
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A system for ω-transaminase mediated (R)-amination using L-alanine as an amine donor†
N.
Richter
a,
J. E.
Farnberger
b,
D.
Pressnitz
b,
H.
Lechner
b,
F.
Zepeck
c and
W.
Kroutil
*b
aACIB GmbH, c/o Heinrichstraße 28, 8010 Graz, Austria
bDepartment of Chemistry, Organic and Bioorganic Chemistry, NAWI Graz, University Graz, Heinrichstraße 28, 8010 Graz, Austria. E-mail: wolfgang.Kroutil@uni-graz.at; Tel: +43-(0)-316-380-5350
cSandoz GmbH, Biocatalysis Lab, Biochemiestraße 10, 6250 Kundl/Tirol, Austria
First published on 19th March 2015
Abstract
Chiral amines are important building blocks for fine chemicals and pharmaceuticals. Consequently, various biocatalytic routes in particular using ω-transaminases (ω-TAs) have been developed recently. Although catalysts for the synthesis of both enantiomers are available, the application of alanine dependent (R)-selective ω-TAs is less favourable due to the requirement of the more expensive D-alanine as an amine donor. Here we describe an efficient method for (R)-amination using ω-TAs in combination with an alanine racemase (AlaR). In this case, the readily available L-alanine can be used as an amine donor leading to improved atom efficiency and significantly reduced costs.
Introduction
ω-Transaminases (ω-TAs) have been proven to be powerful catalysts frequently used in the synthesis of chiral amines.1–8 As an alternative to classical chemical strategies,9–12 they offer a green and sustainable synthetic route and can provide access to both enantiomers of the amine. This is enabled by the recent identification of numerous ω-TAs with (S)- as well as (R)-stereoselectivity.13–19 However, due to the unfavoured reaction equilibrium of the reductive amination of ketones employing alanine as an amine donor as well as co-product inhibition, the reaction requires in situ techniques to remove the co-product pyruvate to set-up efficient processes. As a consequence, many studies addressed these limitations of ω-TAs over the last decade, and a variety of methods have been developed to efficiently shift the equilibrium of the desired reductive amination reaction.2,3,6 This progress enabled the development of various interesting applications employing ω-TAs20–30 and their exploitation on an industrial scale.15 Nevertheless, the selection of the amine donor is a crucial parameter, while a donor such as 2-propylamine seems to be a good choice with respect to economic efficiency, its use is restricted to certain ω-TAs.3,15,17,18,29,31,32 Therefore, a natural donor like alanine might be preferred; however, while L-alanine is readily available to be employed with (S)-selective enzymes, (R)-selective enzymes require the far more expensive D-alanine. This limitation makes their application less efficient, compared to their (S)-selective counterparts, and alternative strategies are required, in particular when amine donors such as 2-propylamine are not an option.Results and discussion
To overcome this limitation for applications with (R)-selective ω-TAs we report here the combination of an alanine racemase (AlaR) with the amination system. Thus, by using L-alanine as the starting material the required more expensive D-alanine is provided in situ by racemisation (Scheme 1). This racemisation concept has been reported for the synthesis of optically pure α-amino acids using α-transaminases; however, the strategy has not been reported for the asymmetric amination of ketones employing ω-TAs.33 In particular, the combination of the AlaR with the frequently used alanine dehydrogenase (AlaDH) to remove the co-product of the amination, pyruvate, by reductive amination to L-alanine (AlaDH system)34 enables the recycling of the amine donor also in the case of (R)-selective ω-TAs.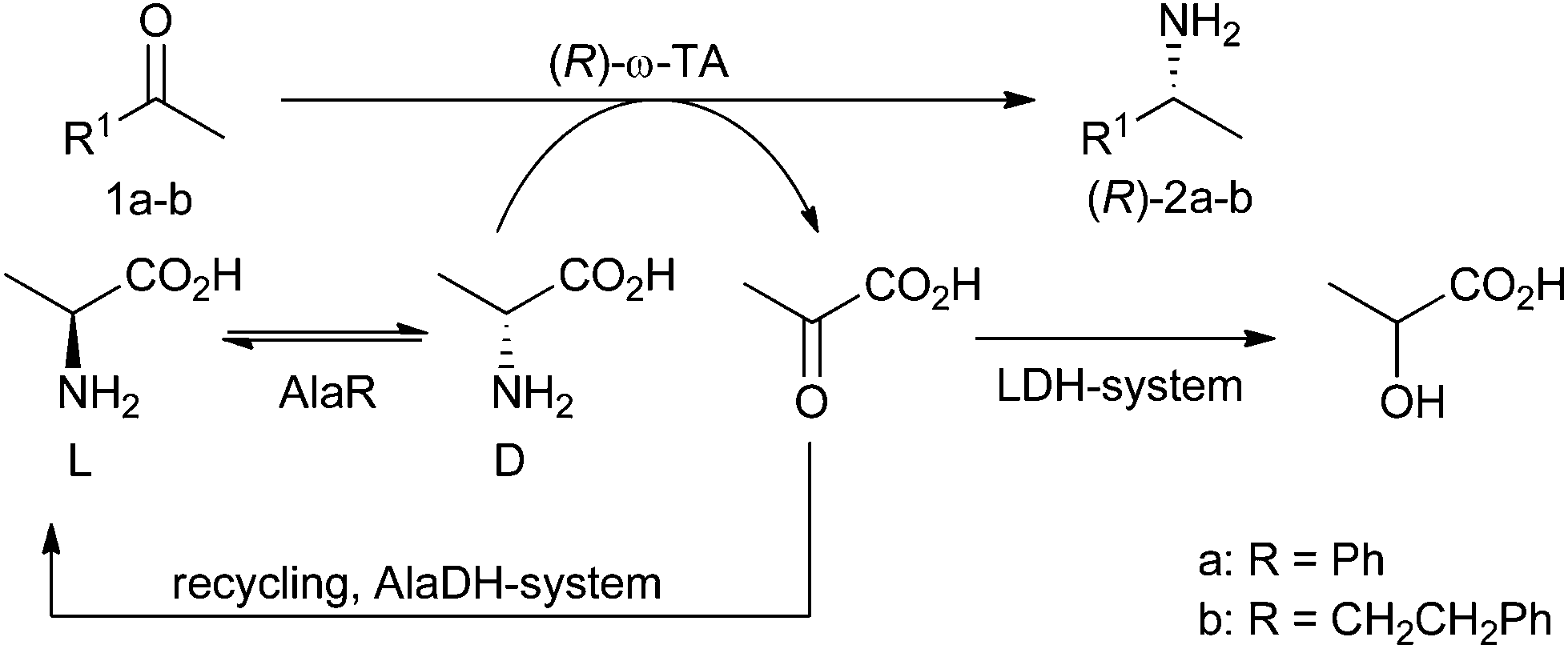 | ||
| Scheme 1 Amination employing (R)-selective ω-TAs and L-alanine as donors. D-Alanine is provided by an alanine racemase (AlaR) by racemisation of L-alanine. The co-product pyruvate is recycled/removed using either an alanine dehydrogenase (AlaDH) or lactate dehydrogenase (LDH). | ||
Cloning and recombinant expression of the SeAlaR and PpAlaR
For the racemisation of L-alanine the PLP-dependent AlaR from Streptomyces coelicolor (SeAlaR)35 was selected as well as an AlaR from Pseudomonas putida (PpAlaR). Both enzymes were cloned in suitable expression vectors and heterologously expressed in E. coli.While the PpAlaR mainly accumulated as insoluble inclusion bodies, good expression was achieved for SeAlaR. However, initial activity studies showed that both enzymes were active towards the racemisation of L-alanine. Based on the better heterologous expression, lyophilised cell free extracts of SeAlaR were used for all further experiments.
Characterisation of the SeAlaR
The SeAlaR was characterised to identify optimal conditions for its application in combination with the other required enzymes in the desired amination reaction (Scheme 1).First the racemisation of 250 mM L-alanine was studied over a period of 30 min, using different amounts of the enzyme (mg mL−1). The results indicated that an efficient racemisation within that time was achieved using 5 mg as well as 2.5 mg of the lyophilised cell free extract (Fig. 1a). In addition to the enzyme loading the influence of DMSO, a co-solvent commonly used with ω-TAs, was studied. For this purpose, activities were determined in the presence of 10 vol% DMSO. As depicted in Fig. 1a the racemisation rate in the absence of DMSO was slightly higher than the reaction in the presence of DMSO. Nevertheless, the results indicate that DMSO can be employed in a reaction with SeAlaR, when the addition of DMSO is required to improve the solubility of lipophilic substrates. As a further crucial parameter the stability of the SeAlaR under ω-TA reaction conditions was investigated. The enzyme was incubated under process conditions (30 °C, in the presence of pyruvate, acetophenone, 1-phenylethylamine and PLP) and the activity in the racemisation of L-alanine after different incubation times was monitored (Fig. 1b, c). The study showed that AlaR is a rather stable catalyst and even after an incubation time of 24 h the racemisation still proceeds equally well as at the start of the reaction (Fig. 1b). The same is true for the racemisation in the presence of DMSO (10 vol%). Here the racemisation still proceeded well although slightly slower than the reaction without the co-solvent (Fig. 1c).
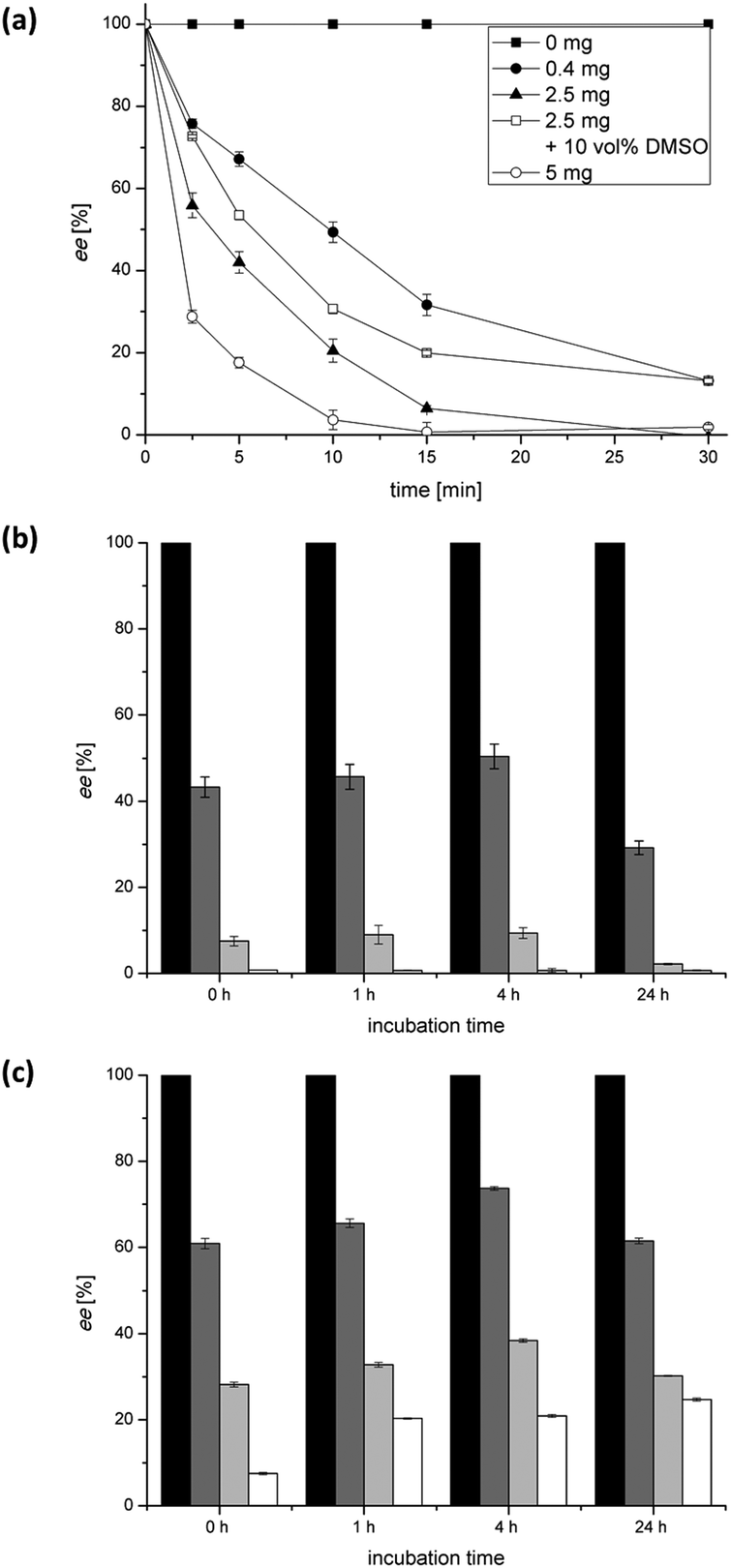 | ||
| Fig. 1 Activity and stability of the SeAlaR. (a) Racemisation profile of L-alanine catalysed by the SeAlaR. The racemisation of 250 mM L-alanine was monitored over a period of 30 min using different amounts of AlaR (mg mL−1) in the presence and absence of DMSO as the co-solvent. (b, c) Racemisation profile of SeAlaR after incubation in the absence (b) and in the presence of DMSO (c) under process conditions (30 °C, in the presence of pyruvate, acetophenone, 1-phenylethylamine and PLP). Samples were taken after 0 min (black bars); 5 min (dark grey bars); 15 min (light grey bars) and 30 min (white bars). | ||
Combination of racemisation and transamination reaction
For the amination employing (R)-selective ω-TAs and D-alanine as amine donors, two systems were used to remove the co-product pyruvate in order to shift the equilibrium: an AlaDH to transform pyruvate to L-alanine (AlaDH system, Scheme 1) or a LDH to reduce pyruvate to lactate (LDH system, Scheme 1).2,3 A comparative study was performed using either only D-alanine or L-alanine in combination with the SeAlaR.SeAlaR and the AlaDH system
The amination of acetophenone (1a) employing the ω-TA from Aspergillus terreus (AT ω-TA)13,14,17–19 and five equivalents of D-alanine (Fig. 2a, black bars) was compared to the reaction with L-alanine and SeAlaR. For the biotransformation using L-alanine 10 equivalents were used to provide five equivalents of the desired D-enantiomer after racemisation by the SeAlaR (Fig. 2a, grey bar). In addition, the same reaction was performed using rac-alanine (10 eq., white bar) and L-alanine (5 eq., light grey bar) without the addition of SeAlaR. The highest conversion was achieved using five equivalents of D-alanine in the absence of SeAlaR (78%). The addition of L-alanine to produce five equivalents of D-alanine in situ led to a conversion of 61%. Since rac-alanine in the control reaction led to a conversion in a similar range (64%) it seemed that inhibition occurs to a certain extent due to the presence of the L-enantiomer. However, when providing only one equivalent of D-alanine both systems led to the same conversion and at 0.5 equivalents even a higher conversion was achieved starting with L-alanine and SeAlaR. This clearly demonstrated that by employing the SeAlaR-catalysed racemisation of L-alanine the efficiency of the desired amination reaction was improved.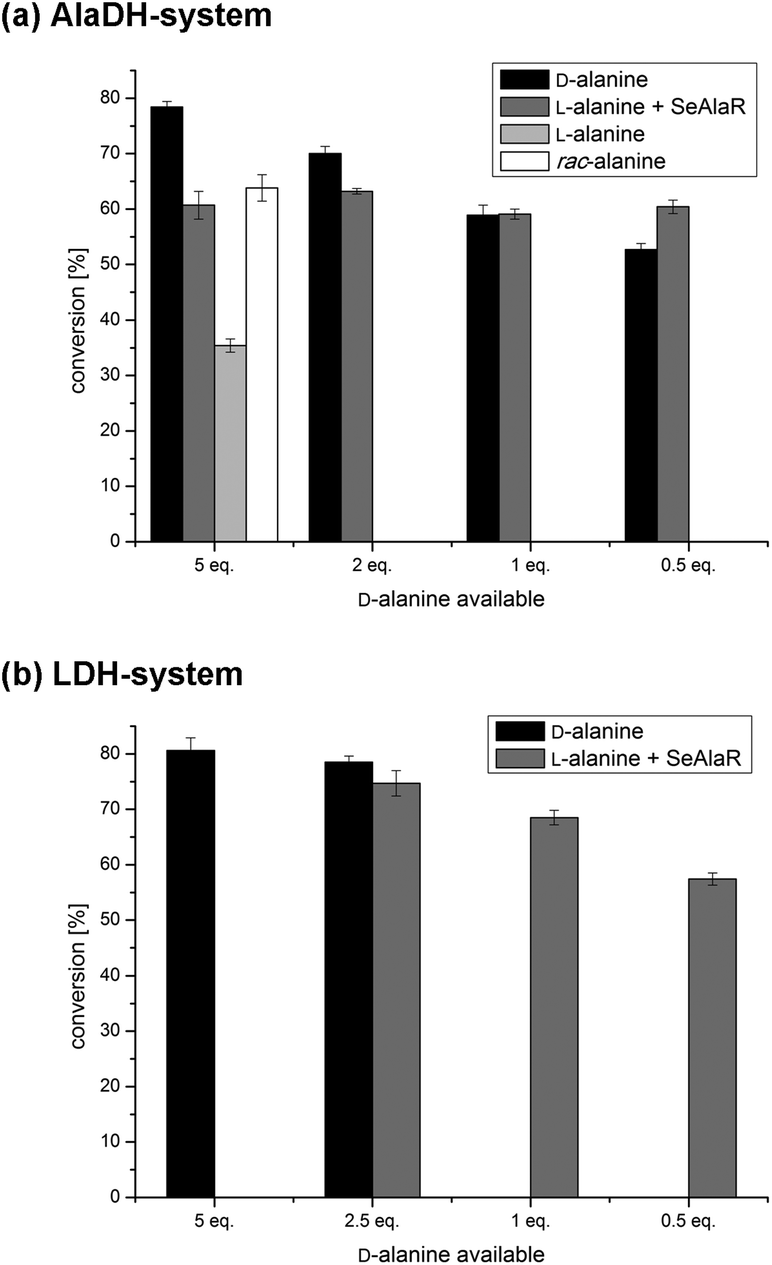 | ||
| Fig. 2 Conversions of the ω-TA-catalysed amination of 1a using AT ω-TA with and without SeAlaR. (a) Conversions for the AlaDH system after 24 h in the presence (grey bars) and in the absence (black bars) of SeAlaR at different equivalents of D-alanine. (b) Conversions for the LDH system in the presence (grey bars) and in the absence (black bars) of SeAlaR at varied amounts of alanine. With the exception of the control reaction where L-alanine was used (a, light grey bar), results are always correlated to the equivalents of D-alanine available in the reaction mixture. | ||
Interestingly, the control reaction with L-alanine (5 eq.) in the absence of SeAlaR also led to a moderate conversion. Additional experiments using purified ω-TA for the transformation could prove that this conversion is due to the presence of an AlaR in the E. coli host.36,37 Thus, the ω-TA employed was highly specific for D-alanine not accepting L-alanine and the E. coli AlaR slowly racemised L-alanine consequently leading to the observed conversion (for results see ESI section 2†). However, the studies also showed that the racemisation by the E. coli AlaR was insufficient, in particular with respect to the reaction time and conversion. This becomes obvious when comparing the reaction rates of the transformations with D-alanine and L-alanine within the first ten hours: while the reaction was almost completed using D-alanine (72%) as the amine donor, a conversion of only 16% was reached in the case of L-alanine (ESI Fig. S3†).
SeAlaR and the LDH system
Similar to the AlaDH system, the LDH system was studied in the presence and absence of SeAlaR. Again the influence of different alanine concentrations was investigated. A maximal conversion of 81% for the amination of 1a was reached using five equivalents of D-alanine without racemisation by the SeAlaR. The best conversion (75%) was obtained for the racemisation system using 5 equivalents of L-alanine leading to 2.5 equivalents of D-alanine after racemisation by the SeAlaR (Fig. 2b). Actually, when comparing both systems (AlaDH and LDH) comparable conversions (78% and 81%) were obtained using five equivalents of D-alanine in the absence of the SeAlaR (Fig. 2a and b, black bar). However, in the presence of SeAlaR-catalysed racemisation slightly different results were obtained. A combination of SeAlaR with the LDH system led to a conversion of 75%, whereas the AlaDH/SeAlaR system led to a conversion of 63%. This variation might be caused by the fact that D-alanine is consumed when using the LDH system and not recycled as in the AlaDH system, hence the concentration of both alanine enantiomers is reduced over time. The decreasing amount of L-alanine present might have a positive effect on the reaction assuming an inhibition by this enantiomer.Since a reduction of alanine equivalents had no significant effect in the case of the AlaDH system (Fig. 2a) the influence of lower alanine concentrations was investigated also with the LDH system. However, in this case the conversion decreased with lower amounts of alanine in the presence of the SeAlaR (Fig. 2b). This decrease might be due to the constantly decreasing alanine concentration in the LDH system, while the concentration in the AlaDH system remained constant.
As a further example, the amination of 4-phenyl-2-butanone (1b) was studied initially using the AT ω-TA. Fig. 3 shows that the amination of 1b led to conversions close to 100% using both the LDH and the AlaDH systems. Even when employing only one equivalent of D-alanine a conversion of 95% was obtained. It is important to note that the additional racemisation catalyst (SeAlaR) did not have any influence on the optical purity of the product which was still very high (ee >99%). This was also true for the amination of 1a where exclusively (R)-1-phenylethylamine (2a) was detected (ee >99%).
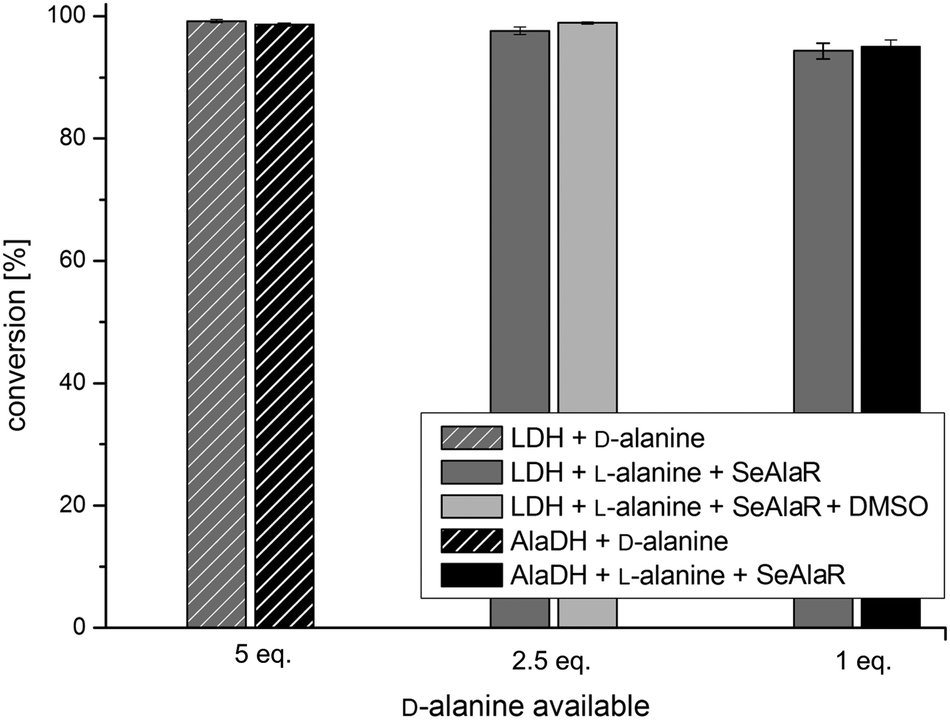 | ||
| Fig. 3 Amination of 1b applying the best conditions (see text) of both the AlaDH and LDH systems with and without the SeAlaR using the AT ω-TA. Conversions after 24 h for the AlaDH system (black bars) and LDH system (grey bars). Control reactions using D-alanine as an amine donor are indicated by the hatched bars. | ||
Finally, the system was tested with a second ω-TA originating from Arthrobacter sp. (ArR)17,38 and 4-phenyl-2-butanone 1b as the substrate. Transaminations with five equivalents of D-alanine were compared to reactions with SeAlaR using the identified optimal conditions for both pyruvate removal systems [AlaDH/FDH (1 eq.) and LDH/GDH (2.5 eq.)]. Again, it was clearly shown that L-alanine can be used successfully as an amine donor in the presence of the SeAlaR. Moreover, comparable conversions were obtained with respect to the use of D-alanine without the SeAlaR (Fig. 4).
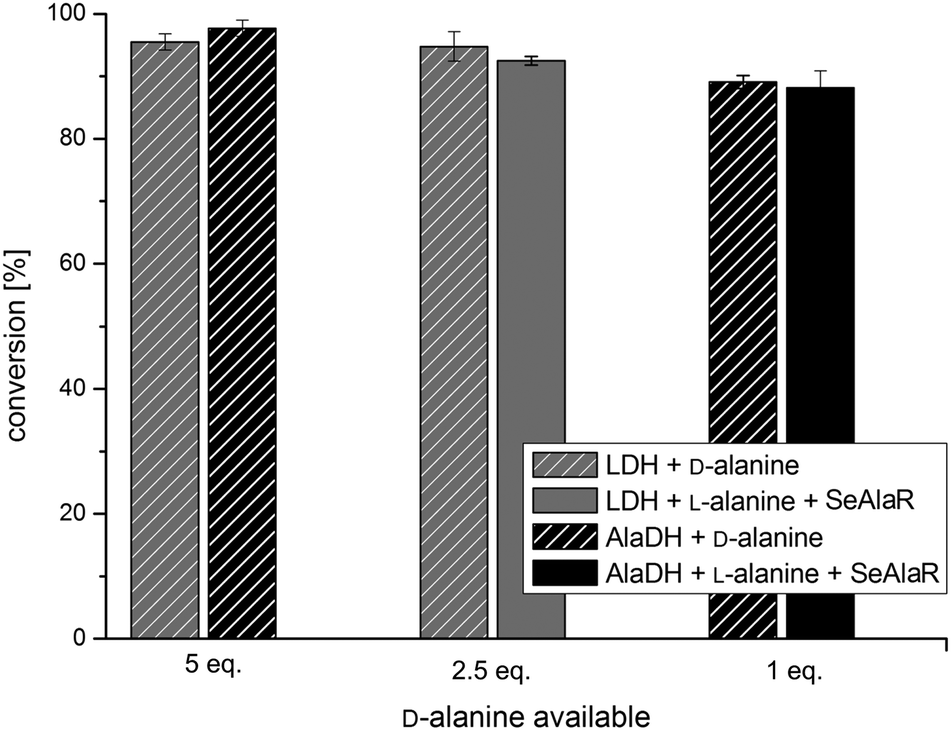 | ||
| Fig. 4 Results of the amination of 1b employing the ArR ω-TA comparing both systems with and without the addition of SeAlaR. Conversions are given for the LDH (grey bars) and AlaDH systems (black bars) after 24 h. Control reactions using D-alanine as an amine donor are indicated by the hatched bars. | ||
Comparison of efficiency
In summary, it was clearly demonstrated that L-alanine can be applied for (R)-selective ω-TAs when used in combination with an AlaR. The most prominent advantage of this method is that the cost of the process using (R)-selective ω-TAs can be significantly reduced by using the much cheaper L-alanine as an amine source as well as being able to reduce the equivalents of the amine donor employed. Table 1 outlines the relative cost for the production of (R)-4-phenyl-2-butylamine [(R)-2b] for the systems applied. By just employing 10 equivalents of L-alanine instead of 5 equivalents of D-alanine the cost of the reaction can be reduced 6.8-fold. Moreover, since an efficient production was also possible using less equivalents of L-alanine the price could be even reduced by 97%, with respect to the amine source, compared to the previously applied reaction conditions.| Process | Equivalents D-alaa | Alanine | Relative costb [%] | E-factor |
|---|---|---|---|---|
| a Equivalents of D-alanine available; for L-alanine + AlaR double equivalents of L-alanine were used to provide comparable equivalents of D-alanine. b Prices for the calculation were taken from Sigma Aldrich (D-alanine, # A7377) and Carl Roth (L-alanine, # 3076) (26.10.2014). | ||||
| AlaDH/FDH system | 5 | D | 100 | 13.75 |
| LDH/FDH system | 5 | D | 100 | 13.78 |
| LDH/GDH/SeAlaR system | 2.5 | L + AlaR | 5.2 | 14.57 |
| 1 | L + AlaR | 2.1 | 13.21 | |
| AlaDH/FDH/SeAlaR system | 1 | L + AlaR | 2.1 | 12.99 |
In a recently published multi-step synthesis of a dual orexin receptor antagonist a ω-TA catalysed amination was performed on a preparative scale (22.3 mol) using D-alanine as an amine source.39 The desired intermediate (piperidone) was produced in 74% yield and excellent stereoselectivity. However, due to its solubility limit only 3.6 equivalents of D-alanine were used, prolonging the reaction time to 31 h. Also, 7.2 kg of D-alanine used on this scale is rather expensive. In theory, by applying the described concept the cost of the amine donor could be reduced by 85.5% when using equal equivalents of d-alanine (7.2 eq. of L-alanine). Moreover, when using the optimised AlaDH system and consequently reducing the equivalents a reduction of even 96% of the cost is possible, demonstrating the impact for amination reactions on an industrial scale.
In addition to the significant reduction in cost, the E-factor was also calculated to address the environmental impact of the different systems (Table 1, for calculations see ESI†). The process without the additional racemisation step using the AlaDH system can be considered as a rather green process (E-factor 13.75). Nevertheless, by introducing the racemisation step a further reduction of the E-factor by 5.5% was possible when employing the AlaDH/SeAlaR system (E-factor 12.99). The additional fermentation, which is required to produce the SeAlaR, has to be taken into account, too. However, it has to be considered that the possibility to use L-alanine instead of D-alanine is also a significant improvement towards a greener process which is unfortunately not considered in the calculation of the E-factor, since using an amine source from the readily available chiral pool of nature makes the whole transformation more environmentally friendly. In particular, a bio-based production of L-alanine using organisms such as Corynebacterium glutamicum as well as E. coli at the expense of glucose is possible.40,41
In addition, the introduced racemisation step improves the efficiency by enabling the recycling of the amine donor when using the AlaDH system. Based on the L-selectivity of the employed AlaDH, L-alanine is produced which can then be racemised to provide the amine donor for the (R)-selective ω-TAs. Theoretically, only one equivalent of L-alanine should be sufficient to aminate the starting ketone with complete conversion and indeed, studies starting with 1 eq. of L-alanine (providing 0.5 eq. of D-alanine after racemisation) led to similar results compared to those observed with 10 times more the amine donor (Fig. 2). The benefit of recycling the amine donor in the case of the AlaDH system becomes even more obvious when comparing the atom efficiency of the AlaDH process, whereby atom efficiency is increased from 50% without to 71% with the addition of AlaR (for calculations see ESI†).
Conclusion
The application of (R)-selective ω-TAs requiring D-alanine as an amine donor is limited by the 13.8-times more expensive D-enantiomer compared to L-alanine. The here introduced racemisation step catalysed by an AlaR from Streptomyces coelicolor (SeAlaR) led to an economically improved and greener (R)-amination reaction system using L-alanine as an amine donor. The experiments showed that comparable conversions were achieved using either D-alanine as an amine donor or L-alanine with the SeAlaR. In addition, it could be shown that the equivalents of the amine donor can be significantly reduced using the described system. In particular, when the AlaR is used in combination with an AlaDH for the removal of the co-product pyruvate, a significantly improved process was established due to the recycling of alanine. As a consequence, the cost for the amine donor alanine can be reduced by up to 97% while simultaneously decreasing the E-factor of the process as well as increasing the atom efficiency.Experimental
Chemicals
Commercial grade chemicals were purchased from different suppliers and used without further purification. DNA-modifying enzymes were obtained from Thermo Scientific or New England Biolabs. L-Lactate dehydrogenase (LDH, evo-1.1.150), formate dehydrogenase (FDH, evo-1.1.230) and glucose dehydrogenase (GDH, evo-1.1.060) (all from evocatal, Germany) are commercially available. The L-alanine dehydrogenase (AlaDH) was used as a purified enzyme which was prepared as described previously.23 Both the used ω-transaminases were employed as lyophilised whole cell preparations prepared as previously reported.17Cloning and recombinant protein expression of SeAlaR and PpAlaR
The previously reported AlaR from Streptomyces coelicolor (SeAlaR, pEG238)35 and the novel AlaR from Pseudomonas putida (PpAlaR, pEG236) were designed as codon-optimised genes (both sequences can be found in ESI†) (GeneArt, Germany). Both genes were subsequently cloned into the expression vector pET21a (Invitrogen, USA) using the restriction sites NdeI/NotI to construct a variant containing a C-terminal His-tag.The AlaRs were expressed in E. coli BL21(DE3)pLysS in lysogeny broth medium containing ampicillin (100 mg mL−1). Cultures were grown at 37 °C until an OD600 of 0.5–0.7 was reached. Enzyme expression was induced by the addition of isopropyl-β-D-thiogalactopyranoside (0.5 mM), and the temperature was reduced to 25 °C. After 16–20 h cells were harvested by centrifugation and stored at −20 °C. To prepare a cell free crude extract, cells (10% v/w) were suspended in sodium phosphate buffer (10 mM, pH 8), and disrupted by ultrasonication (2 × 1 min, 0.1 s pulse on and 0.4 s pulse off, 40% output). The crude extract was cleared by centrifugation (20 min, 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm), freeze dried and stored at −20 °C.
000 rpm), freeze dried and stored at −20 °C.
Determination of the racemisation rate
To test the activity of both AlaRs the racemisation of L-alanine was followed over time. Different amounts of AlaR (0.4–5 mg mL−1) were incubated with L-alanine (250 mM) in sodium phosphate buffer (50 mM, pH 8) containing pyridoxal-5′-phosphate (PLP; 1 mM) at 30 °C and 500 rpm. For the determination of the co-solvent tolerance of the AlaR, DMSO (10 vol%) was added to the reaction. After 2.5, 5, 10 and 30 min samples were withdrawn (10 μL), quenched with MeOH (10 μL) and subsequently diluted 500-fold with a HPLC-MS eluent (MeOH–H2O = 65:35 with 0.1% of formic acid). Denatured protein was removed by centrifugation (3 min, 13000 rpm) and the supernatant was analysed by HPLC-MS (1260 Infinity equipped with a quadrupole 6120, both from Agilent, USA) using a chiral Chirobiotic-T column (150 × 4.5 mm, Astec, Sigma-Aldrich, Switzerland) under the following conditions: MeOH–H2O–HCO2H (65:35:0.1) at a flow rate of 1 mL min−1; elution times; L- (2.3 min) and D-alanine (2.7 min).
Stability of the SeAlaR
For the determination of the SeAlaR stability, the enzyme was incubated under process conditions (30 °C, in the presence of pyruvate, acetophenone, 1-phenylethylamine and PLP) and the activity was determined after different incubation times. For this study the SeAlaR (2 mg mL−1) was incubated in sodium phosphate buffer (50 mM, pH 8) containing sodium pyruvate (12.5 mM), acetophenone (12.5 mM), 1-phenylethylamine (12.5 mM) and PLP (1 mM) at 30 °C and 600 rpm. After 0 h, 1 h, 4 h and 24 h L-alanine (250 mM) was added to start the reaction and samples were taken over 30 min and analysed as described before.ω-TA-catalysed amination of acetophenone and 4-phenyl-2-butanone
For the determination of enantiomeric excess samples were derivatised to the corresponding acetamides by adding pyridine (2 μL) and acetic anhydride (6 μL) to the extracted organic phase. Samples were incubated at 40 °C and 800 rpm for 2 h and quenched by the addition of aqu. sat. NaHCO3 (25 μL). Then the organic phase was analysed via gas chromatography (Agilent 7890 A system with a FID detector) equipped with a chiral Varian Dex CB column (25 m, 0.32 mm, 0.25 μm). The following method was used: 120 °C, 5 °C min−1 to 180 °C, hold 2 min, retention times: 1-methyl-3-phenylpropylamine: (S) 9.7 min, (R) 9.9 min, 1-phenylethylamine: (S) 6.4 min, (R) 6.7 min.
Acknowledgements
N.R. has been supported by the Austrian BMWFJ, BMVIT, SFG, Standortagentur Tirol and ZIT through the Austrian FFG-COMET-Funding Program. D.P. has been funded by the European Union's seventh framework program FP7/2007-2013 under grant agreement # 289646 (KyroBio). H.L. acknowledges financial support by the FWF for the DK “Molecular Enzymology” (Project W9). Support by NAWI Graz is acknowledged.Notes and references
- H. Kohls, F. Steffen-Munsberg and M. Höhne, Curr. Opin. Chem. Biol., 2014, 19, 180–192 CrossRef CAS PubMed
.
- M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42–51 CrossRef
- D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS PubMed
- M. S. Malik, E. S. Park and J. S. Shin, Appl. Microbiol. Biotechnol., 2012, 94, 1163–1171 CrossRef CAS PubMed
- S. Mathew and H. Yun, ACS Catal., 2012, 2, 993–1001 CrossRef CAS
- R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129–143 CrossRef CAS
- W. Kroutil, E. M. Fischereder, C. S. Fuchs, H. Lechner, F. G. Mutti, D. Pressnitz, A. Rajagopalan, J. H. Sattler, R. C. Simon and E. Siirola, Org. Process Res. Dev., 2013, 17, 751–759 CrossRef CAS PubMed
- J. Ward and R. Wohlgemuth, Curr. Org. Chem., 2010, 14, 1914–1927 CrossRef CAS
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed
- T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS
- A. Bartoszewicz, N. Ahlsten and B. Martin-Matute, Chem. – Eur. J., 2013, 19, 7274–7302 CrossRef CAS PubMed
- M. Rueping, J. Dufour and F. R. Schoepke, Green Chem., 2011, 13, 1084–1105 RSC
- M. Höhne, S. Schätzle, H. Jochens, K. Robins and U. T. Bornscheuer, Nat. Chem. Biol., 2010, 6, 807–813 CrossRef PubMed
- S. Schätzle, F. Steffen-Munsberg, A. Thontowi, M. Höhne, K. Robins and U. T. Bornscheuer, Adv. Synth. Catal., 2011, 353, 2439–2445 CrossRef
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed
- J. Jiang, X. Chen, D. Zhang, Q. Wu and D. Zhu, Appl. Microbiol. Biotechnol., 2015, 99, 2613–2621 CrossRef CAS PubMed
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, J. H. Sattler and W. Kroutil, Adv. Synth. Catal., 2011, 353, 3227–3233 CrossRef CAS
- D. Pressnitz, C. S. Fuchs, J. H. Sattler, T. Knaus, P. Macheroux, F. G. Mutti and W. Kroutil, ACS Catal., 2013, 3, 555–559 CrossRef CAS
- C. S. Fuchs, M. Hollauf, M. Meissner, R. C. Simon, T. Besset, J. N. H. Reek, W. Riethorst, F. Zepeck and W. Kroutil, Adv. Synth. Catal., 2014, 356, 2257–2265 CrossRef CAS
- K. Smithies, M. E. B. Smith, U. Kaulmann, J. L. Galman, J. M. Ward and H. C. Hailes, Tetrahedron: Asymmetry, 2009, 20, 570–574 CrossRef CAS PubMed
- N. Richter, R. C. Simon, W. Kroutil, J. M. Ward and H. C. Hailes, Chem. Commun., 2014, 50, 6098–6100 RSC
- M. Fuchs, D. Koszelewski, K. Tauber, W. Kroutil and K. Faber, Chem. Commun., 2010, 46, 5500–5502 RSC
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, N. G. Turrini, J. H. Sattler, A. Lerchner, A. Skerra and W. Kroutil, Eur. J. Org. Chem., 2012, 1003–1007 CrossRef CAS
- I. K. Mangion, B. D. Sherry, J. J. Yin and F. J. Fleitz, Org. Lett., 2012, 14, 3458–3461 CrossRef CAS PubMed
- R. C. Simon, B. Grischek, F. Zepeck, A. Steinreiber, F. Belaj and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 6713–6716 CrossRef CAS PubMed
- T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed
- C. Molinaro, P. G. Bulger, E. E. Lee, B. Kosjek, S. Lau, D. Gauvreau, M. E. Howard, D. J. Wallace and P. D. O'Shea, J. Org. Chem., 2012, 77, 2299–2309 CrossRef CAS PubMed
- E. Busto, R. C. Simon, B. Grischek, V. Gotor-Fernández and W. Kroutil, Adv. Synth. Catal., 2014, 356, 1889–1889 CrossRef
- E. S. Park, J. Y. Dong and J. S. Shin, ChemCatChem, 2013, 5, 3538–3542 CrossRef CAS
- E. Siirola, F. G. Mutti, B. Grischek, S. F. Hoefler, W. M. F. Fabian, G. Grogan and W. Kroutil, Adv. Synth. Catal., 2013, 355, 1703–1708 CrossRef CAS
- E. S. Park, J. Y. Dong and J. S. Shin, Org. Biomol. Chem., 2013, 11, 6929–6933 CAS
- K. E. Cassimjee, C. Branneby, V. Abedi, A. Wells and P. Berglund, Chem. Commun., 2010, 46, 5569–5571 RSC
- A. Galkin, L. Kulakova, T. Yoshimura, K. Soda and N. Esaki, Appl. Environ. Microbiol., 1997, 63, 4651–4656 CAS
- D. Koszelewski, I. Lavandera, D. Clay, G. M. Guebitz, D. Rozzell and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 9337–9340 CrossRef CAS PubMed
- S. C. Willies, J. L. White and N. J. Turner, Tetrahedron, 2012, 68, 7564–7567 CrossRef CAS PubMed
- E. Wang and C. Walsh, Biochemistry, 1978, 17, 1313–1321 CrossRef CAS
- U. Strych and M. J. Benedik, J. Bacteriol., 2002, 184, 4321–4325 CrossRef CAS
-
Y. Yamada, A. Iwasaki and N. Kizaki, Kaneka Corporation, EP0987332 A1, 2000 Search PubMed
- M. Girardin, S. G. Ouellet, D. Gauvreau, J. C. Moore, G. Hughes, P. N. Devine, P. D. O'Shea and L. C. Campeau, Org. Process Res. Dev., 2013, 17, 61–68 CrossRef CAS
- J. Becker and C. Wittmann, Curr. Opin. Biotechnol., 2012, 23, 631–640 CrossRef CAS PubMed
- X. Zhang, K. Jantama, J. C. Moore, K. T. Shanmugam and L. O. Ingram, Appl. Microbiol. Biotechnol., 2007, 77, 355–366 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc02363c |
| This journal is © The Royal Society of Chemistry 2015 |