
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
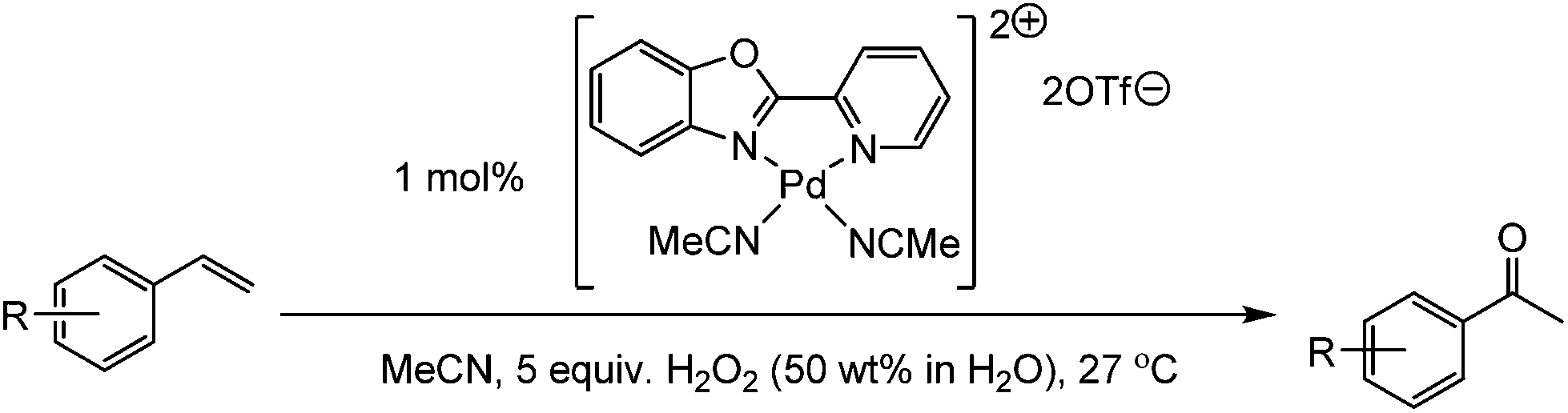
Cationic palladium(II) complexes as catalysts for the oxidation of terminal olefins to methyl ketones using hydrogen peroxide†
Qun
Cao
,
David S.
Bailie
,
Runzhong
Fu
and
Mark J.
Muldoon
*
School of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, Belfast, BT9 5AG, UK. E-mail: m.j.muldoon@qub.ac.uk
First published on 9th March 2015
Abstract
Ligated Pd(II) complexes have been studied for the catalytic oxidation of terminal olefins to their corresponding methyl ketones. The method uses aqueous hydrogen peroxide as the terminal oxidant; a sustainable and readily accessible oxidant. The choice of ligand, counterion and solvent all have a significant effect on catalytic performance and we were able to develop systems which perform well for these challenging oxidations.
The oxidation of a terminal olefin to a methyl ketone is a very useful transformation in organic chemistry, one which can be employed for the preparation of fine chemicals, agrichemicals and pharmaceuticals. The Wacker–Tsuji oxidation is a well-known method for this reaction and generally employs PdCl2, CuCl2 and O2 in a DMF–H2O solvent system.1 There are a number of drawbacks to this approach however, for example, there is a need to improve both the rate and selectivity for many types of substrates. Although the original Wacker Process utilised copper salts for the oxidation of ethylene2 it has been highlighted that copper salts often do more than just reoxidise Pd(0) back to the active Pd(II) species, and their presence can lead to unwanted side-reactions and/or reduced selectivity when trying to prepare ketones.3 Consequently, many groups have investigated alternative approaches, developing improved methods for the oxidation of both terminal and internal olefins to their corresponding ketones. In an attempt to replace the copper co-catalyst, alternative electron transfer-mediator systems, such as polyoxometalates,4 metal macrocycle/quinone combinations,5 or benzoquinone/NaNO2/HClO4,6 have been reported. Some researchers have also utilised oxidants such as benzoquinone7 iron(III) sulphate,8 chromium trioxide,9 and potassium bromate.10 Using O2 as the sole oxidant is attractive, and a number of methods have been developed which utilise O2 for the direct re-oxidation of Pd(0).11–14 Kaneda and co-workers have developed a system that utilises N,N-dimethylacetamide (DMA) as a solvent and can oxidise both terminal and internal olefins.13 This copper-free method utilises catalyst loadings of 5–10 mol% Pd for internal olefins13b,c and 0.5–1 mol% for terminal olefins (Fig. 1A),13a under O2 atmospheres (1–6 atm of O2 pressure). A limitation of this method is that it is not effective for the oxidation of styrenes to their corresponding acetophenones. In terms of aerobic systems for the oxidation of styrenes, there are two recent notable reports. The first by Reiser and co-workers utilised 5 mol% of a Pd(II) catalyst, which was prepared from a chiral pseudo C2-symmetrical bis(isonitrile) ligand (Fig. 1B).12 More recently, Wang and co-workers reported a ligand free system, which used 10 mol% Pd(OAc)2, along with (1 equiv.) of trifluoroacetic acid (TFA) in a DMSO–H2O (10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent system (Fig. 1C).14
:1) solvent system (Fig. 1C).14
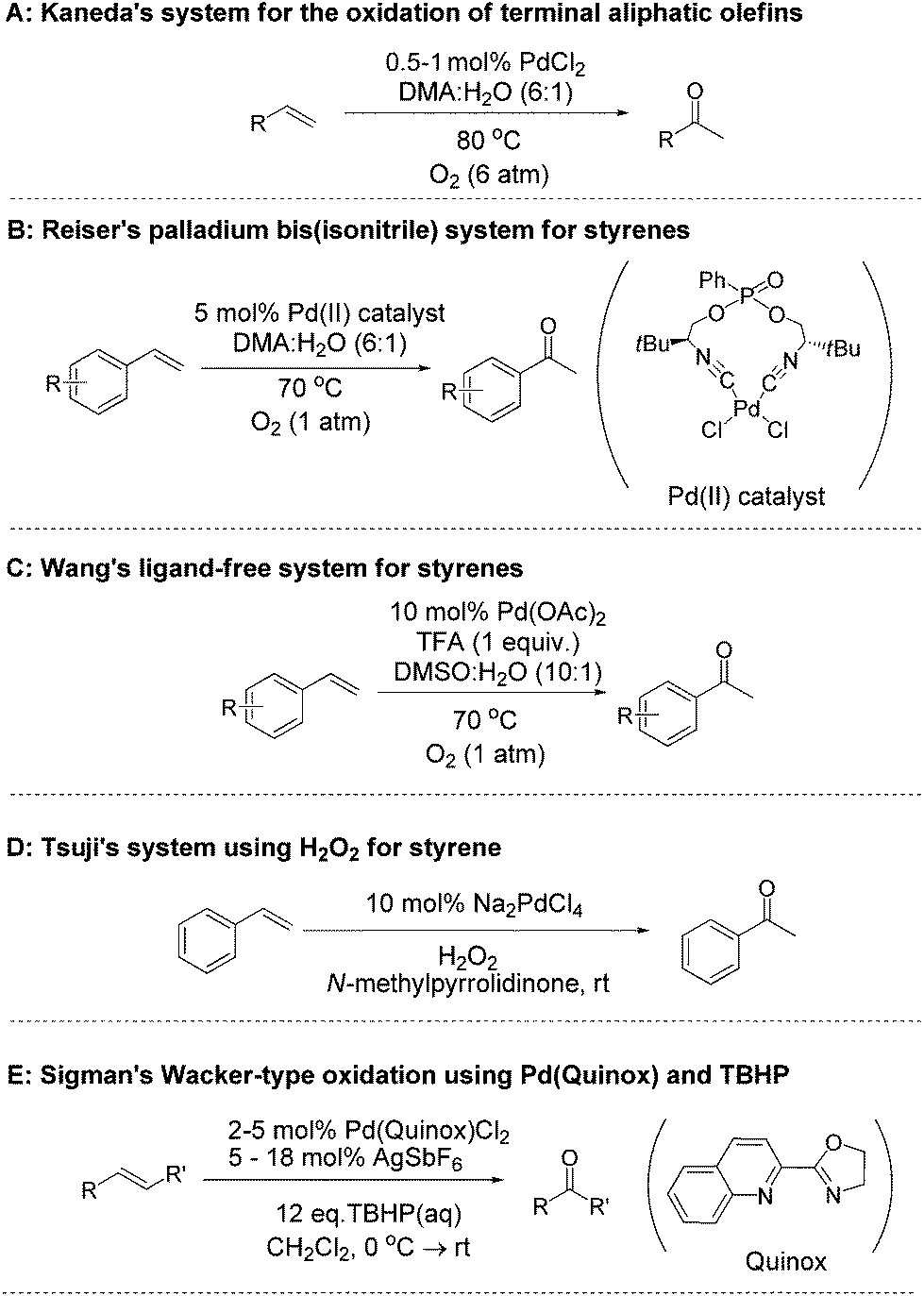 | ||
| Fig. 1 Literature examples of Pd(II) catalysed Wacker oxidations. | ||
Based on the state-of-the-art we were interested in trying to develop new catalysts for Wacker-type oxidation of olefins, and we were attracted to the use of hydrogen peroxide (H2O2) as the terminal oxidant. Aqueous H2O2 is a sustainable oxidant which can be readily used under mild conditions and has a track record of large scale industrial use.15 Although H2O2 has been used for Wacker oxidation chemistry, we felt there was scope for developing more effective methods. Indeed, the use of H2O2 for Wacker oxidations dates back some time, with Moiseev et al. reporting its use for the Pd(II) catalysed oxidation of ethylene in 1960.16 In 1980, both the Tsuji17 and Mimoun18 groups reported the use of H2O2 using simple Pd(II) salts, and in the case of the Tsuji work, this included styrene (Fig. 1D).17 Since then, others have examined various approaches to improve the performance of Pd(II)/H2O2 for Wacker oxidation of styrenes, such as using phase transfer catalysts,19 micelles,20 ionic liquids21,22 and ligands appended to solid supports.23,24 In the work to-date, the reports have not explored a wide range of substrates or studied a wide variety of catalyst structures. Furthermore there is a need to improve catalyst performance (i.e. reduce Pd loading and improve product selectivity).
An alternative peroxide to H2O2 is tert-butyl hydroperoxide (TBHP) which has also been examined for Pd(II) Wacker-type reactions since 1980.17,25 Recent work by Sigman and co-workers has demonstrated the benefits of developing ligand modulated catalysts for improving the performance of these TBHP mediated oxidations.26,27 Their quinoline-2-oxazoline (quinox) based catalyst is shown (Fig. 1E).
Unlike Pd(II)/TBHP systems, there have been very few examples of studies using ligands to improve catalyst performance for Pd(II)/H2O2 systems; therefore we wished to examine a range of ligands for Pd(II)/H2O2 mediated Wacker-type oxidations.
We began our studies by testing Pd(OAc)2 with acetic acid as the solvent, as this system had previously been used by Roussel and Mimoun.18 In their studies they had focussed on aliphatic olefins and when we examined this system for styrene we found that the reaction gave a significant amount of unwanted side products. The selective oxidation to one product is a particular challenge with styrene based substrates. Styrenes can polymerise and it is known that with Pd(II) catalysts and H2O2 several products can be formed. For example, products such as styrene oxide, benzaldehyde, benzoic acid, 1-phenylethane-1,2-diol, phenylacetaldehyde and 1,3-diphenyl-1-butene can all be produced.19,22,24,28 It is difficult to quantify every side product by GC and so, we focussed on accurately determining the yield of the desired acetophenone and the conversion of the substrate using an internal standard. Acetic acid is not ideal, as H2O2 and acetic acid will lead to the formation of peracetic acid, which can oxidise styrene to styrene oxide, and styrene oxide can then react further in acetic acid to form a number of products.29 GC-MS and NMR analysis confirmed that we were producing such side-products (see ESI†). Unfortunately we found that replacing acetic acid with other organic solvents led to poor catalyst activity (see ESI†). Therefore, we used acetic acid as the solvent and screened a number of ligands. Pleasingly we found that the presence of a ligand could improve both the rate of conversation and the selectivity towards the desired acetophenone. We then discovered that if we altered the counterion from acetate and prepared the cationic analogue using weakly coordinating anions then the resultant complex delivered good catalytic performance in acetonitrile; a solvent that had previously been poor for Pd(OAc)2 complexes (see ESI†). We screened a number of ligands in acetonitrile, forming cationic complexes in situ using bis(acetonitrile)dichloropalladium(II), ligand and silver triflate. The ligands screened are shown in Fig. 2.
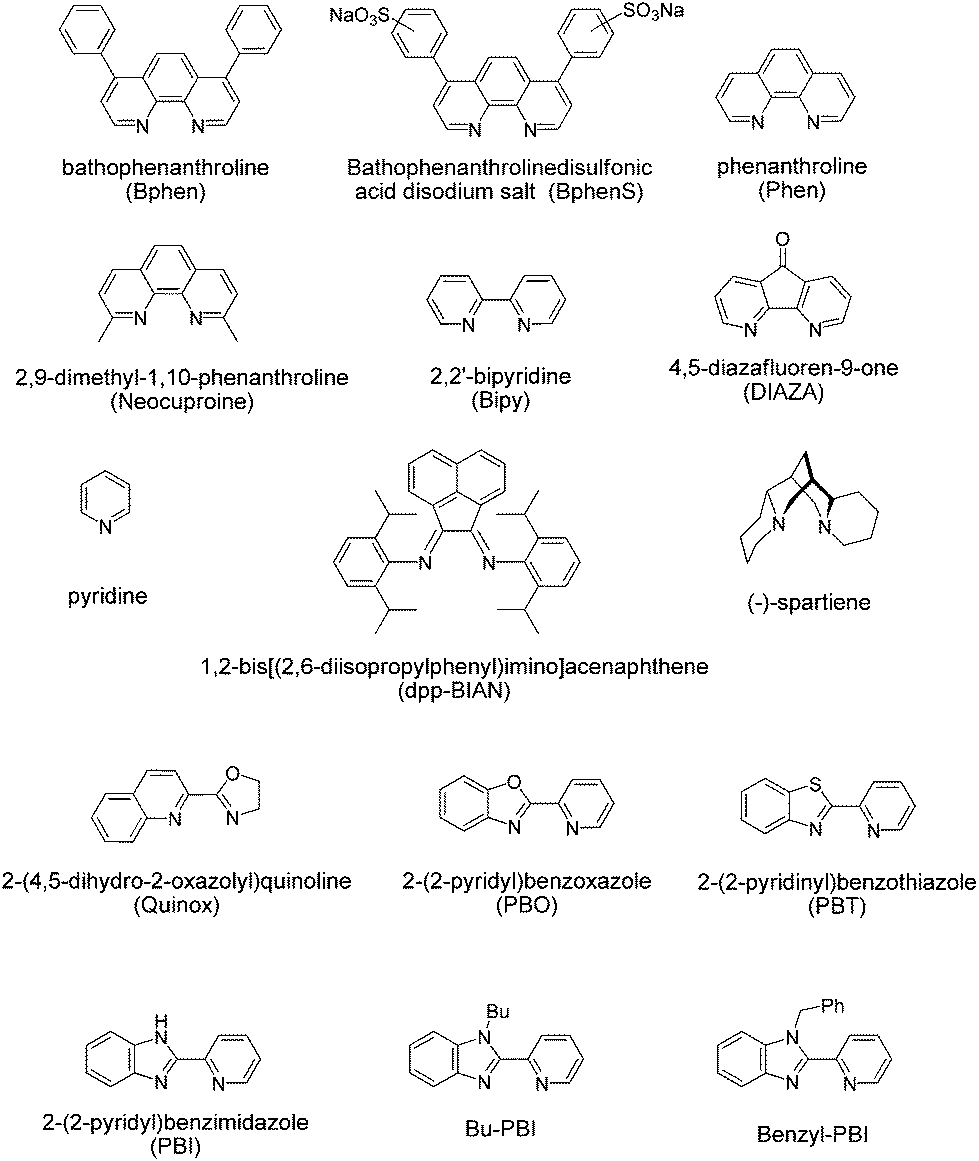 | ||
| Fig. 2 Structures and acronyms of the ligands that were tested for Pd(II)/H2O2 oxidation of olefins. | ||
As shown in Fig. 3, ligands had a substantial impact on both the conversion of styrene and the yield of the desired acetophenone. Further work will be needed to understand the observed ligand effects, as even relatively similar ligands had quite different results. We found that the 2-(2-pyridyl)benzoxazole (PBO) ligand resulted in the best catalytic performance for the oxidation of styrene to acetophenone. This ligand has similarity to the “Quinox” ligand used by Sigman and co-workers for their TBHP mediated oxidations.26 As shown in Fig. 3, the Quinox ligand did not perform as well as PBO under our reaction conditions. Although not commercially available, PBO is an attractive ligand as it can be prepared in high yields from inexpensive commercially available starting materials, using a simple procedure that was reported by Hayashi and co-workers (Fig. 4).30
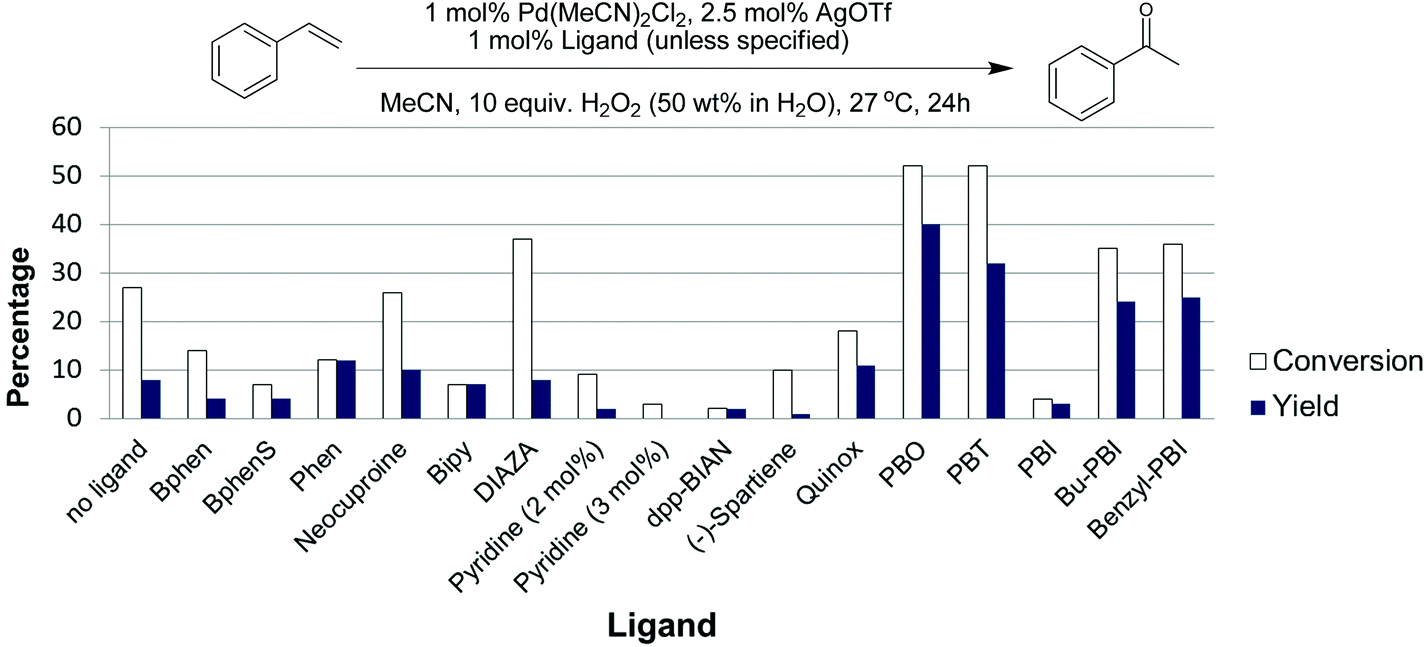 | ||
| Fig. 3 Evaluation of ligands for the Pd(II)/H2O2 oxidation of styrene in acetonitrile. Experimental details in ESI.† | ||
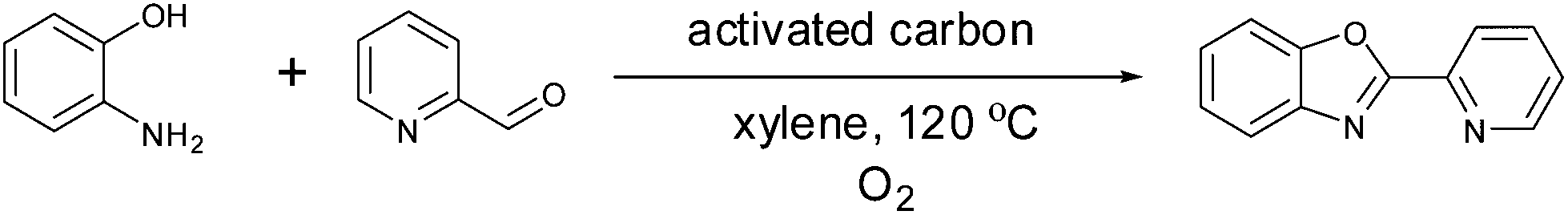 | ||
| Fig. 4 Synthesis of the 2-(2-pyridyl)benzoxazole (PBO) ligand. | ||
Given the effect of switching from acetate to triflate, we tested a number of other counter ions, as shown in Fig. 5.
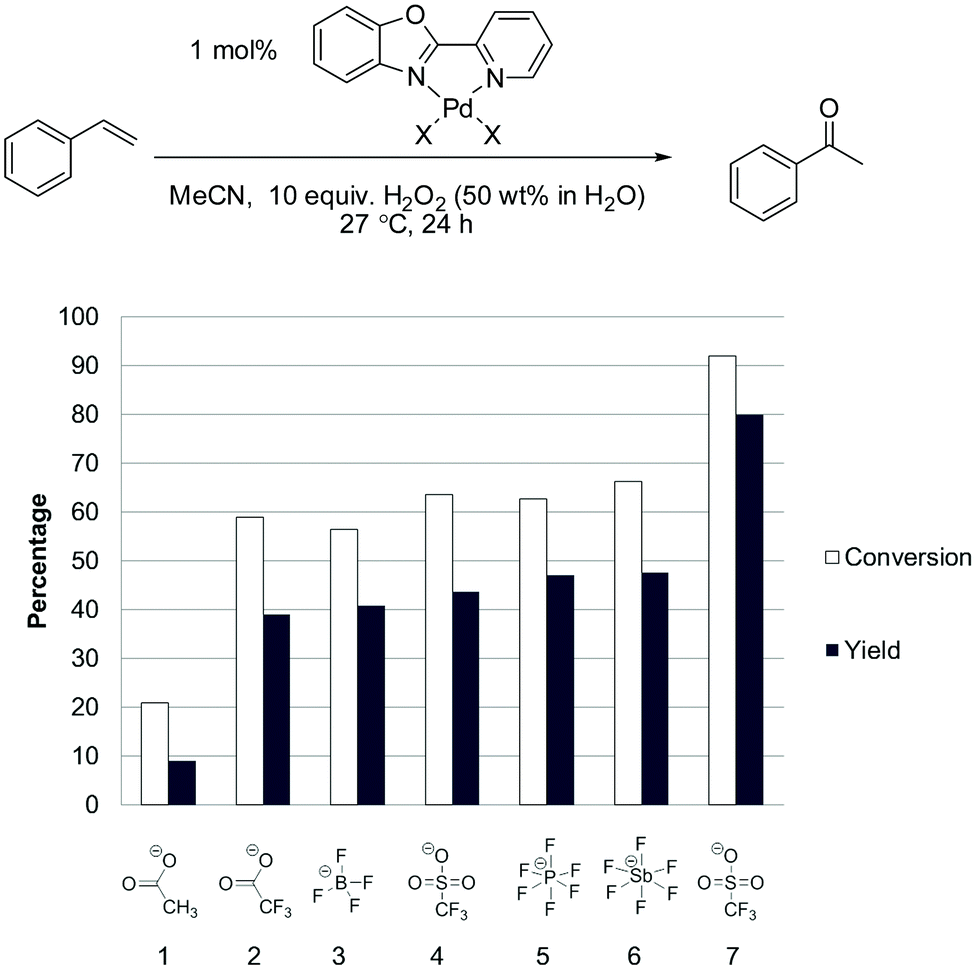 | ||
| Fig. 5 Evaluation of the counter ion in the Pd(II)/H2O2 oxidation of styrene in acetonitrile. Experimental details in ESI.† Entry 1 = Pd(OAc)2; entry 2 = Pd(CF3COO)2; entries 3–6 = 1 mol% pre-made & isolated (PBO)PdCl2 complex and 2.5 mol% of the corresponding silver salt. Entry 7 = pre-made & isolated (PBO)Pd(MeCN)2(OTf)2 complex. | ||
Using the silver salt method there was little difference between [OTf]− and weaker anions such as [PF6]− or [SbF6]−. Furthermore we found that better catalytic performance could be obtained when complexes were prepared without using this in situ silver salt method. In the case of triflate, the (PBO)Pd(MeCN)2(OTf)2 complex can be readily prepared and isolated using a facile procedure with triflic acid (see ESI†).
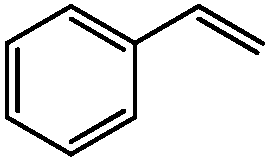
We then utilised the (PBO)Pd(MeCN)2(OTf)2 complex for a range of styrene substrates (Table 1). We found that with 1 mol% catalyst we could obtain good yields in many cases. As mentioned earlier, we utilised an internal standard to accurately determine the substrate conversion and yield of acetophenone. We further validated this method by obtaining isolated yields of several of the less volatile products and found that the isolated yields were in good agreement with those obtained via the GC method. Both steric and electronic effects can be seen in Table 1. Having a substituent in the 2-position reduces the yield, for example compare entries 2, 3, 4 and 14 and also entries 7, 8 and 9. Electron withdrawing substituents reduce the yield, with the nitro substituted styrene the worse affected, but this is in-line with what was previously found with Sigman's TBHP system.27a The lowest yield was obtained for the sterically hindered 2,4,6-trimethylstyrene (entry 14), but tests showed the yield could be increased with higher catalyst loadings.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Time | Product | Conv.a [%] | Yielda [%] |
| General reaction conditions (further details in ESI): (PBO)Pd(MeCN)2(OTf)2 (1 mol%, 0.0150 g, 0.0221 mmol), solvent (4 mL), 5 equiv. H2O2 (50 wt% in H2O) (0.645 mL), styrene (2.208 mmol).a Conversion and yield determined by GC using biphenyl as an internal standard. Examples of isolated yields are in parenthesis.b 10 equiv. H2O2 (50 wt% in H2O).c 50 °C.d 2 mol% catalyst.e 5 mol% catalyst. | |||||
| 1 |
|
24 h |
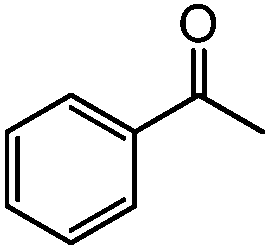
|
92 | 80 |
| 2 |
|
48 h |
|
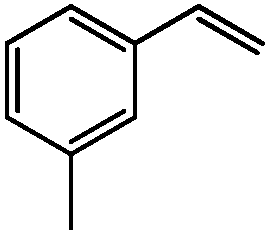
99 | 65 |
| 3 |
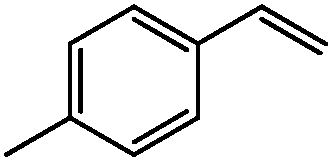
|
24 h |
|
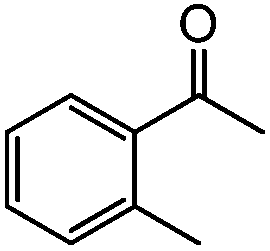
96 | 92 (90) |
| 4 |
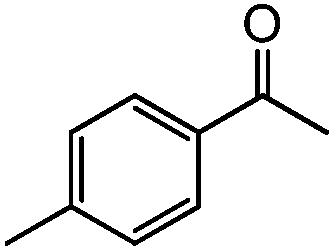
|
24 h |
|
91 | 90 |
| 5b |
|
24 h |
|
92 | 78 |
| 6 |
|
24 h |
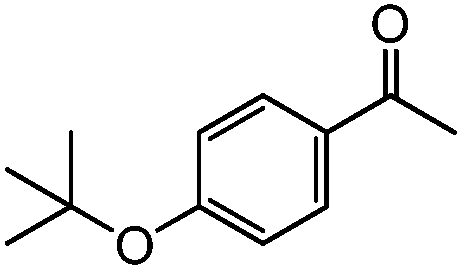
|
95 | 70 |
| 7b,c |
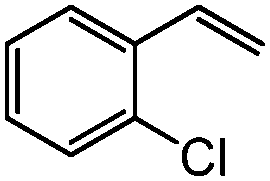
|
48 h |
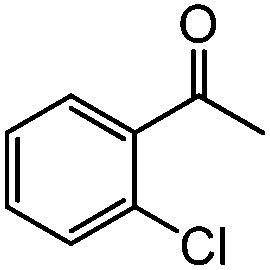
|
92 | 50 |
| 8c |
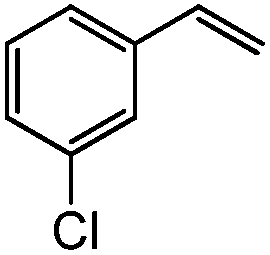
|
24 h |
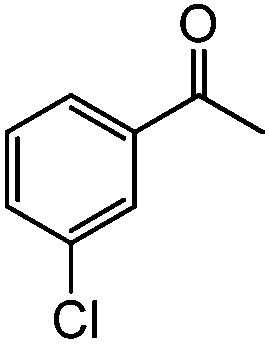
|
98 | 71 |
| 9c |
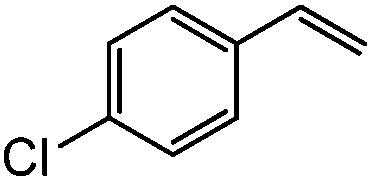
|
24 h |
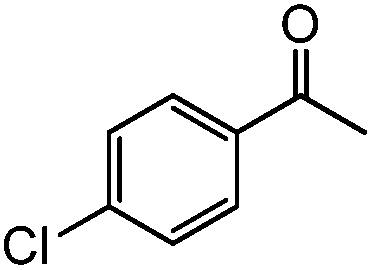
|
100 | 79 (77) |
| 10c |
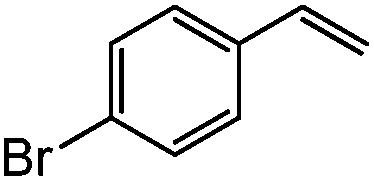
|
24 h |
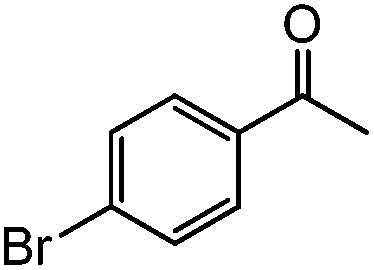
|
99 | 64 |
| 11c |
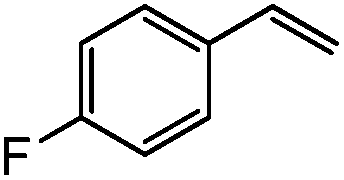
|
24 h |
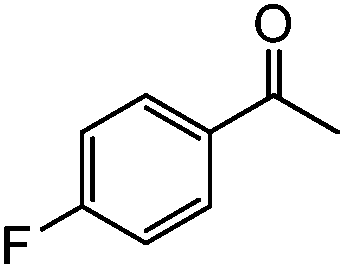
|
100 | 90 |
| 12 |
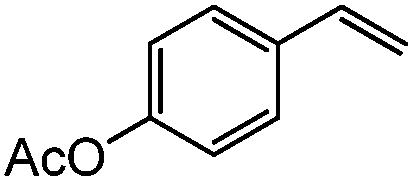
|
30 h |
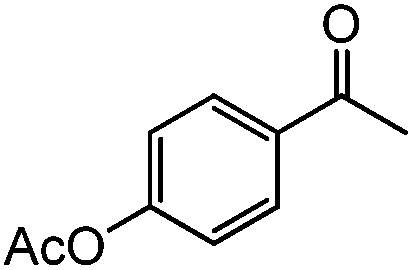
|
89 | 85 (82) |
| 13c |
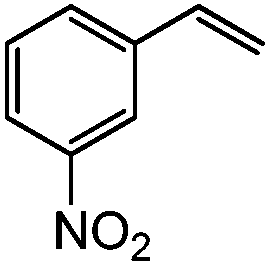
|
48 h |
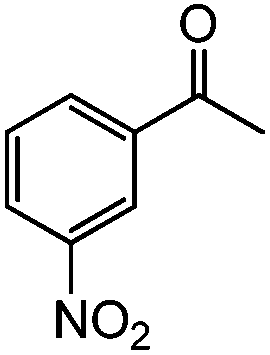
|
92 | 54 |
| 14c |
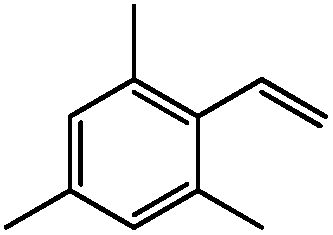
|
48 h |
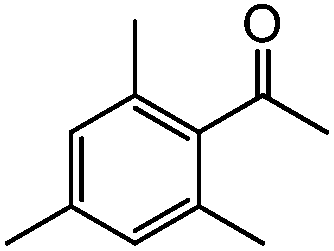
|
77 | 30 |
| 24 h | 88d | 53d | |||
| 24 h | 100e | 68e | |||
We then examined the oxidation of aliphatic olefins, using 1-octene as a model substrate. Aliphatic substrates are challenging because of the competing isomerisation of the double bond, something that Pd(II) complexes can readily facilitate.31 We found that the (PBO)Pd(MeCN)2(OTf)2 complex in acetonitrile was not an effective catalyst system as it resulted in a high degree of isomerisation and only a small amount of the desired 2-octanone was produced. Furthermore, negligible amounts of the internal isomers were oxidised. We investigated a number of avenues to try and improve catalyst performance and Table 2 shows some selected examples, with further examples detailed in the ESI.†
|
|
|||||
|---|---|---|---|---|---|
| Entry | Time | Solvent | Ligand | Conversiona [%] | Yielda [%] |
| General reaction conditions (further details in ESI): (Ligand)PdCl2(MeCN)2 (1 mol%, 0.0221 mmol), solvent (10 mL), 10 equiv. H2O2 (50 wt% in H2O) (1.29 mL), 1-octene (0.23 g, 2.208 mmol), 27 °C.a Conversion of substrate and yield of product determined by GC using biphenyl as an internal standard. | |||||
| 1 | 1 h | MeCN | PBO | 99 | 7 |
| 2 | 3 h | DMA | PBO | 15 | 1 |
| 3 | 1 h | Acetone | PBO | 97 | 25 |
| 4 | 24 h | MeCN | Phen | 89 | 23 |
| 5 | 24 h | EtOAc | Phen | 12 | 12 |
| 6 | 24 h | Acetone | Phen | >99 | 67 |
| 7 | 24 h | Acetone | Bphen | >99 | 80 |
| 8 | 24 h | 2-Butanone | Bphen | >99 | 74 |
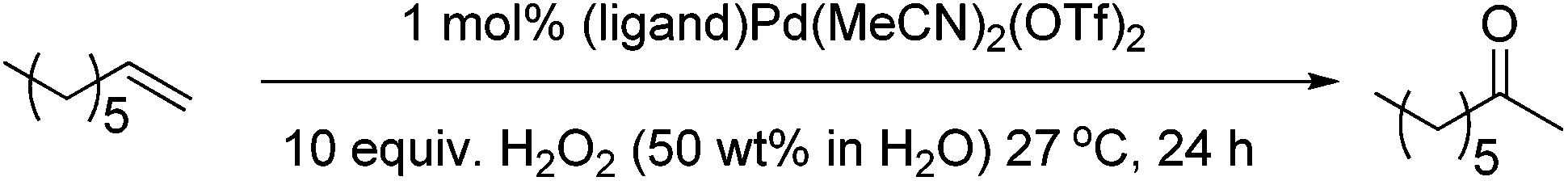
During these studies we found that acetone was the best solvent and that other ligands gave better results than PBO. We then re-examined our series of ligands in acetone (Fig. 6) and found that bathophenanthroline (Bphen) was the best ligand for this substrate. Although screening studies identified acetone as the best solvent we felt that perhaps this was not an ideal solvent (particularly if these reactions were to be scaled up), because acetone and H2O2 can lead to the production of dangerous peroxides such as triacetone triperoxide and diacetone diperoxide.32 We thought that perhaps the higher yields of desired product were due to the formation of such peroxides and these could be responsible for increasing the rate of olefin oxidation, allowing the catalyst to compete with the undesired isomerisation pathway. Previous work by the Alper33 and Sigman26 groups has shown that using THF as a solvent can promote aerobic Wacker oxidation reactions. This is thought to be due to the in situ formation of 2-tetrahydrofuryl hydroperoxide. More recently, Weinstein and Stahl reported that peroxide species generated in situ from 1,4-dioxane and O2 acted as the oxidant in palladium catalysed aryl C–H amination reactions.34
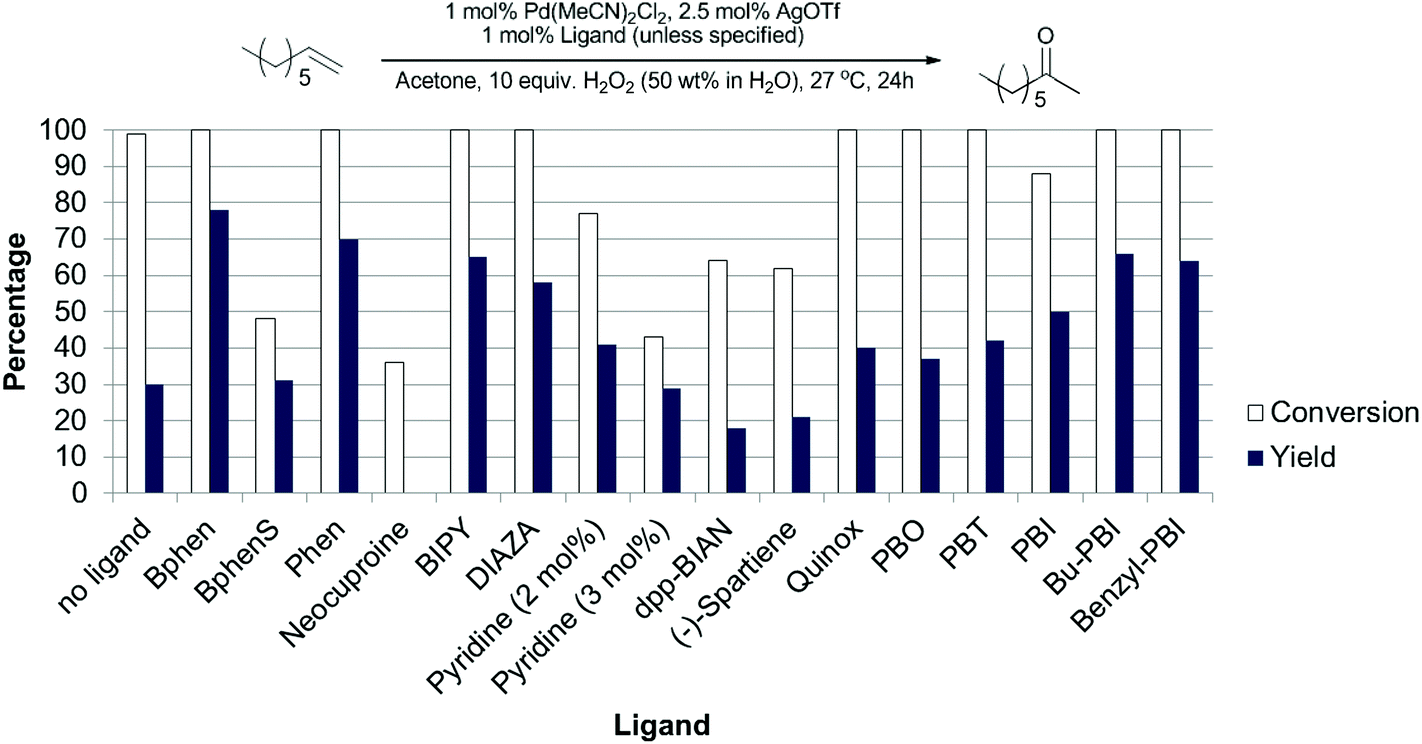 | ||
| Fig. 6 Evaluation of ligands for the Pd(II)/H2O2 oxidation of 1-octene in acetone. Experimental details in ESI.† | ||
We also tested 2-butanone as a solvent and found that this also resulted in a good yield (entry 8 Table 2). We presumed that this further supported the idea of in situ formation of organic peroxides that are then acting as oxidants, as 2-butanone and H2O2 can form 2-butanone peroxide (often referred to as MEKP (methyl ethyl ketone peroxide)). Although 2-butanone peroxide is not without its dangers,35 it is safer than acetone peroxide and is used industrially (e.g. in the preparation of polymers). 2-Butanone peroxide is commercially available so we tested it as an oxidant and were surprised to find that very little 2-octanone was produced (Table 3). So if 2-butanone peroxide is being produced in situ it would appear that it is not responsible for the improved oxidation of 1-octene.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Conv.a [%] | Yielda [%] | Isomersa [%] |
| Reaction conditions: (Bphen)Pd(MeCN)2(OTf)2 (1 mol%, 0.0180 g, 0.0221 mmol), solvent as specified (10 ml), 2-butanone peroxide (3.12 g), 1-octene (0.25 g, 2.208 mmol), 27 °C.a Conversion of 1-octene, yield of 2-octanone and percentage of internal isomers produced were determined by GC using biphenyl as internal standard.b 1.29 mL deionised water was added. | ||||
| 1 | MeCN | 8 | 3 | 2 |
| 2 | 2-Butanone | 10 | 7 | 2 |
| 3b | 2-Butanone–H2O | 13 | 6 | 2 |
| 4b | MeCN–H2O | 4 | 0 | 3 |
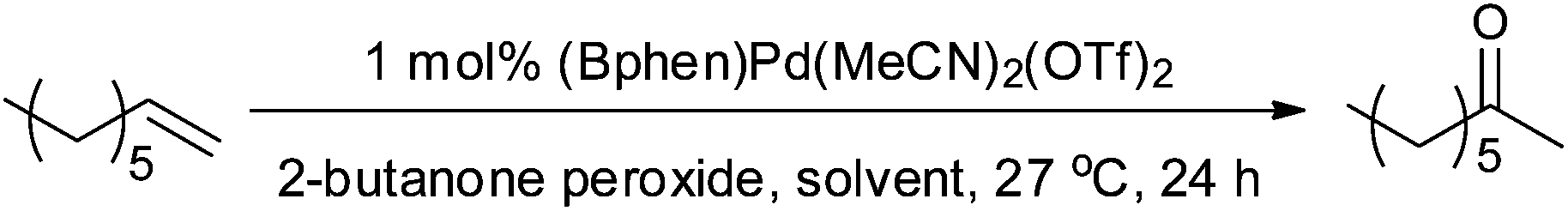
We then looked at the influence of the solvent on the isomerisation of 1-octene to internal olefins in the absence of H2O2 (Table 4). It can be seen from entries 1–3 in Table 4 that isomerisation is faster in acetone and 2-butanone compared to in acetonitrile. Of course in the oxidation reactions, water is present as we utilise aqueous solutions of H2O2 and when we added water we found that it had a significant influence. In acetonitrile (c.f. entries 1 and 4) the addition of water increased the amount of isomerisation. However, when water is added to acetone (c.f. entries 2 and 5) and 2-butanone (c.f. entries 3 and 6) there is a dramatic reduction in the amount of isomerisation taking place. This reduction in isomerisation helps explain why these solvents are good for oxidation of aliphatic olefins. Given that ketones and water have such a dramatic solvent effect we wonder if the presence of hydrates (geminal diols) could possibly be playing a role in reducing isomerisation. However, further studies will be required to understand the solvent effects on these reactions as the solvent will undoubtedly influence other steps in the catalytic cycle. For example in catalytic studies, ethyl acetate did not promote isomerisation but the degree of oxidation was also low (see Table 2 entry 5 and ESI† for more examples).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time | 1-Octenea [%] | Isomersa [%] |
| Reaction conditions: (Bphen)Pd(MeCN)2(OTf)2 (1 mol%, 0.0221 mmol), solvent (10 mL), 1-octene (0.25 g, 2.208 mmol), 27 °C.a Yield of internal isomers and percentage of 1-octene remaining was determined by GC using biphenyl as internal standard.b 1.29 mL deionised water was added into the reaction. | ||||
| 1 | MeCN | 0.5 h | 88 | 12 |
| 1 h | 83 | 17 | ||
| 3 h | 73 | 27 | ||
| 2 | Acetone | 0.5 h | 10 | 90 |
| 1 h | 2 | 97 | ||
| 3 h | 2 | 98 | ||
| 3 | 2-Butanone | 0.5 h | 11 | 89 |
| 1 h | 4 | 96 | ||
| 3 h | 3 | 97 | ||
| 4b | MeCN–H2O | 0.5 h | 87 | 13 |
| 1 h | 70 | 29 | ||
| 3 h | 28 | 72 | ||
| 5b | Acetone–H2O | 0.5 h | 82 | 18 |
| 1 h | 77 | 23 | ||
| 3 h | 72 | 28 | ||
| 6b | 2-Butanone–H2O | 0.5 h | 96 | 4 |
| 1 h | 96 | 4 | ||
| 3 h | 93 | 7 | ||
Conclusions
In this study we have examined a number of ligands and reaction conditions and this has enabled us to develop catalytic methods for the oxidation of olefins to their corresponding methyl ketones. The catalytic performance of these systems compares favourably to those previously reported in the literature. Further work needs to be carried out to understand the influence of ligand structure and solvent on the performance of these reactions. On a small lab scale we believe that the methods are convenient and accessible with limited hazards. Using acetone or 2-butanone with H2O2 would be more hazardous on a larger scale, but we believe that systems could be safely engineered. For example, Siegel and co-workers recently described the advantages of using a continuous flow system when phthaloyl peroxide was used as an oxidant.36 Flow systems would ensure that the volumes remain small and all peroxides could be easily destroyed at the end of the reaction. Indeed, it is also worth pointing out that on an industrial scale, regardless of the solvent, there needs to be precautions taken when H2O2 is used.37Acknowledgements
For support we thank the Department of Employment and Learning, Northern Ireland, Queen's University Belfast Ionic Liquid Laboratories (QUILL) and the Centre for the Theory and Application of Catalysis (CenTACat). We also thank Prof. Paul Davey for insightful discussions.References
- (a) W. H. Clement and C. M. Selwitz, J. Org. Chem., 1964, 29, 241 CrossRef CAS; (b) J. Tsuji, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1991, vol. 7, p. 449 Search PubMed; (c) J. Tsuji, Synthesis, 1984, 369 CrossRef CAS.
- (a) J. Smidt, W. Hafner, R. Jira, S. Sedlmeier, R. Sieber, R. Rttinger and H. Kojer, Angew. Chem., 1959, 71, 176 CrossRef CAS; (b) Review: J. Smidt, W. Hafner, R. Jira, R. Sieber, S. Sedlmeier and A. Sabel, Angew. Chem., Int. Ed. Engl., 1962, 1, 80 CrossRef.
- Review: C. N. Cornell and M. S. Sigman, Inorg. Chem., 2007, 46, 1903 CrossRef CAS PubMed.
- (a) H. Ogawa, H. Fujinami, K. Taya and S. Teratani, J. Chem. Soc., Chem. Commun., 1981, 1274 RSC; (b) H. Ogawa, H. Fujinami, K. Taya and S. Teratani, Bull. Chem. Soc. Jpn., 1984, 57, 1908 CrossRef CAS; (c) A. W. Stobbe-Kreemers, M. van der Zon, M. Makkee and J. J. F. Scholten, J. Mol. Catal. A: Chem., 1996, 107, 247 CrossRef CAS; (d) A. Kishi, T. Higashino, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2000, 41, 99 CrossRef CAS; (e) T. Yokota, A. Sakakura, M. Tani, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2002, 43, 8887 CrossRef CAS; (f) E. G. Zhizhina, M. V. Simonova, V. F. O. Klavdii and I. Matveev, Appl. Catal., A, 2007, 319, 91 CrossRef CAS PubMed.
- (a) J.-E. Bäckvall, R. B. Hopkins, H. Grennberg, M. Mader and A. K. Awasthi, J. Am. Chem. Soc., 1990, 112, 5160 CrossRef; (b) Review: J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506 CrossRef CAS PubMed.
- G. Zhang, X. Xie, Y. Wang, X. Wen, Y. Zhao and C. Ding, Org. Biomol. Chem., 2013, 11, 2947 CAS.
- B. Morandi, Z. K. Wickens and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 9751 CrossRef CAS PubMed.
- R. A. Fernandes and D. A. Chaudhari, J. Org. Chem., 2014, 79, 5787 CrossRef CAS PubMed.
- R. A. Fernandes and V. Bethi, Tetrahedron, 2014, 70, 4760 CrossRef CAS PubMed.
- M. G. Kulkarni, Y. B. Shaikh, A. S. Borhade, S. W. Chavhan, A. P. Dhondge, D. D. Gaikwad, M. P. Desai, D. R. Birhade and N. R. Dhatrak, Tetrahedron Lett., 2013, 54, 2293 CrossRef CAS PubMed.
- (a) M. Higuchi, S. Yamaguchi and T. Hirao, Synlett, 1996, 1213 CrossRef CAS PubMed; (b) G.-J. ten Brink, I. W. C. E. Arends, G. Papadogianakis and R. A. Sheldon, Appl. Catal., A, 2000, 194–195, 435 CrossRef; (c) G.-J. ten Brink, I. W. C. E. Arends, G. Papadogianakis and R. A. Sheldon, Chem. Commun., 1998, 2359 RSC; (d) C. N. Cornell and M. S. Sigman, Org. Lett., 2006, 8, 4117 CrossRef CAS PubMed; (e) J. Wang, L.-N. He, C.-X. Miao and Y.-N. Li, Green Chem., 2009, 11, 1317 RSC.
- A. Naik, L. Meina, M. Zabel and O. Reiser, Chem. – Eur. J., 2010, 16, 1624 CrossRef CAS PubMed.
- (a) T. Mitsudome, T. Umetani, N. Nosaka, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Angew. Chem., Int. Ed., 2006, 45, 481 CrossRef CAS PubMed; (b) T. Mitsudome, K. Mizumoto, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2010, 49, 1238 CrossRef CAS PubMed; (c) T. Mitsudome, S. Yoshida, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2013, 52, 5961 CrossRef CAS PubMed.
- Y.-F. Wang, Y.-R. Gao, S. Mao, Y.-L. Zhang, D.-D. Guo, Z.-L. Yan, S.-H. Guo and Y.-Q. Wang, Org. Lett., 2014, 16, 1610 CrossRef CAS PubMed.
- (a) R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC; (b) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943 CrossRef CAS PubMed.
- I. I. Moiseev, M. N. Vargaftik and Y. K. Syrkin, Dokl. Akad. Nauk SSSR, 1960, 130, 820 CAS.
- J. Tsuji, H. Nagashima and K. Hori, Chem. Lett., 1980, 257 CrossRef CAS.
- M. Roussel and H. Mimoun, J. Org. Chem., 1980, 45, 5387 CrossRef CAS.
- G. Barak and Y. Sasson, J. Chem. Soc., Chem. Commun., 1987, 1266 RSC.
- N. Alandis, I. Ricolattes and A. Lattes, New J. Chem., 1994, 18, 1147 CAS.
- V. V. Namboodiri, R. S. Varma, E. Sahle-Demessie and U. R. Pillai, Green Chem., 2002, 4, 170 RSC.
- C. Chiappe, A. Sanzone and P. J. Dyson, Green Chem., 2011, 13, 1437 RSC.
- Y. V. Subba Rao, S. S. Rani and B. M. Choudary, J. Mol. Catal., 1992, 75, 141 CrossRef.
- P. Borah and Y. Zhao, J. Catal., 2014, 318, 43 CrossRef CAS PubMed.
- H. Mimoun, R. Charpentier, A. Mitschler, J. Fischer and R. Weiss, J. Am. Chem. Soc., 1980, 102, 1047 CrossRef CAS.
- C. N. Cornell and M. S. Sigman, J. Am. Chem. Soc., 2005, 127, 2796 CrossRef CAS PubMed.
- (a) B. W. Michel, A. M. Camelio, C. N. Cornell and M. S. Sigman, J. Am. Chem. Soc., 2009, 131, 6076 CrossRef CAS PubMed; (b) B. W. Michel, J. R. McCombs, A. Winkler and M. S. Sigman, Angew. Chem., Int. Ed., 2010, 49, 7312 CrossRef CAS PubMed; (c) B. W. Michel, L. D. Steffens and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 8317 CrossRef CAS PubMed; (d) J. R. McCombs, B. W. Michel and M. S. Sigman, J. Org. Chem., 2011, 76, 3609 CrossRef CAS PubMed; (e) R. J. DeLuca, J. L. Edwards, L. D. Steffens, B. W. Michel, X. Qiao, C. Zhu, S. P. Cook and M. S. Sigman, J. Org. Chem., 2013, 78, 1682 CrossRef CAS PubMed.
- B. Feng, Z. Hou, X. Wang, Y. Hu, H. Li and Y. Qiao, Green Chem., 2009, 11, 1446 RSC.
- T. Cohen, M. Gughi, V. A. Notaro and G. Pinkus, J. Org. Chem., 1962, 27, 814 CrossRef CAS.
- Y. Kawashita, N. Nakamichi, H. Kawabata and M. Hayashi, Org. Lett., 2003, 5, 3713 CrossRef CAS PubMed.
- Examples of isomerisation by Pd(II): (a) J. F. Harrod and A. J. Chalk, J. Am. Chem. Soc., 1964, 86, 1776 CrossRef CAS; (b) D. B. Dahl, C. Davies, R. Hyden, M. L. Kirova and W. G. Lloyd, J. Mol. Catal. A: Chem., 1997, 123, 91 CrossRef CAS; (c) H. J. Lim, C. R. Smith and T. V. RajanBabu, J. Org. Chem., 2009, 74, 4565 CrossRef CAS PubMed; (d) M. S. Winston, P. F. Oblad, J. A. Labinger and J. E. Bercaw, Angew. Chem., Int. Ed., 2012, 51, 9822 CrossRef CAS PubMed.
- (a) J. C. Oxley, J. L. Smith, P. R. Bowden and R. C. Rettinger, Propellants, Explos., Pyrotech., 2013, 38, 244 CrossRef CAS; (b) J. C. Oxley, J. L. Smith, L. Steinkamp and G. Zhang, Propellants, Explos., Pyrotech., 2013, 38, 841 CrossRef CAS.
- M. Sommovigo and H. Alper, J. Mol. Catal., 1994, 88, 151 CrossRef CAS.
- A. B. Weinstein and S. S. Stahl, Catal. Sci. Technol., 2014, 4, 4301 CAS.
- J.-H. Chia, S.-H. Wub and C.-M. Shu, J. Hazard. Mater., 2009, 171, 1145 CrossRef PubMed.
- A. M. Eliasen, R. P. Thedford, K. R. Claussen, C. Yuan and D. Siegel, Org. Lett., 2014, 16, 3628 CrossRef CAS PubMed.
- G. R. Astbury, Org. Process Res. Dev., 2002, 6, 893 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Optimisation experiments and experimental procedures. See DOI: 10.1039/c4gc02465f |
| This journal is © The Royal Society of Chemistry 2015 |