
Recent progress on supported polyoxometalates for biodiesel synthesis via esterification and transesterification
Nilesh
Narkhede
,
Sukriti
Singh
and
Anjali
Patel
*
Polyoxometalate and Catalysis Laboratory, Department of Chemistry, Faculty of Science, The M. S. University of Baroda, Vadodara-390002, Gujarat, India. E-mail: aupatel_chem@yahoo.com
First published on 29th September 2014
Abstract
Biodiesel is now recognized as a “green fuel” that has several advantages over conventional diesel. In the present review we discuss the catalytic esterification and transesterification reactions to the clean synthesis of biodiesel, over most readily investigated supported polyoxometalates to meet future societal demands. Here for the first time, we are reviewing biodiesel synthesis using supported lacunary polyoxometalates; also how the prevailing reaction conditions like reaction time, alcohol content, temperature, and catalyst amount affect the catalytic activity of the catalyst is discussed in detail. The new results for supported lacunary polyoxometalates are included where the effect of catalysts and supports was correlated with the catalytic activity.
 Nilesh Narkhede | Nilesh Narkhede received his MSc degree in Inorganic Chemistry from The M. S. University of Baroda, Vadodara (India) in 2008. He joined the Department of Chemistry as a research fellow under UGC, New Delhi sanctioned project in 2011 in the same university. Presently he is a Senior Research Fellow, CSIR, New Delhi and pursuing his PhD at Polyoxometalate and Catalysis Laboratory in the same university under the supervision of Prof. (Dr) Anjali Patel. His research interest focuses on synthesis and characterization of silicotungstate based catalysts for acid catalyzed biodiesel production as well as conversion of glycerol to value-added chemicals. |
 Ms. Sukriti Singh | Ms Sukriti Singh received her MSc degree in Analytical Chemistry from The M. S. University of Baroda, Vadodara (India) in 2011. She joined the Department of Chemistry as a research fellow under DST, New Delhi sanctioned project in 2011 in the same university. She is presently a Senior Research Fellow, DST, New Delhi and pursuing her PhD under the guidance of Prof. (Dr) Anjali Patel at the same university. Her research interest lies in the design of heterogeneous phosphotungstate based catalysts for green chemical synthesis via oxidation and acid catalyzed reactions. |
 Anjali Patel | Anjali Patel received her PhD degree in 1993 from Chemistry Department, The M. S. University of Baroda, Vadodara, India and she has pursued post-doctoral from IRC, CNRS, Lyon, France during 1993–94. Since 1997 she has been working as a faculty member at the same university. At present she is a Professor and her current research interests cover materials science, polyoxometalates, heterogeneous catalysis, and green chemistry. She is leading a research group focusing on tailoring of new catalytic materials based on polyoxometalates, lacunary as well as transition metal substituted polyoxometalates and their applications for organic transformations such as esterification, alkylation, acylation, hydrogenation, oxidation, C–C coupling and bio-diesel production. |
1. Introduction
In the last few years, biodiesel has emerged as one of the most potential renewable energy replacing the existing petrol-derived diesel. It is a renewable, biodegradable and non-toxic fuel which can be easily produced through esterification or transesterification reaction.1 Several methodologies are available which utilize homogeneous or heterogeneous catalysts for the synthesis of biodiesel. The most used commercial technology for biodiesel production is the transesterification of triglycerides with methanol under basic conditions. Generally, alkaline catalysts are used for biodiesel production, as they are cheaper, but the major issue is the saponification, which is more pronounced in feedstock with large amounts of free fatty acids (FFAs), further they suffer from traditional separation problems.2,3 Therefore much attention has been paid to easily reusable solid acid catalysts. Polyoxometalates (POMs) are excellent candidates among solid acid catalysts. POMs are discrete early transition metal–oxide cluster anions and comprise a class of inorganic complexes of unrivaled versatility and structural variation in both symmetry and size, with applications in many fields of science.4Free acidic forms of POMs are known as heteropolyacids (HPAs).5 The catalytic properties of HPAs have drawn wide attention in the preceding two decades owing to the versatility of these compounds as catalysts, which has been demonstrated both by successful large-scale applications and by promising laboratory results.6 Strong acidity of HPAs is one of the significant points that are exploited for biodiesel production. Supported HPAs are the best alternative, as they can carry out simultaneously esterification and transesterification. In the present review the contributions of supported HPAs have been reviewed, especially based on the different supports as it is known that a ‘support’ does not play always merely a mechanical role but it can also modify the catalytic properties. Hence we have incorporated this aspect in the present review, where the focus is laid on the morphology and advantages of the various supports such as metal oxides (silica, alumina, zirconia, niobia, etc.), clays, zeolites and mesoporous silica (Scheme 1).
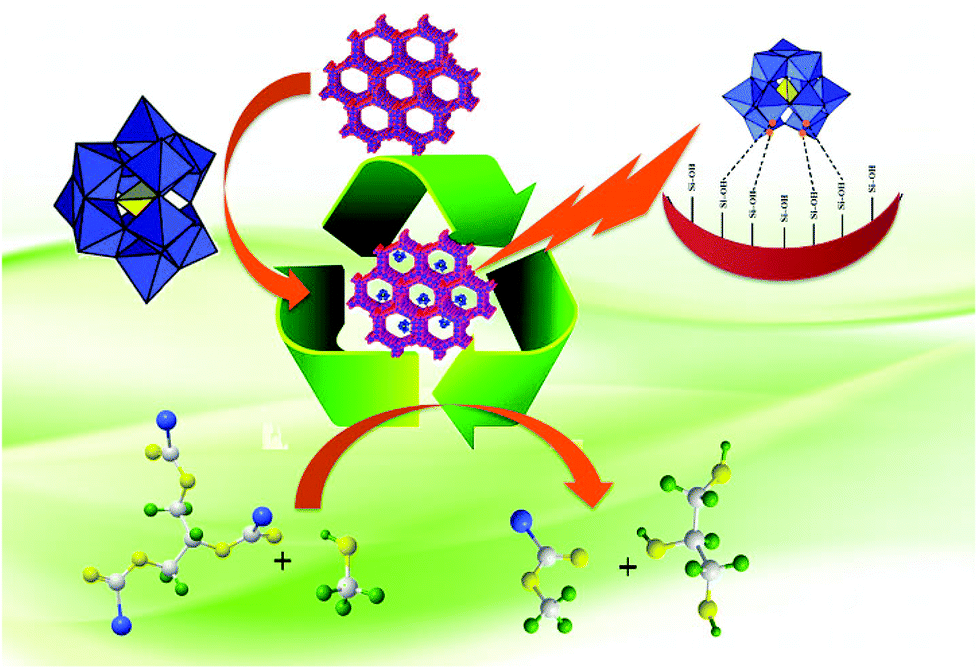 | ||
| Scheme 1 . Biodiesel synthesis over supported LPOMs. | ||
Lacunary polyoxometalates (LPOMs) are a subclass of POMs and important due to their unique structural as well as chemical properties.4 Even though LPOMs are equally important as POMs, no review for biodiesel synthesis is available in the literature. Here for the first time, we are reviewing biodiesel synthesis using supported LPOMs.
The present review describes the following points: what are heteropolyacids? Contribution of supported HPAs in biodiesel production, and transesterification over HPAs supported onto mesoporous silica (MCM-41, MCM-48). A particular focus of this review will be on the synthesis of supported LPOMs as well as their contribution towards biodiesel production. Further catalytic activity was correlated with the acidity as well as surface area of the catalysts.
2. Biodiesel
Biodiesel is a non-petroleum based fuel that consists of alkyl esters derived from either the transesterification of triglycerides (TGs) or the esterification of free fatty acids (FFAs) with low molecular weight alcohols.1–3 Biodiesel can be used in neat form or as a mixture with conventional diesel fuel, which is stable and compatible in all ratios, termed as biodiesel blends. A blend of 80% petroleum diesel and 20% biodiesel (known as B20) can be used in unmodified diesel engines.3 The use of biodiesel instead of conventional fuel does not require any large-scale technical interventions on the engine. Biodiesel is biodegradable, less toxic and with lower exhaust emission of smoke, dust, COx (x = 1 or 2) and hydrocarbon compared to fossil diesel which makes it an environmentally friendly fuel.7,8 Biodiesel can also be used as a heating fuel in domestic and commercial boilers, a mix of heating oil and biofuel which is standardized and taxed slightly differently from diesel fuel used for transportation. It is sometimes known as “bioheat” (which is a registered trademark of the National Biodiesel Board [NBB] and the National Oilheat Research Alliance [NORA] in the U.S., and Columbia Fuels in Canada).Biodiesel finds applications as fuel for domestic vehicular use, railways as well as in aircrafts in many countries. In 2007, McDonalds of UK announced that it would start producing biodiesel from the waste oil by-product of its restaurants. This fuel would be used to run its fleet.9 The British train operating company Virgin Trains claimed to have run the UK's first “biodiesel train”, which was converted to run on 80% petroleum based diesel and 20% biodiesel.10 Also in 2007, Disneyland began running the park trains on B98 (98% biodiesel). The program was discontinued in 2008 due to storage issues, but in January 2009, it was announced that the park would then be running all trains on biodiesel manufactured from its own used cooking oils.11 This is a change from running the trains on soy-based biodiesel. On November 7, 2011 United Airlines flew the world's first commercial aviation flight on a microbially derived biofuel using Solajet, Solazyme's algae-derived renewable jet fuel. This shows the progress in applications of biodiesel worldwide.11
2.1 What is biodiesel chemically?
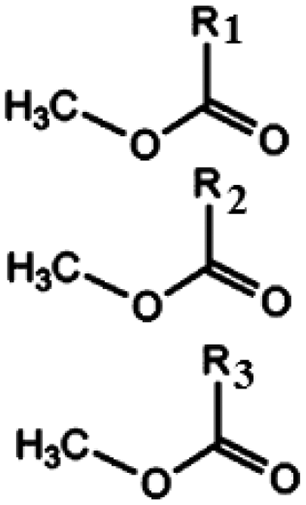
Oils and fats are a wide group of compounds that are generally soluble in organic solvents and insoluble in water. Chemically, they are tri-esters of glycerol and any of several fatty acids. Fats may be either solid or liquid at room temperature, depending on their structure and composition. Although the words “oils” and “fats” are all used to refer to fats, in reality “oils” is usually used to refer to fats that are liquids at normal room temperature, while “fats” is usually used to refer to fats that are solids at normal room temperature.There are two kinds of fatty acids: saturated and unsaturated fatty acids. Examples of common fatty acids are stearic, oleic, linolenic and palmitic. Table 1 shows the chemical structure of mono, di and triglycerides as well as biodiesel (alkyl esters). Table 2 indicates the chemical structure of fatty acids generally present in most oils.
| Triglyceride | Diglyceride | Monoglyceride | Biodiesel (alkyl esters) |
|---|---|---|---|
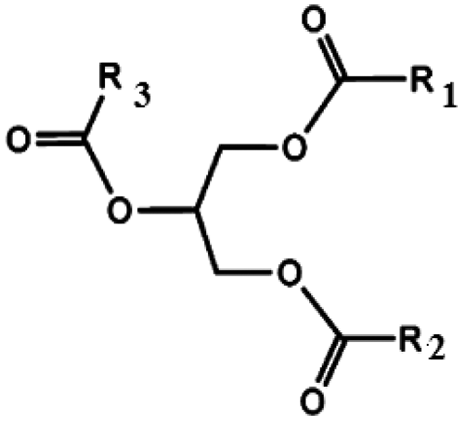
|
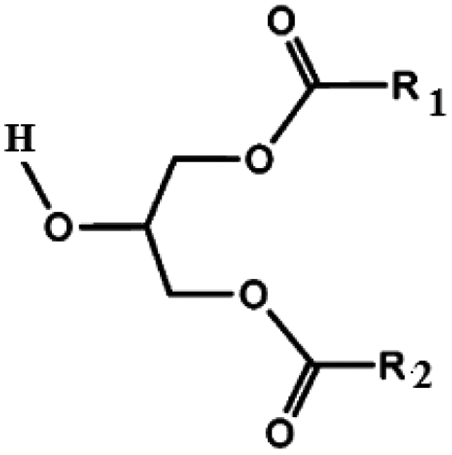
|
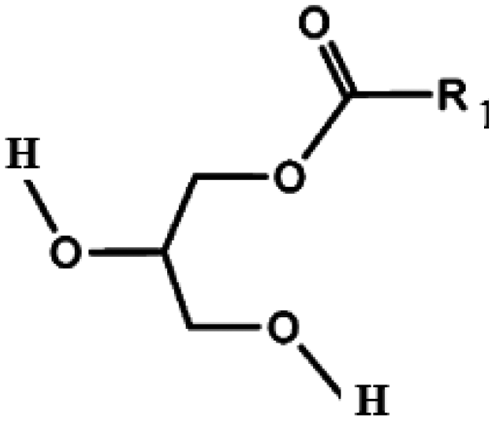
|
|
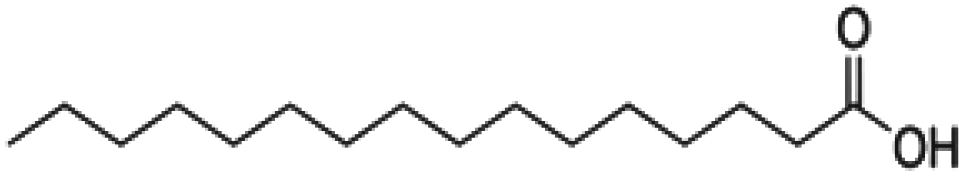
| Fatty acid | Structure | Common acronym |
|---|---|---|
| Palmitic acid |
|
C16:0 |
| Stearic acid |

|
C18:0 |
| Oleic acid |
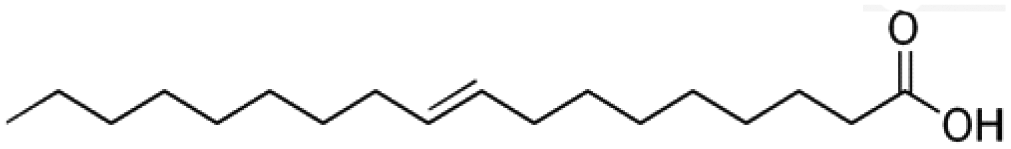
|
C18:1 |
| Linoleic acid |

|
C18:2 |
| Linolenic acid |
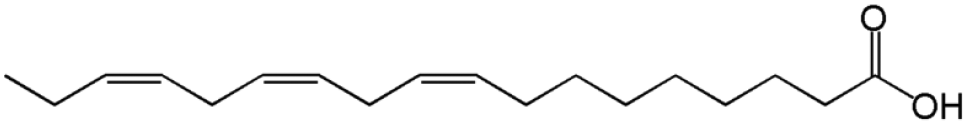
|
C18:3 |
2.2 Feedstocks for biodiesel production
In the biodiesel production process, raw materials account for almost 75% of the total biodiesel cost. The major obstacle to the commercialization of biodiesel, in comparison with petroleum-based diesel, is primarily the high raw material cost.12–16Basically all vegetable oils and animal fat can be used as feedstock for biodiesel production. Most of these oils and fats have a similar chemical composition; they consist of triglycerides with different amounts of individual fatty acids. The major fatty acids are those with a chain length of 16 and 18 carbons, whereas the chain could be saturated or unsaturated. Methyl esters produced from these fatty acids have very similar combustion characteristics in a diesel engine, because the major components in fossil diesel fuel are also straight chain hydrocarbons with a chain length of about 16 carbons (hexadecane, ‘cetane’). The major feedstocks for the biodiesel production (Fig. 1) today are rape seed oil (Canola), soybean oil, coconut oil, sunflower oil and palm oil.17–21
 | ||
| Fig. 1 Different feedstocks in biodiesel production. | ||
Particularly in Asian countries like India and China the use of non-edible seed oils for biofuel production is very popular; in that case there would be no competition with the food production, especially when these oil plants are grown on marginal areas not suitable for food production. Particularly Jatropha curcas L. has attracted enormous attention in the last few years, especially in India, Indonesia and in the Philippines. As there would be no competition with the food production and also with the traditional agricultural areas, Jatropha could fill the gap between actual vegetable oil production and demand for biofuels.22,23
An interesting alternative for low cost biodiesel production is the utilization of low quality raw materials as feedstocks such as waste cooking oil obtained from canteens, restaurants and from houses which are rich in free fatty acids.24–28
2.3 Conventional catalysts for biodiesel production
Several methodologies are available, which utilize homogeneous, heterogeneous as well as bio-catalysts. Although enzymatic catalysts are very selective and present high conversions using low oil to alcohol molar ratios,29 they are very expensive and show unstable activities.30 Supercritical conditions require high temperatures and hence increasing the overall cost of biodiesel production processes.Table 3 summarizes the catalyst type and the optimal reaction conditions for some homogeneously catalyzed (base and acid) transesterification for biodiesel production. Sulfuric acid and alkali hydroxides (NaOH and KOH) are the most-used catalysts in transesterification processes.31–35 Base catalysts are highly catalytically active and low cost while biodiesel of high quality is obtained in a short reaction time. Almost in all reported studies the ester yield higher than 90% is achieved, independently of the type of the used catalyst, alcohol and plant oil. Beside a lot of advantages, the use of homogeneous base catalysts has many drawbacks. The main disadvantages of base catalysts are the impossibility of converting FFA to alkyl esters and soap formation in the presence of FFA, which reduces the biodiesel yield and prevents glycerol separation. However this problem can be overcome by the use of homogeneous acid catalysts which prevents soap formation. But corrosion of reaction vessels and problem of recycling are the key issues with traditional liquid acids. Therefore commercialization of biodiesel production is difficult due to the technological drawbacks such as separation and purification steps that increase the cost factor to maximum.
| Medium of reaction | Catalyst | Reaction conditionsa | Yield/conversion (%) | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|---|
| a Reaction conditions: amount of catalyst wt%/mole ratio of alcohol to substrate/reaction temperature °C/reaction time. | ||||||
| Homogeneous bases | NaOH | 1.1 wt%/7/70/20 min | 94 | High catalytic activity, low cost, mild operation conditions | Low FFA content requirement, anhydrous conditions, saponification, emulsion formation, more wastewater generated from purification steps | 31–33 |
| CH3ONa, CH3OK | (0.4–1.5 wt%)/5/70/3 | <90 | ||||
| KOH | 1 wt%/6/65/60 min | 99 | ||||
| Homogeneous acids | H2SO4 | 0.32 wt%/24/60/1 h | 98 | Catalyze esterification and transesterification simultaneously, no soaponification | Corrosive, more waste generation by neutralization, difficult to recycle, higher reaction temperature | 34–35 |
| CF3COOH | 2 wt%/20/120/5 h | 98 | ||||
| Heterogeneous bases | Li/CaO | 2 wt%/12/65/8 h | 94 | Noncorrosive, environmentally benign, recyclable, fewer disposal problems, easy separation | Low FFA content requirement, anhydrous conditions, more wastewater from purification, high molar ratio and reaction temperature and pressure, diffusion limitations, high cost | 36–39 |
| KF/Al2O3 | 4 wt%/12/65/3 h | 90 | ||||
| K3PO4 | 4 wt%/6/60/2 h | 97 | ||||
| Mg–Ln mixed oxide | 5 wt%/53/65/30 min | 100 | ||||
| Heterogeneous acids | TiO2/SO42− | 2 wt%/12/230/8 h | 90 | Catalyze esterification and transesterification simultaneously, recyclable, eco-friendly greener catalysts | Low acid site concentrations, low microporosity, diffusion limitations | 40–44 |
| KNO3/Al2O3 | 6 wt%/12/70/6 h | 84 | ||||
| SO4/SnO2 | 3 wt%/6/200/- | 80 | ||||
| S-ZrO2 | 5 wt%/20/120/1 h | 98 | ||||
| Al2O3/ ZrO2/ WO3 | 4 wt%/40/250/20 h | 90 | ||||
Additionally, in order to meet the specified product quality, both the processes involve a number of washing and purification steps producing a large amount of wastewater, which is environmentally unfavorable and requires appropriate treatment. The high amount of water used in washing and consequent treatment of the resulting large effluent increases the overall process cost. For these reasons, alternative methods have been developed for biodiesel synthesis.
2.4 Heterogeneous catalysts for biodiesel production
The heterogeneous catalysis process is expected to be an effective biodiesel production process with low cost and minimal environmental impact because of the possibility of simplifying the production and purification processes under mild conditions. Therefore, many heterogeneous alkali catalysts for the transesterification of oils have been developed, such as basic alkali and alkaline metal oxides and mixed oxides.36–39 The solid base catalysts are easily regenerated and have a less corrosive nature, leading to safer, cheaper and more environment-friendly operations. However these processes suffer from low FFA content requirement as well as anhydrous conditions.Heterogeneous acids catalyzing transesterification reactions have strong potential to replace liquid acid catalysts because of their obvious advantages: the solid acid catalysts are insensitive to the FFA content, esterification and transesterification occur simultaneously, the washing step of biodiesel can be avoided, separation of the catalyst from the reaction medium is easy, low product contamination, easy regeneration, the catalyst can be recycled and the corrosion problem can be reduced even in the presence of acid species.40–44 The ideal solid acid catalyst for transesterification reaction should have characteristics such as an interconnected system of large pores, moderate to high concentrations of strong acid sites and the hydrophobic surface.
Enormous literature is available on biodiesel production over a variety of heterogeneous acid catalysts and it would be quite difficult to summarize all the literature. Therefore in the present review we restrict ourselves to the biodiesel synthesis over supported POMs. To begin with let us have a brief overview on characteristic structural features and properties of POMs (parent HPAs as well as LPOMs).
3. What are heteropolyacids?
HPAs are acidic forms of POMs. POMs are discrete early transition metal–oxide cluster anions and comprise a class of inorganic complexes of unrivaled versatility and structural variation in both symmetry and size, with applications in many fields of science.4They have the general formula [XxMmOy]q−, in which X is the hetero atom, usually a main group element (e.g. P, Si, Ge, As), and M is the addenda atom, a d-block element in high oxidation state, usually VIV,V, MoVI or WVI. These compounds are always negatively charged although the negative density is widely variable depending on the elemental composition and the molecular structure.
The polyoxometalates have been known since the work of Berzelius on the ammonium 12-molybdophosphate in 1826.45 With the development of polyoxometalate chemistry various types of structures were discovered (Keggin, Silverton, Dawson, Anderson), amongst them Keggin type POMs are most stable and more acidic.
Keggin type POMs are polymeric oxoanions formed from different mononuclear oxoanions. These oxoanions tend to polymerize by dehydration at low pH forming polyanions and water as shown in the following equation.
| 12MO4m− + XO4p− + 23H+ → [XM12O40]n− + H2O |
The structure of [PW12O40]3− (12-tungstophosphoric acid, TPA) was first reported by Keggin in 1933. The ideal α type Keggin anion has Td symmetry and consists of a central XO4 tetrahedron surrounded by twelve MO6 octahedra. The twelve MO6 octahedra comprise four groups of three edge-shared octahedra, the M3O13 triplet,46,47 which has a common oxygen vertex connected to the central heteroatom. The oxygen atoms in this structure fall into four classes of symmetry-equivalent oxygens: X–Oa–(M)3, M–Ob–M, connecting two M3O13 units by corner sharing; M–Oc–M, connecting two M3O13 units by edge sharing; and Od–M, where M is the addenda atom and X is the heteroatom. The schematic representation of Keggin type POM is shown in Fig. 2.
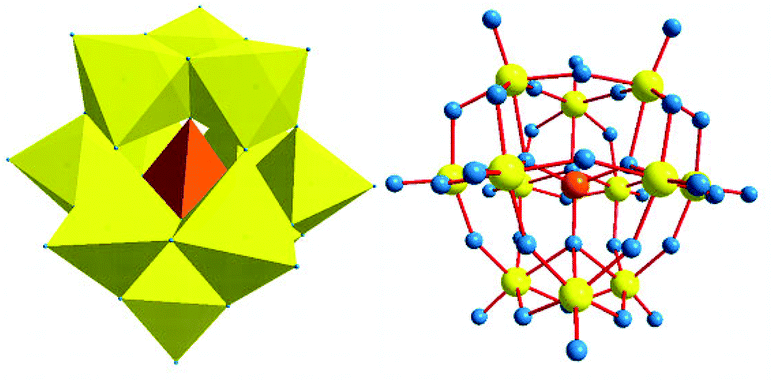 | ||
| Fig. 2 Keggin type heteropoly anion [XM12O40]3−. | ||
It was proved that HPAs in the solid state are pure Brønsted acids and are stronger acids than the conventional solid acids (Fig. 3) such as SiO2–Al2O3, H3PO4/SiO2, HX and HY zeolites.48,49
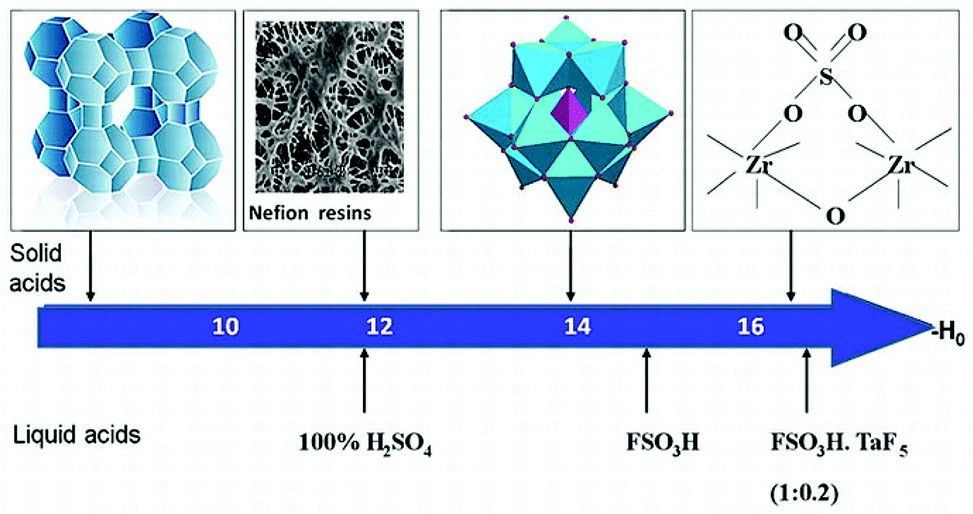 | ||
| Fig. 3 HPAs as stronger solid acids than the conventional solid acids. | ||
3.1 Advantages of HPAs over traditional mineral acids
HPAs have a number of advantages over traditional homogeneous catalysts. (a) They catalyze a wide variety of reactions in the homogeneous liquid phase offering strong options for more efficient and cleaner processing compared to conventional mineral acids.50–53 (b) Being stronger acids, they display significantly higher catalytic activity than mineral acids. In particular in organic media, the molar catalytic activity of HPA is often 100–1000 times higher than that of H2SO4.5,54 (c) This makes it possible to carry out the catalytic process at a lower catalyst concentration and/or at a lower temperature. (d) HPA based catalysis lacks side reactions such as sulfonation, chlorination, nitration, etc., which occur with mineral acids.5 (e) As stable, relatively nontoxic crystalline substances, HPAs are also preferable with regard to safety and ease of handling.In spite of having these advantages, they suffer from disadvantages such as high solubility in polar solvents, low surface area, low thermal stability, and difficulty of separation at the end of the reaction.6
LPOMs are a sub-class of POMs with a set of unique properties such as multidenticity, rigidity, thermal and oxidative stability. They represent an important class of compounds due to their unique structural as well as chemical properties. Controlled treatment of POM species with a base can produce “lacunary” POM species wherein one or more addenda atoms have been eliminated from the structure along with the oxygen atoms.4 Due to the structural diversity as well as the unique electronic properties, LPOMs are of potential importance in catalysis.
Removal of one or two MO units from the fully occupied POMs, [XMVI12O40]n−, gives rise to mono- or di-lacunary POMs, [XMVI11O39](n+4)− and [XMVI10O36](n+5)−. When a solution of [XM12O40]n− is treated with a base (pH 4–5), a series of hydrolysis reactions occurs leading to the formation of mono- and di-lacunary POMs (Scheme 2).
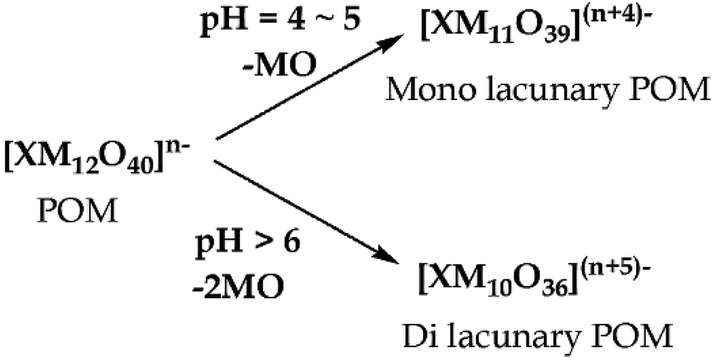 | ||
| Scheme 2 Formation of lacunary polyoxometalate. | ||
The structures of the lacuna obtained with the LPOMs are mainly dependent on the number of vacancies generated in the parent ‘saturated’ structure.55 The classical examples of geometries observed in lacunary polyoxometalates are shown in Fig. 4.
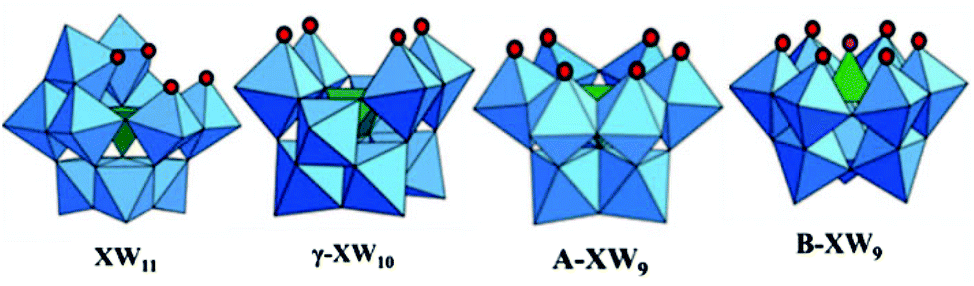 | ||
| Fig. 4 Different types of lacunary polyoxometalate structures derived from the parent Keggin unit. | ||
Among the large variety of LPOMs, the mono-lacunary Keggin type POMs form the most versatile class of lacunary polyoxometalates. As mentioned earlier, the removal of one MO at suitable pH from parent POMs leads to the formation of mono-lacunary polyoxometalates (Fig. 5).
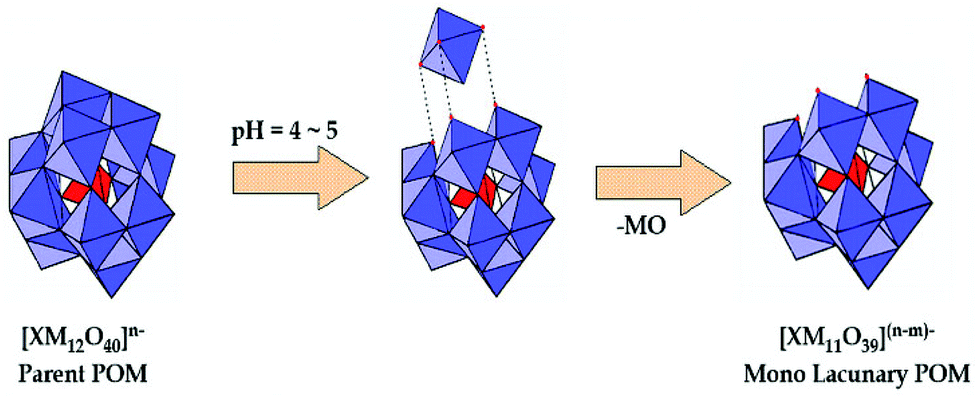 | ||
| Fig. 5 Formation of mono-lacunary polyoxometalate. | ||
Like parent POMs, the LPOMs are good homogeneous catalysts but suffer from the traditional disadvantages such as high solubility, low surface area, recovery and recycling. These traditional problems can be overcome by development of heterogeneous catalysts. This can either be done by supporting them onto suitable supports or by converting them into insoluble salts. The supporting of LPOMs onto the suitable supports is a better way to make heterogeneous catalysts.
The advantages of supported POMs are (a) increased thermal stability and surface area, (b) high catalytic activity and selectivity, (c) separation from a reaction mixture is easy and (d) repeated use is possible.
3.2 Different methods of supporting
One of the important steps in designing a catalyst is supporting of POMs onto the supports.56 Either the support can be in a preformed state or both can be formed together from the solution simultaneously.Commonly used methods of supporting are as follows: (a) co-precipitation, (b) deposition precipitation, (c) pore filling/dry impregnation/incipient wetness and (d) equilibrium adsorption/ion exchange/wet impregnation.
As impregnation is the most accepted method for the preparation of supported catalysts, we are focusing on this method only.
- transport of the solute to the pore system of the support bodies;
- diffusion of the solute within the pore system;
- uptake of the solute by the pore wall.
In the case of wet impregnation, a fourth process is operative, viz. transport of the solute to the outer particle surface. Depending on the processing conditions, different profiles of the active phase over the support body will be obtained. For instance, depending on the pH, the interaction with the support can be strong or weak, and even repulsion can exist.
The literature survey shows that there are many reviews reporting the contribution of solid acid catalysts for biodiesel production,57–66 but there are no reviews which solely highlight the contribution of POMs (HPAs as well as LPOMs) in biodiesel production. Recently Fang Su et al. reported a review entitled “Advancements in solid acid catalysts for biodiesel synthesis” in Green Chemistry.67 In the mentioned review, the authors have discussed homogeneously catalyzed biodiesel synthesis using Keggin type HPAs (H3PW12O40, H4SiW12O40, H3PMo12O40 and H4SiMo12O40). Further they have reviewed the heterogeneously catalyzed synthesis of biodiesel using Cs salt of H3PW12O40 and supported H3PW12O40. Supported catalysts mainly include ZrO2, SBA-15, SiO2, Al2O3, activated carbon, Nb2O5 and zeolite Hβ supports. Even though the authors have made significant efforts to include work done over H3PW12O40 supported onto different supports, there are other important reports on the same which need consideration. Hence, in the present review we have tried to fill the gap between the previous review and the present one.
The present review focuses on the research work that has already been carried out in the area of biodiesel production by esterification of free fatty acids and transesterification of triglycerides (TGs) over supported HPAs. This review also reports for the 1st time synthesis of supported LPOMs and biodiesel production as well as some recent results.
4. Contribution of supported HPAs in biodiesel production
As mentioned earlier supported HPAs have gained tremendous interest in biodiesel production. There are a number of reports available for the same; for the sake of simplicity, we have categorized them on the basis of the following class of supports.(a) metal oxides (silica, alumina, zirconia, niobia, etc.)
(b) clays
(c) carbons
(d) zeolites
(e) mesoporous silica
4.1 HPAs supported on metal oxide supports (SiO2, ZrO2, Nb2O5, Al2O3 and Ta2O5)
Dalai and co-workers have reported the use of TPA impregnated onto four different supports such as hydrous zirconia, silica, alumina and activated carbon. The synthesized catalysts were used for the biodiesel production from low quality canola oil. The TPA supported on ZrO2 showed the best activity, but the reaction temperature highly affected the cost factor in biodiesel production.68Castanheiro and co-workers synthesized a series of H3PW12O40, H4Si12O40, H3PMo12O40 supported onto silica.69 It was observed that the immobilization of HPA on silica increased the specific surface area and the microporous volume of the material, which decrease continuously with the increase of heteropolyacid loading of silica. The synthesized materials were used in the esterification of palmitic acid with methanol. It was observed that the catalytic activity decreases in the series: PW-silica > SiW-silica > PMo-silica. The amount of HPA on the catalyst, after the methanol contact, was measured by ICP and it was found that the catalyst lost 3% of the PW from the support.
Hamad and his group have reported H3PW12O40/SiO2 and Cs2HPW12O40 for transesterification of rapeseed oil. These catalysts possessed the Brønsted acidity of high strength and catalytic activity, better than H2SO4 and H3PO4, however the acid strength did not necessarily correlates with the catalytic activity.70
Zirconia is another metal oxide which has attracted much attention because of its thermal stability, stability under oxidizing and reducing conditions and amphoteric character of its surface hydroxyl groups. Halligudi and his group71 have carried out a comparative study of zirconia supported isopoly and heteropoly tungstate in transesterification of sunflower oil with methanol. The isopoly tungstate based catalysts were found to be more active than the heteropoly tungstate one.
Guo et al. have reported transesterification of soybean oil to biodiesel catalyzed by mesostructured Ta2O5-based hybrid catalysts functionalized with both alkyl-bridged organosilica moieties and Keggin-type heteropolyacid. Different H3PW12O40 loadings, ranging from 3.6 to 20.1%, were obtained by a one-step sol–gel hydrothermal route in the presence of a triblock copolymer surfactant. The catalyst showed a promising activity in the esterification of lauric acid and myristic acid, in the transesterification of tripalmitin, as well as soybean oil. The catalyst also shows high activity and selectivity towards simultaneous esterification and transesterification under mild conditions. The enhanced acid-catalytic reactivity after the introduction of both acidic and hydrophobic functionalities within the Ta2O5 matrix is discussed. Finally, the reusability of the hybrid materials was evaluated for three catalytic runs.72–74
Dias and co-workers have reported the synthesis, characterization and application of the H3PW/ZrO2 catalyst for esterification of oleic acid with ethanol as a model reaction to produce long chain esters.75 The zirconia support used was a commercial one and had a monoclinic phase for all experiments carried out. The recycling study was carried out up to four cycles. The conversion was found to be dropped from 88% to 45% after the 1st cycle and at the fourth cycle it was about 20%. Better results were obtained with a sequence of washing with n-hexane, drying at 100 °C and calcination at 300 °C for 4 h and recovered conversion values were as high as 70% and up to 40% for the fourth cycle. The authors reported that the initial loss of H3PW by leaching was not the only cause for the lower conversion and deactivation.
Sai Prasad and co-workers reported TPA supported onto hydrous zirconia for the esterification of palmitic acid with methanol.76 The catalyst with 20 wt% TPA calcined at 300 °C exhibited the highest activity. The pseudo-homogeneous (PH), Eley–Rideal (ER) and Langmuir–Hinshelwood–Hougen–Watson (LHHW) kinetic models were applied to correlate the experimental kinetic data and kinetic parameters were evaluated. The activation energy comes out to be as low as 34.2 kJ mol−1 for 20% TPA/ZrO2, presenting potential of the catalyst to replace traditional liquid acid catalysts.
The same group has reported TPA supported onto SnO2 for the esterification of palmitic acid with methanol.77 15 wt% TPA/SnO2 was reported as the best composition of the catalyst, and calcined at the temperature of 400 °C. Among the catalysts, 15 wt% TPA/SnO2 was the most promising one with the highest fatty acid conversion. The catalyst was recycled and reused with consistent activity. They reported a pseudo first order model to correlate the experimental kinetic data and kinetic parameters were evaluated. The activation energy of the catalysts is comparable to that of mineral acids and lower than usual solid acid catalysts. Apparent activation was calculated to be 8.68 kcal mol−1.
Niobium is an interesting and noteworthy catalyst as well as a support for different catalytic reactions. The catalytic applications of niobium compounds have increased in recent years.78–80 Niobia can also be used as a promoter and also as a solid acid catalyst. Since it is reducible over a wide temperature range, niobia is also known as a typically strong metal support interacting with (SMSI) oxide. Sai Prasad and his group have reported the catalytic activity of TPA impregnated on niobium oxide for esterification of palmitic acid78 and transesterification of used cooking oil.79,80 The esterification activity was correlated with the acidity of the catalysts, which is dependent on the presence of intact Keggin ions on the support. The catalyst 25 wt% TPA/Nb2O5 showed the maximum activity and the total acidity. The effect of calcination temperature was studied and it was observed that the new phase WO3 was observed for the catalyst calcined above 500 °C. The catalyst was recycled and reused with a negligible loss in activity up to five cycles. The kinetic studies suggested that esterification of palmitic acid follows the first-order kinetics with an activation energy of 13.68 kcal mol−1.
Biodiesel synthesis from crude Jatropha oil was carried out by Badday et al. using an ultrasound-assisted process.81 Several γ-alumina supported TPA catalysts were synthesized and characterized to elucidate their catalytic behaviours. The catalyst with 25% loading achieved the highest yield. Different design models such as full factorial design, Taguchi's algorithm and response surface methodology have been established for optimizing the data. The significant role of ultrasound in decreasing the required reaction temperature due to the generated heat during the collapse of the cavitation bubbles was also proved.
Sheikh and co-workers have synthesized a series of 12-tungstophosphoric acid (HPW) and Cs exchanged HPW supported on different supports ZrO2 and SiO2.82 The leaching study revealed that CsHPW showed least leaching. The leaching of the active species during 1 h reaction, based upon the final conversion under specified reaction conditions, was in the following order: 20%HPW/Al2O3 = 20%HPW/ZrO2 > 20%WO3/ZrO2 > 20%HPW/SiO2 > CsHPW.
4.2 HPAs supported on clays
Yadav and co-workers reported the use of different lower and higher alcohols, viz methanol, ethanol, n-propanol and n-octanol, for the synthesis of respective fatty acid esters by transesterification of vegetable oil (triglycerides).83 The activity with different supports like clay (K-10), activated carbon, ZSM-5, H-beta and TS-1 was compared. The superacids TPA as well as NH4+ salt of H3PMo12O40 were used to increase the acidity and so the activity by loading onto K-10. The activity data were compared at the best-optimized identical set of operating reaction conditions for all the catalysts and the best catalyst was proposed. The best catalyst (10% TPA/clay) was further investigated for the different refined, crude, cooked vegetable oil. The same group has also reported the use of TPA supported onto K-10 montmorillonite clay for transesterification of edible and non-edible oil at very high temperatures and pressure.84 The activity decreases significantly during the fourth recycle and any post-treatment cannot be given to regenerate the catalysts due to the alteration in the clay pore structure, because of a collapse resulting in a severe reduction in surface area.The esterification of FFA, oleic acid, was carried out over H3PW12O40 immobilized onto 3-aminopropyltriethoxysilane (H2N(CH2)3Si(OC2H5)3) functionalized palygorskite by Wang and his group. 15% (w/w) H3PW12O40 catalyst, HPW/APTES-Pa, showed excellent activity and steady reusability up to four cycles.85
Amazon kaolin flint was also successfully utilized by Barros and co-workers as a support for incorporation of 12-tungstophosphoric acid (HPW) in different proportions, 20, 40 and 60 wt%, via impregnation in aqueous solution (HCl 0.1 and 0.5 mol L−1) and acetonitrile.86 The catalytic results showed that HPW supported on metakaolins with acid treatment promoted good conversion of oleic acid esterification, followed by a gradual increase with the added support in HPW. The kinetic parameters such as activation energy (Ea) and Arrhenius constant (A) obtained were 44.75 kJ mol−1 and 1.06 × 10−5 L mol−1 min, respectively.
Very recently Bokade and his group87 have reported the synthesis of methyl oleate biodiesel over H3PW12O40 anchored montmorillonite K10 by response surface methodology and kinetic modeling. The 20% (w/w) H3PW12O40/K10 follows green principles with potential advantages of complete oleic acid conversion, under milder operating conditions and catalyst reusability up to four cycles. The second-order pseudohomogeneous kinetic model indicated that the esterification of oleic acid with methanol is kinetically controlled with an activation energy of 43.7 kJ mol−1.
4.3 HPAs supported on carbon
Activated carbon fibre (ACF) was also used as a support for heterogenizing 12-molybdophosphoric acid (HPMo) and (12-tungstophosphoric acid) HPW by Alcaniz-Monge et al.88 HPW/A20 was a more active catalyst, furthermore it also showed more tolerance to leaching. They further demonstrated that washing with ethanol and H2SO4 provides an acidic medium which reduces HPA leaching and H+ coming from the H2SO4 regenerates the active sites and thereby maintains the catalytic activity for a higher number of cycles.Badday et al. have reported optimization of the biodiesel production process from Jatropha oil catalyzed by the activated carbon-supported TPA catalyst under application of ultrasonic energy.89 The influence of ultrasonic energy on different processes was elucidated. Also they have not observed any leaching which revealed that the reaction was predominately heterogeneous in nature. Very recently the same group reported ultrasound-assisted transesterification of crude Jatropha oil by activated carbon-supported heteropolyacid catalysts.90 They have elucidated the effect of water and FFA contents in the Jatropha oil on the reaction. The catalysts showed moderate water tolerance to a limit of 1% w/w water content.
4.4 HPAs supported on zeolites
Even though zeolite based catalysts are excellent shape selective acid catalysts, no reports are available for HPAs supported onto zeolites for biodiesel production.Recently, our group has reported biodiesel production by esterification of oleic acid with methanol over a heterogeneous acid catalyst comprising TPA/TSA supported onto zeolite Hβ.91,92 The effects of various reaction parameters were studied to optimize the conditions for the maximum conversion. Both the catalysts showed excellent activity as well as potential for being used as a recyclable catalytic material after simple regeneration without a significant loss in conversion. The activation energy for TPA3/Hβ was found to be 45.2 kJ mol−1 whereas for 30% SiW12/Hβ it was 49.8 kJ mol−1.
Table 4 summarizes all these data which give idea about the feedstocks used and operational reaction conditions for the maximum conversion/yield.
| Catalyst | Feedstock | Reaction conditionsa | Conversion/yield ( %) | Ref. |
|---|---|---|---|---|
| a Reaction conditions: amount of catalyst wt%/ mole ratio of alcohol to substrate/reaction temperature °C/reaction time. | ||||
| TPA/HZ, TPA/SiO2, TPA/Al2O3 | Low quality canola oil | 3 wt%/6/200/10 (pressure 200 psi) | 77 | 68 |
| PW-silica, SiW-silica, PMo-silica | Fatty acids (palmitic, oleic, stearic acid) | 4.2 wt%/10/60/30 | 100 (palmitic acid) | 69 |
| H3PW12O40/SiO2, Cs2HPW12O40 | Rapeseed oil | 0.6 g/6/80/3 | 14.5 | 70 |
| 15 PZ-750 | Sunflower oil | 3 wt%/20/180/5 | 71 | 71 |
| 15 SZ-750 | Sunflower oil | 3 wt%/20/180/5 | 80 | 71 |
| H3PW12O40/Ta2O5 | Soybean oil + 20 wt% myristic acid | 2 wt%/90/65/24 | 90 | 72–74 |
| 20%H3PW/ZrO2 | Oleic acid | 1/6/100/4 | 88 | 75 |
| 20%TPA/ZrO2 | Palmitic acid | 0.054 g cm−3/15/65/3 | 90 | 76 |
| 15 wt% TPA/SnO2 | Palmitic acid | 1 g/14/65/200 min | 81.2 | 77 |
| 25 wt% TPA/Nb2O5 | Palmitic acid | 10 wt%/300/65/4 | 97.5 | 78 |
| 25 wt% TPA/Nb2O5 | Used cooking oil | 1.65 wt%/14/65/5 | 94 | 79 |
| 25 wt % TPA/Nb2O5 | Used cooking Oil | 3 wt%/18/200/20 (pressure 600 psi) | 92 | 80 |
| TPA25–Al | Jatropha oil | 4 wt%/19/65/50 min | 85 | 81 |
| CsHPW | 10% Oleic acid in soybean oil | 3 wt%/20/200/10 | 90.4 | 82 |
| TPA supported on K-10 montmorillonite | Waste cooking oil | 5 wt%/15/170/8 (pressure 250 psi) | 94 | 83–84 |
| HPW/APTES-Pa | Oleic acid | 1.5 wt%/8/100/4 | 90 | 85 |
| PW(20)MFS | Oleic acid | 5 wt%/30/130/2 | 97 | 86 |
| 20% H3PW12O40/K10 | Oleic acid | 5 wt%/8/165/5 | 100 | 87 |
| TPA20-AC | Jatropha oil | 4 wt%/20/65/40 min | 87 | 88 |
| Activated carbon-supported HPAs | Jatropha oil | 4.42/25/65/40 min | 91 | 89 |
| HPW/A20 | Palmitic acid | 52 wt%/15/60/6 | 90 | 90 |
| TPA3/Hβ | Oleic acid | 0.1/20/60/6 | 84 | 91 |
| 30% SiW12/Hβ | Oleic acid | 0.1/20/60/10 | 86 | 92 |
4.5. HPAs supported onto mesoporous silica
A literature survey shows that biodiesel production has been carried out over HPAs supported onto supports such as silica, zirconia, niobia and clays. At the same time reports on the use of heteropolyacids anchored to mesoporous materials for biodiesel production are scanty.The discovery of the M-41S family of mesoporous materials has greatly increased the range of supports for preparing heterogeneous catalysts. The advantage of using ordered mesoporous silica especially MCM-41 and SBA-15 as supports in heterogeneous catalysis is their relatively large pores which facilitate mass transfer and very high surface area which allows a high concentration of active sites per mass of material.93–95 Furthermore the amorphous pore walls give mesoporous silica a great deal of flexibility in terms of their composition and pore channel structure and allow post-synthesis modifications which may be performed for pore size control framework stabilisation, compositional modifications or the formation of mesoporous/zeolite composite materials.96
Castanheiro et al. reported a detailed study on esterification of palmitic acid for biodiesel production over 12-tungstophosphoric acid (PW) immobilized on SBA-15.97 They studied esterification of palmitic acid in detail. They have also reported esterification of oleic acid and stearic acid under optimized conditions. They observed small leaching of the TPA from SBA-15 to the liquid phase during the reaction. The catalyst was reused for the next cycle and the overnight dried sample (at 110 °C) showed no decrease in the conversion.
Hameed et al. studied single-step esterification of crude non-edible karanj (Pongamia pinnata) oil to fatty acid methyl esters over the mesostructured SBA-16 supported 12-molybdophosphoric acid (MP) catalyst.98 The catalyst with 15 wt% MP exhibited a peerless catalytic activity. The effect of different operational parameters was investigated over the MP-S-16(15) catalyst toward the maximum FAME yield. The catalyst could be reused up to four cycles without any loss in the conversion.
Yan et al. have recently reported mesoporous H4SiW12O40-SiO2 for production of methyl and ethyl levulinate biodiesel. The catalyst 20 wt% H4SiW12O40-SiO2 exhibited the best activity in the synthesis of both methyl and ethyl levulinate biodiesel.99 ICP analysis showed 1.0% leaching of H4SiW12O40 during the 1st run, but for the subsequent 4 runs no further leaching of tungsten was detected.
Dalai and coworkers have reported TPA supported using an organic functional group and its incorporation into the SBA-15 structure. The catalytic activity of these catalysts was tested for esterification and transesterification of canola oil.100 They observed that organic functional group anchored TPA on SBA-15 shows higher catalytic activity for esterification of FFA, whereas TPA incorporated into SBA-15 shows higher catalytic activity for transesterification of waste green seed canola oil. A 45 wt% TPA incorporated SBA-15 gives the maximum yield. From the kinetic investigations, they observed that the formation of methyl oleate was the slowest, compared to other fatty acid methyl ester formation.
Recently our group has reported biodiesel production by esterification of palmitic acid and oleic acid with methanol over a heterogeneous acid catalyst comprising TSA/TPA and mesoporous silica.101–105 The catalysts comprising TPA supported onto MCM-41101 and SBA-15102 have been used for esterification of palmitic acid and oleic acid, respectively, with methanol. Very high TOF values (8.3 min−1 for TPA3/MCM-41 and 9.3 min−1 for TPA3/SBA-15) have been observed for both the catalysts. The kinetic studies show that both the reactions follow first order kinetics with respect to the substrate and the rates are not mass transfer limited. The activation energy was found to be 38 kJ mol−1 for TPA3/MCM-41 and 44.6 kJ mol−1 for TPA3/SBA-15. The transesterification reaction of waste cooking oil was also carried out with very high conversions. Both the catalysts showed good recyclability up to four cycles without any loss in the conversion. The excellent catalytic performance was attributed to the large surface area and pore diameter of the mesoporous supports as well as the Brønsted acid strength of TPA.
A catalyst comprising TPA supported onto MCM-48 was used for the first time for biodiesel synthesis via esterification of oleic acid.103 The kinetic studies showed that esterification of oleic acid follows first order kinetics with an activation energy of 40.3 kJ mol−1. The excellent catalytic activity over TPA3/MCM-48 was extended to transesterification reaction for biodiesel production from waste cooking oil, WCO and Jatropha oil, JO, as low cost feedstocks with very high conversion of 95% and 93%, respectively. One of the substantial achievements was the scale up procedure in which excellent conversion for WCO (91%) and JO (87%) with 25 g oil was obtained. A single step production process for heterogeneous acid catalysis was also suggested for biodiesel production.
Further the next acidic species in the Keggin family, TSA was used to synthesize an active heterogeneous catalyst TSA3/MCM-41.104 The application of the catalyst was carried out for esterification of oleic acid as well as transesterification of JO. The activity was compared with that of TSA3/SBA-15. The higher conversions for both esterification and transesterification were obtained with TSA3/SBA-15 than that with TSA3/MCM-41 indicating that the former is more active. The large surface area, high value of total acidity and availability of more Brønsted acid sites, all together imply the excellent catalytic activity of TSA3/SBA-15 as compared to that of TSA3/MCM-41. Catalytic results demonstrated that the size of the support channels as well as the pore diameter play a crucial role in the catalyst performance.
Very recently, we published a work on transesterification of WCO and esterification of oleic acid over TSA supported onto SBA-15.105 Esterification of oleic acid was also carried out and a detailed kinetic study shows an activation energy of 50 kJ mol−1. Furthermore the catalytic activity of the catalyst in the study was compared with TSA3/Hβ to see the effect of support (SBA-15 and Hβ). It was reported that the high surface area of TSA3/SBA-15 as compared to TSA3/Hβ should result in higher conversion, but no significant difference in the conversion of WCO was observed. Hence, it was concluded that the catalytic system was not a surface type heterogeneous one, but it is a pseudo-liquid type (I) heterogeneous catalyst where the activity is directly proportional to the acidity of the catalyst. The catalyst was recycled up to four times after simple work up without any significant loss in the activity. A detailed mechanism for transesterification reaction was proposed over TSA3/SBA-15.
The results of mesoporous material supported HPAs are summarized in Table 5.
| Catalyst | Feedstock | Reaction conditionsa | Conversion/ yield (%) | Ref. |
|---|---|---|---|---|
| a Reaction conditions: amount of catalyst wt%/mole ratio of alcohol to substrate/reaction temperature °C/reaction time h. | ||||
| PW3-SBA-15 | Palmitic acid | 0.2/95/60/6 | 96 | 97 |
| MP-S-16(15) | Karanj oil | 2.5%/8/140/5 | 81.8 | 98 |
| H4SiW12O40-SiO2 | Levulinic acid | 0.104 g/50/65/6 (hexane) | 79 | 99 |
| TPA supported SBA-15 (functionalized) | Canola oil | 3 wt%/20/150/10 | 97 | 100 |
| TPA3/MCM-41 | Palmitic acid | 0.1 g/40/40/6 | 99 | 101 |
| TPA3/SBA-15 | Oleic acid | 0.1 g/40/40/4 | 90 | 102 |
| TPA3/MCM-48 | Oleic acid | 0.1 g/20/60/8 | 95 | 103 |
| TSA3/MCM-41 | Oleic acid | 0.1 g/40/60/10 | 99 | 104 |
| TSA3/SBA-15 | WCO | 0.3 g/8/65/8 | 86 | 105 |
| 30% SiW11/MCM-41 | Oleic acid | 0.1 g/40/65/16 | 81 | 106 |
Literature survey suggests that there is only one report on biodiesel synthesis using supported LPOMs and that too by our group.106 The catalyst 30% SiW11/MCM-41 was found to show excellent activity for the esterification of oleic acid with methanol under mild conditions. A kinetic study shows that esterification of oleic acid follows a first-order rate law with an activation energy of 49.8 kJ mol−1. Studies also reveal that the catalyst can be used for biodiesel production via transesterification of different oils such as Jatropha oil, sunflower oil, cotton seed oil and mustard oil. No pre-treatment was necessary for transesterification of these oils except for simple filtering to remove sludge and food particles.
As the present review's main focus is on LPOMs, here we are describing synthesis of the catalyst as well as its basic characterization in order to enrich reader's knowledge. From past six years our group has been working on the synthesis and characterization of LPOMs, the synthesis and detail characterization of some catalysts such as PW11/ZrO2,107 SiW11/ZrO2,108 PW11/MCM-41109 and SiW11/MCM-41106 can be found in mentioned references. However for the readers’ convenience, here we have included the main characterization of PW11/MCM-41 and SiW11/MCM-41 wherever it is required. This review also describes some of our new results of PW11/MCM-48.
5. Synthesis and characterization of the supported catalyst (PW11/MCM-48)
The synthesis was carried out in three steps.a. Synthesis of MCM-48
Synthesis and characterization of MCM-48 can be found in our earlier report.103
b. Synthesis of PW11
The sodium salt of PW11 was successfully synthesized and characterized by the method reported by us.109 Sodium tungstate dihydrate (0.22 mol, 72.5 g) and anhydrous disodium hydrogen phosphate (0.02 mol, 2.84 g) were dissolved in 150–200 mL of double distilled water and heated to 80–90 °C followed by the addition of concentrated nitric acid in order to adjust the pH to 4.8. The volume was then reduced to half by evaporation and the heteropoly anion was separated by liquid–liquid extraction with 80–100 mL of acetone. The extraction was repeated until the acetone extract showed the absence of nitrate ions. The extracted sodium salt was dried in air.
c. Synthesis of 30% PW11/MCM-48
A catalyst containing 30% of PW11 supported onto MCM-48 was synthesized by an incipient impregnation method. One gram of MCM-48 was impregnated with an aqueous solution of PW11 (0.3/30 g mL−1 of distilled water) and dried at 100 °C for 10 h. The obtained material was designated as 30% PW11/MCM-48 and was characterized by FT-Raman and XRD analysis.
Similarly, SiW11 supported onto the MCM-41 catalyst was prepared following the same process.106
5.1 Characterization of the synthesized catalysts
The textural properties and n-butyl amine acidity of the synthesized catalysts are presented in Table 6.| Catalyst | Surface area (m2 g−1) | Pore width d (Å) | Acidic strength Ei (mV) | Strength of acid sites (meq g−1) | Total acidity (meq g−1) | |
|---|---|---|---|---|---|---|
| Very strong | Strong | |||||
| MCM-41 | 659 | 47.90 | 48 | 0 | 2.0 | 2.0 |
| 30%SiW11/MCM-41 | 536 | 39.60 | 260 | 0.9 | 2.4 | 3.3 |
| MCM-48 | 755 | 30.0 | 60 | 0 | 2.2 | 2.2 |
| 30%PW11/MCM-48 | 319 | 27.5 | 290 | 1.2 | 2.1 | 3.4 |
Fig. 6 indicates the different types of acidic sites on the catalyst surface measured by the potentiometric titration method. The acidity of the catalyst measured by this technique allows us to evaluate the total number of acid sites as well as their acidic strength. In order to interpret the results, it is suggested that the initial electrode potential (Ei) indicates the maximum acid strength of the surface sites and the range where the plateau is reached (meq g−1 solid) indicates the total number of acid sites. The acidic strength of surface sites can be assigned according to the following ranges: very strong site, Ei > 100 mV; strong site, 0 < Ei < 100 mV; weak site, −100 < Ei < 0 mV and very weak site, Ei < −100 mV.110
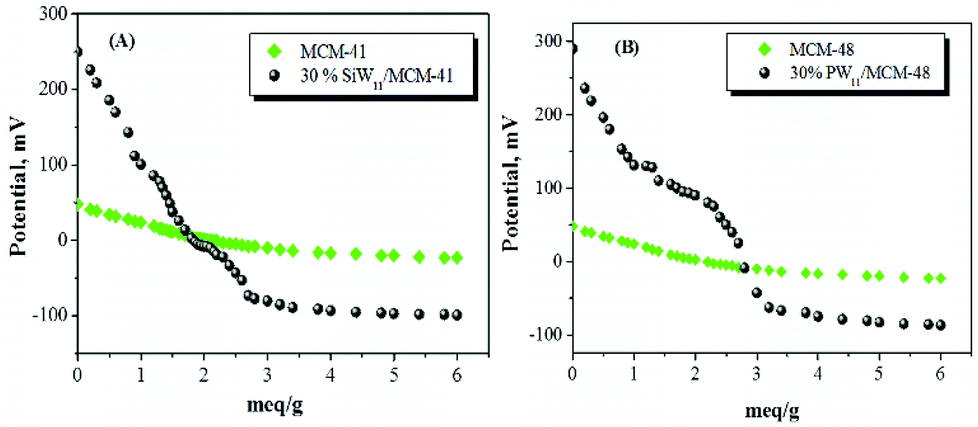 | ||
| Fig. 6 Potentiometric titration curves of (A) MCM-41 based catalyst and (B) MCM-48 based catalysts (taken from ref. 110). | ||
The plot of the electrode potential as a function of meq n-butylamine g−1 of the catalysts is shown in Fig. 6. It is observed that the catalysts, 30% PW11/MCM-48 and 30% SiW11/MCM-41, contain very strong acid sites. The strength of acidic sites in terms of the initial electrode potential is shown in Table 6. It is clear that the incorporation of species PW11/SiW11 increases the strength of the acid sites of catalysts to a great extent. The acidic strength of 30% SiW11/MCM-41 (260 mV) is lower than that of 30% PW11/MCM-41 (290 mV). This is mainly due to the highly strong acidic nature of the phosphotungstates relative to silicotungstate counterparts.
Raman spectra of PW11 show bands at 990, 985, 909, 513, and 225 cm−1, corresponding to νs (W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Od), νas (W–Od), νas (W–Ob–W), νs (W–Oc–W), and νs (W–Oa), respectively (Fig. 7) where Oa, Ob, Oc, and Od are the oxygen atoms linked to phosphorous, oxygen atoms bridging two tungsten (from two different triads for Ob and from the same triad for Oc), and linked to the terminal oxygen WO, respectively. The spectra of 30% PW11/MCM-48 display all the bands for PW11 confirming the retainment of the structure. The Raman spectrum of SiW11 shows typical bands at 971, 890, 814, 521 and 231 cm−1 corresponding to νs (W–Od), νas (W–Od), νas (W–Ob–W), νs (W–Oc–W), and νs (W–Oa), respectively (Fig. 7c). The presence of these bands confirms the formation of lacunary SiW11 species. The catalyst 30% SiW11/MCM-41 showed Raman bands at 969, 879, 793 and 224 cm−1 with respect to νs (W–Od), νas (W–Od), νas (W–Ob–W) and νs (W–Oa), respectively (Fig. 7d). The presence of these bands confirms the intact SiW11 species in 30% SiW11/MCM-41.110
Od), νas (W–Od), νas (W–Ob–W), νs (W–Oc–W), and νs (W–Oa), respectively (Fig. 7) where Oa, Ob, Oc, and Od are the oxygen atoms linked to phosphorous, oxygen atoms bridging two tungsten (from two different triads for Ob and from the same triad for Oc), and linked to the terminal oxygen WO, respectively. The spectra of 30% PW11/MCM-48 display all the bands for PW11 confirming the retainment of the structure. The Raman spectrum of SiW11 shows typical bands at 971, 890, 814, 521 and 231 cm−1 corresponding to νs (W–Od), νas (W–Od), νas (W–Ob–W), νs (W–Oc–W), and νs (W–Oa), respectively (Fig. 7c). The presence of these bands confirms the formation of lacunary SiW11 species. The catalyst 30% SiW11/MCM-41 showed Raman bands at 969, 879, 793 and 224 cm−1 with respect to νs (W–Od), νas (W–Od), νas (W–Ob–W) and νs (W–Oa), respectively (Fig. 7d). The presence of these bands confirms the intact SiW11 species in 30% SiW11/MCM-41.110
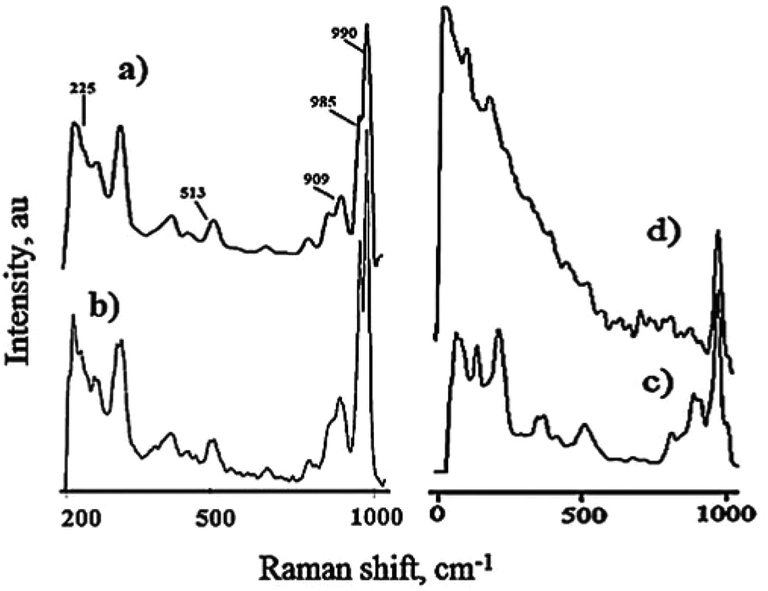 | ||
| Fig. 7 FT-Raman spectra of (a) PW11, (b) 30% PW11/MCM-48 (c) SiW11 and (d) 30% SiW11/MCM-41. (taken from ref. 110). | ||
Furthermore, the slight shift in the Raman bands for both the catalysts indicates the chemical interaction of the active species PW11/SiW11 with the surface silanol groups of supports.
The XRD pattern of MCM-48 (Fig. 8) exhibits a main characteristic peak corresponding to the 211 plane at 2.3° along with a broad shoulder peak from 220 at 3.0°. Several peaks in the range 4–5° of diffraction angles were also observed, corresponding to the reflections of the 400, 321 and 420 planes of a typical MCM-48 mesostructure. The XRD pattern of 30% PW11/MCM-48 (Fig. 8) reveals that the MCM-48 structure is intact even after the PW11 loading, however broadening and decrease in the intensity of the 211 plane at 2.3° for the catalyst was observed. Furthermore, the absence of some characteristic peaks of the crystalline phase of PW11 indicates that the active species is highly dispersed inside the channels of MCM-48.
 | ||
| Fig. 8 X-ray diffraction patterns of (a) MCM-48, (b) PW11, (c) 30% PW11/MCM-48, (d) MCM-41, (e) SiW11, (f) 30% SiW11/MCM-41 (taken from ref. 106). | ||
XRD patterns of MCM-41 and 30% SiW11/MCM-41 are shown in Fig. 8. The XRD pattern of the MCM-41 shows a sharp reflection around 2θ = 2° corresponding to the (100) plane indicating a well-ordered hexagonal structure of MCM-41. The comparison of the XRD patterns of MCM-41 and the catalysts reveals that the mesoporous structure of MCM-41 is rather intact even after supporting of SiW11 species. Furthermore the absence of characteristic peaks of the crystalline phase of SiW11 in the catalyst indicates that the active species are highly dispersed inside the hexagonal channels of MCM-41.106
6. Catalytic reaction (transesterification of soybean oil)
Soybean oil (SO) was used as a feedstock without any pre-treatment. The properties such as the acid value, saponification value, iodine value and average molecular weight for soybean oil are given in Table 7. Acid values and saponification values were determined by acid–base titration and from these values, the average molecular weight was determined using the following equation,where SV denotes the saponification value and AV denotes the acid value.
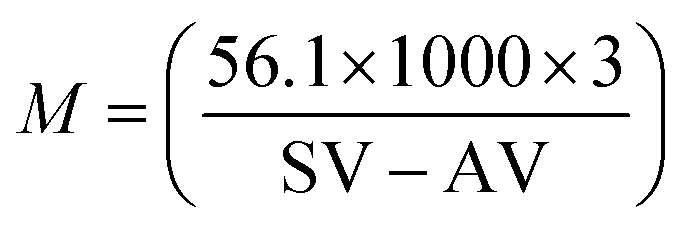
| Properties | Value |
|---|---|
| Acid value/ mg KOH g−1 | 0.467 |
| Saponification value/ mg KOH g−1 | 178.3 |
| Iodine value/ g I2 per 100 g oil | 121.4 |
| Average molecular weight/ g mol−1 | 946.5 |
| Color | Pale yellow |
6.1 Transesterification over 30% PW11/MCM-48
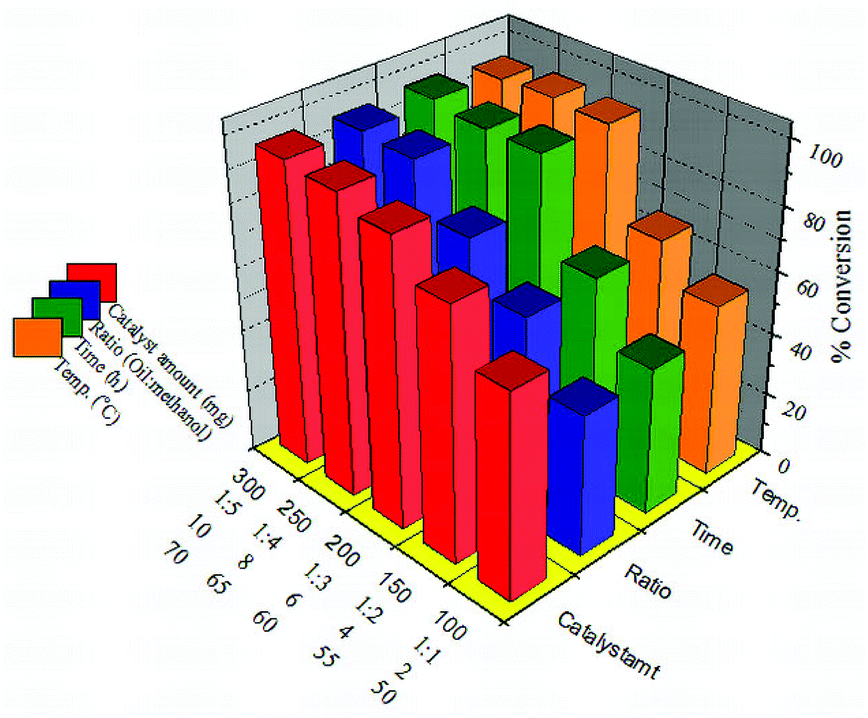
In order to overcome the equilibrium limitation of the transesterification of SO, the reaction was carried out by taking methanol in excess. The effect of different reaction variables such as the oil/alcohol ratio, the amount of catalyst, reaction time and temperature was studied to optimize the conditions for the maximum conversion.The effect of the amount of catalyst on oil conversion was investigated and the catalyst amount was varied in the range of 100–300 mg. The conversion increased with the increase in the amount of 30% PW11/MCM-48 (Fig. 9) and reached a maximum of 95% with 250 mg catalyst. The increase in the conversion can be attributed to an increase in the number of available catalytically active sites. However, with a further increase in the amount of catalyst, the conversion remains constant due to attainment of saturation/equilibrium conversion.
 | ||
| Fig. 9 Different reaction parameter optimization for transesterification of SO over 30% PW11/MCM-48. | ||
Transesterification reaction consists of a series of consecutive reversible reactions to produce three moles of esters and one mole of glycerol. The effect of the w/w ratio of oil to methanol was studied in the range of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:5 (Fig. 9) and it was seen that by increasing the methanol ratio the viscosity of the reaction mixture decreases. This promotes better mixing between reactants and catalyst, which enhances the rate of mass transfer. This eventually results in a higher conversion within a fixed reaction time. The percentage conversion accordingly increases with the increase in methanol ratio up to 1:4. On further increasing the molar ratio no significant increase in conversion was observed, which might be due to the saturation of active sites with methanol molecules rather than oil and hindering the completion of oil being protonated at the active sites. At the optimum 1:4 w/w ratios, conversion achieved was 95%.
:1 to 1:5 (Fig. 9) and it was seen that by increasing the methanol ratio the viscosity of the reaction mixture decreases. This promotes better mixing between reactants and catalyst, which enhances the rate of mass transfer. This eventually results in a higher conversion within a fixed reaction time. The percentage conversion accordingly increases with the increase in methanol ratio up to 1:4. On further increasing the molar ratio no significant increase in conversion was observed, which might be due to the saturation of active sites with methanol molecules rather than oil and hindering the completion of oil being protonated at the active sites. At the optimum 1:4 w/w ratios, conversion achieved was 95%.
The reaction time always plays a significant role in the transesterification reaction, especially in reactions catalyzed by heterogeneous catalysts, mainly due to mass transfer limitations. Therefore, long reaction time has become one of the vital factors to drive the reaction to completion instead of the reaction temperature and oil to alcohol w/w ratio. The conversion of soybean oil for 30% PW11/MCM-48 increases with increase in the reaction time from 2 to 10 h (Fig. 9). At 6 h the conversion is maximum (95%), but on further increasing the reaction time there is no significant increase in the percentage conversion. On further increasing the reaction time there was no significant increase in conversion, due to the blocking of active sites by glycerol molecules formed during the reaction.
The effect of temperature on the reaction was studied and it was seen that the temperature evidently affects both the reaction rate and conversion of soybean oil into biodiesel (Fig. 9). At 50 °C, 42% conversion was observed. But with a subsequent increase in temperature, percentage conversion was also increased. It can be seen that a maximum conversion of 95% was obtained at 65 °C. On the other hand, the conversion decreased for temperatures beyond 65 °C which can be ascribed to attainment of boiling point of methanol. Thus, methanol remained in the vapour phase in the reactor and was less available for the reaction.
The optimized conditions for transesterification reaction over 30% PW11/MCM-48 are as follows: w/w ratio of oil to alcohol 1:4; amount of catalyst 250 mg; and reaction temperature 65 °C; reaction time 6 h. Under these optimized conditions 30% PW11/MCM-48 gives the maximum conversion of 95%.
6.2 Transesterification over 30% SiW11/MCM-41
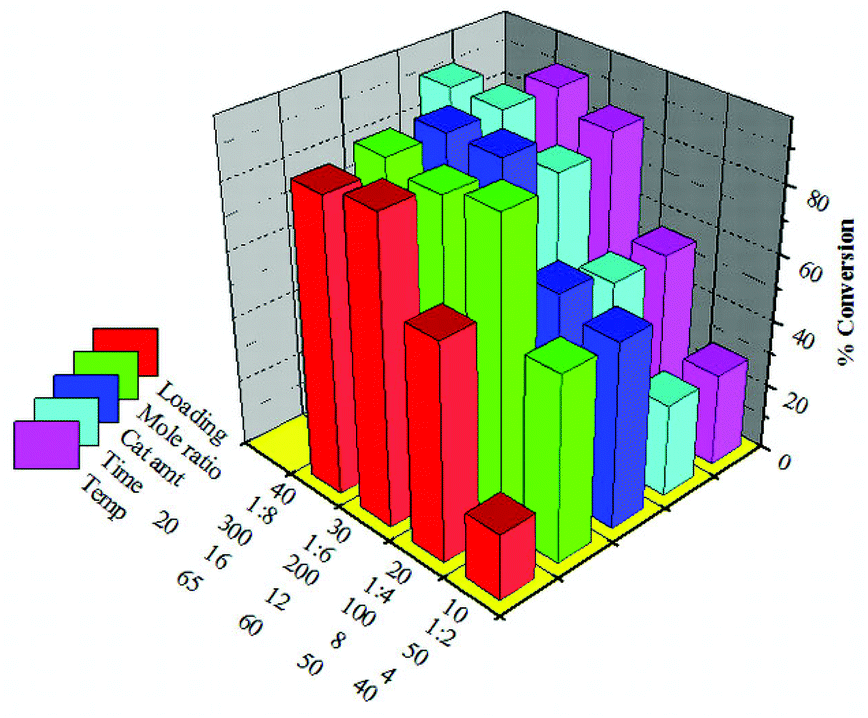
The detailed optimization of reaction parameters for the maximum conversion of SO over 30% SiW11/MCM-41 was carried out as described in the previous section. The optimized conditions for 30% SiW11/MCM-41 (Fig. 10) are as follows: w/w ratio of oil to alcohol 1:4; amount of catalyst 200 mg; reaction time 16 h and reaction temperature 65 °C. Under these conditions the maximum conversion of 91% was achieved.
 | ||
| Fig. 10 Different reaction parameter optimization for transesterification of SO over 30% SiW11/MCM-41. | ||
6.3 Effect of active species (POMs/LPOMs)
In order to see the effect of active species transesterification of SO was carried out over 30% PW12/MCM-48 and 30% SiW12/MCM-41 and the results are presented in Fig. 11. The details on synthesis and characterization of both the catalysts can be found in our earlier reports.103,104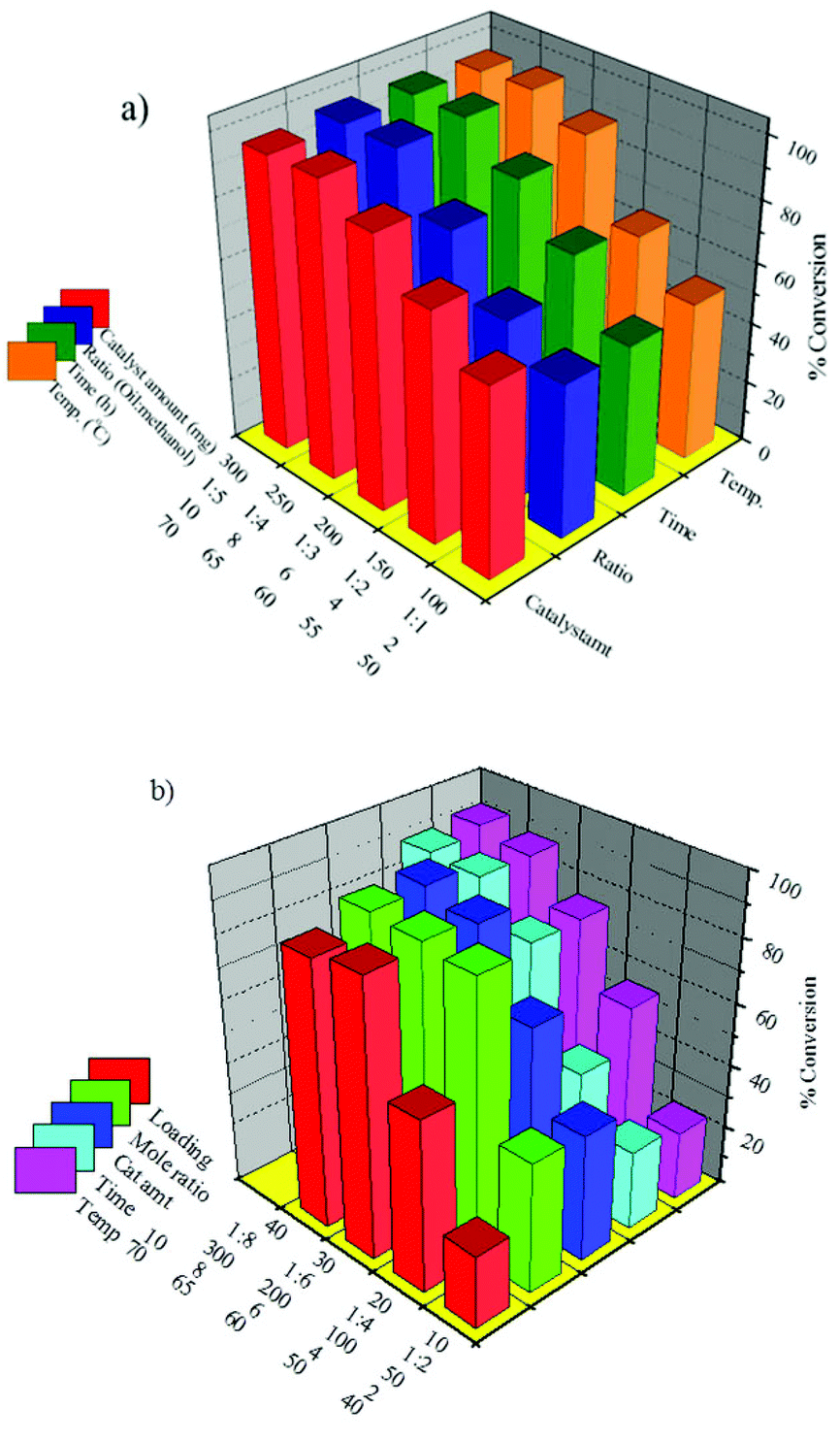 | ||
| Fig. 11 Different reaction parameter optimization for transesterification of SO over (a) 30% PW12/MCM-48 and (b) 30% SiW12/MCM-41. | ||
The optimized conditions for the maximum conversion of SO transesterification reaction over 30% PW12/MCM-48 (maximum conversion 97%) are as follows: w/w ratio of oil to alcohol 1:4; amount of catalyst 250 mg; and reaction temperature 65 °C; reaction time 8 h. And those for 30% SiW12/MCM-41 (maximum conversion 88%) are as follows: w/w ratio of oil to alcohol 1:4; amount of catalyst 200 mg; and reaction temperature 65 °C; reaction time 8 h.
Table 8 describes the activity of parent as well as lacunary POM based catalysts under the same conditions. It is clear from Table 8 that the activity of 30% PW12/MCM-48 is higher than that of 30% PW11/MCM-48. This can be explained as follows.
| Catalyst | Reaction conditionsa | Conv. (%) | TON | TOF (min−1) |
|---|---|---|---|---|
| a Reaction conditions: amount of catalyst wt%/mole ratio of alcohol to substrate/reaction temperature °C/reaction time h. | ||||
| 30% PW12/MCM-48 | 0.2 g/4/65/8 | 89 | 305 | 0.63 |
| 30% PW11/MCM-48 | 0.2 g/4/65/8 | 81 | 283 | 0.58 |
| 30% SiW12/MCM-41 | 0.2 g/4/65/8 | 88 | 290 | 0.60 |
| 30% SiW11/MCM-41 | 0.2 g/4/65/8 | 58 | 186 | 0.38 |
The reason being the acidic character of polyoxometalates is mainly due to the acidic addenda atoms, i.e. tungsten in the present case and removal of one tungsten-oxygen unit from the parent PW12 is expected to decrease the acidity and as a result the activity of the PW11. This can be seen by the change in the values of acidic strength (Table 6). The acidic strength of 30% PW12/MCM-48 (490 mV) is higher than that of 30% PW11/MCM-48 (290 mV).
The surface area of 30% PW11/MCM-48 (319 m2 g−1) is higher than 30% PW12/MCM-48 (286 m2 g−1). This suggests that the catalytic activity of both the catalysts is not directly proportional to the surface area confirming that the present catalytic systems are not surface type heterogeneous in which the catalytic activity is directly proportional to the surface area. Hence, it can be concluded that the present catalytic system is the pseudo-liquid type (I) heterogeneous catalyst where the activity is directly proportional to the acidic strength of the catalyst.105 The order of the catalytic activity for other two systems (30% SiW12/MCM-41 > 30% SiW11/MCM-41) can be explained in a similar manner.
6.4 Effect of the support
It is well known that the ‘support’ does not play always merely a mechanical role but it can also modify the catalytic properties of the obtained catalyst. The nature of the support such as structural mesoporosity and high specific surface area is an important factor towards the formation of a successful anchoring platform and for better catalytic activity. In order to see the effect of the support, we have carried out synthesis of catalysts comprising PW12/PW11 supported onto two different supports, MCM-41 and MCM-48. It can be seen from Table 9 that 30% PW12/MCM-48 is more active than 30% PW12/MCM-41. The trend in the activity of the catalysts 30% PW12/MCM-48 and 30% PW12/MCM-41 is not consistent with the surface area of the catalysts. The better activity of MCM-48 based catalyst can be correlated with the acidic nature of the support and 3-dimentional pore network where entering and leaving-off of the reactant and a product molecule is more feasible.| Catalyst | Surface area (m2 g−1) | Reaction conditionsa | Conv. (%) | TON |
|---|---|---|---|---|
| a Reaction conditions: amount of catalyst wt%/mole ratio of alcohol to substrate/reaction temperature °C/reaction time h. | ||||
| 30% PW12/MCM-48 | 286 | 0.25 g/4/65/8 | 97 | 266 |
| 30% PW12/MCM-41 | 360 | 0.25 g/4/65/8 | 93 | 247 |
| 30% PW11/MCM-48 | 319 | 0.25 g/4/65/8 | 91 | 267 |
| 30% PW11/MCM-41 | 252 | 0.25 g/4/65/8 | 85 | 239 |
6.5 Recycling study
Transesterification of soybean oil was carried out with the recycled supported catalysts, under the optimized conditions. After the reaction, the catalysts were separated from the reaction mixture by simple centrifugation; the first washing was done with methanol to remove the products, then the subsequent washings were done with distilled water and then dried at 100 °C, and the recovered catalyst was charged for the further run. No appreciable decrease in the conversion was observed after two cycles (Table 10).| Catalyst | Number of cycles (Conv. (%)) | ||
|---|---|---|---|
| Fresh | 1st | 2nd | |
| 30% PW12/MCM-48 | 97 | 96 | 94 |
| 30% PW11/MCM-48 | 95 | 93 | 92 |
| 30% SiW12/MCM-41 | 88 | 86 | 85 |
| 30% SiW11/MCM-41 | 91 | 90 | 88 |
The recycling study was carried out using a single reaction batch and the catalyst was recovered by centrifugation. The catalyst quantity recovered was charged for the next run. Hence there is a handling loss of the catalyst which prevents further use of the catalyst. However, the use of the catalyst can be extended for further runs. Furthermore, in order to evaluate the elution of the polyoxometalates, the leaching test was carried out.
Polyoxometalates can be quantitatively characterized by the heteropoly blue colour, which is observed when it is reacted with a mild reducing agent such as ascorbic acid. In the present study, this method was used for determining the leaching of PW12, PW11, SiW12 and SiW11 from the support. Standard samples containing 1–5% of PW12 (or subsequent species) in water were prepared. To 10 mL of the above samples, 1 mL of 10% ascorbic acid was added. The mixture was diluted to 25 mL. The resulting solution was scanned at a λmax of 785 nm for its absorbance values. A standard calibration curve was obtained by plotting values of absorbance against % concentration. 1 g of the supported catalyst with 10 mL water was refluxed for 18 h. Then 1 mL of the supernatant solution was treated with 10% ascorbic acid. Development of blue colour was not observed, indicating that there was no leaching. The same procedure was repeated with oil: methanol (1:4 mole ratios) samples and the filtrate of the reaction mixture after completion of the reaction in order to check the presence of any leached POM species. The leaching of tungsten (W) from catalysts was confirmed by carrying out an analysis of the product mixtures (by AAS). Analysis of the product mixtures showed that if any W were present it was below the detection limit, which corresponds to less than 1 ppm. On the basis of these results, it can be concluded that there is no leaching of active species from the supports and the catalysts are truly heterogeneous in nature.
6.6 Characterization of regenerated catalysts
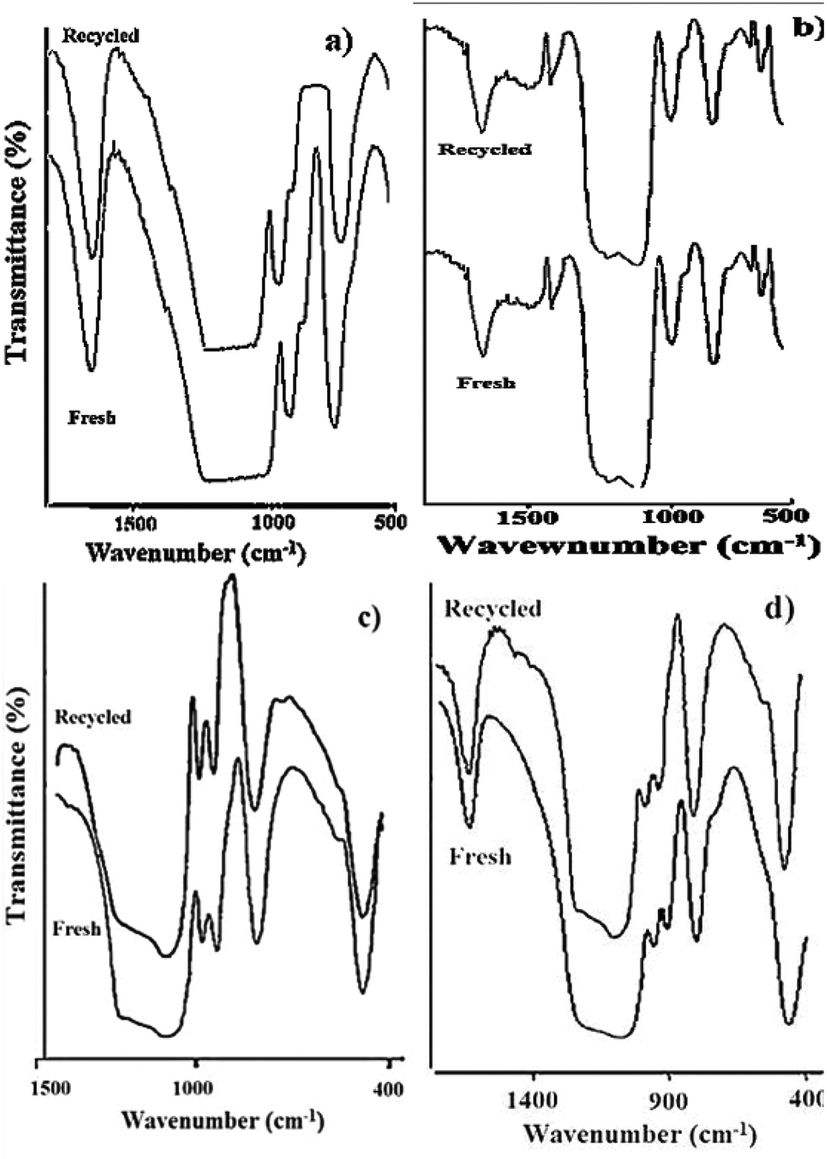
The FT-IR data for the fresh as well as the recycled catalyst are presented in Fig. 12. The typical bands for PW12 at 982 cm−1 corresponding to Vas vibration of WO, and at 897 cm−1 for stretching vibrations of W–Ob–W are clearly observed in recycled 30% PW12/MCM-48 (Fig. 12a). No significant shift in the FT-IR band position of the recycled catalyst compared to the fresh catalyst indicates the retention of the Keggin structure of PW12 on MCM-48. Similarly, in Fig. 12b, no appreciable shift in the FT-IR band position of the recycled 30%-PW11/MCM-41 catalyst compared to the fresh catalyst indicates the retention of the structure of PW11.
 | ||
| Fig. 12 FT-IR spectra of fresh and recycled catalysts (a) 30% PW12-MCM-48, (b) 30% PW11/MCM-48, (c) 30% SiW12/MCM-41 and (d) 30% SiW11/MCM-41 (taken from ref. 104,106). | ||
The FT-IR spectra of recycled 30% SiW12/MCM-41 (Fig. 12c) showed the retention of typical bands for SiW12, at 979 cm−1 and 923 cm−1 corresponding to WOd and Si–Oa symmetric stretching, respectively. The FT-IR spectra of the recycled catalyst 30% SiW11/MCM-41 (Fig. 12d) shows retention of bands at 960 cm−1 (WOd), and 900 cm−1 (Si–Oa) suggesting that the structure of SiW11 in the regenerated catalyst is intact. The above study shows that there is no deactivation of the catalyst after reuse.
6.7 Mechanism of transesterification
The probable transesterification mechanism between triglyceride and methanol is shown in Fig. 13.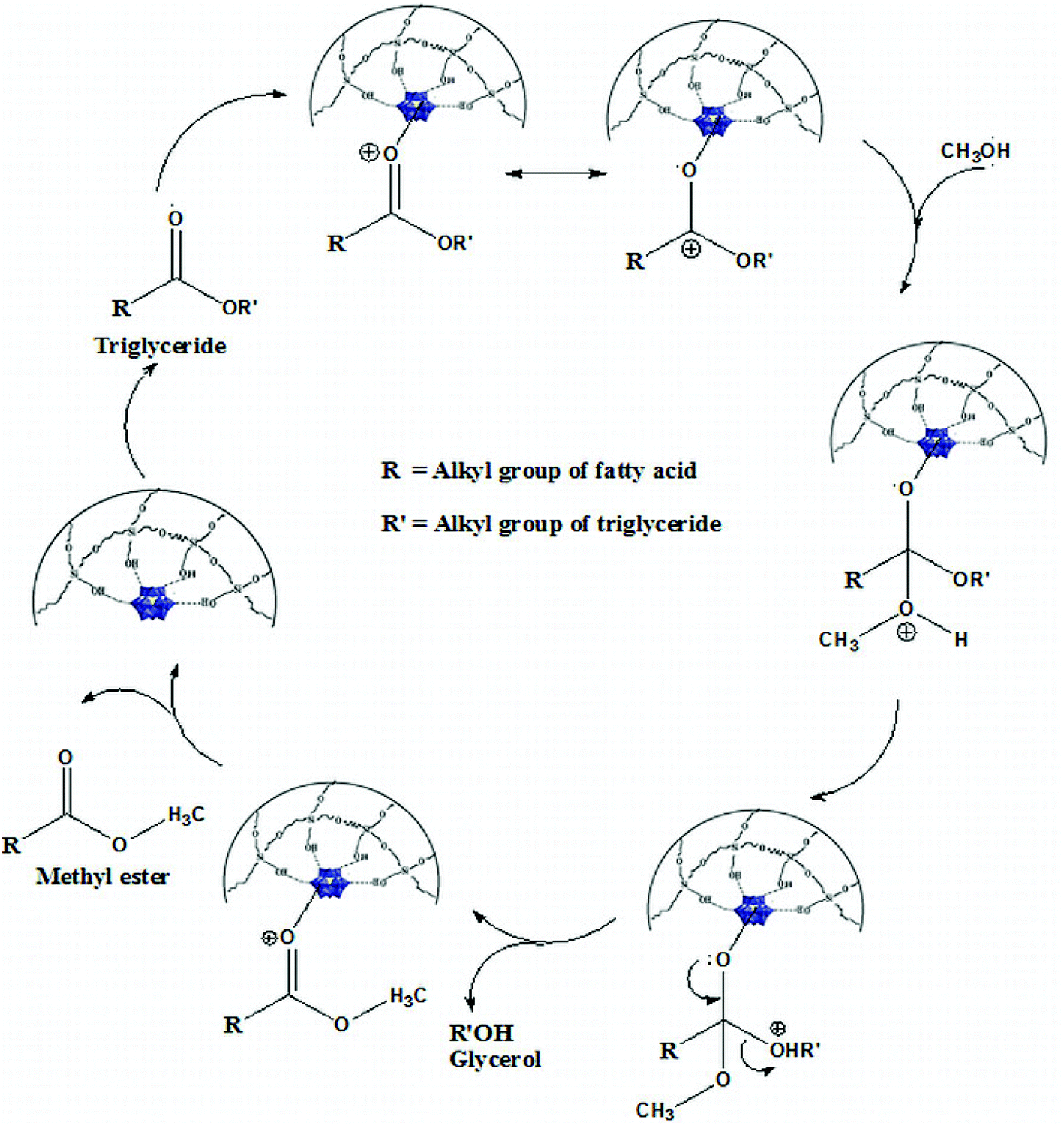 | ||
| Fig. 13 Mechanism of transesterification reaction. | ||
The very high surface area and pore dimension of the present catalyst play an important role in diffusion of large triglyceride molecules onto the surface of the catalyst. The carbocation is formed by acid sites of the catalyst with the carbonyl oxygen. Heteropolyacids are known to stabilise the carbocation intermediate thereby assisting the rate of the reaction.5 A tetrahedral intermediate is formed by nucleophilic attack of methanol to the carbocation (Fig. 13). The tetrahedral intermediate eliminates H2O to form biodiesel. This mechanism can be extended to di-glycerides or tri-glycerides. The leaving off of the product molecule is again a steric effect in the heterogeneous catalysis and the present catalyst contains a large pore diameter that facilitates the escape of the product molecule.
6.8 Possible industrial scheme for biodiesel production
In our previous study, transesterification of WCO and JO was successfully scaled up for biodiesel synthesis using up to 25 g feedstock over PW12/MCM-48 with high conversion and identical optimization conditions as compared to the batch scale.103 Similar results can be expected for other feedstocks, including soybean oil without changing any mechanistic requirement of the system. Depending on the requirements, the size of the unit can be scaled up to get higher production capacity. A possible scheme of this integrated process using heterogeneous acid catalysts is shown in Fig. 14. After completion of transesterification reaction settling can be used to separate the two products, i.e. crude glycerol and biodiesel. After settling, crude glycerol is washed with water to remove impurities and the traces of the catalyst. In heterogeneous catalysis, the reaction mainly takes place on the surface of active sites. Hence after the completion of the reaction, the unreacted as well as the by-product molecules must be removed from the surface in order to activate the catalyst for the next run.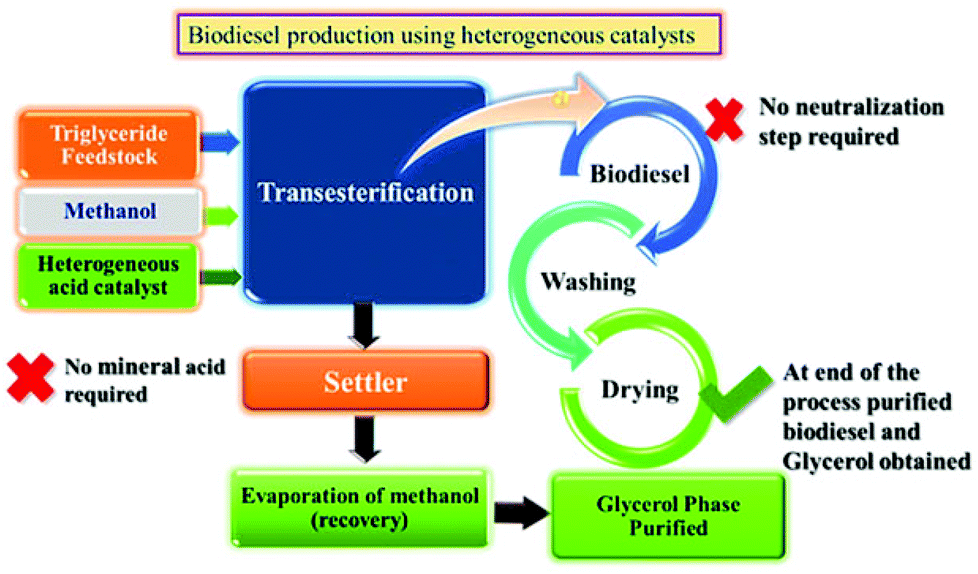 | ||
| Fig. 14 Process for biodiesel production over heterogeneous acid catalysts. | ||
7. Conclusions and outlook
The present review exclusively focuses on the application of supported HPAs for the biodiesel synthesis. The literature survey shows that key advantages of using supported HPAs are (a) the ability to carry out esterification and transesterification reaction simultaneously (b) tolerance to FFA and water, (c) high acidity and (d) reusability. In addition, various methods such as microwave conditions, and ultrasonic treatment can reduce the reaction time, production costs, catalyst and alcohol requirements for biodiesel synthesis. These methods can increase the quality of FAME for applications to diesel engines without any kind of engine modification. In the present review, new results for transesterification of soybean oil have been discussed over parent PW12 and SiW12 supported onto two supports (MCM-41 and MCM-48).The review was also extended for the first time on the application of LPOMs towards biodiesel synthesis by observing excellent activity for our earlier reported work on lacunary SiW11 supported to MCM-41 for the esterification of oleic acid with methanol under mild conditions. In light of these results transesterification of soybean oil was efficiently carried out over PW11 and SiW11 supported onto MCM-41 as well as MCM-48. All the catalysts showed very promising results for the transesterification reaction under mild conditions. Also, the catalyst shows potential for being used as a recyclable catalytic material after simple regeneration without any significant loss in the conversion. Furthermore the activities were correlated with the acidity as well as structural morphology of the supports. Among all the catalysts PW12 was found to be the best active species among POMs, PW11 as the best active species in the case of LPOMs and MCM-48 was the best support.
The use of homogeneous mineral acids in the traditional biodiesel production process can corrode the system and hence reduce the life span of the reactor. Also the use of mineral acid requires an additional neutralization step and hence increases the overall cost of the process. However, in the present process, a heterogeneous acid catalyst would allow easy separation and reuse of the catalyst from the reaction medium and avoids the neutralization step necessary while using the acidic or alkaline homogeneous catalyst. The transesterification reaction is followed by evaporation to purify the crude biodiesel and to recycle the excess methanol.
Biodiesel production using heterogeneous acid catalysts can definitely improve the overall energy and economic efficiency. Also a higher grade, salt free glycerol can be obtained as a byproduct, hence a reduction of the purification costs will be achieved. In addition, environmental and economic benefits can be achieved as the glycerol which is a byproduct of biodiesel production can undergo organic transformations leading to a variety of products via acetalization, acetylation, carboxylation and oxidation reactions in order to reduce the biodiesel production cost.
The present system has great potential for the practical large scale up process for biodiesel synthesis using a heterogeneous acid catalyst in a single step (Fig. 14). Also by looking at the future perspective for biodiesel synthesis over POMs, advancement can be done by studying the different combinations of POMs as well as LPOMs and supports.
The overall production process will be economical as the catalyst can be used up to many cycles, total steps can be reduced and methanol can be recycled and reused for subsequent runs. Furthermore, it minimizes the production cost as obtained glycerol can be converted to value added products via various organic transformations. Thus the proposed process of low cost biodiesel production along with the value added product is a good environmentally viable option for the future.
Acknowledgements
Anjali Patel and Sukriti Singh are thankful to the Department of Science and Technology, New Delhi, India (SR/S5/GC-01/2009) for the financial support and the award of research fellowship, respectively. AP is also thankful to Dr Varsha Brahmkhatri for her valuable help in preparation of the manuscript, at the initial stage.Notes and references
- Z. Helwani, M. R. Othman, N. Aziz, W. J. N. Fernando and J. Kim, Fuel Process. Technol., 2009, 90, 1502–1514 CrossRef CAS PubMed.
- A. Demirbas, Energy Convers. Manage., 2009, 50, 14–34 CrossRef CAS PubMed.
- B. Buczek and L. Czepirski, Inform. AOCS, 2004, 15, 186–188 Search PubMed.
- M. T. Pope, Inorganic Chemistry Concepts Vol. 8: Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 Search PubMed.
- T. Okuhara, N. Mizuno and M. Misono, Adv. Catal., 1996, 41, 113–252 CAS.
- I. V. Kozhevnikov, Catalysts for fine chemical synthesis, Catalysis by polyoxometalates, Wiley, Chichester, 2002, vol 2 Search PubMed.
- A. Demirbas, Energy Convers. Manage., 2003, 44, 2093–2109 CrossRef CAS.
- C. Stavarache, M. Vinatoru, R. Nishimura and Y. Maeda, Ultrason. Sonochem., 2005, 12, 367–372 CrossRef CAS PubMed.
- P. V. Tekade, O. A. Mahodaya, G. R. Khandeshwar and B. D. Joshi, Sci. Rev. Chem. Commun, 2012, 2(3), 208–211 CAS.
- G. Pahl, Biodiesel: Growing a New Energy Economy, Chelsea Green Publication Company, US, 2004 Search PubMed.
- Biodiesel, http://en.wikipedia.org/wiki/Biodiesel#cite_note-26 (accessed July 2014).
- G. Knothe, J. V. Gerpen and J. Krahl, The Biodiesel Handbook, AOCS Press, USA, 2005 Search PubMed.
- M. J. Haas, Fuel Process. Technol., 2005, 86, 1087–1096 CrossRef CAS PubMed.
- M. J. Haas, A. J. McAloon, W. C. Yee and T. A. Foglia, Bioresour. Technol., 2006, 97, 671–678 CrossRef CAS PubMed.
- M. G. Kulkarni and A. K. Dalai, Ind. Eng. Chem. Res., 2006, 45, 2901–2913 CrossRef CAS.
- Y. Zhang, M. A. Dube, D. D. McLean and M. Kates, Bioresour. Technol., 2003, 90, 229–240 CrossRef CAS.
- S. Peter and E. Weidner, Eur. J. Lipid Sci. Technol., 2007, 109, 11–16 CrossRef CAS.
- Z. Tang, L. Wang and J. Yang, Eur. J. Lipid Sci. Technol., 2008, 110, 747–753 CrossRef CAS.
- N. Dizge, C. Aydiner, D. Y. Imer, M. Bayramoglu, A. Tanriseven and B. Keskinler, Bioresour. Technol., 2009, 100, 1983–1991 CrossRef CAS PubMed.
- V. B. Veljkovic, O. S. Stamenkovic, Z. B. Todorovic, M. L. Lazic and D. U. Skala, Fuel, 2009, 88, 1554–1562 CrossRef CAS PubMed.
- C. C. M. Silva, N. F. P. Ribeiro, M. V. M. Souz and D. A. G. Aranda, Fuel Process. Technol., 2010, 91, 205–210 CrossRef CAS PubMed.
- H. J. Berchmans and S. Hirata, Bioresour.. Technol., 2008, 99, 1716–1721 CrossRef CAS PubMed.
- S. A. Raja, D. S. R. Smart and C. L. R. Lee, Res. J. Chem. Sci., 2011, 1, 81–87 Search PubMed.
- P. Patil, S. Denga, J. I. Rhodes and P. J. Lammers, Fuel, 2010, 89, 360–364 CrossRef CAS PubMed.
- J. M. N. van Kasteren and A. P. Nisworo, Resour., Conserv. Recycl., 2007, 50, 442–458 CrossRef PubMed.
- X. Meng, G. Chen and Y. Wang, Fuel Process. Technol., 2008, 89, 851–857 CrossRef CAS PubMed.
- J. Yan, Y. Yan, S. Liu, J. Hu and G. Wang, Bioresour. Technol., 2010, 102, 4755–4758 CrossRef PubMed.
- Z. Wen, X. Yu, S.-T. Tu, J. Yan and E. Dahlquist, Bioresour. Technol., 2010, 101, 9570–9576 CrossRef CAS PubMed.
- M. A. Jackson and J. W. King, J. Am. Oil Chem. Soc., 1996, 73, 353–356 CrossRef CAS.
- Y. Zhang, M. A. Dube, D. D. McLean and M. Kates, Bioresour. Technol., 2003, 89, 1–16 CrossRef CAS.
- D. Y. C. Leung and Y. Guo, Fuel Process. Technol., 2006, 87, 883–890 CrossRef CAS PubMed.
- M. Y. Koh and T. I. M. Ghazi, Renewable Sustainable Energy Rev., 2011, 15, 2240–2251 CrossRef CAS PubMed.
- G. Mendow, N. S. Veizaga and C. A. Querini, Bioresour. Technol., 2011, 102, 6385–6391 CrossRef CAS PubMed.
- S. V. Ghadge and H. Raheman, Biomass Bioenergy., 2005, 28, 601–605 CrossRef CAS PubMed.
- X. Miao, R. Li and H. Yao, Energy Convers. Manage., 2009, 50, 2680–2684 CrossRef CAS PubMed.
- L. C. Meher, M. G. Kulkarni, A. K. Dalai and S. N. Naik, Eur. J. Lipid Sci. Technol., 2006, 108, 389–397 CrossRef CAS.
- B. Xu, G. Xiao, L. Cui, R. Wei and L. Gao, Energy Fuels, 2007, 21, 3109–3112 CrossRef.
- S. N. Babu, S. Rekha, S. P. S. Prasad and N. Lingaiah, Energy Fuels, 2008, 22, 1965–1971 CrossRef.
- E. M. Shahid and Y. Jamal, Renewable Sustainable. Energy Rev., 2008, 12, 2484–2494 CrossRef CAS PubMed.
- C. He, B. Peng, D. Wang and J. Wang, Front. Chem. Eng. China, 2007, 1, 11–15 CrossRef.
- V. P. Amish, N. Subrahmanyam and P. A. Payal, Fuel, 2009, 88, 625–628 CrossRef PubMed.
- S. H. Shuit, Y. T. Ong, K. T. Lee, B. Subhash and S. H. Tan, Biotechnol. Adv., 2012, 30, 1364–1380 CrossRef CAS PubMed.
- G. C. Martins, T. Sergio, M. L. Ledo and U. Schuchardt, Bioresours. Technol., 2008, 99, 6608–6613 CrossRef PubMed.
- S. Furuta, H. Matsuhashi and K. Arata, Biomass Bioenergy, 2006, 30, 870–873 CrossRef CAS PubMed.
- J. J. Berzelius, Pogg. Ann. Chem., 1826, 6, 369–392 CrossRef.
- J. F. Keggin, Nature, 1933, 131, 908–909 CrossRef CAS.
- J. F. Keggin, Proc. R. Soc. London, Ser. A, 1934, 144, 75–100 CrossRef CAS.
- M. Misono, N. Mizuno, K. Katamura, A. Kasai, Y. Konishi, K. Sakata, T. Okuhara and Y. Yoneda, Bull. Chem. Soc. Jpn., 1982, 55, 400–406 CrossRef CAS.
- M. Furuta, K. Sakata, M. Misono and Y. Yoneda, Chem. Lett., 1979, 31–34 CrossRef CAS.
- M. Misono, Catal. Rev. Sci. Eng., 1987, 29, 269–321 CAS.
- C. Rocchiccioli-Deltcheff, M. A. Amirouche, G. Herve, M. Fournier, M. Che and J. M. Tatiboute, J. Catal., 1990, 126, 591–599 CrossRef CAS.
- P. G. Vazquez, M. N. Blanco and C. V. Caceres, Catal. Lett., 1999, 60, 205–215 CrossRef CAS.
- S. Swanmi, N. Shin-ichi, T. Okuhara and M. Misono, J. Catal., 1997, 166, 263–271 CrossRef.
- I. V. Kozhevnikov, Russ. Chem. Rev., 1987, 56, 811–825 CrossRef PubMed.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed.
- J. H. Clark, A. P. Kybett and D. J. Maequarrie, Supported reagents: preparation, analysis and application, VCH Inc., New York, 2000 Search PubMed.
- Y. C. Sharma and B. Singh, Biofuels, Bioprod. Biorefin., 2011, 5, 69–92 CrossRef CAS.
- J. A. Melero, J. Iglesias and G. Morales, Green Chem., 2009, 11, 1285–1308 RSC.
- I. M. Atadashi, M. K. Aroua, A. R. Abdul Aziz and N. M. N. Sulaiman, J. Ind. Eng. Chem., 2013, 19, 14–26 CrossRef CAS PubMed.
- M. Zabeti, W. M. A. W. Daud and M. K. Aroua, Fuel Process. Technol., 2009, 90, 770–777 CrossRef CAS PubMed.
- J. S. Lee and S. Saka, Bioresour. Technol., 2010, 101, 7191–7200 CrossRef CAS PubMed.
- A. K. Endalew, Y. Kiros and R. Zanzi, Biomass Bioenergy, 2011, 35, 3787–3809 CrossRef CAS PubMed.
- A. P. Singh Chouhan and A. K. Sarma, Renewable Sustainable Energy Rev., 2011, 15, 4378–4399 CrossRef PubMed.
- M. E. Borges and L. Díaz, Renewable Sustainable Energy Rev., 2012, 16, 2839–2849 CrossRef CAS PubMed.
- A. Islama, Y. H. T. Yap, C. M. Chu, E. S. Chan and P. Ravindra, Process Saf. Environ. Prot., 2013, 91, 131–144 CrossRef PubMed.
- C. Komintarachat and S. Chuepeng, Ind. Eng. Chem. Res., 2009, 48, 9350–9353 CrossRef CAS.
- F. Su and Y. Guo, Green Chem., 2014, 16, 2934–2957 RSC.
- M. G. Kulkarni, R. Gopinath, L. C. Meher and A. K. Dalai, Green Chem., 2006, 8, 1056–1062 RSC.
- C. S. Caetano, I. M. Fonseca, A. M. Ramos, J. Vital and J. E. Castanheiro, Catal. Commun., 2008, 9, 1996–1999 CrossRef CAS PubMed.
- B. Hamad, R. O. L. Souza de, G. Sapaly, M. G. C. Rocha, P. G. P. Oliveira de and W. A. Gonzalez, Catal. Commun., 2008, 10, 92–97 CrossRef CAS PubMed.
- G. Sunita, B. M. Devassy, A. Vinu, D. P. Sawant, V. V. Balasubramanian and S. B. Halligudi, Cat. Commun., 2008, 9, 696–702 CrossRef CAS PubMed.
- L. Xu, W. Li, J. Hu, X. Yang and Y. Guo, Appl. Catal., B, 2009, 90, 587–594 CrossRef CAS PubMed.
- L. Xu, Y. Wang, X. Yang, J. Hu, W. Li and Y. Guo, Green Chem., 2009, 11, 314–317 RSC.
- L. Xu, W. Li, J. Hu, K. Li, X. Yang, F. Ma, Y. Guo, X. Yu and Y. Guo, J. Mater. Chem., 2009, 19, 8571–8579 RSC.
- C. F. Oliveira, L. M. Dezaneti, F. A. C. Garcia, Julio L. de Macedo, J. A. Dias, S. C. L. Dias and K. S. P. Alvim, Appl. Catal., A, 2010, 372, 153–161 CrossRef CAS PubMed.
- K. Srilatha, N. Lingaiah, P. S. Sai Prasad, B. L. A. Prabhavathi Devi and R. B. N. Prasad, React. Kinet., Mech. Catal., 2011, 104, 211–226 CrossRef CAS.
- K. Srilatha, Ch. Ramesh Kumar, B. L. A. Prabhavathi Devi, R. B. N. Prasad, P. S. Sai Prasada and N. Lingaiah, Catal. Sci. Technol., 2011, 1, 662–668 CAS.
- K. Srilatha, N. Lingaiah, B. L. A. P. Devi, R. B. N. Prasad, S. Venkateswar and P. S. S. Prasad, Appl. Catal., A, 2009, 365, 28–33 CrossRef CAS PubMed.
- K. Srilatha, B. L. A. Prabhavathi Devi, N. Lingaiah, R. B. N. Prasad and P. S. Sai Prasad, Bioresour. Technol., 2012, 119, 306–311 CrossRef CAS PubMed.
- K. Srilatha, T. Issariyakul, N. Lingaiah, P. S. Sai Prasad, J. Kozinski and A. K. Dalai, Energy Fuels, 2010, 24, 4748–4755 CrossRef CAS.
- A. S. Badday, A. Z. Abdullah and K. T. Lee, Appl. Energy, 2013, 105, 380–388 CrossRef CAS PubMed.
- R. Sheikh, M. S. Choi, J. S. Im and Y. H. Park, J. Ind. Eng. Chem., 2013, 19, 1413–1419 CrossRef CAS PubMed.
- V. V. Bokade and G. D. Yadav, Process Saf. Environ. Prot., 2007, 85(B5), 372–377 CrossRef CAS.
- V. V. Bokade and G. D. Yadav, Ind. Eng. Chem. Res., 2009, 48, 9408–9415 CrossRef CAS.
- L. X. Zhang, Q. Z. Jin, J. H. Huang, Y. F. Liu, L. Shan and X. G. Wang, Energy Sources, Part A, 2012, 34, 1037–1045 CrossRef CAS.
- O. da Silva Lacerda, J. R. M. Cavalcanti, T. M. de Matos, R. S. Angélica, G. N. da Rocha Filho and I. de C. Lopes Barros, Fuel, 2013, 108, 604–611 CrossRef PubMed.
- K. Y. Nandiwale and V. V. Bokade, Ind. Eng. Chem. Res., 2014 DOI:10.1021/ie500672v.
- J. Alcaniz-Monge, G. Trautwein and J. P. Marco-Lozar, Appl. Catal., A, 2013, 468, 432–441 CrossRef CAS PubMed.
- A. S. Badday, A. Z. Abdullah and K. T. Lee, Renewable Energy, 2013, 50, 427–432 CrossRef CAS PubMed.
- A. S. Badday, A. Z. Abdullah and K. T. Lee, Renewable Energy, 2014, 62, 10–17 CrossRef CAS PubMed.
- A. Patel and N. Narkhede, Energy Fuels, 2012, 26, 6025–6032 CrossRef CAS.
- N. Narkhede and A. Patel, Ind. Eng. Chem. Res., 2013, 52, 13637–13644 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- T. Vralstad, G. Oye, J. Sjoblom and M. Stocker, J. Dispersion Sci. Technol., 2006, 27, 489–496 CrossRef.
- A. Tuel, Microporous Mesoporous Mater., 1999, 27, 151–169 CrossRef CAS.
- I. Tropecelo, M. H. Casimiro, I. M. Fonseca, A. M. Ramos, J. Vital and J. E. Castanheiro, Appl. Catal., A, 2010, 390, 183–189 CrossRef PubMed.
- M. S. Khayoon and B. H. Hameed, Fuel Process. Technol., 2013, 114, 12–20 CrossRef CAS PubMed.
- K. Yan, G. Wu, J. Wen and A. Chen, Catal. Commun., 2013, 34, 58–63 CrossRef CAS PubMed.
- C. Baroi and A. K. Dalai, Catal. Today, 2013, 207, 74–85 CrossRef CAS PubMed.
- V. Brahmkhatri and A. Patel, Ind. Eng. Chem. Res., 2011, 50, 6620–6628 CrossRef CAS.
- V. Brahmkhatri and A. Patel, Appl. Catal., A, 2011, 403, 161–172 CrossRef CAS PubMed.
- S. Singh and A. Patel, J. Cleaner Prod., 2014, 72, 46–56 CrossRef CAS PubMed.
- V. Brahmkhatri and A. Patel, in Environmentally Benign Catalysts Based on Heteropolyacids for Clean Organic Transformations, ed. A. Patel, Springer Inc., New York, 2013, pp. 189–208 Search PubMed.
- N. Narkhede, V. Brahmkhatri and A. Patel, Fuel, 2014, 135, 253–261 CrossRef CAS PubMed.
- A. Patel and N. Narkhede, Catal. Sci. Technol., 2013, 3, 3317–3325 CAS.
- P. A. Shringarpure and A. Patel, Dalton Trans., 2010, 39, 2615–2621 RSC.
- S. Pathan and A. Patel, J. Coord. Chem., 2010, 63, 4041–4049 CrossRef CAS.
- A. Patel and S. Singh, Microporous Mesoporous Mater., 2014, 195, 240–249 CrossRef CAS PubMed.
- N. Narkhede and A. Patel, RSC Adv., 2014, 4, 19294–19301 RSC.
| This journal is © The Royal Society of Chemistry 2015 |