182 | 2.5 | 883 | 0.161 | 3.3 | 0.741 | 0.13 | 1.0 | |
| Losses | 0.016 | 0.7 | — | 0.010 | 0.2 | — | 0.004 | 0.03 | |
| Freeze-dried extracts | 0.403 | 5.5 | 998 | 0.403 | 8.4 | 0.963 | 0.39 | 2.9 | |
| SCO2 + EtOH | Raw pomace | 7.260 | 660 | 4.792 | 76 | 1.809 | 13.13 | 100 | |
| EtOH | 1.500 | 1000 | 1.500 | 23.8 | — | — | — | ||
| Total | 8.760 | 100 | 6.292 | 100 | |||||
| Exaust pomace | 4.390 | 60.5 | 947 | 4.157 | 66.1 | 1.611 | 7.07 | 53.8 | |
| Extracts | |||||||||
| Emulsion (Fat + EtOH + water) | 4.230 | 58.3 | 487 | 2.059 | 32.7 | 1.420 | 6.01 | 45.7 | |
| Losses | 0.140 | 1.9 | — | 0.075 | 1.2 | — | 0.06 | 0.4 | |
| Freeze-dried extracts | 0.565 | 7.7 | 989 | 0.559 | 8.9 | 10.62 | 6.01 | 45.7 | |
SCO2 extractions from OMW
The mass balance of FM, DM and TP of the two extractions performed are reported in Table 1. SCO2 resulted in two separate phases: one aqueous and the second fat-like. The transport of water by the CO2 stream is related to a physical phenomenon (induced by pressure, heat and flux), while the fat fraction transport is both physical and chemical in nature (SCO2 behaves like a non-polar solvent), as indicated by Adani.36 Together, these two fractions accounted for 16.3% of initial FM and for 8.4% of initial DM (Table 1). Over 83% and 91% of initial FM and DM, respectively, were left in the exhaust OMW, while a negligible fraction was lost within the circuit (Table 1). The OMW was left relatively humid after extraction (Table 1).In SCO2 + EtOH, the aqueous and fat fractions were extracted as a homogeneous emulsion, probably attributed to the polar action of ethanol. For the same reason, the extraction and transport of the aqueous phase was more efficient (over 85% removal of initial moisture content) and the exhaust OMW remained almost dry at the end of the SCO2 + EtOH (947 gDM kg−1 FM, Table 1); the emulsion weighted almost 50% of the initial mass (including ethanol), with negligible losses (Table 1). At the same time, similarly to SCO2, the extracted DM represented 11.7% of the initial DM, while 86.8% of it remained in the exhaust.
Notable differences between SCO2 and SCO2 + EtOH were observed for TP extraction; it exceeded 45% yield in the extract of SCO2 + EtOH, while very weak extraction yields were achieved by SCO2, with 97% of the TP left in the exhaust OMW (Table 1). Both extracts, when freeze-dried, accounted for 5–6% by mass of the initial OMW and, while the SCO2 extract showed a TP concentration of 0.967 gGAE kg−1, the SCO2 + EtOH reached 10.86 gGAE kg−1.
Phenolic compound speciation indicated that di-hydroxytyrosol accounted for over 50% of TP in both extracts (Table 2). At the same time, the relative percentages of single phenolic compounds were similar in both extractions, though in SCO2 + EtOH all of them were nearly 10 times more concentrated than SCO2 alone (Table 2).
| Phenolic compounds | SCFE | SCFE + EtOH | FAMEa | SCFE | SCFE + EtOH | ||
|---|---|---|---|---|---|---|---|
| mg kg−1 DM | % of TP | mg kg−1 DM | % of TP | g kg−1 DM | g kg−1 DM | ||
| a FAME = fatty acids methyl esters; PUFA = poly-unsaturated fatty acids; MUFA = mono-unsaturated fatty acids; SAFA = saturated fatty acids. | |||||||
| Gallic acid | 44 | 4.7 | 312 | 3.1 | Methyl elaidate | 451 | 521 |
| 2,4-Diidroxybenzoic | 2 | 0.3 | 47 | 0.5 | Methyl palmitate | 148 | 210 |
| 4-Hydroxybenzoic | 10 | 1.1 | 117 | 1.2 | cis-Vaccenic acid | 69 | 64 |
| Tyrosol | 29 | 3.1 | 157 | 1.6 | Methyl 10-ketostearate | 59 | 0 |
| Caffeic acid | 5 | 0.6 | 85 | 0.8 | Methyl stearate | 58 | 51 |
| Chlorogenic acid | 78 | 8.3 | 759 | 7.6 | Methyl eicosanoate | 38 | 19 |
| Vanillic acid | 44 | 4.7 | 353 | 3.5 | Methyl palmitoleate | 17 | 17 |
| Syringic acid | 34 | 3.6 | 246 | 2.4 | Ethyl Oleate | 15 | 0 |
| Di-hydroxytyrosol simil | 512 | 54.1 | 6046 | 60.1 | Methyl linoleate | 13 | 10 |
| Ferulic acid | 42 | 4.4 | 281 | 2.8 | Methyl behenate | 8 | 7 |
| trans-p-Coumaric acid | 8 | 0.8 | 104 | 1.0 | Heptadecenoic acid, methyl ester | 4 | 12 |
| Luteolin7p-glucoside | 15 | 1.5 | 221 | 2.2 | Others | 29 | 16 |
| Oleuropein-glicone | 21 | 2.2 | 83 | 0.8 | |||
| Oleuropein | 18 | 1.9 | 193 | 1.9 | Total FAME | 908 | 925 |
| Cinnammic acid | 61 | 6.4 | 772 | 7.7 | PUFAa | 34 | 20 |
| Luteolin | 22 | 2.3 | 274 | 2.7 | MUFAa | 551 | 601 |
| Others | 21 | 2.3 | 808 | 8.0 | SAFAa | 315 | 299 |
| Squalene | 21 | 10 | |||||
Both extracts were substantially composed of fats (as all fatty acids and esters were converted into FAMEs before analysis, total FAME represented over 90% of DM, Table 2) and both of them composed of over 60% elaidate (i.e. a trans-isomer of oleate) and palmitate, while the rest was composed of 10 main compounds, as shown by the FAME speciation (Table 2). Mono-unsaturated (MU) and poly-unsaturated (PU) fatty acids (FA) were a considerable fraction of the fats, i.e. nearly 600 g kg−1 DM in both extracts (Table 2).
Among them, some compounds of particular interest were found in relevant concentrations: linoleate and cis-vaccenic acid were found in similar concentrations in both extracts (around 20 and 70 g kg−1 DM, respectively, Table 2). Squalene (2,6,10,15,19,23-hexamethyl tetracosahexaene) was also detected at concentrations of 21 and 10 g kg−1 DM in SCO2 and SCO2 + EtOH, respectively.
Of the two options of SCO2 investigated, coupling EtOH to CO2 resulted in enhancing, by a factor of 10, the extraction of phenolic compounds (Tables 1 and 2), while no significant differences were observed in extracting the fat fractions (Table 2). This was expected; a supercritical CO2 stream alone is known to possess the capability of extracting and transporting non-polar compounds, such as fats, and to have less of an effect on polar molecules, such as phenols.21 The addition of EtOH as a co-solvent optimized phenolic transport to the supercritical fluid phase. On the other hand, EtOH addition simultaneously resulted in a massive transport of the initial moisture of OMW, with the exhaust OMW left nearly dried and the extract with over 50% water content (Table 1). This, in a scaled-up process, would impose higher energy requirement to dry or freeze-dry the extract, as compared to SCO2, where the moisture content of the extract was around 20% (Table 1). However, SCO2 alone was not sufficient to achieve satisfactory extraction of the polyphenols (Table 1).
The compositions of both extracts in terms of FAME were found similar to typical olive oil.37 Elaidate is the trans-isomer of oleate and, together with palmitate, represented the large majority of both extracts. Squalene, well known for showing important anti-tumor, anti-oxidant and functional activity in the human body,12 was also found in relatively high concentrations (10–20 g kg−1 DM, Table 2), when compared with typical concentrations found in literature for olive oils (4–10 g kg−1 of olive oil).38 This is regardless of the use of EtOH in the extraction, Squalene being soluble in solvents like hexane or SCO2 (Table 2).
Kinetics of oxidation and pyrolysis of raw and treated OMW
As seen in Table 3, the impact of extraction on total elemental composition of carbon, hydrogen and nitrogen, by mass, was minimal and close to standard experimental errors of ±0.4%. This suggests that the thermal reactivities of the materials should be similar.| Sample | %Carbon | %Nitrogen | %Hydrogen |
|---|---|---|---|
| Raw | 47.59 ± 0.061 | 0.14 ± 0.001 | 6.25 ± 0.042 |
| SCO2 | 47.08 ± 0.061 | 0.14 ± 0.006 | 6.35 ± 0.006 |
| SCO2 + EtOH | 46.82 ± 0.055 | 0.14 ± 0.006 | 6.34 ± 0.006 |
Experiments were conducted to determine the impact of extraction treatment on the pyrolysis and oxidation kinetics; two particle sizes (125–300 μm and 300–500 μm) were analyzed to further probe the effect of particle size on the apparent activation energy. It was observed by Van de Velden39 that there are mass transfer limitations in the pyrolysis of larger particles as larger particles and higher heating rates cause a temperature gradient from the outside to the center of the particle. Fig. 2 is a representative thermogravimetric (TG) curve with an inset derivative thermogravimetric (DTG) curve for the pyrolysis of raw OMW at each particle size and heating ramp rate used. We clearly see from these results that the kinetics of thermal decomposition is significantly influenced by the heating rate and to a lesser extent by the particle size (Fig. 2). Table 4 presents the peak mass loss temperature and rate (determined through DTG curves of each sample, as shown the ESI in Tables S1–S4 and Fig. S2 and S3†) for each sample. We see that the peak DTG points occur within approximately 600–630 K for pyrolysis, and 550–580 K for oxidation. For both pyrolysis and oxidation, the higher heating rates display higher peak temperatures (on the order of ∼20 K higher for each sample), no matter the particle size. Fig. 3 illustrates the DTG curves of oxidation for the raw and extracted OMW samples at 100 K min−1 for small and larger particles; we note the shapes of the DTG curves are similar for each sample, but that the maximum rate of mass loss is higher for the smaller particle sizes. Both of these observations point to clear heat and mass transport limitations: higher heating rates result in higher peak mass loss temperatures and bigger particles lead to lower peak mass loss rates. Therefore, we note that the activation energies presented herein are “global” or “apparent” activation energies, encompassing these transport limitations within the reaction chemistry to provide “lumped” activation energy of the particles in question and the applied heating rate. Jauhiainen40 presented a thorough discussion on the simultaneous decomposition of biomass in the context of OMW pyrolysis and oxidation in an attempt to explain some of the TG and DTG behavior of OMW conversion. Ounas41 found similar TG behavior for the pyrolysis of olive residue at heating rates ranging from 2 to 50 K min−1 as we observe here. The extraction treatment does not appear to largely impact the DTG results, though we do observe a significant impact of extraction treatment on the global activation energies of pyrolysis and oxidation for the OMW, despite these potential heat and mass transfer limitations.
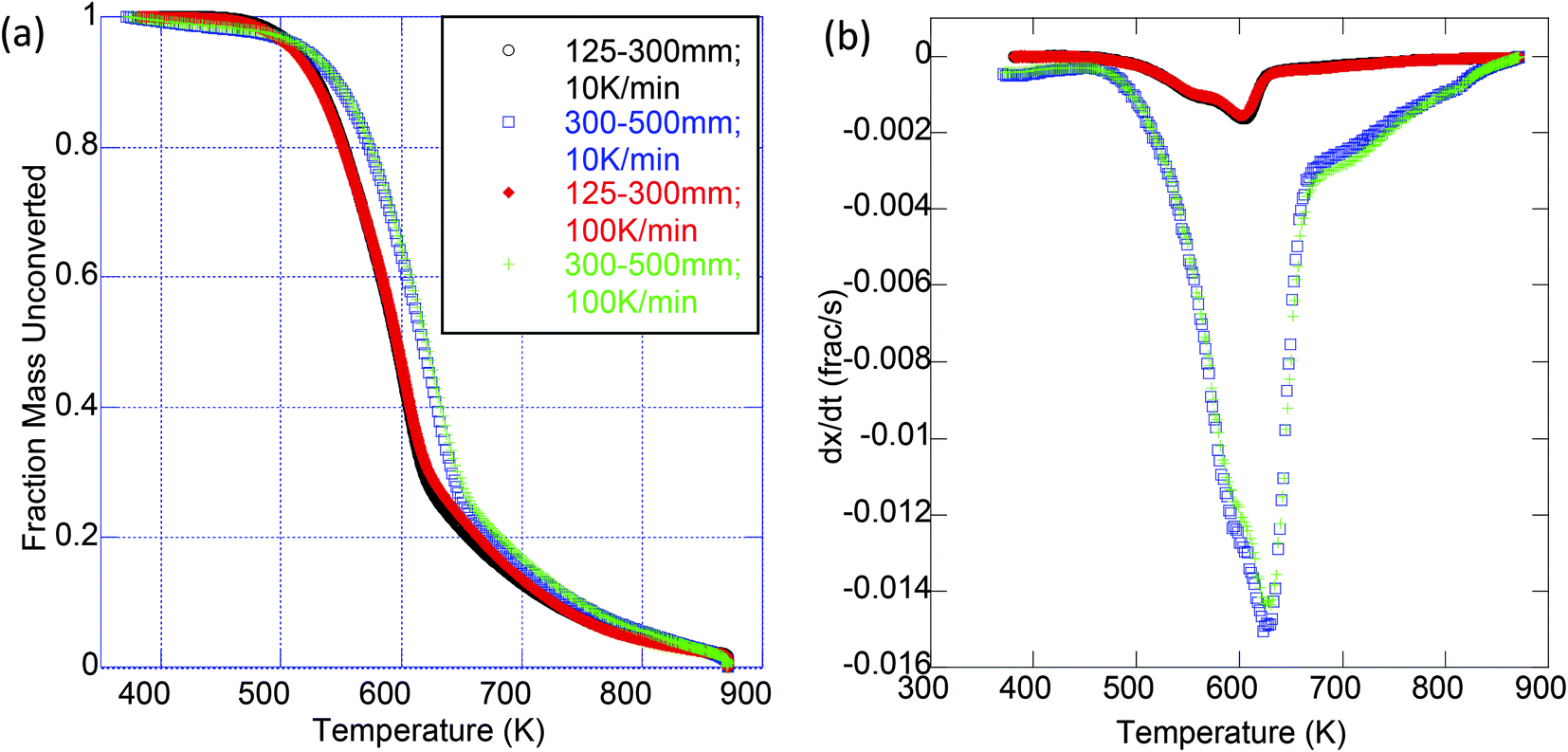 | ||
Fig. 2 Pyrolysis of raw olive mill waste (○) 125–300 μm, 10 K min−1; ( ) 300–500 μm, 10 K min−1; ( ) 300–500 μm, 10 K min−1; ( ) 125–300 μm 100 K min−1; ( ) 125–300 μm 100 K min−1; ( ) 300–500 μm, 100 K min−1. (a) TG curve (fraction of mass remaining as a function of temperature). (b) DTG curve (rate of fractional mass loss/time as a function of temperature). ) 300–500 μm, 100 K min−1. (a) TG curve (fraction of mass remaining as a function of temperature). (b) DTG curve (rate of fractional mass loss/time as a function of temperature). | ||
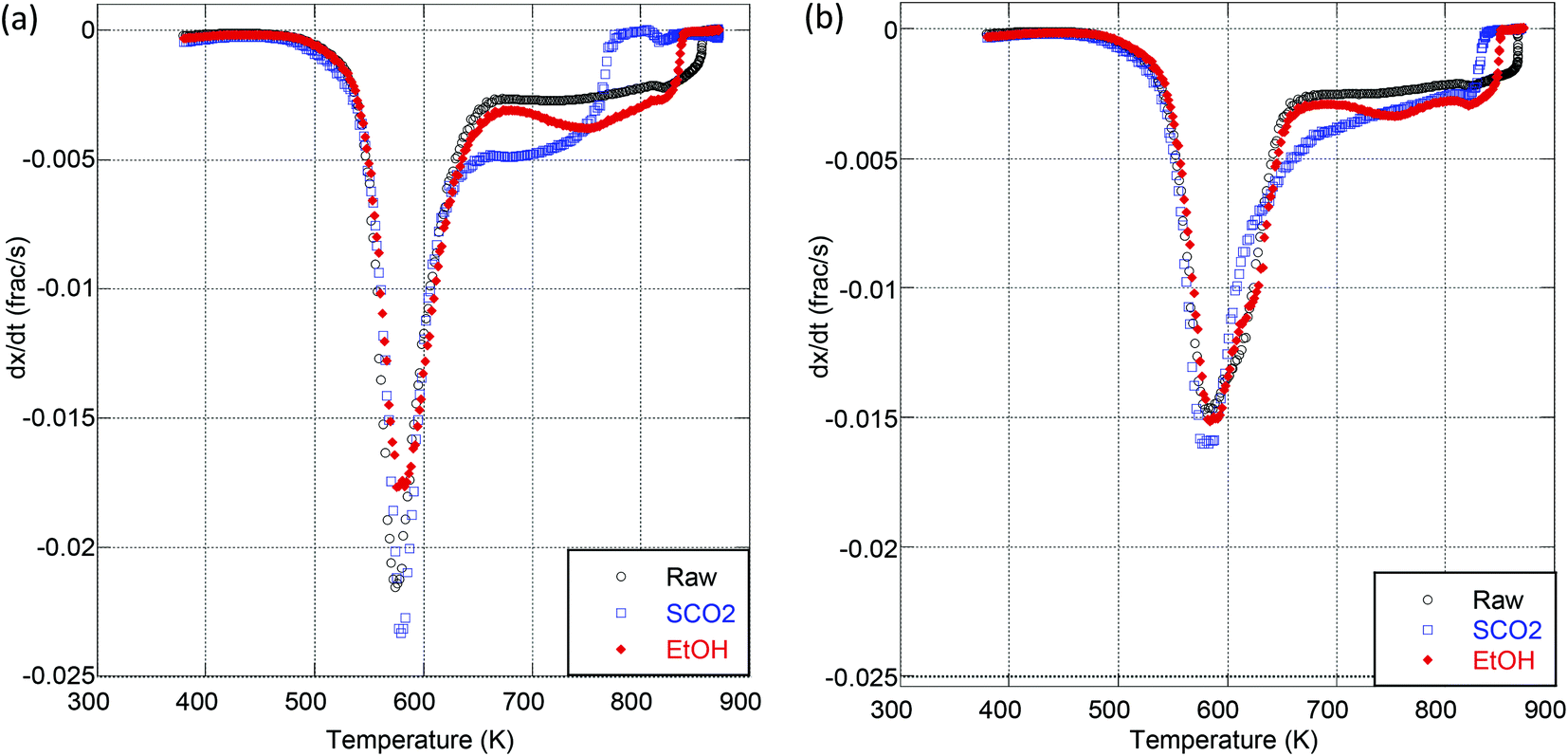 | ||
Fig. 3 DTG curves of oxidation of raw and extracted olive mill waste at 100 K min−1 for (a) 125–300 μm particles and (b) 300–500 μm particles. (a) (○) Raw 125–300 μm, 100 K min−1; ( ) SCO2 125–300 μm, 100 K min−1; ( ) SCO2 125–300 μm, 100 K min−1; ( ) SCO2 + EtOH 125–300 μm 100 K min−1. (b) (○) Raw 300–500 μm, 100 K min−1; ( ) SCO2 + EtOH 125–300 μm 100 K min−1. (b) (○) Raw 300–500 μm, 100 K min−1; ( ) SCFE 300–500 μm, 100 K min−1; ( ) SCFE 300–500 μm, 100 K min−1; ( ) EtOH 300–500 μm 100 K min−1. ) EtOH 300–500 μm 100 K min−1. | ||
| Mass loss regime 1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Thermal treatment | Sample | Particle size range, mm | Heating rate, °C min−1 | Onset temperature (K) | Endset temperature (K) | Activation energy (kJ mol−1) | Pre-exponential factor (s−1) | Mass fraction loss | Peak DTG temperature (K) | Peak DTG rate (g s−1) |
| Pyrolysis (N2) | Raw | 125–300 | 10.0 | 470.7 ± 0.1 | 560.1 ± 0.03 | 74.5 ± 0.7 | 8.30 × 104 ± 1.38 × 104 | 0.30 | 605.4 | −0.00194 |
| 100.0 | 476.5 ± 0.3 | 575.8 ± 0.3 | 71.7 ± 1.1 | 2.17 × 104 ± 5.98 × 103 | 0.27 | 624.4 | −0.01513 | |||
| 300–500 | 10.0 | 470.7 ± 0.05 | 560.1 ± 0.01 | 68.9 ± 0.7 | 2.34 × 104 ± 3.64 × 103 | 0.30 | 607.3 | −0.00163 | ||
| 100.0 | 476.7 ± 0.1 | 576.2 ± 0.2 | 71.5 ± 1.1 | 2.04 × 104 ± 4.63 × 103 | 0.26 | 630.1 | −0.01505 | |||
| SCO2 | 125–300 | 10.0 | 470.8 ± 0.1 | 560.1 ± 0.1 | 58.8 ± 0.6 | 3.94 × 103 ± 2.96 × 103 | 0.30 | 597.8 | −0.00171 | |
| 100.0 | 477.0 ± 0.3 | 576.2 ± 0.5 | 60.0 ± 0.3 | 1.69 × 103 ± 9.32 × 101 | 0.27 | 623.2 | −0.01442 | |||
| 300–500 | 10.0 | 470.8 ± 0.05 | 560.0 ± 0.03 | 60.4 ± 1.8 | 3.40 × 103 ± 1.27 × 103 | 0.28 | 601.0 | −0.00158 | ||
| 100.0 | 487.4 ± 0.5 | 576.2 ± 0.6 | 57.7 ± 0.2 | 9.54 × 102 ± 5.11 × 101 | 0.25 | 623.2 | −0.01302 | |||
| SCO2 + EtOH | 125–300 | 10.0 | 470.6 ± 0.1 | 559.9 ± 0.02 | 63.8 ± 0.9 | 7.22 × 103 ± 1.60 × 103 | 0.28 | 596.6 | −0.00177 | |
| 100.0 | 476.6 ± 0.2 | 575.6 ± 0.4 | 60.1 ± 0.3 | 1.66 × 103 ± 1.16 × 102 | 0.26 | 625.4 | −0.01411 | |||
| 300–500 | 10.0 | 470.7 ± 0.03 | 560.0 ± 0.02 | 64.2 ± 0.9 | 7.18 × 103 ± 1.35 × 103 | 0.26 | 603.2 | −0.00165 | ||
| 100.0 | 477.1 ± 0.4 | 576.2 ± 0.6 | 60.3 ± 0.2 | 1.53 × 103 ± 1.04 × 102 | 0.24 | 629.8 | −0.01315 | |||
| Oxidation (Air) | Raw | 125–300 | 10.0 | 505.0 ± 0.04 | 545.0 ± 0.2 | 138.2 ± 0.9 | 2.14 × 1011 ± 5.18 × 1010 | 0.27 | 553.5 | −0.00190 |
| 100.0 | 529.7 ± 0.4 | 529.7 ± 0.5 | 171.2 ± 1.0 | 6.87 × 1013 ± 1.59 × 1013 | 0.39 | 574.1 | −0.02193 | |||
| 300–500 | 10.0 | 504.9 ± 0.04 | 544.4 ± 0.2 | 135.1 ± 2.0 | 1.18 × 1011 ± 4.94 × 1010 | 0.30 | 557.3 | −0.00200 | ||
| 100.0 | 529.7 ± 0.5 | 529.7 ± 0.6 | 139.2 ± 3.9 | 5.33 × 1010 ± 4.59 × 1010 | 0.34 | 582.0 | −0.01484 | |||
| SCO2 | 125–300 | 10.0 | 505.4 ± 0.1 | 545.2 ± 0.3 | 124.3 ± 0.8 | 9.09 × 109 ± 1.83 × 109 | 0.26 | 577.3 | −0.01020 | |
| 100.0 | 529.7 ± 0.4 | 529.7 ± 0.4 | 138.6 ± 2.1 | 1.86 × 1013 ± 3.21 × 1013 | 0.39 | 578.8 | −0.02375 | |||
| 300–500 | 10.0 | 505.3 ± 0.1 | 544.8 ± 0.1 | 126.3 ± 4.6 | 1.46 × 1010 ± 2.26 × 1010 | 0.27 | 559.9 | −0.00190 | ||
| 100.0 | 529.7 ± 0.1 | 529.7 ± 0.2 | 137.9 ± 5.3 | 4.70 × 1010 ± 3.59 × 1010 | 0.35 | 583.6 | −0.01637 | |||
| SCO2 + EtOH | 125–300 | 10.0 | 505.4 ± 0.1 | 545.6 ± 0.2 | 136.5 ± 1.8 | 1.84 × 1011 ± 8.68 × 1010 | 0.32 | 553.5 | −0.00187 | |
| 100.0 | 529.7 ± 0.4 | 529.7 ± 0.5 | 147.5 ± 4.6 | 4.48 × 1011 ± 4.70 × 1011 | 0.36 | 580.0 | −0.09178 | |||
| 300–500 | 10.0 | 505.3 ± 0.04 | 544.8 ± 0.05 | 143.1 ± 0.3 | 7.30 × 1011 ± 4.20 × 1010 | 0.30 | 551.6 | −0.00211 | ||
| 100.0 | 529.7 ± 0.2 | 529.7 ± 0.3 | 137.6 ± 3.3 | 2.89 × 1010 ± 1.73 × 1010 | 0.34 | 582.0 | −0.01537 | |||
| Mass loss regime 2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Thermal treatment | Sample | Particle size range, mm | Heating rate, °C min−1 | Onset temperature (K) | Endset temperature (K) | Activation energy (kJ mol−1) | Pre-exponential factor (s−1) | Mass fraction loss | Peak DTG temperature (K) | Peak DTG rate (g s−1) |
| Pyrolysis (N2) | Raw | 125–300 | 10.0 | 579.8 ± 0.03 | 607.8 ± 1.0 | 87.6 ± 0.2 | 8.75 × 105 ± 3.49 × 104 | 0.38 | 605.4 | −0.00194 |
| 100.0 | 603.9 ± 0.2 | 628.3 ± 0.2 | 74.4 ± 1.6 | 3.21 × 104 ± 9.71 × 103 | 0.38 | 624.4 | −0.01513 | |||
| 300–500 | 10.0 | 579.8 ± 0.03 | 600.6 ± 2.3 | 85.3 ± 1.9 | 5.52 × 105 ± 2.17 × 105 | 0.33 | 607.3 | −0.00163 | ||
| 100.0 | 609.6 ± 0.3 | 625.3 ± 0.3 | 72.4 ± 4.3 | 2.44 × 104 ± 1.70 × 104 | 0.32 | 630.1 | −0.01505 | |||
| SCO2 | 125–300 | 10.0 | 576.4 ± 0.1 | 596.9 ± 0.04 | 81.4 ± 1.0 | 1.63 × 105 ± 1.42 × 105 | 0.33 | 597.8 | −0.00171 | |
| 100.0 | 604.1 ± 0.5 | 628.4 ± 0.5 | 67.2 ± 0.9 | 7.56 × 103 ± 1.39 × 103 | 0.37 | 623.2 | −0.01442 | |||
| 300–500 | 10.0 | 576.4 ± 0.03 | 596.9 ± 0.03 | 72.8 ± 1.0 | 3.86 × 104 ± 7.07 × 103 | 0.32 | 601.0 | −0.00158 | ||
| 100.0 | 604.2 ± 0.6 | 628.5 ± 0.7 | 58.4 ± 1.8 | 1.21 × 103 ± 3.83 × 102 | 0.35 | 623.2 | −0.01302 | |||
| SCO2 + EtOH | 125–300 | 10.0 | 576.2 ± 0.02 | 596.7 ± 0.02 | 80.8 ± 0.4 | 2.18 × 105 ± 1.54 × 104 | 0.33 | 596.6 | −0.00177 | |
| 100.0 | 603.5 ± 0.5 | 627.7 ± 0.5 | 66.1 ± 1.6 | 6.10 × 103 ± 1.89 × 103 | 0.37 | 625.4 | −0.01411 | |||
| 300–500 | 10.0 | 576.3 ± 0.02 | 596.8 ± 0.02 | 72.3 ± 0.7 | 3.28 × 104 ± 5.34 × 103 | 0.32 | 603.2 | −0.00165 | ||
| 100.0 | 604.0 ± 0.6 | 628.3 ± 0.6 | 56.9 ± 4.2 | 1.04 × 103 ± 9.38 × 102 | 0.35 | 629.8 | −0.01315 | |||
| Oxidation (Air) | Raw | 125–300 | 10.0 | 566.6 ± 0.4 | 580.7 ± 0.6 | 40.5 ± 0.3 | 6.89 × 101 ± 2.24 × 101 | 0.33 | 553.5 | −0.00190 |
| 100.0 | 667.4 ± 0.7 | 809.1 ± 0.8 | 36.7 ± 8.5 | 8.10 × 100 ± 1.16 × 101 | 0.50 | 574.1 | −0.02193 | |||
| 300–500 | 10.0 | 565.5 ± 0.3 | 579.1 ± 0.3 | 17.7 ± 2.0 | 7.24 × 10−1 ± 5.04 × 10−1 | 0.41 | 557.3 | −0.00200 | ||
| 100.0 | 666.6 ± 0.2 | 809.6 ± 0.3 | 20.4 ± 2.1 | 1.67 × 10−1 ± 5.84 × 10−2 | 0.48 | 582.0 | −0.01484 | |||
| SCO2 | 125–300 | 10.0 | 566.7 ± 0.7 | 582.3 ± 3.5 | 50.8 ± 14.0 | 2.28 × 102 ± 3.51 × 102 | 0.35 | 577.3 | −0.01020 | |
| 100.0 | 667.0 ± 1.1 | 803.5 ± 0.6 | 42.8 ± 3.4 | 1.78 × 101 ± 5.45 × 100 | 0.56 | 578.8 | −0.02375 | |||
| 300–500 | 10.0 | 565.9 ± 0.2 | 579.7 ± 0.3 | 29.2 ± 1.7 | 7.03 × 100 ± 2.58 × 100 | 0.41 | 559.9 | −0.00190 | ||
| 100.0 | 666.3 ± 0.5 | 809.1 ± 0.3 | 35.0 ± 3.7 | 4.26 × 100 ± 2.70 × 100 | 0.54 | 583.6 | −0.01637 | |||
| SCO2 + EtOH | 125–300 | 10.0 | 567.0 ± 0.3 | 580.9 ± 0.4 | 36.5 ± 3.4 | 3.17 × 101 ± 3.72 × 101 | 0.34 | 553.5 | −0.00187 | |
| 100.0 | 666.7 ± 0.9 | 807.6 ± 2.0 | 35.2 ± 4.1 | 4.34 × 100 ± 2.09 × 100 | 0.58 | 580.0 | −0.09178 | |||
| 300–500 | 10.0 | 565.9 ± 0.1 | 579.4 ± 0.1 | 16.5 ± 1.8 | 5.42 × 10−1 ± 2.02 × 10−1 | 0.41 | 551.6 | −0.00211 | ||
| 100.0 | 665.9 ± 0.3 | 808.9 ± 0.5 | 35.9 ± 3.9 | 3.22 × 100 ± 1.73 × 100 | 0.54 | 582.0 | −0.01537 | |||
| Mass loss regime 3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Thermal treatment | Sample | Particle size range, mm | Heating rate, °C min−1 | Onset temperature (K) | Endset temperature (K) | Activation energy (kJ mol−1) | Pre-exponential factor (s−1) | Mass fraction loss | Peak DTG temperature (K) | Peak DTG rate (g s−1) |
| Pyrolysis (N2) | Raw | 125–300 | 10.0 | 639.0 ± 0.03 | 737.9 ± 0.04 | 7.5 ± 0.2 | 3.15 × 10−2 ± 9.46 × 10−3 | 0.22 | 605.4 | −0.00194 |
| 100.0 | 683.1 ± 0.3 | 731.9 ± 0.3 | 14.4 ± 1.2 | 1.13 × 10−1 ± 2.39 × 10−2 | 0.18 | 624.4 | −0.01513 | |||
| 300–500 | 10.0 | 639.0 ± 0.03 | 737.8 ± 0.1 | 11.6 ± 1.9 | 8.35 × 10−2 ± 2.72 × 10−2 | 0.27 | 607.3 | −0.00163 | ||
| 100.0 | 683.6 ± 0.4 | 725.8 ± 0.5 | 22.3 ± 1.4 | 4.73 × 10−1 ± 1.22 × 10−1 | 0.19 | 630.1 | −0.01505 | |||
| SCO2 | 125–300 | 10.0 | 639.0 ± 0.04 | 737.8 ± 0.02 | 3.4 ± 0.8 | 1.81 × 10−2 ± 4.73 × 10−3 | 0.27 | 597.8 | −0.00171 | |
| 100.0 | 683.0 ± 0.6 | 731.8 ± 0.6 | 10.0 ± 0.6 | 5.84 × 10−2 ± 5.53 × 10−3 | 0.19 | 623.2 | −0.01442 | |||
| 300–500 | 10.0 | 639.0 ± 0.03 | 737.8 ± 0.03 | 13.8 ± 1.1 | 1.39 × 10−1 ± 2.68 × 10−2 | 0.31 | 601.0 | −0.00158 | ||
| 100.0 | 683.2 ± 0.7 | 732.1 ± 0.7 | 24.5 ± 1.1 | 8.23 × 10−1 ± 1.64 × 10−1 | 0.20 | 623.2 | −0.01302 | |||
| SCO2 + EtOH | 125–300 | 10.0 | 638.9 ± 0.03 | 737.8 ± 0.1 | 13.3 ± 2.6 | 1.31 × 10−1 ± 6.80 × 10−2 | 0.29 | 596.6 | −0.00177 | |
| 100.0 | 682.4 ± 0.7 | 731.2 ± 0.7 | 16.5 ± 0.7 | 1.82 × 10−1 ± 2.01 × 10−2 | 0.19 | 625.4 | −0.01411 | |||
| 300–500 | 10.0 | 639.0 ± 0.01 | 738.0 ± 0.02 | 28.9 ± 3.6 | 2.42 × 100 ± 1.23 × 100 | 0.31 | 603.2 | −0.00165 | ||
| 100.0 | 683.0 ± 0.7 | 731.9 ± 0.7 | 30.4 ± 3.6 | 2.53 × 100 ± 1.28 × 100 | 0.23 | 629.8 | −0.01315 | |||
| Oxidation (Air) | Raw | 125–300 | 10.0 | 641.7 ± 0.3 | 680.3 ± 0.8 | 141.2 ± 5.9 | 3.94 × 109 ± 3.57 × 109 | 0.40 | 553.5 | −0.00190 |
| 100.0 | 810.7 ± 0.8 | 847.9 ± 2.8 | 181.3 ± 9.1 | 1.52 × 1010 ± 1.55 × 1010 | 0.06 | 574.1 | −0.02193 | |||
| 300–500 | 10.0 | 641.1 ± 0.3 | 674.8 ± 0.4 | 180.0 ± 9.0 | 2.37 × 1013 ± 3.00 × 1013 | 0.30 | 557.3 | −0.00200 | ||
| 100.0 | 811.2 ± 0.3 | 850.6 ± 0.01 | 168.4 ± 4.0 | 7.22 × 108 ± 3.72 × 108 | 0.08 | 582.0 | −0.01484 | |||
| SCO2 | 125–300 | 10.0 | 641.1 ± 0.6 | 676.6 ± 2.6 | 160.2 ± 8.6 | 5.19 × 1011 ± 3.71 × 1011 | 0.39 | 577.3 | −0.01020 | |
| 100.0 | 805.0 ± 0.6 | 842.1 ± 0.2 | 118.9 ± 9.4 | 1.44 × 107 ± 1.42 × 107 | 0.01 | 578.8 | −0.02375 | |||
| 300–500 | 10.0 | 641.5 ± 0.2 | 675.3 ± 0.4 | 132.6 ± 7.7 | 2.82 × 109 ± 3.53 × 109 | 0.33 | 559.9 | −0.00190 | ||
| 100.0 | 810.7 ± 0.3 | 830.5 ± 0.8 | 141.2 ± 17.3 | 2.49 × 107 ± 2.05 × 107 | 0.04 | 583.6 | −0.01637 | |||
| SCO2 + EtOH | 125–300 | 10.0 | 642.0 ± 0.2 | 679.3 ± 3.3 | 160.0 ± 3.9 | 2.06 × 1011 ± 2.29 × 1011 | 0.34 | 553.5 | −0.00187 | |
| 100.0 | 809.1 ± 2.1 | 843.6 ± 1.4 | 108.4 ± 4.1 | 8.20 × 105 ± 8.30 × 105 | 0.01 | 580.0 | −0.09178 | |||
| 300–500 | 10.0 | 641.4 ± 0.1 | 674.9 ± 0.2 | 162.7 ± 8.3 | 9.63 × 1011 ± 1.29 × 1012 | 0.29 | 551.6 | −0.00211 | ||
| 100.0 | 810.5 ± 0.5 | 830.9 ± 0.4 | 133.1 ± 7.6 | 9.80 × 106 ± 9.32 × 106 | 0.04 | 582.0 | −0.01537 | |||
For both pyrolysis and oxidation, we see three mass loss regimes (Table 4), or distinct regions on the Arrhenius plots, characterized by linear ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k vs. 1/T portions with abrupt discontinuities. This behavior was observed by many across the biomass literature for both pyrolysis and combustion processes.32,39,42,43 The temperatures at which these discontinuities occur are relatively independent of particle size and heating rate for pyrolysis, and are strongly influenced by heating rate during oxidation, as seen in Table 4 and Fig. 4 and 5. We have labeled each of these discrete sections “Mass Loss Regimes”. For pyrolysis of lignocellulosic materials, these three regimes are often roughly attributed to the decomposition of hemicellulose, cellulose and lignin, and for the oxidation process to the pyrolysis of volatiles, followed by the oxidation of these volatiles and finally resulting char oxidation. The activation energies of pyrolysis ranged from 57.7–74.5 kJ per mole in regime 1, from 56.9–87.6 kJ per mole in regime 2, and 3.4–30.4 in regime 3. In their pyrolysis of solid OMW, Taralas44 reported an overall activation energy of approximately 90 kJ mol−1 for particles between 0.5 and 1 mm up to 975 K. Ounas41 reported activation energies obtained from the Ozawa–Flynn–Wall and Vyazovkin methods for fractional conversions of OMW ranging from 148–219 kJ mol−1 pyrolyzed at 2, 10, 20 and 50 K min−1 with an average particle size of 0.2 mm. Jauhiainen40 find two different mass loss regimes for the pyrolysis under helium at 5, 10 and 20 K min−1 of olive mill stones, ranging from 69.4–181.8 kJ mol−1 using a modified Arrhenius equation that optimizes the pre-exponential factor and kinetic constant at a given temperature using a fourth-order Runge–Kutta method. They do not report a particle size.
k vs. 1/T portions with abrupt discontinuities. This behavior was observed by many across the biomass literature for both pyrolysis and combustion processes.32,39,42,43 The temperatures at which these discontinuities occur are relatively independent of particle size and heating rate for pyrolysis, and are strongly influenced by heating rate during oxidation, as seen in Table 4 and Fig. 4 and 5. We have labeled each of these discrete sections “Mass Loss Regimes”. For pyrolysis of lignocellulosic materials, these three regimes are often roughly attributed to the decomposition of hemicellulose, cellulose and lignin, and for the oxidation process to the pyrolysis of volatiles, followed by the oxidation of these volatiles and finally resulting char oxidation. The activation energies of pyrolysis ranged from 57.7–74.5 kJ per mole in regime 1, from 56.9–87.6 kJ per mole in regime 2, and 3.4–30.4 in regime 3. In their pyrolysis of solid OMW, Taralas44 reported an overall activation energy of approximately 90 kJ mol−1 for particles between 0.5 and 1 mm up to 975 K. Ounas41 reported activation energies obtained from the Ozawa–Flynn–Wall and Vyazovkin methods for fractional conversions of OMW ranging from 148–219 kJ mol−1 pyrolyzed at 2, 10, 20 and 50 K min−1 with an average particle size of 0.2 mm. Jauhiainen40 find two different mass loss regimes for the pyrolysis under helium at 5, 10 and 20 K min−1 of olive mill stones, ranging from 69.4–181.8 kJ mol−1 using a modified Arrhenius equation that optimizes the pre-exponential factor and kinetic constant at a given temperature using a fourth-order Runge–Kutta method. They do not report a particle size.
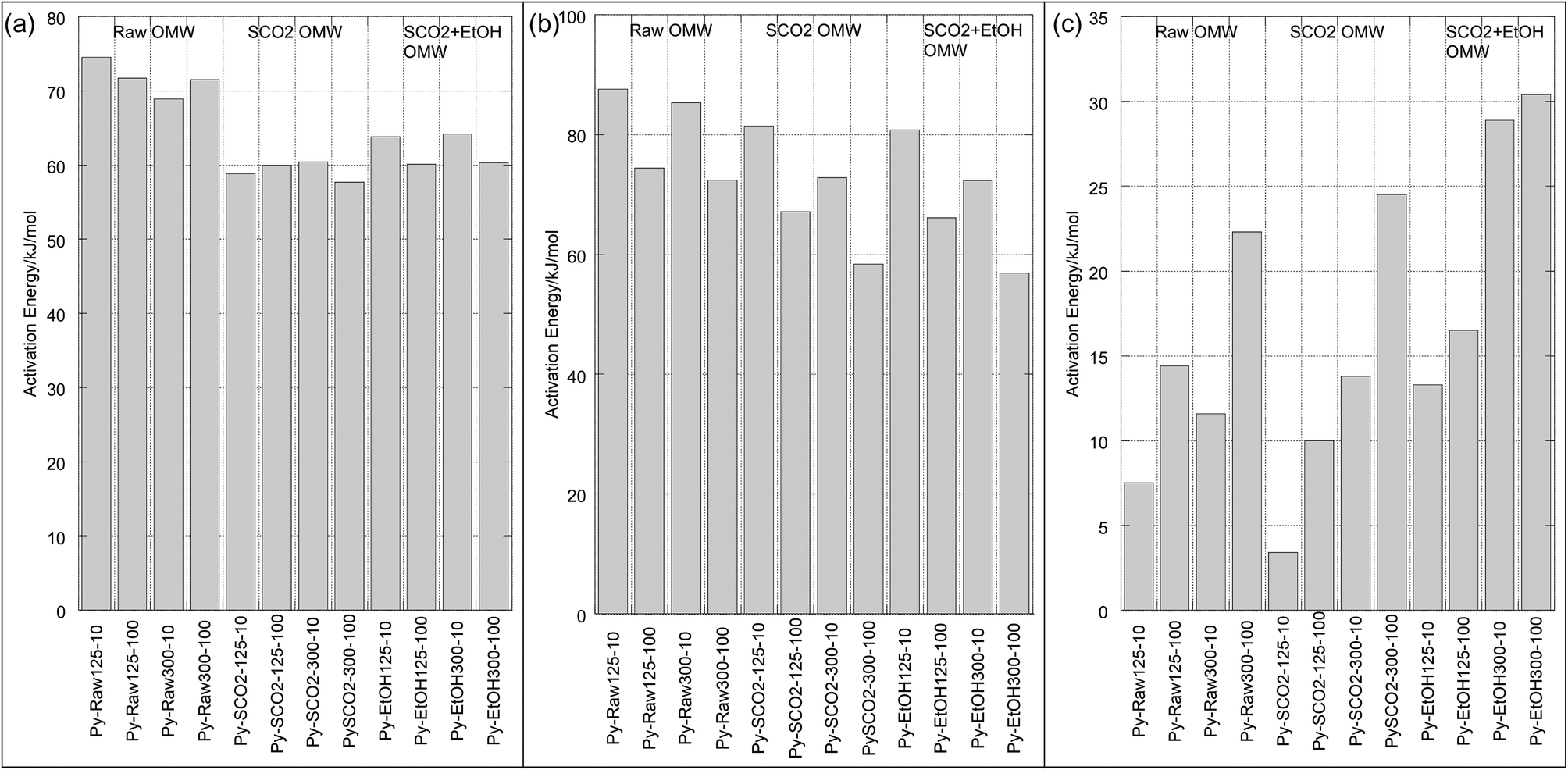 | ||
| Fig. 4 Activation energies of pyrolysis for each mass loss regime [x-axis labels: treatment (pyrolysis or oxidation) – sample (raw, SCO2, SCO2 + EtOH)/size (125–300 or 300–500 μm) – heating rate (10 or 100 K min−1)]. (a) Mass loss regime 1. (b) Mass loss regime 2. (c) Mass loss regime 3. | ||
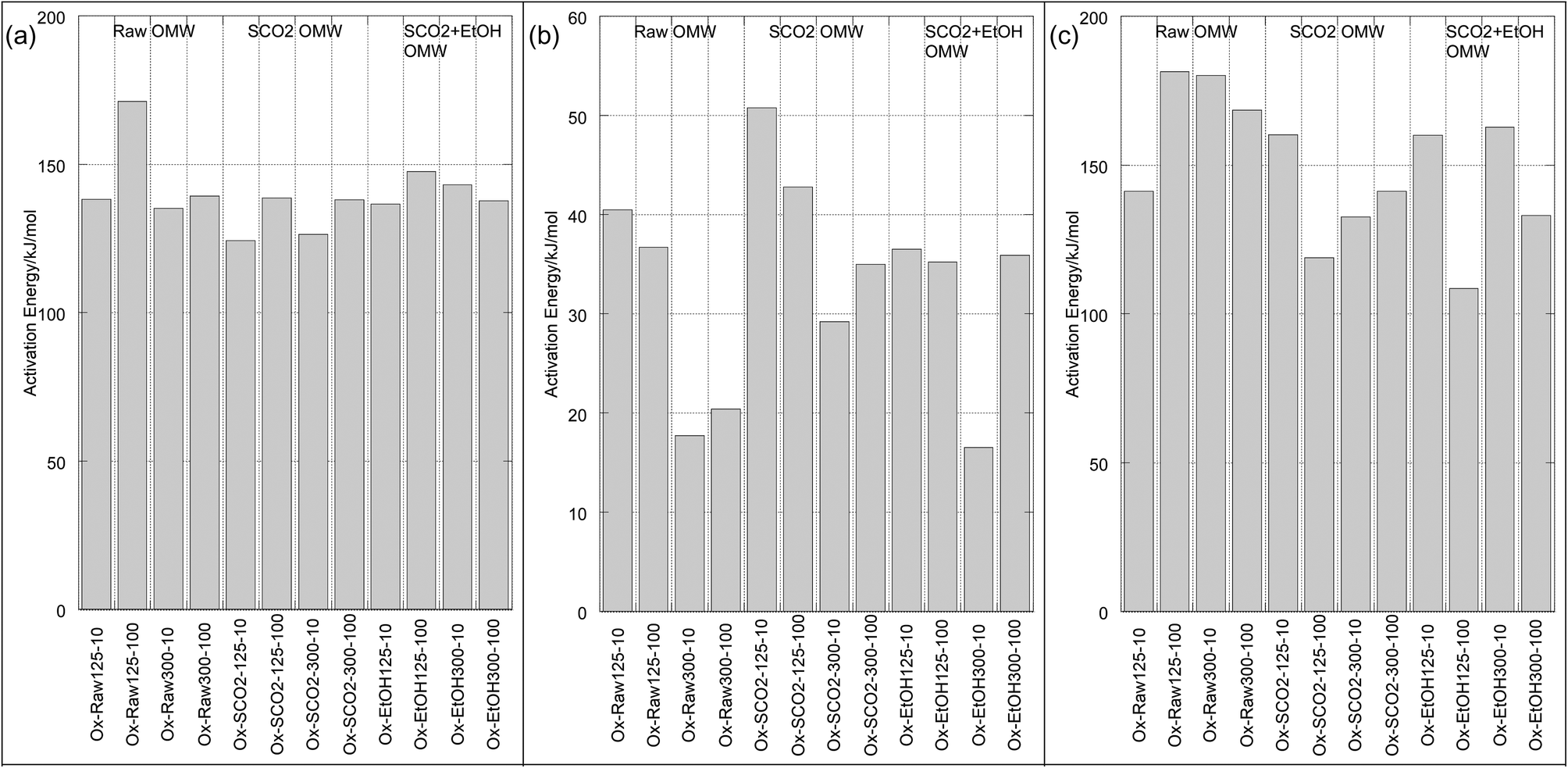 | ||
| Fig. 5 Activation energies of oxidation for each mass loss regime [x-axis labels: treatment (pyrolysis or oxidation) – sample (raw, SCO2, SCO2 + EtOH)/size (125–300 or 300–500 μm) – heating rate (10 or 100 K min−1)]. (a) Mass loss regime 1. (b) Mass loss regime 2. (c) Mass loss regime 3. | ||
In the low temperature mass loss regime, the raw untreated OMW showed approximately 10% higher apparent activation energy for pyrolysis than SCO2 or SCO2 + EtOH in the first and second mass loss regimes (Table 4). It is plausible that the hemicellulosic materials were physically and chemically disrupted, and/or the removal of the fat fractions (see DM balance in Table 1) decreased the apparent activation energies of the treated materials; given scant qualitative evidence on SEM imaging (Fig. 6) this is likely a chemically induced transformation, though as the elemental distribution of C, H, N across samples is similar (Table 3) it seems possible that polymeric cellulose chains were disrupted. In fact, an emerging topic in the biomass to bioenergy conversion literature is the variety of potential pretreatment options to enhance digestibility of lignocellulosic materials45 and CO2 “explosion pretreatment” (SCO2 at approximately 200 °C, 1000–4000 psi, i.e. at higher temperature than here) has been shown to form carbonic acid, which hydrolyzes hemicellulose, and also increases the accessible surface area of the biomass,46 lending credence to the more chemically induced nature of this treatment.
 | ||
| Fig. 6 SEM images of raw and extracted olive mill waste biochars (125–300 μm) before and after pyrolysis at 600 °C. a. Raw OMW. b. SCO2 OMW. c. SCO2 + EtOH OMW. d. Pyrolyzed raw OMW. e. Pyrolyzed SCO2 OWM. f. Pyrolyzed SCO2 + EtOH OMW. | ||
In the third (high temperature) pyrolysis mass loss regime we see a distinct effect of particle size and heating rate on the predominantly lignin decomposition. The apparent activation energies of the larger particles are up to twice as big as the smaller particles, likewise the slower heating rate has substantially lower activation energy for each particle size. In this case, the activation energy (for the same particle size/heating rate) is higher for the SCO2 + EtOH than for the SCO2, which is higher than the raw OMW. This indicates a high likelihood that the SCO2 and SCO2 + EtOH pre-treatment effect only the cellulose and hemicellulose portions of the OMW. Lignin is known to decompose from ∼190 to 600 °C; at lower temperature regimes the raw OMW is likely decomposing more lignin, as it is the “glue” that holds together the cellulose and hemicellulose. This “glue” was likely disrupted by hemicellulose hydrolysis during SCO2 treatment, thereby condensing the lignin together and causing a more energy-intensive, delayed decomposition at higher temperature.
The effect of extraction treatment is somewhat more limited on the oxidation kinetics of the OMW. The activation energies, with the exception of the raw OMW 125–300 μm, 100 °C min−1 sample, were all within ∼25 kJ mol−1 of each other as seen in Table 4. The first and third mass loss regimes – representing devolatilization and char oxidation, respectively – have similarly high apparent activation energies (124.3 to 171.2 and 108.4 to 181.3, respectively), whereas the second regime, representing volatile oxidation and continuing devolatilization is substantially lower (ranging from 16.5 to 50.8 kJ mol−1) for all samples. It is not clear that the extraction pre-treatment has any impact on the energy required to initiate combustion of OMW. Jauhiainen40 report oxidation activation energies of OMW cake of 153.7, 66.4 and 133.3 kJ mol−1 for each of three mass loss regimes, in excellent agreement with our results.
Porosity development via extraction and pyrolysis of OMW
BET adsorption isotherms showed a high degree of linearity within the 0–0.35 P/P0 range, and yielded specific surface areas up to 538 m2 gcarbon−1 for the SCO2 + EtOH OMW (Table 5). The specific surface area of the SCO2 OMW is over 10% greater than the OMW, and the SCO2 + EtOH OWM is over 25% greater than the OMW (Table 5). The pyrolyzed OMW samples are highly mesoporous, conforming to typical type IV isotherms. Interestingly, González47 find BET surface areas of only 53 m2 gchar−1, for olive stones pyrolyzed under nitrogen at 600 °C for 60 min. On a per gram char basis, our surface areas are over an order of magnitude larger; this is likely due to the considerably larger particles (1–2 mm) used by González.47| Carbon content (gcarbon/gsample) | Specific surface area per gram sample (m2 gchar−1) | Specific surface area per gram carbon (m2 gcarbon−1) | |
|---|---|---|---|
| Raw OMW | 0.808 | 341.5 | 422.7 |
| SCO2 OMW | 0.729 | 344.3 | 472.3 |
| SCO2 + EtOH OMW | 0.832 | 447.9 | 538.3 |
In Fig. 6, presenting SEM images of raw and extracted OMW samples, we see some evidence of structural change within these samples, namely that the raw OMW are more morphologically heterogeneous with larger particle agglomerates than either of the two SCO2-extracted samples.
Overview of the proposed biorefinery chain
The over-arching theme of the experimental work was to probe a potential biorefinery chain to improve the possibilities of managing the vast quantities of OMW produced globally. The use of SCO2 coupled with a polar co-solvent (Ethanol) is to be preferred to the sole SCO2, as the polarity ensures maximized extraction of bio-phenols. UFA-rich extracts of potential interest in nutraceutics/pharmaceutics can be obtained, with interesting concentrations of di-hydroxytyrosol, squalene and other high-value compounds. The extraction treatments (especially SCO2 + EtOH) influenced both oxidation and pyrolysis processes: slight decrease in activation energies, consistent increase in specific surface area and evident structural modifications at level of mesopores. Interestingly, the SCO2 + EtOH flux was found to act as physical/chemical carrier for over 85% of the initial moisture content of the raw OMW. This is of fundamental importance for the efficiency of successive pyrolysis/combustion processes. On the other side, as aqueous fraction is transferred to the obtained extract, heat would be required to freeze-dry it and recover ethanol by distillation. Here, this step, as well as an energetic, mass transfer and economic balance of the overall proposed biorefinery chain is left open for future deepening of this study.Experimental
The OMW samples were obtained from an olive oil processing facility in Andria (BA, Italy). Extraction of polyphenols was carried out using a SCO2 pilot plant (details follow), granted by Separeco Srl, Italy. Analytical characterizations were carried out at University of Milan. Thermochemical conversion experiments and analysis of chars were carried out at the University of New Hampshire and Boston University.Supercritical CO2 extractions
The polyphenols were extracted from the OMW using a pilot-scale plant (SFE100 Series Plant – Separeco Srl, Italy; Fig. S1 in ESI†). The plant contains an extractor of 14 dm3, a gravity separator of 5 dm3, 2 cyclonic separator of 3 dm3, a condenser, a heater and two heat exchangers. The extractions were performed on samples of fresh raw OMW of nearly 7 kg FM, with a density of approximately 0.53 kg dm−3. Extracting conditions were set as follows: pressure 250 bar, temperature 70 ± 1 °C, CO2 flow rate 80 kg h−1. The extraction was prolonged until no further weight was extracted from the sample. Two types of SCO2 extractions were tested: one with pure SCO2 and the other using ethanol as a co-solvent (SCO2 + EtOH). Ethanol was added to the biomass in the ratio of 20% w/w, corresponding to a ratio of 0.25% w/w EtOH–CO2. Extraction times resulted of 420 and 480 min for SCO2 and SCO2 + EtOH, respectively. After extractions, concentrated extracts were freeze-dried to concentrate fat and phenolic fractions. All extracts and the exhaust OMW obtained were weighed and analyzed to determine their DM and TP contents, to draw a mass balance around the extraction process.Determination of phenolic compounds and fatty acids methyl esters in OMW and SCO2 extracts
The total phenolic compounds (TP) content was determined colorimetrically at 765 nm using Foline–Ciocalteu reagent, according to Singleton,48 and the results were expressed as gallic acid equivalents (GAE) in g kg−1 FM. The composition of the raw untreated and supercritical fluid extracted olive mill wastes was determined via HPLC on a Finnigan Thermo Surveyor instrument, constituted by a LC Pump Plus, an Autosampler Plus and a PDA Plus diode array detector settled on 280 nm fixed wavelength and in scan mode. A Nova-Pak C18 column (300 mm × 3.9 mm, 4 μm – Waters) was used at room temperature with a 90 min gradient of water–acetic acid (98/2) (solvent A) and 0.5% of acetic acid in water–acetonitrile (solvent B) at a flow rate of 0.8 ml min−1 and 10 μl injection volume. The gradient program was operated from 10% to 15% of B from 0–10 min, held for 3 min and increased in a linear gradient to 100% (10–65 min).Fatty acids profiles were determined after esterification of lipids and detection by GC-MS analysis (Agilent 6850 Series, Agilent Technologies). The chloroform phase, obtained as reported for the lipids extraction, was evaporated at 30 °C using a rotary evaporator under a nitrogen flux. After that, 4 mL of 6% sodium hydroxide dissolved in methanol:dH2O (4:1 v/v) was added to the sample which was maintained at 60 °C for 3 hours in a thermostatic bath. Fatty acids trans-esterification was achieved by adding to the sample 4 mL of a boron trifluoride: methanol solution and by heating the sample for 30 min in a vapor recovery system. Esterified fatty acids were extracted twice with 5 mL of hexane. 1 μl of each extract was then injected in the GC-MS apparatus, using a non-polar column HP-5 (30 m, 0.25 i.d., 0.25 μm film thickness). Total analysis time was 96.75 min. and the flow rate was 1.20 mL min−1 with helium as the carrier gas. Quantification of fatty acids was determined injecting an external multiple standard GRAIN FAME (Supelco).
Activation energies of pyrolysis and oxidation
The exhaust OMW samples were dried and ground in a ball mill and mechanically sieved to yield particles between 125–300 and 300–500 μm. Elemental analysis of carbon, nitrogen and hydrogen contents of each sample was determined by LECO Corporation and reported in Table 3. The apparent activation energies required to pyrolyze and oxidize the raw and extracted OMW samples were measured using non-isothermal thermo-gravimetric analysis over two different heating rates and two particle sizes using a Mettler Toledo TGA/DSC-1. Between 5 and 15 mg of each sample were loaded into a 70 μL alumina crucible to achieve a thin layer on the bottom of the pan to prevent mass transfer limitations. Samples were pyrolyzed (or oxidized) under 50 mL min−1 of N2 to provide an oxygen-free environment (and run in air at the same flow rate for oxidation) with nitrogen as the protective gas at 10 mL min−1. The method started by heating the OMW to 110 °C and holding it at 110 °C for 20 minutes to drive off water and purge the system. The samples were then cooled to 25 °C with continual nitrogen (air) flow. The analytical step was carried out under constant nitrogen (air) flow between 25 °C and 600 °C and held at 600 °C for 15 minutes, with heating rates of 10 °C min−1 and 100 °C min−1 to query the effect of heating rate on the apparent activation energy for each olive waste material. Each sample was repeated 3 times and a standard deviation of the three trials was calculated. The mass of the sample was logged every second to the 10−6 grams, along with time and temperature, accurate to 0.01 °C.Thermo-gravimetric analysis, or TGA, is often criticized for a lack of applicability to industry because it is often run at relatively slow heating rates (10–25 °C min−1). However, slower pyrolysis processes are often used to produce a variety of materials and biosynthetic fuels. As such, we query the effect of heating rate on thermal decomposition up to the experimentally reproducible range of our TGA, 100 °C min−1. While the oxidation reactivities measured here are at lower temperatures compared to small-particle industrial combustion, the particles will likely be within the Zone II kinetics regime at the initial stage of char combustion, shifting to Zone I near 100% burn-out. As such, low temperature measurements are useful in studying the latter stages of burn out for industrial applications, though they cannot be used to describe thermal annealing behavior of the char particles.49,50
Many kinetic studies of biomass thermal decomposition show a reaction order close to one; it is common in the biomass literature to apply this global or apparent reaction order to account for all the reactions occurring simultaneously during pyrolysis.51,52 By assuming an apparent reaction order of one, this enables determination of the pre-exponential factor (A) and apparent activation energy (Ea) via the Arrhenius equation of the form:
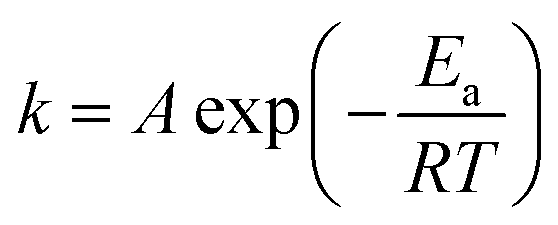 | (1) |
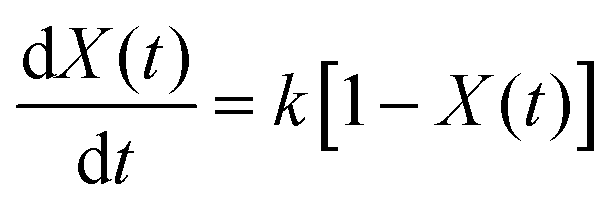 | (2) |
The temperature increases linearly with a constant heating rate seen in eqn (3).
| T = To(1 + αt) | (3) |
Eqn (2) can be rewritten taking the heating rate into account to yield eqn (4).
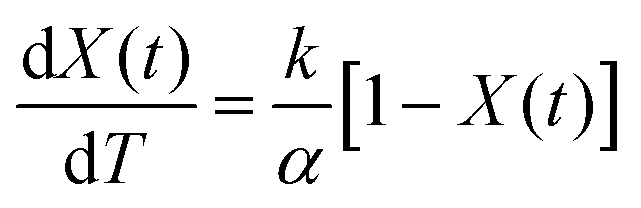 | (4) |
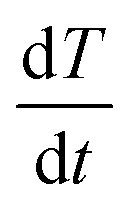 (K s−1) and X(t), the fractional decomposition, is given by:
(K s−1) and X(t), the fractional decomposition, is given by: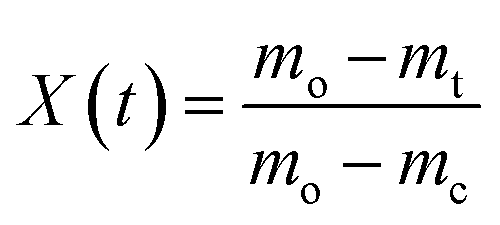 | (5) |
Physical characterization of the materials
Chars of the 125–300 μm particle size samples were prepared in an inert nitrogen environment (100 mL min−1 flow rate) in a 1′′ tube furnace. The samples were heated under nitrogen to and held at 110 °C for 1 hour to remove any moisture. The samples were then heated at a rate of 20 °C min−1 to 600 °C. The specific surface areas of raw OMW, SCO2, and SCO2 + EtOH and pyrolyzed OMW, at particle size fractions of 125–300 μm, were determined using a Micromeritics ASAP 2020 Sorption Analyzer. Approximately 400 mg of sample were degassed at 300 °C for 10 hours under high vacuum to remove any gases and vapors on the surfaces of the sample. The sample was then transferred from the degasser to the analyzer to determine the surface area and porosity through nitrogen adsorption at 77.35 K using the BET equation. The specific surface area on a per-gram of carbon basis was determined using the carbon content of the samples determined in the TGA oxidation experiments. Morphological changes occurring because of devolatilization were examined via scanning electron microscopy (SEM).Conclusions
The proposed biorefinery concept was analyzed in detailed aspects of the extraction and the successive energetic valorization, and useful data were obtained. Future work, to determine mass and energy fluxes and define the integration of the considered processes in the proposed biorefinery concept, will address the following key questions:1. How much energy (both electric power and heat) is needed for the SCO2 + EtOH extraction process, including CO2 recompression, extract freeze-drying and EtOH recovery?
2. How much heat is recovered by oxidation of the treated material?
3. Alternatively, how much and what syngas/bio-oil is obtainable by pyrolysis of the treated material?
4. Is the overall biorefinery energy-efficient? With what configuration?
5. What are the final production costs of the extracts and of the biofuel (syngas or bio-oil) and are they compensated by their economic value?
6. What is the overall energy balance and feasibility of the proposed biorefinery?
7. Given an economic overview, what is the feasibility, unit costs of energy/biofuels and products possible for this bio-refinery concept?
Acknowledgements
The authors appreciate the funding of Veolia Water Solutions and Technologies and the help of Nathalie Martin-Ionesco in promoting this international collaboration. Partial financial support was also granted by Gruppo Ricicla – DISAA, University of Milan, Italy. The authors thank Melissa Lever of Simmons College for assistance with the SEM images. A portion of this material is based upon work supported by the National Science Foundation under grant no. NSF CBET-1127774.References
- Faostat, http://faostat3.fao.org/faostat-gateway/go/to/home/E (accessed November 2014).
- A. G. Vlyssides, M. Loizides and P. K. Karlis, J. Clean Prod., 2004, 12, 603–611 CrossRef.
- L. D. Giovacchino, S. Sestili and D. D. Vincenzo, Eur. J. Lipid Sci. Technol., 2002, 587–601 CrossRef.
- L. Lesage-Meessen, D. Navarro, S. Maunier, J. C. Sigoillot, J. Lorquin, M. Delattre, J. L. Simon, M. Asther and M. Labat, Food Chem., 2001, 75, 501–507 CrossRef CAS.
- M. Niaounakis and C. P. Halvadakis, in Olive Processing Waste Management, Elsevier Inc., San Diego, 2006 Search PubMed.
- S. Martín-Peláez, M. I. Covas, M. Filtó, A. Kušar and I. Pravst, Mol. Nutr. Food Res., 2013, 57, 760–771 Search PubMed.
- R. Capasso, G. Cristinzio, A. Evidente and F. Scognamiglio, Phytochemistry, 1992, 31, 4125–4128 CrossRef CAS.
- G. Ouzounidou, G. I. Zervakis and F. Gaitis, Terr. Aquat. Environ. Toxicol., 2010, 22–34 Search PubMed.
- E. Aranda, I. Sampedro, J. A. Ocampo and I. García-Romera, Appl. Microbiol. Biotechnol., 2004, 64, 132–135 CrossRef CAS PubMed.
- Sigma-Aldrich, https://www.sigmaaldrich.com/sigma-aldrich/home.html, (accessed December 2014).
- Prohealth, https://www.prohealth.com/shop/product.cfm/product__code/PH398, (accessed December 2014).
- R. W. Owen, A. Giacosa, W. E. Hull, B. Spiegelhalder and H. Bartsch, Food Chem. Toxicol., 2000, 38, 647–659 CrossRef CAS.
- A. Shabtay, Y. Hadar, H. Eitam, A. Brosh, A. Orlov, Y. Tadmor, I. Izhaki and Z. Kerem, Bioresour. Technol., 2009, 100, 6457–6464 CrossRef CAS PubMed.
- T. Coskun, E. Debik and N. M. Demir, Desalination, 2010, 259, 65–70 CrossRef CAS PubMed.
- R. Japón-Luján, J. M. Luque-Rodríguez and M. D. Luque de Castro, J. Chromatogr., A, 2006, 1108, 76–82 CrossRef PubMed.
- H. K. Obied, M. S. Allen, D. R. Bedgood, P. D. Prenzler, K. Robards and R. Stockmann, J. Agric. Food Chem., 2005, 53, 823–837 CrossRef CAS PubMed.
- R. Japón-Luján and M. D. Luque de Castro, J. Chromatogr., A, 2006, 1136, 185–191 CrossRef PubMed.
- F. Le Floch, M. T. Tena, A. Ríos and M. Valcárcel, Talanta, 1998, 46, 1123–1130 CrossRef CAS.
- B. Diaz-Reinoso, A. Moure, H. Domingues and J. C. Parajo, J. Agric. Food Chem., 2006, 2441–2469 CrossRef CAS PubMed.
- A. Perez-Galvez, M. Jaren-Galan and M. I. Minguez-Mosquera, in Handbook of Fruits and Fruit Processing, Blackwell Publishing, Ames, Iowa, 2006, pp. 565–579 Search PubMed.
- H. J. Vandenburga, A. A. Clifford, K. D. Bartle, J. Carrol, I. Newton, L. M. Garden, J. R. Dean and C. T. Costley, Analyst, 1997, 122, 101R–115R RSC.
- P. Garcia-Salas, A. Morales-Soto, A. Segura-Carretero and A. Fernández-Gutiérrez, Molecules, 2010, 15, 8813–8826 CrossRef CAS PubMed.
- P. S. Rodis, V. T. Karathanos and A. Mantzavinou, J. Agric. Food Chem., 2002, 596–601 CrossRef CAS PubMed.
- G. A. Baddi, J. Cegarra, G. Merlina, J. C. Revel and M. Hafidi, J. Hazard. Mater., 2009, 1119–1123 CrossRef PubMed.
- International Energy Agency. World Energy Outlook, Paris, 2008. http://www.iea.org/media/weowebsite/2008-1994/WEO2008.pdf (Accessed 27 September 2014).
- K. Celis, I. Van Driessche, R. Mouton, G. Vanhoyland and S. Host, Meas. Sci. Rev., 2001, 1(1), 177–180 Search PubMed.
- C. Di Blasi, Prog. Energy Combust. Sci., 2008, 34, 47–90 CrossRef CAS PubMed.
- A. Saddawi, J. M. Jones, A. Williams and M. A. Wojtowicz, Energy Fuels, 2010, 24, 1274–1282 CrossRef CAS.
- M. J. J. Antal, G. Varhegyi and E. Jakab, Ind. Eng. Chem. Res., 1998, 37, 1267–1275 CrossRef CAS.
- J. Chattopadhyay, C. Kim, R. Kim and D. Pak, Korean J. Chem. Eng., 2008, 25(5), 1047–1053 CrossRef CAS.
- H. Demiral, I. Demiral, B. Karabacakoglu and F. Tümsek, Chem. Eng. Res. Des., 2010, 532, 1–8 Search PubMed.
- J. M. Encinar, J. F. Gonzalez, G. Martinez and S. Roman, J. Anal. Appl. Pyrolysis, 2009, 85, 197–203 CrossRef CAS PubMed.
- P. Galiatsatou, M. Metaxas, D. Arapoglou and V. Kasselouri-Rigopoulou, Waste Manage., 2002, 803–812 CrossRef CAS.
- C. E. Gokce, S. Guneysu, S. Aydin and S. Arayici, Open Environ. Pollut. Toxicol. J., 2009, 43–48 CrossRef CAS.
- B. B. Uzun, A. E. Putun and E. Putun, Energy Fuels, 2007, 21, 1768–1776 CrossRef CAS.
- F. Adani, EU Pat, EP20090709779, University of Milan, 2013 Search PubMed.
- S. Laroussi-Mezghani, P. Vanloot, J. Molinet, N. Dupuy, M. Hammami, N. Grati-Kamoun and J. Artaud, Food Chem., 2015, 173, 122–132 CrossRef CAS PubMed.
- D. Grigoriadou, A. Androulaki, E. Psomiadou and M. Z. Tsimidou, Food Chem., 2007, 105(2), 675–680 CrossRef CAS PubMed.
- M. Van de Velden, J. Baeyens, A. Brems and R. Dewil, Renew Energy, 2010, 232–242 CrossRef CAS PubMed.
- J. Jauhiainen, J. A. Conesa, R. Font and I. Martín-Gullón, J. Anal. Appl. Pyrolysis, 2004, 72, 9–15 CrossRef CAS PubMed.
- A. Ounas, A. Aboulkas, K. El harfi, A. Bacaoui and A. Yaacoubi, Bioresour. Technol., 2011, 102, 11234–11238 CrossRef CAS PubMed.
- L. Buessing and J. L. Goldfarb, J. Anal. Appl. Pyrolysis, 2012, 96, 78–85 CrossRef PubMed.
- A. M. Celaya, A. T. Lade and J. L. Goldfarb, Fuel Process. Technol., 2015, 129, 39–51 CrossRef CAS PubMed.
- G. Taralas and M. G. Kontominas, J. Anal. Appl. Pyrolysis, 2006, 76, 109–116 CrossRef CAS PubMed.
- V. B. Agbor, N. Cicek, R. Sparling, A. Berlin and D. B. Levin, Biotechnol. Adv., 2011, 29, 675–685 CrossRef CAS PubMed.
- Y. Z. Zheng, H. M. Lin and G. T. Tsao, Biotechnol. Lett., 1995, 17, 845–850 CrossRef CAS.
- J. F. González, S. Román, J. M. Encinar and G. Martínez, J. Anal. Appl. Pyrolysis, 2009, 85, 134–141 CrossRef PubMed.
- V. E. Singleton, R. Orthofer and R. M. Lamuela-Ravento's, Methods Enzymol., 1999, 299, 152–178 CAS.
- M. L. Chan, J. M. Jones, M. Pourkashanian and A. Williams, Fuel, 1999, 78, 1539–1552 CrossRef CAS.
- J. M. Jones, T. G. Bridgeman, L. I. Darvell, B. Gudka, A. Saddawi and A. Williams, Fuel Process. Technol., 2012, 101, 1–9 CrossRef CAS PubMed.
- P. Dhepe and R. Sahu, Green Chem., 2010, 12, 2153–2156 RSC.
- P. Parasuraman, R. Singh, T. Bolton, S. Omori and R. Francis, Bioresources, 2007, 459–471 CAS.
- P. A. Webb and C. Orr, in Analytical Methods in Fine Particle Technology, Micromeritics Instrument Corporation, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc00076a |
| This journal is © The Royal Society of Chemistry 2015 |