
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization of transition metal nanoparticle-phosphonium ionic liquid composite catalytic systems for deep hydrogenation and hydrodeoxygenation reactions†
Abhinandan
Banerjee
and
Robert W. J.
Scott
*
Department of Chemistry, University of Saskatchewan, Saskatoon, SK S7N 5C9, Canada. E-mail: robert.scott@usask.ca; Tel: +1-306-966-4730
First published on 10th December 2014
Abstract
A variety of metal nanoparticle (NP)/tetraalkylphosphonium ionic liquid (IL) composite systems were evaluated as potential catalysts for the deep hydrogenation of aromatic molecules. Particles were synthesized by reducing appropriate metal salts by LiBH4 in a variety of ILs. Gold NPs were used as probes to investigate the effect of both chain lengths of the alkyl substituents on the phosphonium cation and the nature of anions, on the stability of NPs dispersed in the ILs. The presence of three medium-to-long alkyl chains (such as hexyl) along with one long alkyl chain (such as tetradecyl) in the IL, coupled with highly coordinating anions (such as halides, or to a smaller extent, bis-triflimides) produced the most stable dispersions. These ILs also showed maximum resistance to heat-induced sintering; for example, TEM studies of Pt NPs heated under hydrogen to 120 °C showed only moderate sintering in trihexyl(tetradecyl)phosphonium chloride and bis(triflimide) ILs. Finally, olefinic hydrogenations, aromatic hydrogenations, and hydrodeoxygenation of phenol were carried out with Ru, Pt, Rh and PtRh NPs using hydrogen at elevated pressures. From preliminary studies, Ru NPs dispersed in trihexyl(tetradecyl)phosphonium chloride emerged as the catalyst system of choice. The presence of borate Lewis-acid by-products in the reaction medium (from the borohydride reduction step) allowed for partial phenol hydrodeoxygenation.
Introduction
In recent years, ionic liquids (ILs) have been used extensively for the stabilization of metal nanoparticles (NPs) and/or as reaction media for metal NP-catalysed reactions.1–5 Imidazolium ILs, in particular, have been studied extensively for their remarkable ability to serve as reaction media and/or stabilizers for such NP-catalysed transformations.6–10 However, the imidazolium ILs offer challenges such as base-induced deprotonation and consequent carbene formation,11 hydrolysis of anions such as PF6− and BF4− to generate corrosive acids,12 variations in the efficiency of NP stabilization induced by the presence of trace amounts of impurities such as unreacted IL precursors,7,13 and comparatively higher melting points of imidazolium halides, which are not room-temperature ILs.14 It is surprising, therefore, that tetraalkylphosphonium ILs, which are stable under highly basic conditions, commercially available at high levels of purity, less sensitive to moisture and oxygen than their imidazolium counterparts, and largely present as liquids at room temperature even when combined with highly coordinating anions such as halides, have not been investigated more extensively as potential replacements for imidazolium ILs in catalytic reactions of industrial and environmental importance.15–17 Several groups, including our own, have shown that NP dispersions of catalytically active precious metals in tetraalkylphosphonium ILs are effective and recyclable catalysts for diverse classes of reactions such as oxidations,18 selective hydrogenations,19,20 and C–C cross coupling reactions.21,22One class of reactions that have remained relatively unexplored in the context of metal NP/tetraalkylphosphonium IL composite catalysts is deep hydrogenations, or hydrogenations of aromatic compounds.23 This is a key reaction in processes such as hydro-refining of heavy oil, production of petrochemicals, synthesis of pharmaceutical products, and generation of biofuel from non-edible lignin.24 Synthesis of biofuels from lignin includes both hydrogenation as well as hydrogenolysis steps,25–27 and it is expected that under suitably tuned reaction conditions, a metal NP/IL composite catalyst can perform both reactions simultaneously. NPs of precious metals such as Ir, Rh and Ru have the advantage of high activity for the hydrogenation of aromatic compounds under mild reaction conditions, and their dispersions in functionalized imidazolium ILs have been used in several studies for hydrogenation of mononuclear aromatic species.28–30 Dyson and co-workers, and others, have applied these systems as catalysts in reactions such as hydrodeoxygenation (HDO) of phenol to cyclohexane, and regioselective hydrogenation of toluene to methyl cyclohexene in imidazolium ILs.31–33 Other protocols also exist for similar conversions, using metal or metal oxide catalysts in different IL media, but to the best of our knowledge, tetraalkylphosphonium ILs have not been used for metal NP-catalysed aromatic hydrogenations, despite their NP-stabilizing abilities and chemical inertness to a variety of reagents.34,35
In this work, we show the synthesis and stabilization of catalytically active precious metal NPs in a variety of tetraalkylphosphonium ILs. Initial studies concerning the correlation between structural aspects of the ILs (such as polarizability of anions, lengths of alkyl substituents, etc.) and their NP-stabilizing abilities show that trihexyl(tetradecyl)phosphonium chloride (P[6,6,6,14]Cl and bis(triflimide)) (P[6,6,6,14]NTf2) were promising candidates for catalytic NP/IL nanocomposite fabrication when compared to ‘short-chain’ (P[4,4,4,4]Cl), ‘medium-chain’ (P[8,8,8,8]Br), and poorly-coordinating-anion-containing (P[6,6,6,14]N(CN)2) ILs. Ru, Rh, Pt, and RhPt NPs in P[6,6,6,14]Cl were active catalysts for simple hydrogenations of allylic alcohols. However, the Ru NP/P[6,6,6,14]Cl IL composite proved to be the most effective system for deep hydrogenations of aromatic compounds such as toluene and phenol (a lignin-analogue) at elevated temperatures and high hydrogen pressures. Minimal NP growth was seen to occur even at higher temperatures under hydrogen atmospheres in P[6,6,6,14]Cl or P[6,6,6,14]NTf2 ILs compared to other tetraalkylphosphonium ILs. Interestingly, significant conversion of phenol to the HDO products was seen even in the absence of an added Lewis acid. Further investigations showed the presence of residual borates in the reaction medium (by-products from the initial BH4− reduction) facilitated the conversion of phenol to C-6 hydrocarbons.36,37 This study shows that metal NP/IL composites hold promise for the potential conversion of lignin to C6-alkanes.
Results and discussion
UV-Vis spectroscopic study of Au NP/IL nanocomposites
Au NPs were synthesized by reducing HAuCl4 by LiBH4 in a variety of ILs. Following the synthesis, the stability of the Au NPs over time and after heating under nitrogen at 150 °C for an hour was monitored by UV-Vis spectroscopy, as shown in Fig. 1. Other than the Au NP/P[4,4,4,4]Cl IL system, all systems show an initial Au plasmon band in the 520–600 nm range. It is evident that the Au NPs show maximum resistance to coalescence in P[6,6,6,14]Cl and P[4,4,4,14]Cl, given the absence of any tremendous shifts in the plasmon band. Au NPs were unstable to aggregation over 3 days in P[4,4,4,4]Cl, and P[4,4,4,8]Cl ILs; this is seen by the shift and dampening of the plasmon band to much higher wavelengths, which is typically due to dipole–dipole interactions between aggregated particles.38 This observation is in line with previous literature: namely, longer alkyl chains in a tetraalkylphosphonium IL bestow upon it greater NP stabilizing abilities and the presence of one alkyl chain longer than the other three (and the resultant asymmetry in the molecular structure of the IL) is crucial for the stability of the NPs in the ILs.4,18–21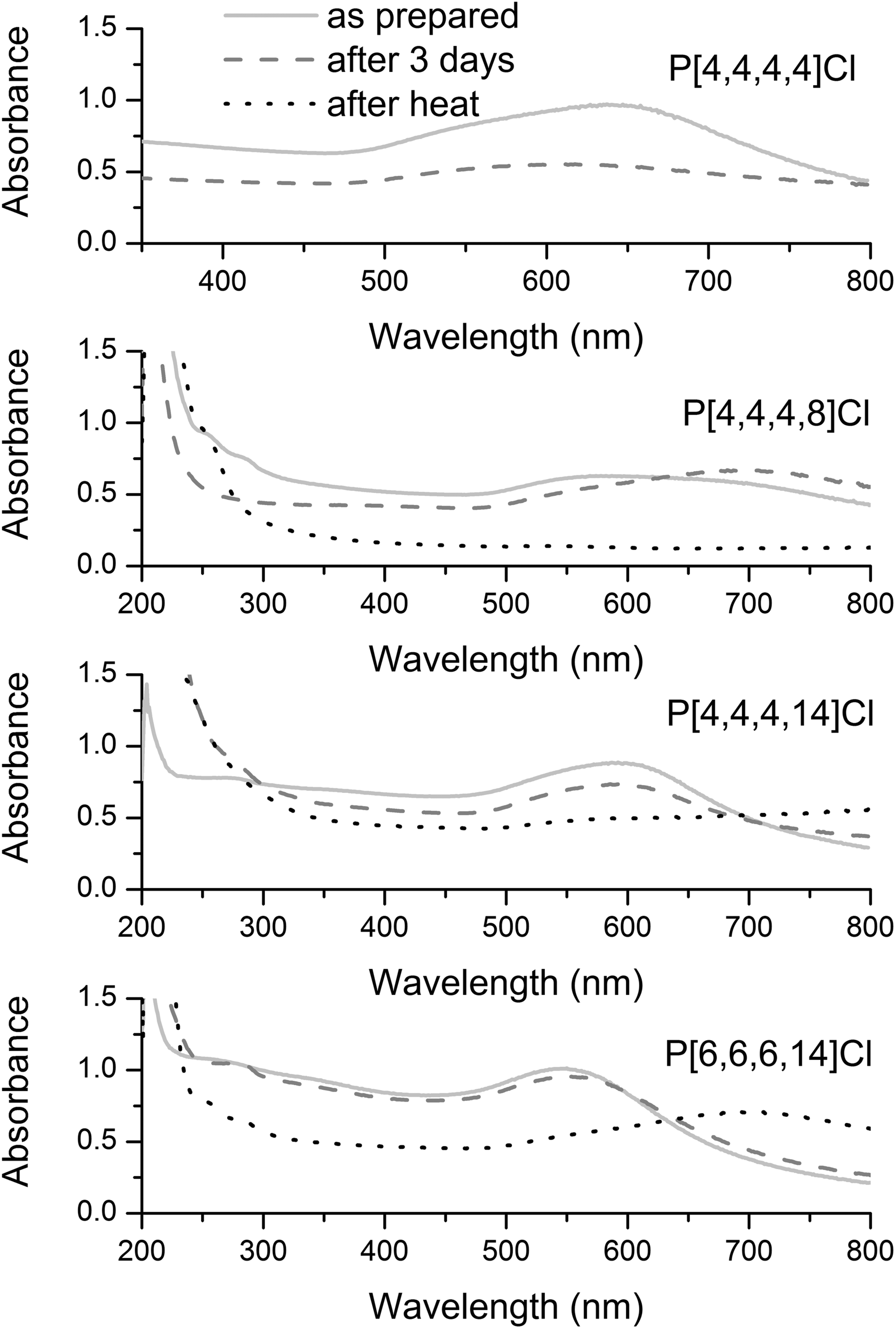 | ||
| Fig. 1 UV-Visible spectra of Au NPs in various representative ILs recorded immediately after synthesis (light grey solid line), after three days (deep grey dashed line), and after heating under N2 at 150 °C for 1 h (black dotted line). | ||
The presence of moderately to strongly coordinating anions (such as halides and bis(triflimide)s) are necessary for the IL to be a good NP stabilizer.20 It is evident, therefore, that P[6,6,6,14]Cl (and to a smaller extent, P[4,4,4,14]Cl) satisfy the criteria for being efficient NP-stabilizers, and could potentially be used in catalytic nanocomposites for hydrogenations. TEM images (Fig. S1, ESI†) support these conclusions: in P[4,4,4,14]Cl before heat treatment, the average particle size is 4.5± 0.6 nm; after heat treatment, there is an average particle size growth of ∼7 nm, with an average final particle size of 11.3 ± 7.1 nm. Similarly, in P[6,6,6,14]Cl, before heat-treatment, the average particle size is 4.1 ± 0.6 nm; after heat-treatment, a bimodal distribution of particle sizes is observed, with mean particle diameters of 3.5 ± 0.6 nm (which corresponds to the as-synthesized particle sizes) and 12.2 ± 3.5 nm (which corresponds to the sintered NP sizes). Although even the longer-chained, asymmetric ILs failed to protect Au NPs completely from heat-induced sintering (as indicated by a blue shift of the Au NP plasmon bands after heating), it was noted that no visible NP precipitation occurred in any of these systems.
Pt NPs in ILs after hydrogen treatment
It is known that presence of hydrogen at elevated temperatures can lead to the formation of surface metal hydrides in Pt NPs. This could potentially destabilize the NPs, leading to their agglomeration and precipitation as other stabilizing species get displaced. Thus it was essential for us to study the a system under hydrogenation reaction conditions in the absence of substrates.39,40 Pt NPs were synthesized in the ILs and exposed to 1 atm hydrogen at 150 °C for 12 h to study their stability. TEM images of Pt NPs in P[4,4,4,8]Cl and P[6,6,6,14]Cl before and after hydrogen treatment are shown in Fig. 2. | ||
| Fig. 2 TEM images of Pt NPs in P[4,4,4,8]Cl (before heating: A, after heating: B), P[6,6,6,14]Cl (before heating: C, after heating: D), and P[6,6,6,14]NTf2 (before heating: E, after heating: F). Average sizes of NPs are as follows: (A): 4.7 ± 1.2 nm, (B): 12.2 ± 6.1 nm, (C): 2.1 ± 0.4 nm, (D): 3.8 ± 0.9 nm, (E): 5.2 ± 1.2 nm, and (F): 9.2 ± 2.1 nm. Heating was performed under 1 atm hydrogen at 150 °C for 12 h. | ||
The Pt NPs grew in size from 4.7 ± 1.2 nm to 12.2 ± 6.1 nm upon heating under hydrogen in P[4,4,4,8]Cl. On the other hand, in P[6,6,6,14]Cl, both the initial size of the Pt NPs and the increase in Pt NP size was much smaller (<2 nm increase). The only promising non-halide IL tested, P[6,6,6,14]NTf2, showed a moderate increase in particle size upon exposure to hydrogen under heat (from 5.2 ± 1.2 nm to 9.2 ± 2.1 nm). Other non-halide ILs such as P[6,6,6,14]N(CN)2 showed spontaneous NP aggregation and precipitation at much lower temperatures (ca. 80 °C). On the basis of these observations, we selected P[6,6,6,14]Cl (as a representative halide IL) and P[6,6,6,14]NTf2 (as a representative non-halide IL) for subsequent experiments.
Hydrogenation of 2-methylbute-3-en-2-ol by NP/IL composites
The hydrogenation of 2-methylbute-3-en-2-ol to 2-methylbutan-2-ol was selected as an initial test reaction, using Pt, Ru, Rh, and Pt/Rh bimetallic NPs. At 30 bar hydrogen pressure and at moderately elevated temperatures (70 °C), high conversions were the norm with almost all the NPs under investigation, with conversions ranging from 88–97% (Table 1). Both P[6,6,6,14]Cl and P[6,6,6,14]NTf2 proved to be capable of metal NP stabilization under reaction conditions. Table 1 summarizes the results.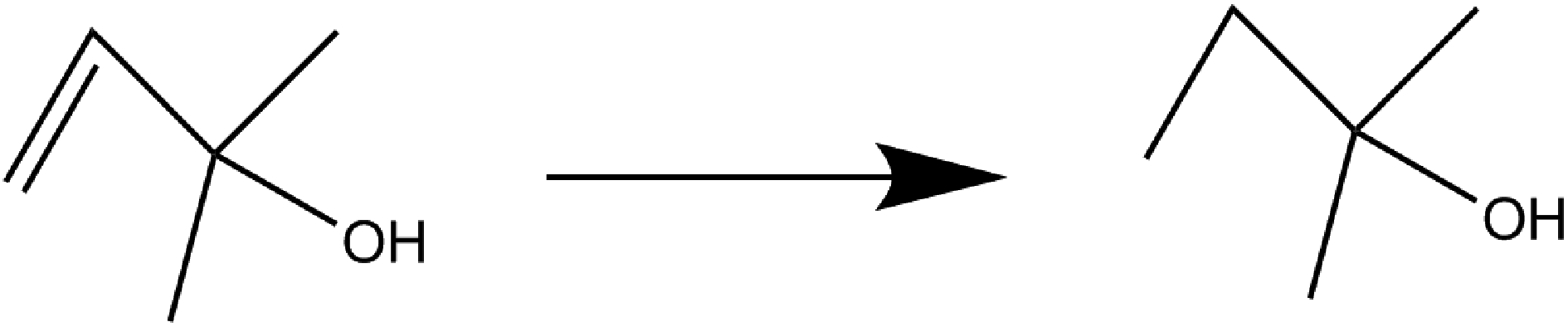
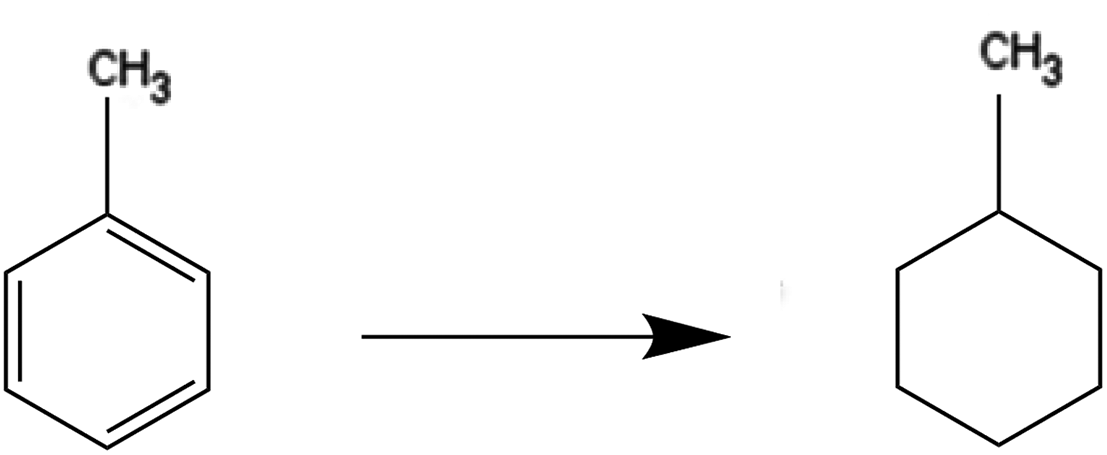
Deep hydrogenation of toluene by NP/IL composites
The deep hydrogenation of toluene to methylcyclohexane requires a high pressure of hydrogen and an efficient catalyst to go to completion, owing to the aromatic stability of the benzene ring.41 It is, therefore, a suitable trial reaction for testing the efficacy of metal NP/IL nanocomposite hydrogenation catalysts. The results obtained during the course of these trials have been summarized in Table 2, for one halide-containing and one halide-free IL. It can be seen that Pt NPs and Pt–Rh NPs were unsuccessful in catalysing this reaction. When Rh2(OAc)6 was used as a precursor for the Rh NPs, poor conversions were obtained; use of Ru NPs led to higher yields and TONs of ca. 70. In all cases, methylcyclohexane was the only product; no partially hydrogenated products could be detected. This is in agreement with previous work by Dupont and co-workers, who used Ru NPs in ILs such as 1-butyl-3-methylimidazolium and 1-decyl-3-methylimidazolium N-bistriflimides (–NTf2) and tetrafluoroborates to give TONs of ca. 170 after 18 hours of reaction.42 Similarly, a Ru-cluster catalyst, used by Dyson and co-workers for hydrogenation of toluene in 1-butyl-3-methylimidazolium tetrafluoroborate, gave an average TON of 240.43 The presence of halides, which are known to inhibit catalytic activities of nanoparticles via active-site blocking, could be responsible for reduced yields. For example, Ir nanoclusters have been shown by others to be poisoned by Cl− for benzene hydrogenation, but remained active for hydrogenation of simple olefinic moieties.44 However, this would not explain why Ru NPs in P[6,6,6,14]NTf2 (a non-halide IL) show similar catalytic activities as the P[6,6,6,14]Cl IL in this study. We are not certain why the deep hydrogenation activity of NPs in tetraalkylphosphonium ILs is lower than for other classes of ILs; possibilities for reduced activity include growth or aggregation of the NP catalysts, polydispersity of catalytic sites in this system and/or presence of different catalytic sites on NP surfaces.45–47 Moderate NP growth for the Ru NP system after catalysis is documented in the next section.|
|
|||
|---|---|---|---|
| Entry | IL | NP system | Conversionc |
a Conditions: substrate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :catalyst = 188; 30 bar Hydrogen; 120 °C; 24 h.
b Reaction performed in a Parr reactor equipped with a mechanical stirrer.
c % conversion derived from GC-FID analysis (see Experimental). :catalyst = 188; 30 bar Hydrogen; 120 °C; 24 h.
b Reaction performed in a Parr reactor equipped with a mechanical stirrer.
c % conversion derived from GC-FID analysis (see Experimental).
|
|||
| 1 | P[6,6,6,14]NTf2 | Ru | 22% |
| 2 | P[6,6,6,14]NTf2 | Pt | ∼2% |
| 3 | P[6,6,6,14]NTf2 | Rh | 5% |
| 4 | P[6,6,6,14]Cl | Ru | 34% |
| 5 | P[6,6,6,14]Cl | Pt | ∼2% |
| 6 | P[6,6,6,14]Cl | Pt/Rh | 4% |
| 7b | P[6,6,6,14]Cl | Ru | 30% |
HDO of phenol by NP/IL composites
We selected 10 mM Ru NP/P[6,6,6,14]Cl as our composite catalyst of choice for the deep hydrogenation of phenol under elevated hydrogen pressures. UV-Vis spectroscopy suggests full reduction of the RuCl3 precursor to Ru NPs (Fig. S8, ESI†); and TEM images (Fig. S9, ESI†) indicated the average particle size was 2.9 ± 1.2 nm. The hydrogenation of phenol and substituted phenols have been carried out over group VIII metals by others; hydrogenation of phenol produces two important compounds, cyclohexanone and cyclohexanol, both of which are utilized in the manufacture of a large number of industrial products.48–53 A tentative pathway for phenol HDO has been depicted in Fig. 3.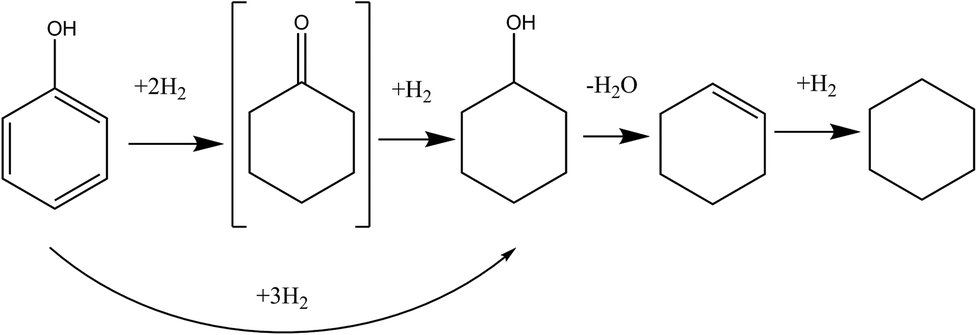 | ||
| Fig. 3 Reaction pathway for phenol HDO by Ru NP in IL. | ||
Phenol was found to be highly soluble in the NP/IL composite solution, and showed moderate conversions under specified reaction conditions. Conversions, product distributions, etc. of various trials have been summarized in Table 3; 1H and 13C NMR spectra can be found in the ESI.† Higher temperatures and greater H2 pressures led to a greater degree of conversion of phenol. Cyclohexanone was not present in any of our product isolates, indicating that it is rapidly converted to cyclohexanol; this is similar to results obtained by some researchers, while others have identified it as a key intermediate, possibly indicating that the nature of the catalyst determines the relative rates of the various steps.54–56 It was surprising, however, that instead of cyclohexanol, the expected hydrogenation product of phenol, a mixture of cyclohexanol, cyclohexene and cyclohexane was isolated. In the presence of a Lewis acid that had been added deliberately to the reaction mixture to catalyse the dehydration of the alcohol, this would be in accordance with previous reports: in fact, steps leading to each of these products has been summarised in Fig. 3.57–59 However, our system did not contain any intentionally-added Lewis acids. In the following section, we identify the species responsible for this serendipitous dehydration.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | T (°C) | PH2 (bar) | Conversionc | A | B | C |
|
a Conditions: substrate:catalyst = 230; 24 h; all reactions performed in a Parr reactor equipped with a mechanical stirrer.
b Recyclability test: RuNP/IL composite recovered after removal of reactants and products via vacuum stripping used for a second catalytic cycle.
c % conversion and selectivities of A, B, and C derived from GC-FID analysis (see Experimental).
|
||||||
| 1 | 120 | 25 | 23% | 27% | 52% | 21% |
| 2 | 120 | 25 | 28% | 38% | 48% | 14% |
| 3 | 110 | 22 | 20% | 60% | 3% | 37% |
| 4 | 135 | 25 | 20% | 62% | 20% | 18% |
| 5 | 65 | 20 | 10% | 77% | 11% | 12% |
| 6b | 110 | 25 | 15% | 41% | 44% | 15% |
In a representative experiment, the TON for the phenol hydrogenation/HDO was ∼65 after 24 h at 120 °C under 25 bar hydrogen. This TON is modest compared to those reported in polymeric IL systems, as well as in the Lewis acid-functionalized ILs,31,32,53,59 (TONs of the order of 200–500) but comparable with the TONs reported for other systems such as Pt nanowires in water (TON = 50), Pd/C/lanthanide triflates in dichloromethane and ILs (TON = 10) and Rh NPs in ILs (TON = 100).60–63 The involvement of the hydroxyl group attached to the ring in the reaction mechanism has previously been found to diminish catalytic activity via active-site blocking,64–67 which might account for reduced TONs. Ru has long since been known for its ability to form cyclohexene selectively during processes such as benzene hydrogenation using Ru NPs.67,68 Possible phase-separation of cyclohexene during the reaction is a potential cause for incomplete conversion of cyclohexene to cyclohexane. Heat-induced changes in particle morphology and/or chemical nature under high H2 pressures during the course of the reaction could also account for this. Two back-to-back catalytic cycles were carried out in order to investigate the recyclability of the system (entry 6); it was seen that the conversion dropped slightly upon recycling with cyclohexene becoming the major product. TEM images (see ESI, Fig. S10†) of Ru NPs in P[6,6,6,14]Cl after one catalytic cycle show slight growth of the average NP size to 4.8 ± 2.0 nm. The presence of a very small population of larger NPs can also be seen in these studies, which may be sintered Ru NPs or Ru NPs in IL droplets.
HDO of phenol by MNP/IL composites: role of borates
The generation of cyclohexane and cyclohexene products suggested that the initially generated cyclohexanol underwent dehydration promoted by a Lewis acid entity present in the reaction mixture. A similar observation was made in the past by Kobayashi and coworkers using polymer-stabilized AuPd catalysts in water, who showed that advantageous borate species in the reaction medium were Lewis acid catalysts.69 Similarly, Riisager and co-workers showed that boric acid, in the presence of salts such as NaCl, is an efficient catalyst for the dehydration of fructose to 5-hydroxymethylfurfural.70 Boric acid, being cheap, non-toxic, and less corrosive than traditional dehydrating agents (such as concentrated sulphuric acid), is a more benign option for acid-catalysed dehydrations.71 Others have demonstrated the conversion of chitin into a nitrogen-containing furan derivative under optimized conditions by using boric acid as a dehydrating agent.72 However, it was still essential for us to rule out other species present in the reaction mixture as possible catalysts. Therefore, control experiments were devised to eliminate the IL itself as the Lewis acid responsible for the dehydration. Details of these control experiments have been summarized in Fig. 4. The IL itself showed no dehydration activity (control experiment A in Fig. 4), while Ru NPs generated in the IL by hydrazine reduction also did not convert cyclohexanol to C6 hydrocarbons (control experiment B). Ru NPs prepared by borohydride reduction converted cyclohexanol to cyclohexane and cyclohexene (control experiment C). Addition of sodium tetraborate (100 mM) to the IL generated ca. 15% C6 hydrocarbons from cyclohexanol. Higher conversions (ca. 35%) for the dehydration of cyclohexanol were seen when lithium borohydride was added to a neat IL sample, followed by exposure to air and quenching, thereby generating a soluble borate species (control experiment D). An additional control experiment, in which the NP/IL composite was used in the absence of H2 to estimate the effect of unreacted borohydride on the product distribution pattern, led to less than 1% conversion, with traces of the alkene as the only identifiable product. Further confirmation of the presence of borate species was obtained by 11B NMR spectroscopy (see ESI†), which indicated the presence of unreacted borohydride in a freshly prepared unquenched Ru NP/IL composite. Upon being quenched with acidified methanol or by prolonged exposure to air, the borohydride peak vanished, and a new peak appeared, which could be assigned to a borate species.73,74 Thus, one of the advantages of synthesizing metal NPs in ILs via borohydride reduction is the generation of Lewis acidic borate by-products in the reaction mixture that can catalyse subsequent reaction steps in a tandem fashion.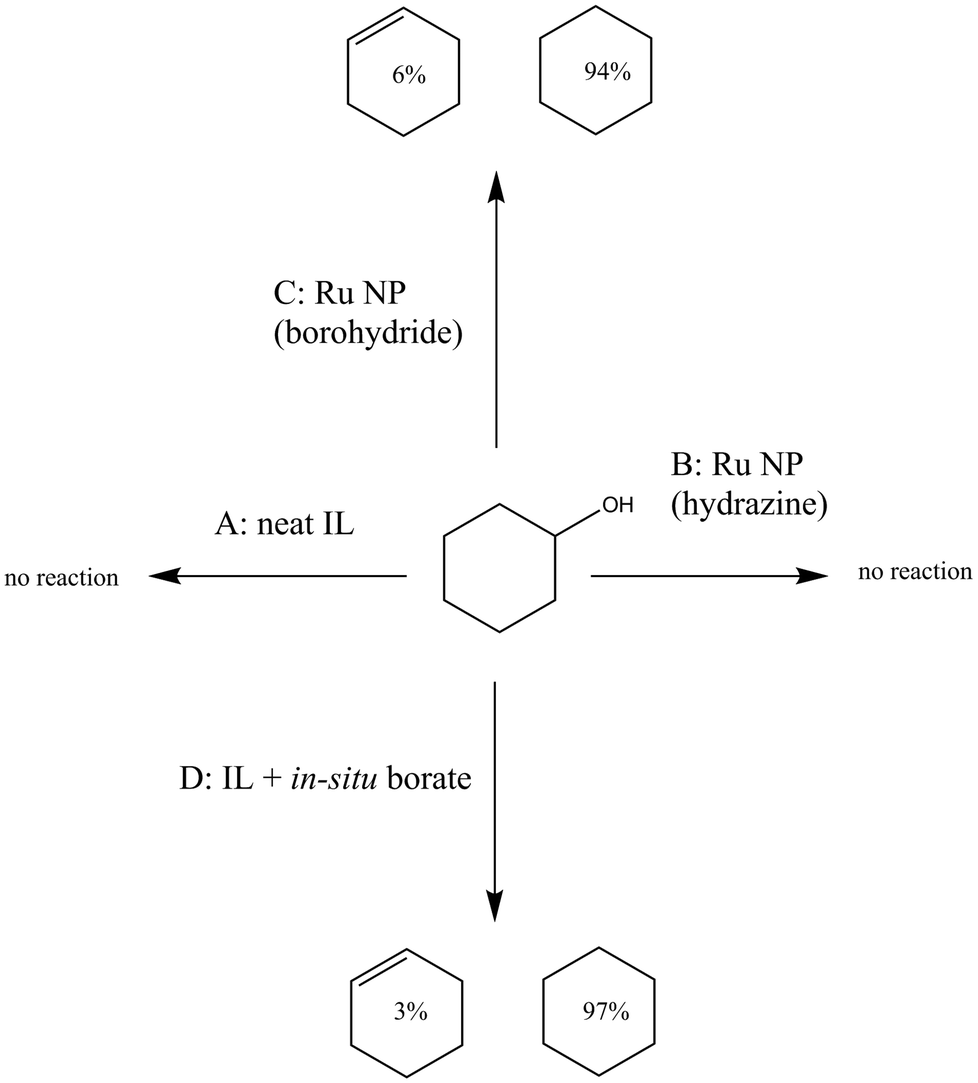 | ||
| Fig. 4 Summary of control experiments performed to evaluate the role of borohydride side products present within the composite catalyst matrix in phenol HDO product distribution. Reaction conditions common to all reactions were as follows: 24 bar Hydrogen pressure; IL P[6,6,6,14]Cl; 120 °C, 24 hours. (A) in neat P[6,6,6,14]Cl, (B) in P[6,6,6,14]Cl containing 10 mM Ru NPs synthesized via hydrazine reduction; (C) in P[6,6,6,14]Cl containing 10 mM Ru NPs synthesized via borohydride reduction; (D) in P[6,6,6,14]Cl containing ca. 150 mM soluble borate generated from lithium borohydride. | ||
Conclusions
A new composite system, transition metal NPs dispersed in tetraalkylphosphonium ILs, was investigated for hydrogenation and phenol HDO catalysis at elevated temperatures and pressures. The following conclusions were drawn from the study:1. Tetraalkylphosphonium ILs bearing three medium-to-long alkyl chains (such as hexyl) along with one long alkyl chain (such as tetradecyl) in the cation, along with a coordinating anion (such as halides, or to a smaller extent, bis-triflimides) are able to offer maximum stabilization for metal NPs.
2. Ru NPs in P[6,6,6,14]Cl and P[6,6,6,14]NTf2 can catalyse the hydrogenation of aliphatic as well as aromatic molecules at high temperatures under high hydrogen pressures; yields and TONs are higher for the aliphatic substrate.
3. Ru NPs in P[6,6,6,14]Cl can catalyse the HDO of phenol at 110–125 °C under 20–25 bar hydrogen pressures, with moderate yields.
4. The product distribution pattern for the nanocomposite-catalysed phenol HDO indicates that the residual borates present within the catalyst as a by-product of the borohydride reduction step actually serve as a Lewis acid catalyst leading to dehydration of cyclohexanol, thus generating C6 hydrocarbons.
It is expected that further research into metal NP/IL nanocomposites, with or without co-catalytic additives, would offer greater insight into the chemistry of these promising catalytic materials.
Experimental section
General information
All chemicals except for the ones listed below were purchased from Sigma Aldrich and used as received. Tetrachloroauric acid, HAuCl4·4H2O and potassium tetrachloropalladate, K2PdCl4, (both 99.9%, metals basis) were all obtained from Alfa Aesar. Other metal precursors, such as rhodium acetylacetonate ([CH3COCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(O–)CH3]3Rh), ruthenium chloride (RuCl3·xH2O), sodium hexachlororhodate (Na3RhCl6), platinum acetylacetonate ([CH3COCHC(O–)CH3]2Pt), and rhodium acetate ([Rh(CH3COO)2]2·2H2O) were purchased from Sigma Aldrich and used without purification. All metal salts were stored under vacuum and flushed with nitrogen after every use. Commercial samples of all the tetraalkylphosphonium ILs mentioned in this work were generously supplied by Cytec Industries Ltd. Commercial samples of liquid ILs were dried under vacuum at 70 °C for 5–6 hours with stirring before use. P[4,4,4,14]Cl, which was a solid at room-temperature, was melted via heating, and used without preceding purification steps, while P[4,4,4,4]Cl, which was obtained as a solution in toluene, was heated under vacuum for 6 hours at 70 °C for solvent removal. Deuterated solvents were purchased from Cambridge Isotope Laboratories. 18 MΩ cm Milli-Q water (Millipore, Bedford, MA) was used throughout. UV-Vis spectra were obtained using a Varian Cary 50 Bio UV-Vis spectrophotometer with a scan range of 200–800 nm and an optical path length of 1.0 cm. 1H and 13C NMR spectra were obtained using a Bruker 500 MHz Avance NMR spectrometer; chemical shifts were referenced to the residual protons of the deuterated solvent. TEM analyses of the NPs in IL were conducted using a Philips 410 microscope operating at 100 kV, with magnifications ranging from 55000 to 145000× (see scale bars in individual TEM images). The samples in IL were prepared by ultrasonication of a 5% solution of the NP/IL solution in THF followed by drop-wise addition onto a carbon-coated copper TEM grid (Electron Microscopy Sciences, Hatfield, PA). To determine average particle diameters, a minimum of 100 particles from each sample were measured from several TEM images using the ImageJ program. Conversion and selectivity for the catalytic reactions were obtained by gas-chromatography (GC) using a flame ionization detector (FID, Agilent Technologies 7890A) and a HP-Innowax capillary column.
C(O–)CH3]3Rh), ruthenium chloride (RuCl3·xH2O), sodium hexachlororhodate (Na3RhCl6), platinum acetylacetonate ([CH3COCHC(O–)CH3]2Pt), and rhodium acetate ([Rh(CH3COO)2]2·2H2O) were purchased from Sigma Aldrich and used without purification. All metal salts were stored under vacuum and flushed with nitrogen after every use. Commercial samples of all the tetraalkylphosphonium ILs mentioned in this work were generously supplied by Cytec Industries Ltd. Commercial samples of liquid ILs were dried under vacuum at 70 °C for 5–6 hours with stirring before use. P[4,4,4,14]Cl, which was a solid at room-temperature, was melted via heating, and used without preceding purification steps, while P[4,4,4,4]Cl, which was obtained as a solution in toluene, was heated under vacuum for 6 hours at 70 °C for solvent removal. Deuterated solvents were purchased from Cambridge Isotope Laboratories. 18 MΩ cm Milli-Q water (Millipore, Bedford, MA) was used throughout. UV-Vis spectra were obtained using a Varian Cary 50 Bio UV-Vis spectrophotometer with a scan range of 200–800 nm and an optical path length of 1.0 cm. 1H and 13C NMR spectra were obtained using a Bruker 500 MHz Avance NMR spectrometer; chemical shifts were referenced to the residual protons of the deuterated solvent. TEM analyses of the NPs in IL were conducted using a Philips 410 microscope operating at 100 kV, with magnifications ranging from 55000 to 145000× (see scale bars in individual TEM images). The samples in IL were prepared by ultrasonication of a 5% solution of the NP/IL solution in THF followed by drop-wise addition onto a carbon-coated copper TEM grid (Electron Microscopy Sciences, Hatfield, PA). To determine average particle diameters, a minimum of 100 particles from each sample were measured from several TEM images using the ImageJ program. Conversion and selectivity for the catalytic reactions were obtained by gas-chromatography (GC) using a flame ionization detector (FID, Agilent Technologies 7890A) and a HP-Innowax capillary column.
Synthesis of NPs in ILs
In a representative synthesis of 5 mM Au NPs in IL, 20 mg of HAuCl4 was added to a 10 mL sample of the trinbutyl(noctyl)phosphonium chloride (P[4,4,4,8]Cl) IL at 60 °C, and vigorously stirred. To this solution, a stoichiometric excess of LiBH4 reagent (1.0 mL, 2.0 M in THF) was injected drop-wise over a period of five minutes. A brisk effervescence followed, and the entire solution turned wine-red, indicating NP formation. After the addition of LiBH4, volatile impurities were removed by vacuum-stripping the system at 70 °C. The Au NP solution thus obtained was stored under nitrogen in a capped vial until use. For the synthesis of Pt and Ru NPs, platinum acetylacetonate and ruthenium chloride, respectively, were used as precursors. Rh NPs, on the other hand, were difficult to synthesize, possibly owing to the limited solubility of most of our Rh precursors of choice. Rhodium acetate had a very high degree of solubility in our ILs, and subsequently, we used that as our precursor of choice. For the synthesis of co-reduced Pt–Rh bimetallic NPs, both precursors were simultaneously dissolved in the IL, and reduced at the same time. Generally, after NP synthesis, excess LiBH4 was quenched with acetone, and volatiles were subsequently removed by vacuum-stripping at 70 °C.Stability tests for NP/IL composites
As mentioned previously, the NPs were subjected to conditions similar to those they would experience during the course of reactions in order to evaluate their stability under such conditions. Briefly, Au NPs formed in short-alkyl-chained, medium-alkyl-chained, and long-alkyl-chained ILs were examined by UV-Vis spectroscopy (after dilution with MeOH to ∼0.1 mM) directly after synthesis, after 3 days of storage, and after being heated to ∼100 °C under nitrogen for a period of 15 minutes to a half hour. Rh and Pt NPs, similarly, were heated under hydrogen at 150 °C for 12 hours, and their TEM images recorded before and after the heat treatment. The results from these experiments assisted us in selecting an optimal metal NP/IL catalytic composite for high pressure hydrogenations.General procedure for hydrogenation reactions
Hydrogenation of organic substrates were carried out in hermetically sealed stainless-steel Parr high-pressure reactors. For the hydrogenations of 2-methylbute-3-en-2-ol and toluene (except for one experiment; see Table 2), a Parr 4790 high-pressure static reactor (without stirring facilities) was used. For phenol HDO, a dynamic Parr reactor equipped with a temperature control system, a mechanical stirrer, and a pressure meter (Parr 4560) was selected. In a general procedure, 10 mL of the NP solution in IL was mixed with the precursor, transferred to the reaction chamber inside the reactor, flushed with hydrogen at moderate pressures to ensure removal of dissolved oxygen, and heated to the requisite temperature under a high pressure of hydrogen inside the sealed reactor (typically, 30 bar). After reaction, the mixture was allowed to cool to ambient temperatures, the hydrogen pressure was released, and the reaction mixture was transferred to a Schlenk flask and vacuum stripped to remove any products formed and/or unreacted starting materials. The products extracted were subsequently characterized by 1H NMR, 13C NMR, and GC-FID. Conversion and selectivity were obtained from GC-FID. For GC-FID analysis, 100 μL of a neat extract was mixed with enough ethyl acetate to make a final volume of 10 mL. 1 mL of this solution was then taken in a GC-vial, and subjected to analysis. The percentage conversion and product selectivities were calculated from two identical measurements, using the GC-FID peak areas and calibration curves for each species.Acknowledgements
The authors acknowledge financial assistance from the National Sciences and Engineering Research Council of Canada (NSERC) in the form of an NSERC Engage grant. In addition, we thank Al Robertson and Jeffrey Dyck of Cytec Industries, Inc. for providing samples of the ILs surveyed as well as providing valuable input into the project design.Notes and references
- M. Ali, A. Gual, G. Ebeling and J. Dupont, ChemCatChem, 2014, 6, 2224 CAS.
- J. Dupont and M. R. Meneghetti, Curr. Opin. Colloid Interface Sci., 2013, 18, 54 CrossRef CAS PubMed.
- G. S. Fonseca, A. P. Umpierre, P. F. Fichtner, S. R. Teixeira and J. Dupont, Chem. – Eur. J., 2003, 9, 3263 CrossRef CAS PubMed.
- A. Banerjee, R. Theron and R. W. J. Scott, Chem. Commun., 2013, 49, 3227 RSC.
- P. Migowski and J. Dupont, Chem. – Eur. J., 2007, 13, 32 CrossRef CAS PubMed.
- P. Dash, N. A. Dehm and R. W. J. Scott, J. Mol. Catal. A: Chem., 2008, 286, 114 CrossRef CAS PubMed.
- P. Dash and R. W. J. Scott, Chem. Commun., 2009, 812 RSC.
- M. T. Kessler, M. K. Hentschel, C. Heinrichs, S. Roitsch and M. H. Prechtl, RSC Adv., 2014, 4, 14149 RSC.
- L. Foppa, J. Dupont and C. W. Scheeren, RSC Adv., 2014, 4, 16583 RSC.
- L. Luza, A. Gual, C. P. Rambor, D. Eberhardt, S. R. Teixeira, F. Bernardi, D. L. Baptista and J. Dupont, Phys. Chem. Chem. Phys., 2014, 16, 18088 RSC.
- L. S. Ott, M. L. Cline, M. Deetlefs, K. R. Seddon and R. G. Finke, J. Am. Chem. Soc., 2005, 127, 5758 CrossRef CAS PubMed.
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361 RSC.
- L. L. Lazarus, C. T. Riche, N. Malmstadt and R. L. Brutchey, Langmuir, 2012, 28, 15987 CrossRef CAS PubMed.
- M. Atilhan, J. Jacquemin, D. Rooney, M. Khraisheh and S. Aparicio, Ind. Eng. Chem. Res., 2013, 52, 16774 CrossRef CAS.
- R. E. Del Sesto, C. Corley, A. Robertson and J. S. Wilkes, J. Organomet. Chem., 2005, 690, 2536 CrossRef CAS PubMed.
- K. J. Fraser and D. R. MacFarlane, Aust. J. Chem., 2009, 62, 309 CrossRef CAS.
- C. J. Bradaric, A. Downard, C. Kennedy, A. J. Robertson and Y. Zhou, Green Chem., 2003, 5, 143 RSC.
- A. Maclennan, A. Banerjee and R. W. Scott, Catal. Today, 2013, 207, 170 CrossRef CAS PubMed.
- K. L. Luska and A. Moores, Green Chem., 2012, 14, 1736 RSC.
- A. Banerjee, R. Theron and R. W. J. Scott, ChemSusChem, 2012, 5, 109 CrossRef CAS PubMed.
- H. A. Kalviri and F. M. Kerton, Green Chem., 2011, 13, 681 RSC.
- V. Ermolaev, D. Arkhipova, L. S. Nigmatullina, I. K. Rizvanov, V. Milyukov and O. Sinyashin, Russ. Chem. Bull., 2013, 62, 657 CrossRef CAS.
- S.-C. Qi, L. Zhang, X.-Y. Wei, J.-i. Hayashi, Z.-M. Zong and L.-L. Guo, RSC Adv., 2014, 4, 17105 RSC.
- S. P. Srivastava and J. Hancsók, Fuels and Fuel-Additives, John Wiley and Sons, Hoboken, New Jersey, 2014 Search PubMed.
- T. Q. Hu, C.-L. Lee, B. R. James and S. J. Rettig, Can. J. Chem., 1997, 75, 1234 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147 CrossRef CAS.
- S. W. Eachus and C. W. Dence, Holzforschung, 1975, 29, 41 CrossRef CAS.
- J. Zhong, J. Chen and L. Chen, Catal. Sci. Technol., 2014, 4, 3555 CAS.
- X.-D. Mu, J.-Q. Meng, Z.-C. Li and Y. Kou, J. Am. Chem. Soc., 2005, 127, 9694 CrossRef CAS PubMed.
- A. P. Umpierre, G. Machado, G. H. Fecher, J. Morais and J. Dupont, Adv. Synth. Catal., 2005, 347, 1404 CrossRef CAS.
- J. Chen, J. Huang, L. Chen, L. Ma, T. Wang and U. I. Zakai, ChemCatChem, 2013, 5, 1598 CrossRef CAS.
- N. Yan, Y. Yuan, R. Dykeman, Y. Kou and P. J. Dyson, Angew. Chem., Int. Ed., 2010, 49, 5549 CrossRef CAS PubMed.
- F. Schwab, M. Lucas and P. Claus, Angew. Chem., Int. Ed., 2011, 50, 10453 CrossRef CAS PubMed.
- T. Ramnial, D. D. Ino and J. A. C. Clyburne, Chem. Commun., 2005, 325 RSC.
- T. Ramnial, S. A. Taylor, M. L. Bender, B. Gorodetsky, P. T. K. Lee, D. A. Dickie, B. M. McCollum, C. C. Pye, C. J. Walsby and J. A. C. Clyburne, J. Org. Chem., 2008, 73, 801 CrossRef CAS PubMed.
- M. W. Drover, K. W. Omari, J. N. Murphy and F. M. Kerton, RSC Adv., 2012, 2, 4642 RSC.
- E. A. Khokhlova, V. V. Kachala and V. P. Ananikov, ChemSusChem, 2012, 5, 783 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212 CrossRef CAS.
- S. Kishore, J. A. Nelson, J. H. Adair and P. C. Eklund, J. Alloys Compd., 2005, 389, 234 CrossRef CAS PubMed.
- T. Hawa and M. Zachariah, J. Chem. Phys., 2004, 121, 9043 CrossRef CAS PubMed.
- P. A. Rautanen, J. R. Aittamaa and A. O. I. Krause, Ind. Eng. Chem. Res., 2000, 39, 4032 CrossRef CAS.
- M. H. G. Prechtl, M. Scariot, J. D. Scholten, G. Machado, S. R. Teixeira and J. Dupont, Inorg. Chem., 2008, 47, 8995 CrossRef CAS PubMed.
- P. Dyson, D. Ellis, T. Welton and D. Parker, Chem. Commun., 1999, 25 RSC.
- E. Bayram, M. Zahmakıran, S. Özkar and R. G. Finke, Langmuir, 2010, 26, 12455 CrossRef CAS PubMed.
- E. E. Finney and R. G. Finke, Inorg. Chim. Acta, 2006, 359, 2879 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS PubMed.
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310 CrossRef CAS.
- N. Mahata, K. Raghavan, V. Vishwanathan, C. Park and M. Keane, Phys. Chem. Chem. Phys., 2001, 3, 2712 RSC.
- J.-F. Zhu, G.-H. Tao, H.-Y. Liu, L. He, Q.-H. Sun and H.-C. Liu, Green Chem., 2014, 16, 2664 RSC.
- Y. Wang, J. Yao, H. Li, D. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362 CrossRef CAS PubMed.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250 CrossRef CAS PubMed.
- A. Chen, Y. Li, J. Chen, G. Zhao, L. Ma and Y. Yu, ChemPlusChem, 2013, 78, 1370 CrossRef CAS.
- H. Xu, K. Wang, H. Zhang, L. Hao, J. Xu and Z. Liu, Catal. Sci. Technol., 2014, 4, 2658 CAS.
- H. Ohta, H. Kobayashi, K. Hara and A. Fukuoka, Chem. Commun., 2011, 47, 12209 RSC.
- C. Zhao and J. A. Lercher, ChemCatChem, 2012, 4, 64 CrossRef CAS.
- A. L. Jongerius, R. Jastrzebski, P. C. Bruijnincx and B. M. Weckhuysen, J. Catal., 2012, 285, 315 CrossRef CAS PubMed.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L.-T. Liu and Y. Kou, ChemSusChem, 2008, 1, 626 CrossRef CAS PubMed.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Chem. Commun., 2010, 46, 412 RSC.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987 CrossRef CAS PubMed.
- Z. Guo, L. Hu, H.-H. Yu, X. Cao and H. Gu, RSC Adv., 2012, 2, 3477 RSC.
- T. Yu, J. Wang, X. Li, X. Cao and H. Gu, ChemCatChem, 2013, 5, 2852 CrossRef CAS.
- A. L. Maksimov, S. N. Kuklin, Y. S. Kardasheva and E. A. Karakhanov, Pet. Chem., 2013, 53, 157 CrossRef CAS.
- J. Y. Shin, D. J. Jung and S.-G. Lee, ACS Catal., 2013, 3, 525 CrossRef CAS.
- D. K. Lee, S. J. Ahn and D. S. Kim, 9th International Symposium on Catalyst Deactivation, Lexington, KY, USA, 2001 Search PubMed.
- A. Popov, E. Kondratieva, L. Mariey, J. M. Goupil, J. El Fallah, J.-P. Gilson, A. Travert and F. Maugé, J. Catal., 2013, 297, 176 CrossRef CAS PubMed.
- E. Furimsky, Catal. Rev., 1983, 25, 421 CAS.
- M. F. F. Rodrigues and A. J. G. Cobo, Catal. Today, 2010, 149, 321 CrossRef CAS PubMed.
- C. Fan, Y.-A. Zhu, X.-G. Zhou and Z.-P. Liu, Catal. Today, 2011, 160, 234 CrossRef CAS PubMed.
- W.-J. Yoo, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 3095 CrossRef CAS PubMed.
- T. Ståhlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Chem. – Eur. J., 2011, 17, 1456 CrossRef PubMed.
- M. Walia, U. Sharma, V. K. Agnihotri and B. Singh, RSC Adv., 2014, 4, 14414 RSC.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204 RSC.
- T. P. Onak, H. Landesman and R. E. Williams, J. Phys. Chem., 1959, 63, 1533 CrossRef CAS.
- C. D. Good and D. M. Ritter, J. Am. Chem. Soc., 1962, 84, 1162 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of phenol HDO products, 11B NMR spectra of borohydride to borate conversion, TEM images of Au NPs in various ILs before and after heating, TEM images and UV-Visible spectra of Ru NPs before and after catalysis. See DOI: 10.1039/c4gc01716a |
| This journal is © The Royal Society of Chemistry 2015 |