
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron-catalyzed olefin hydrogenation at 1 bar H2 with a FeCl3–LiAlH4 catalyst†
Tim N.
Gieshoff
a,
Matteo
Villa
a,
Alice
Welther
a,
Markus
Plois
b,
Uttam
Chakraborty
b,
Robert
Wolf
b and
Axel
Jacobi von Wangelin
*a
aInstitute of Organic Chemistry, University of Regensburg, 93040 Regensburg, Germany. E-mail: axel.jacobi@ur.de
bInstitute of Inorganic Chemistry, University of Regensburg, 93040 Regensburg, Germany
First published on 7th January 2015
Abstract
The scope and mechanism of a practical protocol for the iron-catalyzed hydrogenation of alkenes and alkynes at 1 bar H2 pressure were studied. The catalyst is formed from cheap chemicals (5 mol% FeCl3–LiAlH4, THF). A homogeneous mechanism operates at early stages of the reaction while active nanoparticles form upon ageing of the catalyst solution.
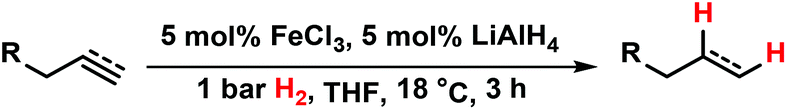
Catalytic hydrogenations of olefins constitute one of the strongholds of transition metal catalysis within organic synthesis and technical processes.1 The majority of methods involve noble metal catalysts based on Pd, Pt, Rh, Ir or toxic metals such as Ni or Co. Iron-catalyzed hydrogenations of olefins have only recently attracted great interest due to their expedient economic and environmental qualities.2 Homogeneous iron catalysts were mostly reported with phosphine and pyridyl-2,6-diimine ligands, sometimes requiring high pressures of H2.3,4 Nanoparticle Fe catalysts could be prepared by reduction of iron salts with Grignard reagents in the absence of a suitable ligand or by decomposition of iron carbonyls.5 Fe-catalyzed reductions of olefins were recently reported with cheap ferrous salt pre-catalysts FeX2 in the presence of an excess of lithium N,N-dimethylaminoborohydride (10 equiv.) or sodium triethylborate (4 equiv.) and required a high catalyst loading or the addition of tetra-dentate ligands.6 Reductions of alkenes and alkynes with LiAlH4 in the presence of various transition metal halides (NiCl2, TiCl2, CoCl2, FeCl3) were already reported in the 1960s and postulated to involve metal hydride species that engage in formal hydrometalations of the olefin.7 Here, we wish to present a synthetic and mechanistic study on a hydrogenation protocol using catalytic amounts of a cheap Fe salt and catalytic amounts of lithium aluminiumhydride (LiAlH4) as catalyst activator under an atmosphere of 1 bar H2 as stoichiometric hydrogen source (Scheme 1).7e This method allows the use of standard (ambient pressure) equipment. H2 is an abundant raw material; LiAlH4 is an easy-to-handle reductant with numerous applications.8
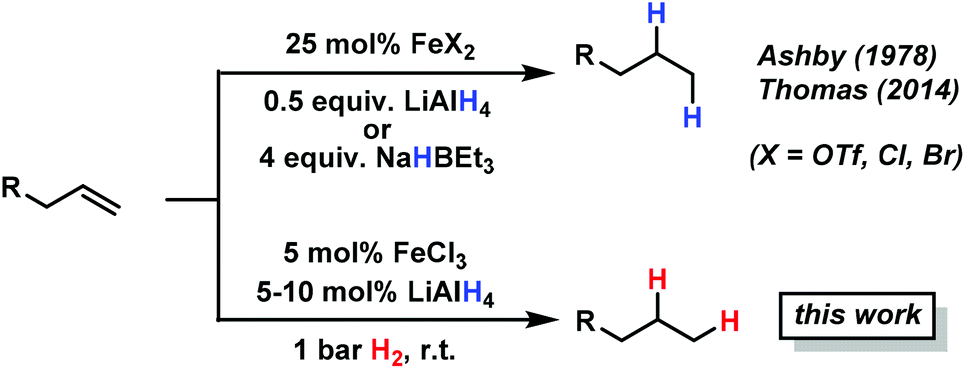 | ||
| Scheme 1 Iron-catalyzed reductions of olefins: hydride vs. hydrogen methods. | ||
Reaction conditions and substrate scope
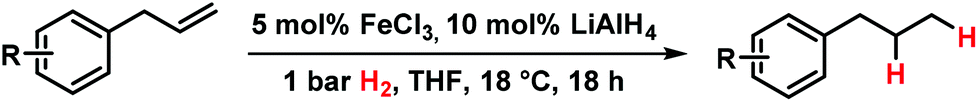
Initial experiments with the model substrate allylbenzene (1) aimed at the identification of a suitable catalytic reductant which assists the formation of a low-valent iron catalyst (with dark brown colour) from the commercial pre-catalyst FeCl3 (Table 1).9 LiAlH4 displayed excellent selectivity which exceeded that of earlier protocols with Grignard reagents.5 Isomerization of the terminal double bond into conjugation – which occurred in the related EtMgCl-mediated protocols (entries 2, 4) – was effectively suppressed.10 NaBH4 was far less active even at elevated temperature and pressure (entries 6, 7). Interestingly, low ratios of LiAlH4–FeCl3 (1/1 to 2/1) fared optimal in the hydrogenation of 1 at 1 bar H2. When employing a larger excess of LiAlH4 (>2/1), the catalytic activity collapsed.7e This stoichiometry differs from literature reports where large excess amounts of hydride reagents effected clean hydrogenations of olefins.6,7a–c At 60 °C, the FeCl3–LiAlH4 catalyst decomposed upon decolorization. The catalyst system comprises of cheap and easy-to-handle reagents (FeCl3 or FeCl2, LiAlH4, THF); the reaction operates under ambient conditions (1 bar H2, 20 °C), which make the general protocol practical for every-day use in standard synthesis laboratories. The optimized conditions were applied to functionalized allylbenzenes and styrenes (Tables 2 and 3).9|
|
||||
|---|---|---|---|---|
| Reductant (mol%) | Deviation from conditions | 2 (%) | 3 , (%) | |
| a Conditions: 5 mol% FeCl3 in THF (0.5 mL) under argon, addition of reductant at r.t., after 10 min addition of 1, after 1 min exchange of Ar with 4 bar H2; b Quantitative GC-FID (internal reference); c During catalyst preparation. | ||||
| 1 | — | — | 0 | 2 |
| 2 | EtMgCl (30) | — | 42 | 56 |
| 3 | EtMgCl (30) | —b | 16 | 0 |
| 4 | EtMgCl (30) | 1 bar H2, 20 h | 60 | 36 |
| 5 | Et2Zn (20) | 30 bar H2, 80 °C, 12 h | 4 | 1 |
| 6 | NaBH4 (100) | 50 bar H2, 24 h | 8 | 0 |
| 7 | NaBH4 (100) | MeOH–THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 50 bar H2, 50 °C, 20 h :1), 50 bar H2, 50 °C, 20 h |
45 | 38 |
| 8 | LiAlH4 (10) | — | 97 | 3 |
| 9 | LiAlH4 (10) | 1 bar H2, 20 h | 98 | 1 |
| 10 | LiAlH4 (30) | As entry 9, open to airc | 95 | 3 |
| 11 | LiAlH4 (10) | FeCl2 | 96 | 1 |
| 12 | LiAlH4 (10) | Fe(acac)3 | 20 | 15 |
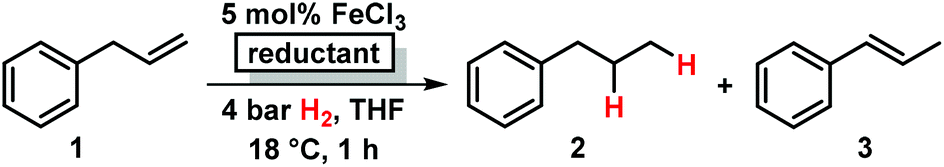
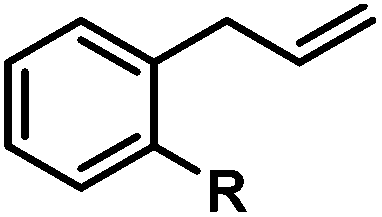
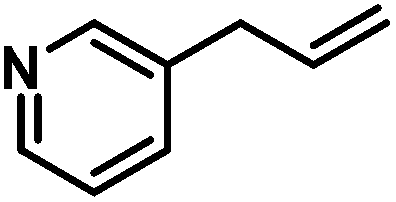
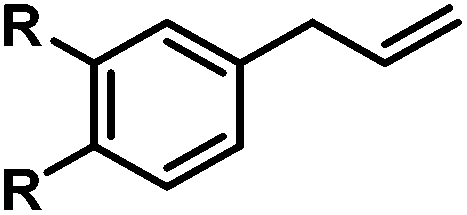
|
|
|||
|---|---|---|---|
| Styrene | R | Yielda (%) | |
| a Quantitative GC-FID (internal reference); conversion (%) in parentheses if <95%. b <12% ethylbenzene. c 20 h, 10 bar H2. d 5 mol% LiAlH4, 3 h. e 20 h. f <5% cumene. g Mixture of partial and full hydrogenation products (∼6/1). | |||
| 1 |
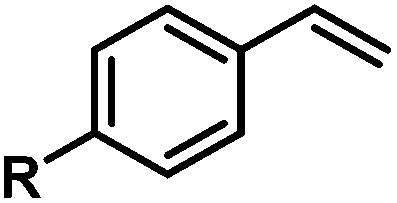
|
H | 100 |
| 2 | Me | 98 | |
| 3 | OMe | 98 | |
| 4 | Cl | 93b | |
| 5 | Br | 94b | |
| 6 | F | 77c | |
| 7 | OBn | 100d | |
| 8 | NH2 | 97c | |
| 9 | CO2Me | 97 | |
| 10 |
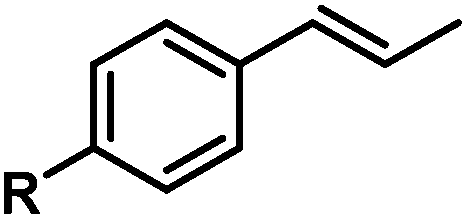
|
H | 100e |
| 11 | OMe | 84 | |
| 12 |
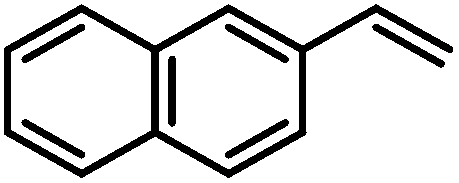
|
100 | |
| 13 |
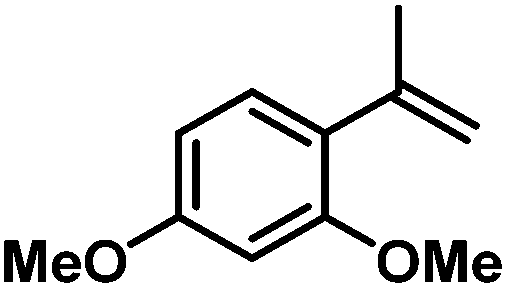
|
86d | |
| 14 |
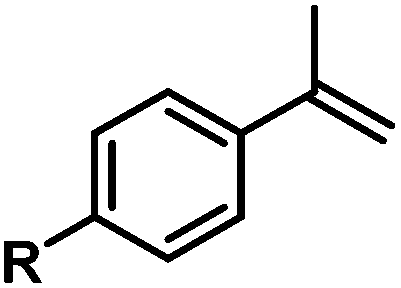
|
H | 100d |
| 15 | Cl | 85 (89)f | |
| 16 | Br | 44 (56)f | |
| 17 | Br | 92c,f | |
| 18 | OMe | 100d | |
| 19 |
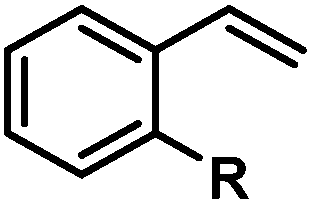
|
Cl | 74 (86)b |
| 20 | OBn | 100d | |
| 21 |
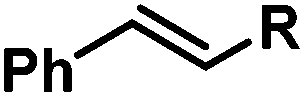
|
Ph | 100d |
| 22 | Bn | 100d | |
| 23 | CO2Et | 58 (68)c | |
| 24 |
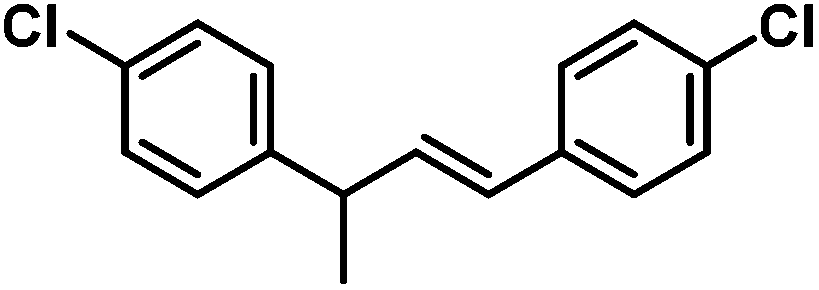
|
48 (54) | |
| 25 | 88c | ||
| 26 |
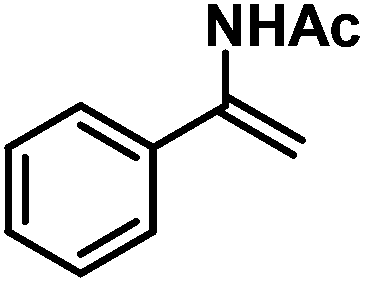
|
33 (58)e | |
| 27 |
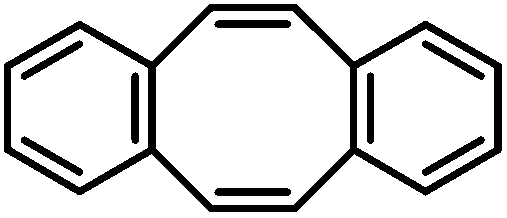
|
75d,g | |
| 28 |
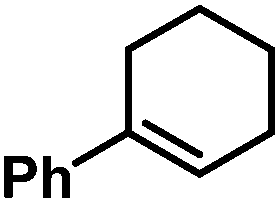
|
18 (18)c | |
Allylbenzenes underwent only minimal olefin isomerization.10 Styrenes exhibited low propensity to undergo polymerization (entry 13, Table 3). The general protocol is compatible with several functional groups including F, Cl, Br, allyl and benzyl ethers, esters, carboxamides, pyridines and anilines. Clean hydrogenation was achieved with bulky, ortho-substituted, and electron-rich styrenes. For comparison, the FeCl3/EtMgCl-derived catalyst effected undesired dehalogenation (Cl, Br)11 and allylether cleavage,12 and showed no activity in the presence of carboxylates or cinnamates. Catalyst decomposition was effected by nitro groups, iodides, nitriles, ketones, and acidic protons (e.g. alkanols, pKa ∼ 17), presumably by oxidation to catalytically incompetent Fe(II) species (decolorization). Tri-substituted styrenes gave low conversions. In general, bulky and functionalized substrates were more reactive at elevated pressures (10 bar H2).13
Hydrogenations of aliphatic alkenes were also catalyzed by FeCl3–LiAlH4 under similar conditions (Table 4).9 Terminal olefins were only slowly isomerized (∼10%).10 Surprisingly, substrates containing moderately acidic protons (pKa ∼ 25)14 underwent hydrogenation with high selectivity (entries 10–13).15 Alkynes underwent Z-selective semi-hydrogenation,16 whereas complete hydrogenation to the alkanes was observed at longer reaction times or elevated pressures.
|
|
||||
|---|---|---|---|---|
| Substrate | Product | Yielda (%) | ||
| a Quantitative GC-FID (internal reference); conversion (%) in parentheses if <95%. b Alkene isomers. c 20 h. d 10 bar, 20 h. e 10 mol% LiAlH4. f 60 °C. g By NMR (vs. CH2Br2). | ||||
| 1 |

|
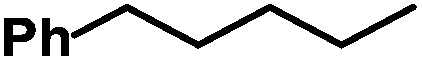
|
82b | |
| 2 |
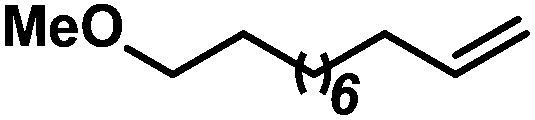
|
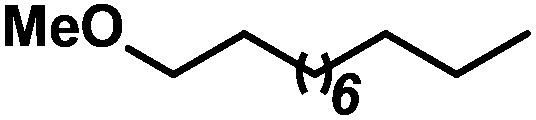
|
69b | |
| 3 |

|

|
82c | |
| 4 |
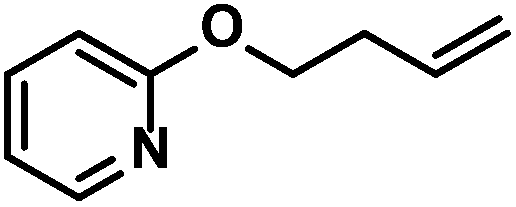
|
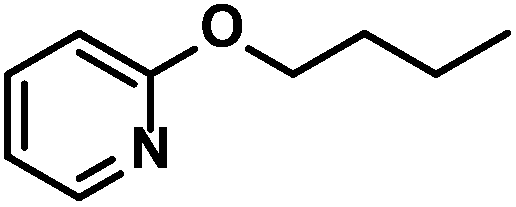
|
89 (89)c | |
| 5 |
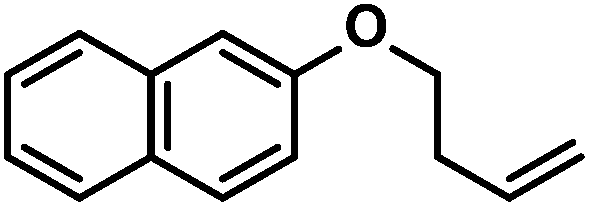
|
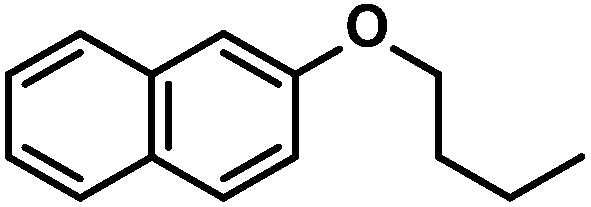
|
65 (65)c | |
| 6 |
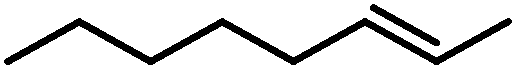
|
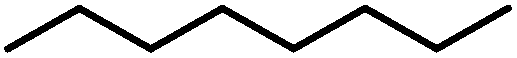
|
21 (41)b | |
| 7 |
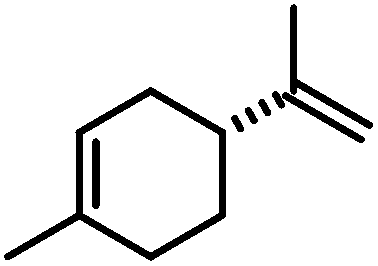
|
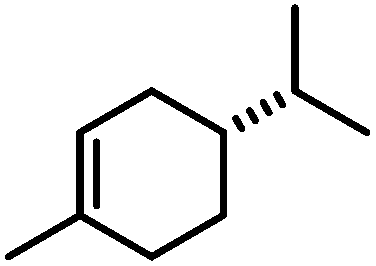
|
100d | |
| 8 |
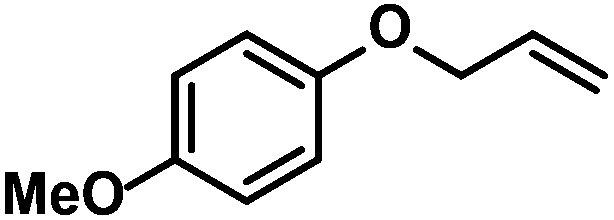
|
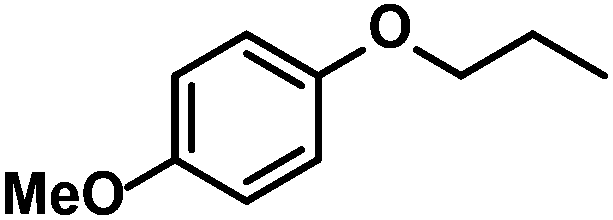
|
2 (10) | |
| 9 | 64 (65)d | |||
| 10 |
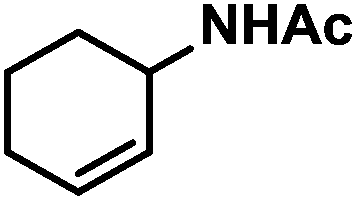
|
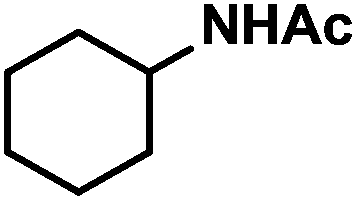
|
12 (16) | |
| 11 | 96d,e | |||
| 12 |
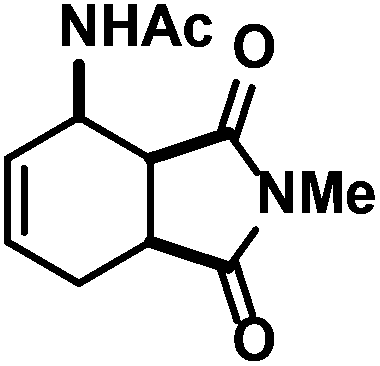
|
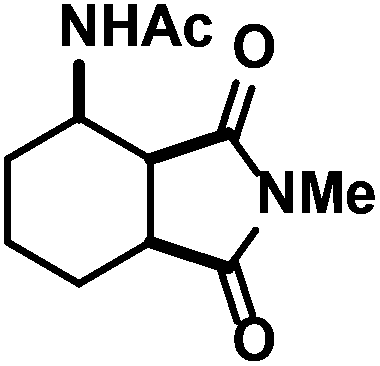
|
38 (38)d,e | |
| 13 | 69 (69)d,e,f | |||
| 14 |
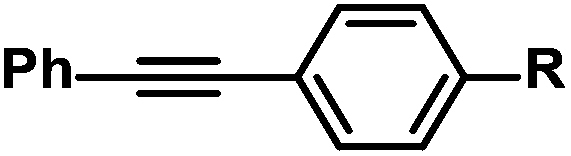
|
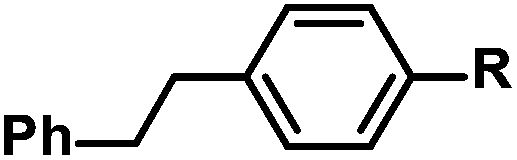
|
R = H | 100c |
| 15 | CO2Me | 92c | ||
| 16 |
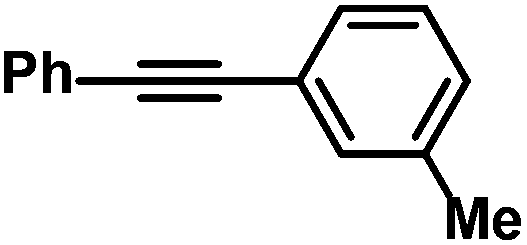
|
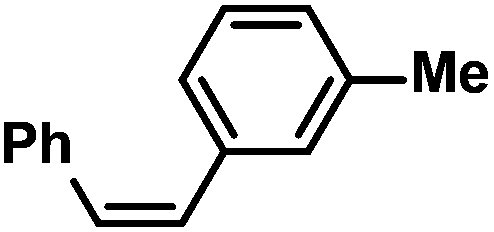
|
75 (80)g | |
| 17 |
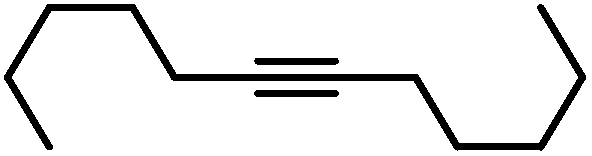
|
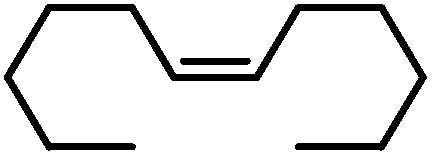
|
90g | |
Mechanistic studies
The distinction between homogeneous and heterogeneous catalysts is a challenging task.17 However, kinetic experiments with selective poisons can provide valuable information on the topicity of the catalyst species. We have performed two sets of poisoning experiments which appear to support a homogeneous mechanism. Dibenzo[a,e]cycloocta-tetraene (dct) is a selective ligand for homogeneous metal species due to its rigid tub-like structure and π-acceptor properties.18 Upon addition of 30 mol% dct (6 equiv. per [Fe]) to the hydrogenation of isopropenylbenzene at 1 bar H2 after 30 min, the catalyst activity was significantly inhibited (Scheme 2, top).9,19 A similar conclusion can be derived from a poisoning experiment with 3 equiv. Hg (60 equiv. Hg per [Fe]). A potential amalgam formation20 was not observed and no significant change of the catalyst activity was observed in comparison with the control reaction (Scheme 2, bottom).9 These results suggest the operation of a homogeneous catalyst species during the early stage of the catalytic hydrogenation. Previous studies showed that the reaction of FeCl3 with an excess of LiAlH4 ultimately leads to the formation of iron metal and AlH3via the intermediate formation of a thermally unstable iron(II) compound with the composition Fe(AlH4)2.21,22 In an attempt to gain deeper insight into the catalyst species operating in homogeneous solution, we treated [FeCl2(tmeda)]2 (tmeda = N,N,N′,N′-tetramethylethylenediamine) with LiAlH4 at −70 °C and obtained dark red crystals of the oligohydride compound [Li(thf)2{Fe(tmeda)}2(AlH5)(Al2H9)] (4, Scheme 3).9 The hexa-metallic macrocyclic cage contains 14 bridging hydrido ligands and two Fe atoms with distorted octahedral coordination geometries. Unfortunately, the thermal instability prevented further spectroscopic characterization.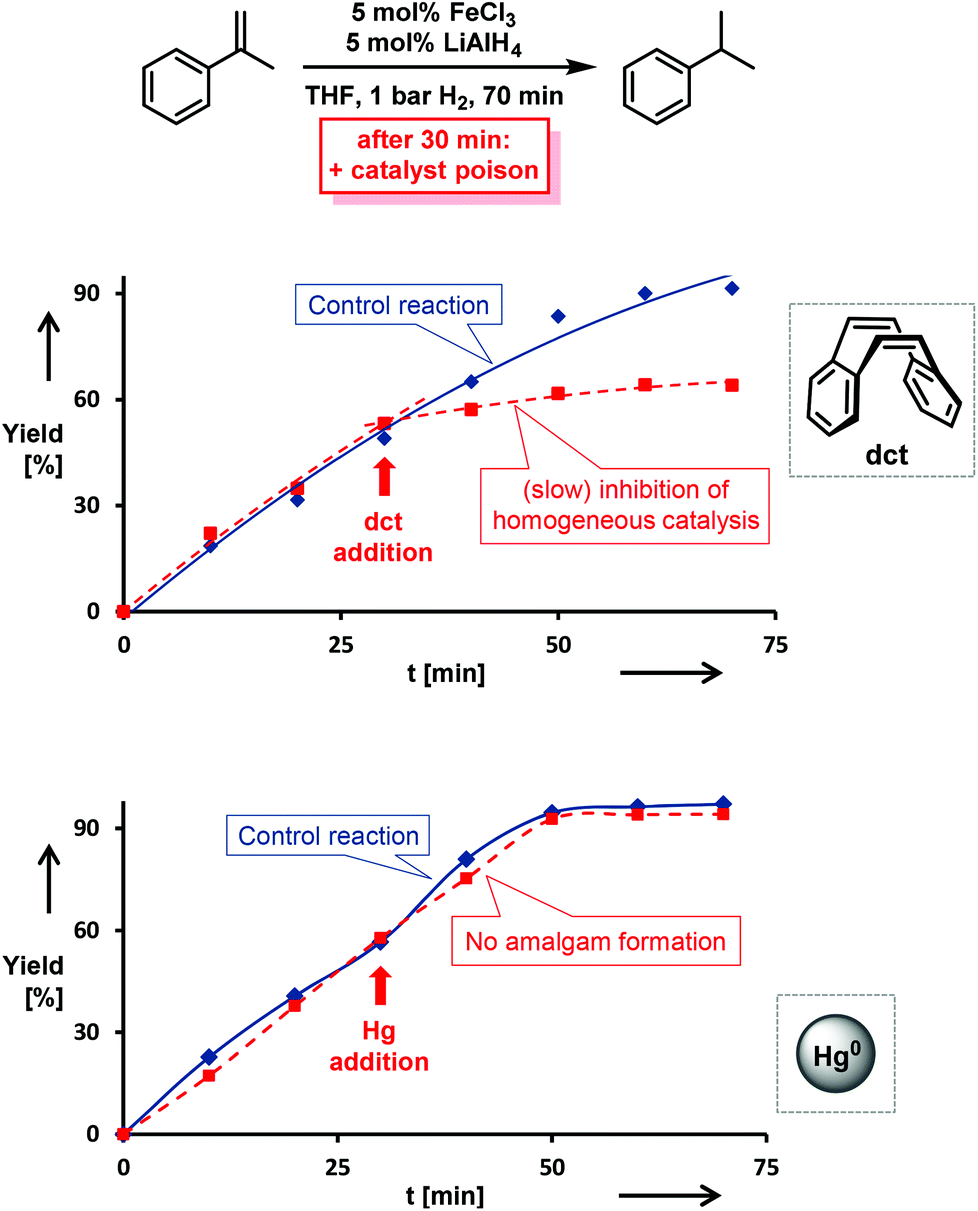 | ||
| Scheme 2 Top: poisoning experiment with 30 mol% dibenzo[a,e]cyclooctatetra-ene (dct, dashed curve) vs. control reaction (solid line). Bottom: poisoning with 3 equiv. Hg (dashed) vs. control reaction (solid line). | ||
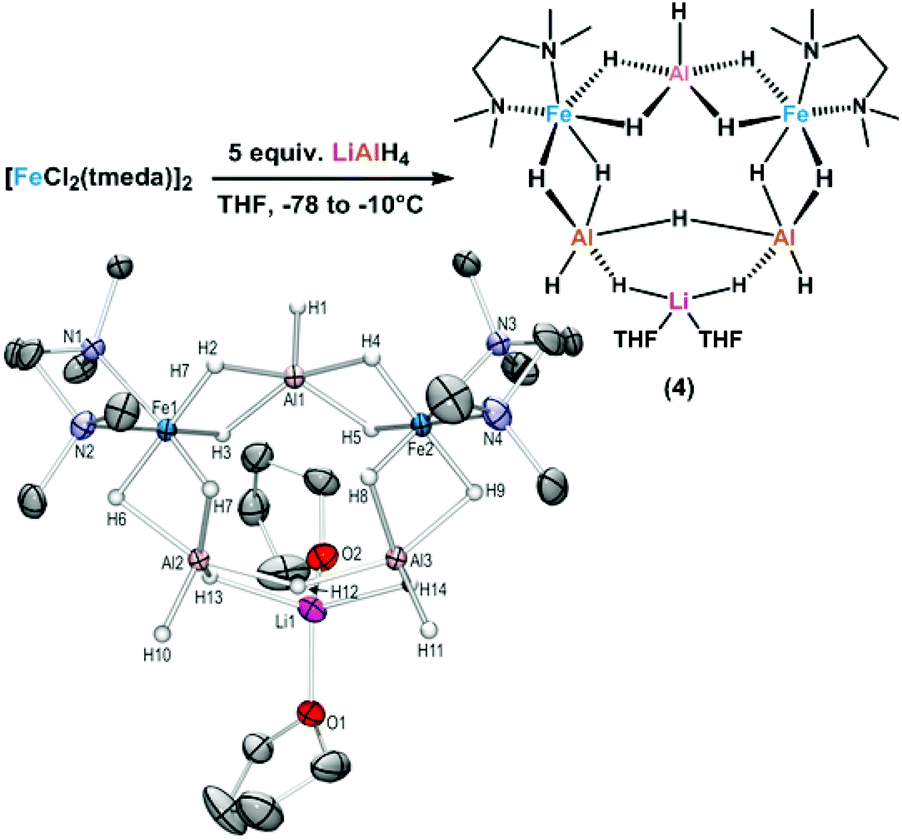 | ||
| Scheme 3 Synthesis of the soluble LiAlFe-oligohydride complex 4. | ||
However, complex 4 showed no activity in hydrogenations of styrenes (1–10 bar H2, −10 °C) and maintained its red colour throughout the reaction. Above −10 °C, the complex rapidly decomposed upon H2 evolution to give a brown paramagnetic species which afforded good yields in hydrogenations at 20 °C and 4 bar H2. The crystallographic characterization of 4 documents that this or similar oligonuclear Fe(II) aluminohydride complexes may be intermediates en route to the formation of catalytically active low-valent iron species.23
The initially homogeneous dark-brown catalyst species (possibly in the oxidation states 0 and/or +1)23 experience rapid ageing and particle formation after appr. 1 h under the reductive conditions. Several methods of synthesis and characterization techniques of naked Fe(0) nanoparticles (prepared by reduction of ferric and ferrous halides) have been reported.5,7,23,24 DLS measurements (dynamic light scattering) of freshly prepared catalyst solutions (5 mol% FeCl3–LiAlH4, THF, r.t., 10 min, then 100 nm nanofiltration) documented the presence of poly-disperse particles of 250–1500 nm size after 30 min of ageing under anaerobic conditions in the absence of substrates. The aged species are much less catalytically active than their homogeneous counterparts. Catalyst solutions (FeCl3–LiAlH4 (1/1) in THF) stored at 0 °C under argon for 6 h, 24 h, and 48 h afforded 42%, 12%, and 5% conversion of α-methylstyrene under standard conditions (see entry 16 in Table 2), respectively.
We postulate a homogeneous mechanism of soluble, low-valent iron catalyst in the initial stage of the hydrogenation reactions (Scheme 4). Such species form by reduction of FeCl3 (or LnFeCl2) with LiAlH4 at above 0 °C and are typically characterized by the dark brown colour. The absence of suitable ligands leads to the formation of Fe(0) nanoclusters5,22,24 which require higher H2 pressures than the homogeneous species to maintain catalytic activity.
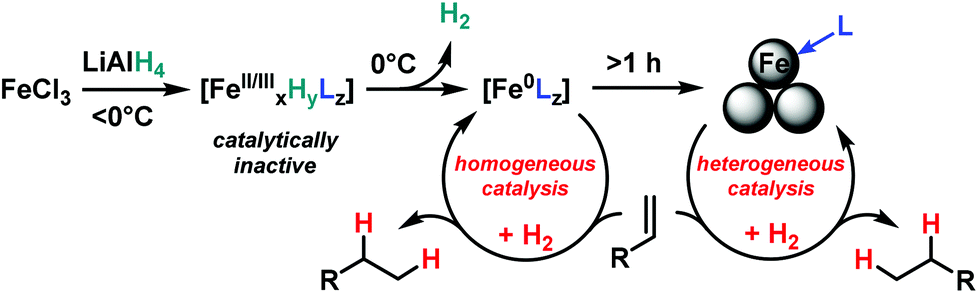 | ||
| Scheme 4 Proposed formation and catalysis of low-valent iron species. | ||
Deuterium incorporation was observed at higher catalyst concentrations (30 mol% FeCl3–LiAlD4) in the absence of H2 which gave ∼55% hydrogenation product (Scheme 5, center).9 Such H2-free conditions can effect H/D scrambling in the starting material and product (via reversible hydroferration) and the formation of radical intermediates (with participation of THF as H donor).9 However, the radical mechanism is very unlikely to operate under hydrogenation conditions in the presence of H2 gas (Scheme 5):9 reaction work-up with deuterium oxide (D2O) and employment of lithium aluminium-deuteride (LiAlD4) showed no deuterium incorporation into the products, respectively (Scheme 5, top right). Further, the intermediacy of free C-radicals is unlikely: employment of the radical probe 1-cyclopropyl-1-phenylethylene25 resulted in less than 2% ring opening (Scheme 5, bottom).9 The hydrogenation of various styrenes (1 bar H2) was unaffected by the presence of 1 equiv. 1,1-diphenylethene. On the other hand, the addition of TEMPO (2,2,6,6-tetramethylpiperidinyloxyl, 1 equiv.) inhibited conversion of α-methylstyrene (no TEMPO adduct detected), possibly by irreversible catalyst oxidation as indicated by the decolorization of the solution.
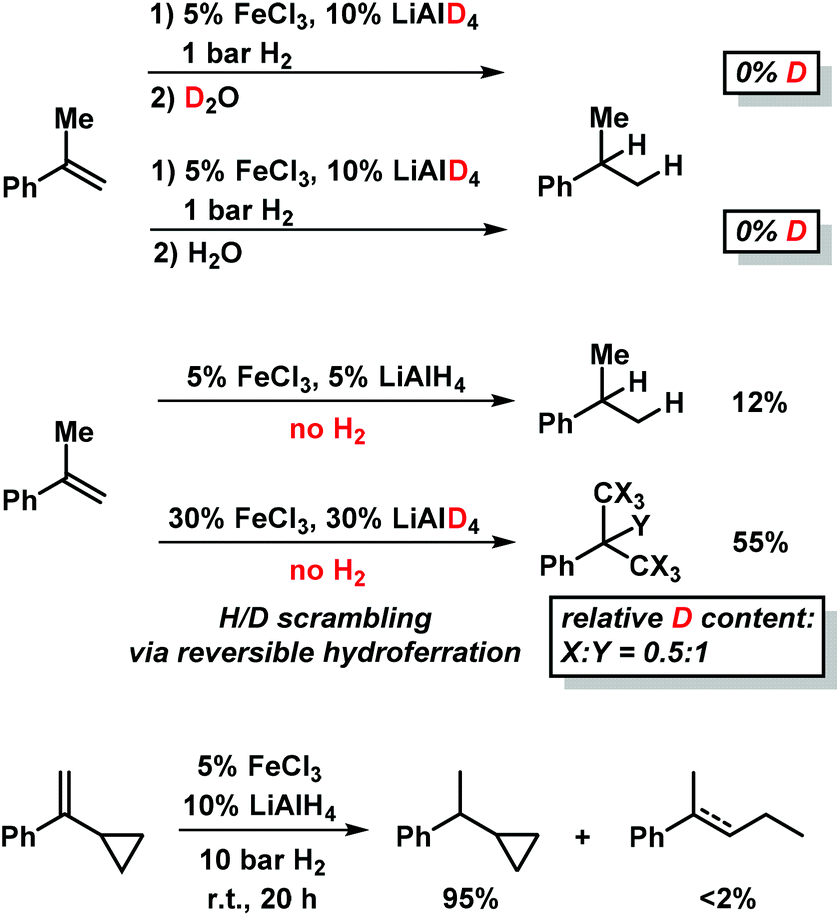 | ||
| Scheme 5 Mechanistic studies with deuterated reagents (top), in the absence of H2 (center), and with radical probe (bottom). | ||
General procedure
A 10 mL vial was charged with a freshly prepared solution of FeCl3 in THF (0.50 mL, 0.05 M) and an aliquot of a vigorously stirred suspension of LiAlH4 in THF (0.50 mL, 0.1 M) under an argon atmosphere. After stirring the dark brown mixture for 10 min (during which H2 evolution can be observed), the alkene (0.50 mmol) was added and the vial purged with dihydrogen gas (1 min). For reactions under higher H2 pressures, the vial was transferred to a high pressure reactor (Parr™), the reactor purged with H2 (1 min), sealed, and the internal pressure adjusted to 1 bar. After 3–20 h at room temperature, the reaction was quenched with saturated aqueous NaHCO3 (1 mL) and extracted with ethyl acetate (2 × 2 mL). The organic phases were dried (Na2SO4) and subjected to flash chromatography (SiO2, pentane/ethyl acetate) or analyzed by quantitative GC-FID analysis vs. pentadecane as internal reference.Conclusions
In summary, we have studied the iron-catalyzed hydrogenation of various styrenes, alkenes, and alkynes under an atmosphere of 1 bar H2. This method uses cheap and easy-to-handle reagents (FeCl3, LiAlH4, THF, H2) which allow facile implementation in standard synthesis labs. Alkynes underwent Z-selective semi-hydrogenation. Sterically hindered and functionalized olefins showed higher conversions at elevated H2 pressures. Mechanistic studies support the notion of a homogeneous catalyst species at the outset of the hydrogenation reactions (<1 h) while catalyst ageing results in the formation of particles which exhibited somewhat lower catalytic activity. The crystallographically characterized homogeneous Fe(II) oligohydride complex 4 can serve as starting point for further model catalyst preparations.Acknowledgements
We thank the Evonik foundation (T. N. G.) and the Fonds der Chemischen Industrie (M. V.) for doctoral fellowships.Notes and references
- (a) Catalytic Hydrogenation, ed. L. Cerveny, Elsevier, Amsterdam, 1986 Search PubMed; (b) The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH. Weinheim, 2007 Search PubMed; (c) S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, New York, 2001 Search PubMed.
- (a) K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849 RSC; (b) B. A. F. Le Bailly and S. P. Thomas, RSC Adv., 2011, 1, 1435 RSC.
- (a) E. J. Daida and J. C. Peters, Inorg. Chem., 2004, 43, 7474 CrossRef CAS PubMed; (b) C. Bianchini, A. Meli, M. Peruzzini, P. Frediani, C. Bohanna, M. A. Esteruelas and L. A. Oro, Organometallics, 1992, 11, 138 CrossRef CAS.
- (a) R. P. Yu, J. M. Darmon, J. M. Hoyt, G. W. Margulieux, Z. R. Turner and P. J. Chirik, ACS Catal., 2012, 2, 1760 CrossRef CAS PubMed; (b) R. J. Trovitch, E. Lobkovsky, E. Bill and P. J. Chirik, Organometallics, 2008, 27, 1470 CrossRef CAS; (c) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794 CrossRef CAS PubMed; (d) T. S. Carter, L. Guiet, D. J. Frank, J. West and S. P. Thomas, Adv. Synth. Catal., 2013, 355, 880 CrossRef CAS.
- (a) P. H. Phua, L. Lefort, J. A. F. Boogers, M. Tristany and J. G. de Vries, Chem. Commun., 2009, 3747 RSC; (b) M. Stein, J. Wieland, P. Steurer, F. Tölle, R. Mülhaupt and B. Breit, Adv. Synth. Catal., 2011, 353, 523 CrossRef CAS; (c) A. Welther, M. Bauer, M. Mayer and A. Jacobi von Wangelin, ChemCatChem, 2012, 4, 1088 CrossRef CAS; (d) R. Hudson, A. Rivière, C. M. Cirtiu, K. L. Luska and A. Moores, Chem. Commun., 2012, 48, 3360 RSC; (e) A. Welther and A. Jacobi von Wangelin, Curr. Org. Chem., 2013, 17, 326 CrossRef CAS; (f) R. Hudson, G. Hamasaka, T. Osako, Y. M. A. Yamada, C. J. Li, Y. Uozumi and A. Moores, Green Chem., 2013, 15, 2141 RSC.
- (a) B. A. F. Le Bailly, M. D. Greenhalgh and S. P. Thomas, Chem. Commun., 2012, 48, 1580 RSC; (b) D. J. Frank, L. Guiet, A. Käslin, E. Murphy and S. P. Thomas, RSC Adv., 2013, 3, 25698 RSC.
- LiAlH4 as hydride source in metal-catalyzed reductions: (a) G. Dozzi, S. Cucinella and A. Mazzei, J. Organomet. Chem., 1979, 164, 1 CrossRef CAS; (b) E. C. Ashby and J. J. Lin, J. Org. Chem., 1978, 43, 2567 CrossRef CAS; (c) E. C. Ashby and J. J. Lin, Tetrahedron Lett., 1977, 51, 4481 CrossRef. For early reports of H2 evolution from iron hydride species and catalytic olefin hydrogenations, see: (d) Y. Takegami, T. Ueno and T. Fujii, Kogyo Kagaku Zasshi (J. Chem. Soc. Jpn., Ind. Chem. Sect.), 1964, 67, 1009 CAS; (e) Y. Takegami, T. Ueno and T. Fujii, Bull. Chem. Soc. Jpn., 1965, 38, 1279 CrossRef CAS.
- (a) Technical production and use of H2; H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 2010 Search PubMed; (b) Selected applications of LiAlH4: S. Yaragorla, Synlett, 2008, 3073 CrossRef CAS.
- Please see ESI† for detailed experimental and analytical data.
- Olefin isomerization with iron carbonyls: (a) J. V. Crivello and S. Kong, J. Org. Chem., 1998, 63, 6745 CrossRef CAS; (b) M. R. Reddy and M. Periasamy, J. Organomet. Chem., 1995, 491, 263 CrossRef; (c) P. A. Tooley, L. W. Arndt and M. Y. Darensbourg, J. Am. Chem. Soc., 1985, 107, 2422 CrossRef CAS; (d) R. Jennerjahn, R. Jackstell, I. Piras, R. Franke, H. Jiao, M. Bauer and M. Beller, ChemSusChem, 2012, 5, 734 CrossRef CAS PubMed; (e) Naked Fe(0) species: M. Mayer, A. Welther and A. Jacobi von Wangelin, ChemCatChem, 2011, 3, 1567 CrossRef CAS. Allylbenzenes were prepared according to: (f) M. Mayer, W. M. Czaplik and A. Jacobi von Wangelin, Adv. Synth. Catal., 2010, 352, 2147 CrossRef CAS.
- (a) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009 CrossRef CAS PubMed; (b) W. M. Czaplik, S. Grupe, M. Mayer and A. Jacobi von Wangelin, Chem. Commun., 2010, 46, 6350 RSC.
- D. Gärtner, H. Konnerth and A. Jacobi von Wangelin, Catal. Sci. Technol., 2013, 3, 2541 Search PubMed.
- (a) α-Methylstyrene was hydrogenated at 4 bar H2 in the presence of various functionalized additives (1 equiv.). No decrease of hydrogenation selectivity was observed when adding PhCl, PhBr, PhCO2Me, PhNH2, PhCONH2, and 1,1-diphenylethylene, respectively. With PhNO2, PhCN, PhI, and MeCN, no conversion of methylstyrene was observed, respectively, neither was the additive significantly consumed (<5%); (b) At 1–2 bar H2, much lower tolerance of functional groups was observed. We assume that the presence of functionalized substrates or polar moieties enhances catalyst ageing and formation of a heterogeneous species which appeared to somewhat less active. It should also be noted that sporadic cases of somewhat lower reproducibilities of reactions at 1 bar H2 pressure were observed with substrates bearing polar functional groups.
- N-Methylacetamide (AcNHMe), pKa 25.9. F. G. Bordwell, J. A. Harrelson and T.-Y. Lynch, J. Org. Chem., 1990, 55, 3337 CrossRef CAS.
- N-Acetylaminocyclohexenes were prepared according to: (a) D. Strübing, H. Neumann, A. Jacobi von Wangelin, S. Klaus, S. Hübner and M. Beller, Tetrahedron, 2006, 62, 10962 CrossRef; (b) S. Hübner, H. Neumann, A. Jacobi von Wangelin, S. Klaus, D. Strübing, H. Klein and M. Beller, Synthesis, 2005, 2084 Search PubMed; (c) S. Klaus, S. Hübner, H. Neumann, D. Strübing, A. Jacobi von Wangelin, D. Gördes and M. Beller, Adv. Synth. Catal., 2004, 346, 970 CrossRef CAS; (d) A. Jacobi von Wangelin, H. Neumann, D. Gördes, A. Spannenberg and M. Beller, Org. Lett., 2001, 3, 2895 CrossRef CAS PubMed.
- Fe-catalyzed Z-selective semihydrogenation of alkynes: (a) S. Enthaler, M. Haberberger and E. Irran, Chem. – Asian J., 2011, 6, 1613 CrossRef CAS PubMed; (b) L. Ilies, T. Yoshida and E. Nakamura, J. Am. Chem. Soc., 2012, 134, 16951 CrossRef CAS PubMed; (c) C. Belger and B. Plietker, Chem. Commun., 2012, 48, 5419 RSC; (d) T. N. Gieshoff, A. Welther, M. T. Kessler, M. H. G. Prechtl and A. Jacobi von Wangelin, Chem. Commun., 2014, 50, 2261 RSC.
- (a) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317 CrossRef CAS; (b) D. Astruc, F. Lu and J. Ruiz Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed; (c) R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed.
- (a) D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 855 CrossRef CAS; (b) G. Franck, M. Brill and G. Helmchen, J. Org. Chem., 2012, 89, 55 CAS; (c) J. F. Sonnenberg and R. H. Morris, Catal. Sci. Technol., 2014, 4, 3426 RSC.
- The observation of a partial inhibition by dct results from a superposition of an effective catalyst inhibition by the presence of dct and (a slower) hydrogenation of dct as substrate itself (see entry 27 in Table 3).
- Iron forms only metastable alloys with mercury (amalgam). The maximum concentration of Fe(0) in Hg was reported to be 2–3 wt%. (a) S. Mørup, S. Linderoth, J. Jacobsen and M. Holmblad, Hyperfine Interact., 1991, 69, 489 CrossRef; (b) S. Linderoth and S. Mørup, J. Phys.: Condens. Matter, 1992, 4, 8627 CrossRef CAS.
- (a) G. W. Schaeffer, J. S. Roscoe and A. C. Stewart, J. Am. Chem. Soc., 1956, 78, 729 CrossRef CAS; (b) H. Neumaier, D. Bückel and G. Ziegelmaier, Z. Anorg. Allg. Chem., 1966, 345, 46 CrossRef CAS; (c) M. E. Kost and A. L. Golovanova, Izv. Akad. Nauk. SSSR, Ser. Khim., 1957, 5, 991 Search PubMed.
- The stable low-spin iron(II) aluminohydride complex [(Me3tacn)2Fe2(μ-AlH6)]+ has recently been reported (tacn = 1,4,7-triazacyclononane): M. Oishi, T. Endo, M. Oshima and H. Suzuki, Inorg. Chem., 2014, 53, 5100 CrossRef CAS PubMed.
- (a) A. Fürstner, R. Martin, H. Krause, G. Seidel, R. Goddard and C. W. Lehmann, J. Am. Chem. Soc., 2008, 130, 8773 CrossRef PubMed; (b) A. Hedström, E. Lindstedt and P.-O. Norrby, J. Organomet. Chem., 2013, 748, 51 CrossRef.
- (a) C. Rangheard, C. de Julian Fernandez, P.-H. Phua, J. Hoorn, L. Lefort and J. G. de Vries, Dalton Trans., 2010, 39, 8464 RSC; (b) R. Schoch, W. Desens, T. Werner and M. Bauer, Chem. – Eur. J., 2013, 19, 15816 CrossRef CAS PubMed; (c) V. Kelsen, B. Wendt, S. Werkmeister, K. Junge, M. Beller and B. Chaudret, Chem. Commun., 2013, 49, 3416 RSC.
- (a) D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317 CrossRef CAS; (b) M. Newcomb, Tetrahedron, 1993, 49, 1151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1034372. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4gc02368d |
| This journal is © The Royal Society of Chemistry 2015 |