
Nanoporous catalysts for biomass conversion
Liang
Wang
and
Feng-Shou
Xiao
*
Key Lab of Applied Chemistry of Zhejiang Province, Institute of Catalysis, Zhejiang University, Hangzhou 310028, China. E-mail: fsxiao@zju.edu.cn
First published on 30th September 2014
Abstract
Biomass conversion is potentially important for producing renewable fuels and chemicals in the future, and the nanoporous materials as acidic, basic, or metallic catalysts play important roles in the biomass conversion. This paper briefly summarizes recent developments on the nanoporous material-based catalysts used for biomass conversion, including mesoporous/macroporous resins, mesoporous metal oxides, microporous/mesoporous carbons, mesoporous silicas, hierarchically mesoporous polydivinylbenzene, and microporous zeolites. Due to their superior thermal and hydrothermal stability, controllable acidity, and relatively easy introduction of mesoporosity into the crystals, compared with other nanoporous catalysts, zeolites have good opportunities for application in biomass conversion.
 Liang Wang | Dr. Liang Wang received his B.S. (2008) and Ph.D. (2013) degrees from the College of Chemistry, Jilin University, China. In July 2013, he joined the Institute of Catalysis in Zhejiang University for postdoctoral work under the guidance of Prof. Feng-Shou Xiao. His current research is mainly focused on heterogeneous catalysis, biomass conversion, and green oxidations. |
 Feng-Shou Xiao | Prof. Feng-Shou Xiao received his B.S. and M.S. degrees from the Department of Chemistry, Jilin University, China. From there he moved to the Catalysis Research Center, Hokkaido University, Japan, where he was involved in collaborative research between China and Japan. He was a Ph.D. student there for two years, and was awarded with a Ph.D. degree at Jilin University in 1990. After postdoctoral work at the University of California at Davis, USA, he joined the faculty at Jilin University in 1994, where he was a full and distinguished professor of Chemistry. He moved to Zhejiang University in 2009, and now he is a full and distinguished professor of Chemistry. For his research in nanoporous materials, Prof. Xiao was recognized with the National Outstanding Award of Young Scientists in the National Science Foundation of China in 1998 and the Thomson Scientific Research Fronts Award in 2008. Currently he is the secretary of the Asia-Pacific Association of Catalysis Societies. |
1. Introduction
Nanoporous materials mainly including micropores (<2 nm), mesopores (2–50 nm), and macropores (>50 nm) have been widely applied in heterogeneous catalysis.1–9 For example, microporous zeolites are excellent catalysts for oil refining, where the acid sites in the framework of zeolite could catalyze the selective formation of desired alkanes because of the micropore size limitation.10–13 However, due to the shortage of fossil resources and environmental concerns related to CO2 emissions,14 much attention has been paid to the catalytic conversion of biomass because there is a huge amount of carbohydrates in biomass (nearly 130 billion metric tons) synthesized with CO2 and H2O using sunlight as the energy source per year, but only about 4% of these compounds are used currently.15 These data confirm the importance of catalytic conversion of biomass as renewable energy.16–23The biomass mainly includes cellulose formed with glucose, hemicellulose formed with polymerization of glucose and xylose, and lignin formed with phenols and derivative phenolic molecules. Besides these sugars and phenol components, the plant can also provide energy storage biomass of triglycerides, lipids, and terpenes.24,25 These compounds can be directly converted into desired fuels and chemicals to replace the existing chemicals related to crude oils. For example, the cellulose can be depolymerized into glucose and cellobiose;18,26–31 the sugars (e.g. glucose, cellulose, and xylose) can be transformed into platform chemicals of 5-hydroxymethylfurfural (HMF), furfural, glycol, and lactic acid;23,32–42 the triglycerides can be transesterificated into biodiesels;43–51 the pyrolysis oil can be hydrodeoxygenated into alkane fuels.52–55
Notably, although these processes start from renewable biomass, much attention should still be focused on waste minimisation and high atom efficiency.56–61 In this field, an important topic is to develop highly efficient catalysts in order to achieve a high yield of the target product under sustainable reaction conditions. Since the end of the 20th century, a large number of catalysts have been confirmed efficiently for transforming the biomass-derived carbohydrate into the desired fuels and chemicals. Especially the past few years have seen an enormous increase in the number of nanoporous catalysts, which usually exhibit exciting catalytic activity, excellent product selectivity, and superior recyclability.15,24,25,62,63 In these cases, the catalyst nanoporosity has obvious advantages for designing efficient heterogeneous catalysts in the following: (1) the nanoporous catalysts generally have high surface areas and large pore volumes, which is very helpful for access of reactants, mass transfer and functionalization of active sites; (2) the catalyst micropore sizes effectively control the product selectivities; and (3) the wettability of the nanoporous catalysts is easily adjusted, which is favourable for adsorption, desorption, and surface diffusion of reactants and products in catalysis.
Although a number of encouraging reviews have already summarized the production of fuels and chemicals from biomass, they are mostly focused on the conversion routes.15,24,64–68 Very differently, the aim of this review is to discuss and evaluate the nanoporous catalysts used in the biomass transformation. Here, the nanoporous materials with acidic, basic, or metallic active sites mainly include functionalized resins, metal oxides, carbons, mesoporous silicas, polydivinylbenzene, and zeolites.
2. Sulfonated resins
The sulfonated resins, such as Amberlyst,69,70 Nafion,71–75 and EBD resins,76 have been widely used in the conversion of biomass because of their commercial synthesis. More importantly, the abundant porosity in the resins is favourable for the functionalization of Brønsted sulfonated groups on the organic framework with high concentration, as well as for the full interaction between substrates and acid sites, leading to high catalytic activity.2.1 Amberlyst resins
Amberlyst-15, synthesized from styrene polymerization and sulfonation, is one of the most famous resins with rich macropores and mesopores (surface area of about 46 m2 g−1), forming a high density of acid sites (4.0 mmol g−1) exposed to the reactants.77 Notably, Amberlyst-15 has been used in almost all acid-catalyzed liquid-phase reactions of biomass, in particular to produce sugars, HMF, and biodiesels. For example, more than 20% yield of sugars could be obtained from the conversion of wood and cellulose over the Amberlyst-15 catalyst in ionic liquid solvents at 100 °C.29,78,79 In the conversion of fructose to HMF, Amberlyst-15 gave HMF yield at nearly 100% in a water-separation reactor.80 For the biodiesel production from esterification of dodecanoic acid with 2-ethylhexanol at 150 °C, Amberlyst-15 is highly active, giving a yield higher than 80%.43However, the nanoporous structure of the Amberlyst-15 catalyst has a relatively low stability. For example, in the transesterification of tripalmitin with methanol to produce biodiesel at 65 °C, the activities of Amberlyst-15 decreased with recycles significantly. N2 sorption analysis indicates that the porous structure of Amberlyst-15 is partially destroyed after recycling in the transesterification.43,81
2.2 Nafion resins
Compared with Amberlyst-15, Nafion resins have relatively high thermal stability in the nanoporous structure.82,83 For example, NR-50, a typical Nafion resin of sulfonated and fluorinated polymers, is stable up to the temperature of 280 °C. When NR-50 was used to catalyze an esterification of acrylic acid with methanol to methyl acrylate, it gave the methyl acrylate yield over 90%, which is even comparable with the homogeneous sulfuric acid catalyst.84 Nafion perfluorinated membrane NR-117 gave furfural yield of 60% in the dehydration of xylose in DMSO solvent.Interestingly, the stability of Nafion resins is significantly improved compared with Amberlyst-15. For example, NR-117 exhibits very good recyclability in the conversion of xylose to furfural at 150 °C;85 SAZ-13 are highly active for long reaction time in the esterification of short-chain carboxylic acids (e.g. acetic, propionic, and butyric acids) with methanol. However, in the esterification of long-chain carboxylic acids (e.g. hexanoic and caprylic acids) with methanol, SAZ-13 shows poor recyclability, because of the accumulation of the long-chain acids on the surface of SAZ-13, which could not be removed even by washing with a large amount of solvents.71,86 This phenomenon is strongly related to its relatively low surface area and pore volume. Therefore, the resins with improved surface area and pore volume are effective for enhancing catalyst activities and recyclability in the biomass conversion.87
2.3 EBD resins
EBD resins are copolymers of styrene–divinylbenzene with functionalized sulfonic acid groups. The copolymerization of styrene with divinylbenzene could effectively increase cross-linking degree, leading to high surface area and pore volume, which is helpful for mass transfer during the reaction process. The typical EBD-100 exhibited high catalytic activity in the esterification of vegetable oils. For example, in the esterification of stearic acid as a model molecule, EBD-100 gave higher catalytic activity than Amberlyst-15, giving a stearic acid conversion higher than 90% in a reaction time of 10 h.2.4 Double-shell resin
Because of the abundant porosity and high surface area in the sulfonated resins (e.g. Amberlyst, Nafion, and EBD resins), they have high concentration of the exposed acidic sites. However, it is still challenging to improve their acidic strength that is very sensitive to the catalytic performances.88–95 Recently, Zhang et al.95 synthesized a polystyrene sulphonic acid with a double-shell nanostructure (PS-SO3H@mesosilica DSNs, Fig. 1), which exhibited higher acid strength than the Amberlyst-15 and Nafion NR50 resins. This phenomenon is assigned to the fact that the sulfonic acid groups in the unique nanoporous structure have strong H-bond interactions, which significantly enhance the acid strength. The DSN catalyst exhibited a conversion of 89.7% in the esterification of lauric acid with ethanol at 80 °C for 6 h, which is even higher than that (71.7%) of the homogeneous H2SO4 catalyst. After treating in DMF to break the H-bond interaction, the obtained catalyst lost most of its activity. This result indicates the importance of the H-bond interaction for the catalytic activity. In the recycling test, 5.4% of the laurate yield dropped in the second run. For further recycling test until the eighth run, no further reduction of yield was observed. After the eighth run, the activity dropped severely, because the resin swelled and caused mesopore blockage on the silicas. This observation suggests that the acid strength of sulfonated resin could also be improved by adjusting the nanoporous structures. | ||
| Fig. 1 (a) Scheme for the synthesis of PS-SO3H@mesosilica DSNs. TEM images of (b) polystyrene (PS) template, (c) PS@mesosilicas with yolk–shell nanostructures (YSNs), and (d) PS-SO3H@mesosilica DSNs. The scale bars in images (b), (c), and (d) are 100, 200, and 200 nm, respectively (reproduced with the permission of ref. 95. Copyright 2014 The Nature Publishing Group). | ||
As shown in this section, it is clear that the sulfonated resins are good acidic catalysts for biomass conversion, where the nanoporous structure is very important for the acid concentration and/or strength on the resins. However, we have to note the fact that the sulfonated resin catalysts still have relatively low stability. For example, even for the most stable Nafion resins, it is still difficult to catalyze the reaction at relatively high temperature, which strongly hinders their wide application in biomass conversion.
3. Nanoporous metal oxides
Metal oxide catalysts play important roles in heterogeneous catalysis.96–98 Apart from the catalytic reactions, the techniques used in catalyst characterization are also developed with the growth of metal oxide-based catalysts.99,100 Compared with the resin catalysts, the nanoporous structures of metal oxides have obvious advantages in thermal stability, because the synthesis of nanoporous metal oxides generally requires calcination at high temperature. In the field of biomass conversion, the nanoporous metal oxides with acidic and basic sites have already been successfully used.3.1 Sulfated or protonated metal oxides
The sulfated metal oxides such as ZrO2, SnO2, TiO2, and Nb2O5 are typical solid acids which have already been used in various reactions.101–114 Notably, compared with the sulfonated resin catalysts with only Brønsted acid sites, a unique feature of sulfated metal oxides is that they have both Brønsted and Lewis acid sites. In these cases, the Lewis acidic sites are metal atoms, while the Brønsted acidic sites are protons on the surface hydroxyl groups of sulfated metal oxides, as shown in Fig. 2.101–114 Usually, the hydroxyl groups on the surface of metal oxides have extremely weak Brønsted acidity. After sulfonation, the S–O bonds on the sulphuric groups are strongly bonded to metal atoms, leading to a coordination of the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond with the hydroxyl groups on metal oxides, which causes the enhancement of Brønsted acidic strength. Obviously, the Brønsted acidic strength is sensitive to the metal type. For example, SO42−/SnO2 and SO42−/ZrO2 exhibit stronger acidic strength than SO42−/TiO2, because the lower electronegativity of Sn and Zr metals than Ti metal causes the proton to be realized more easily.101 It is worth pointing out that the nanoporous structure of metal oxides with high surface areas is very positive for improving the Brønsted acid density.
O bond with the hydroxyl groups on metal oxides, which causes the enhancement of Brønsted acidic strength. Obviously, the Brønsted acidic strength is sensitive to the metal type. For example, SO42−/SnO2 and SO42−/ZrO2 exhibit stronger acidic strength than SO42−/TiO2, because the lower electronegativity of Sn and Zr metals than Ti metal causes the proton to be realized more easily.101 It is worth pointing out that the nanoporous structure of metal oxides with high surface areas is very positive for improving the Brønsted acid density.
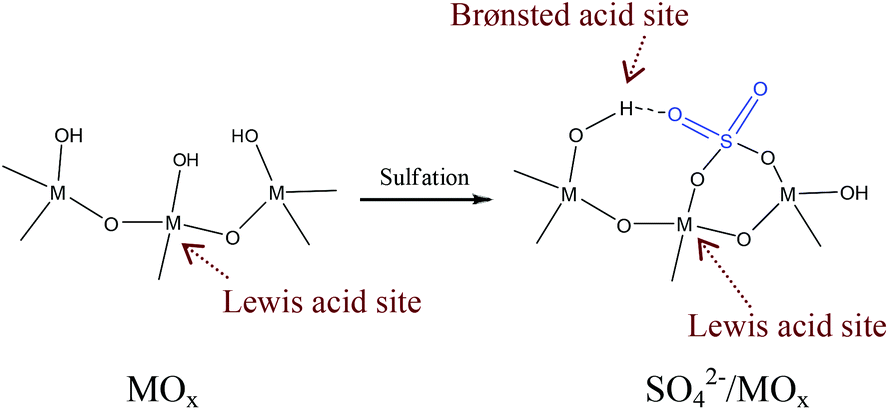 | ||
| Fig. 2 Scheme of the Brønsted and Lewis acid sites on metal oxides and sulfated metal oxides (M = Zr, Sn, Ti, Nb). | ||
SO42−/ZrO2 is a typical sulfated metal oxide used in the conversion of biomass.115–120 Suwannakarn et al.120 reported that SO42−/ZrO2 is catalytically active for transesterification of tricaprylin with methanol, ethanol, and n-butanol at 120 °C. However, they found that the activity loss occurred during the recycling tests because of the leaching of sulfur species from the catalysts. Notably, the conventional synthesis of SO42−/ZrO2 catalysts from ZrO2 is mostly performed by post-sulfonation and calcinations, which might result in a relatively weak interaction between SO42− and ZrO2 species. To solve this problem, Sun et al.121 reported a solvent-free route to synthesize sulfated ZrO2 (S-ZrO2) from a simple mixture of ZrOCl2 and (NH4)2SO4, followed by grinding and calcining, where the interaction between SO42− and ZrO2 could be highly improved. As a result, S-ZrO2 is very active for the depolymerization of cellulose.29
In addition, we also note that the sulfated nanoporous oxides have relatively low surface areas, which strongly limits the access of the acid sites to reactants. In order to solve this problem, Jiménez-López et al.122 reported a supported SO42−/ZrO2 in mesoporous silicas such as MCM-41 (Zr-MCM), which showed the high yield of ethyl esters and good recyclability from ethanolysis of sunflower oil.
Besides the supported metal oxides, introduction of unique nanoporosity into the metal oxides is also an effective route for increasing sample surface areas. A typical example is a TiO2 nanotube with one-dimensional nanopores, which has been widely studied in photocatalysis, sensors, and batteries.123–125 Kitano et al.126 synthesized a TiO2 nanotube with Brønsted acidic sites by hydrothermal treatment of TiO2 in basic aqueous solution, followed by proton exchange. This TiO2 nanotube shows a surface area as high as 400 m2 g−1 and a pore size of about 5 nm. The protonated TiO2 nanotube is highly efficient for catalyzing the conversion of glucose to HMF in water solvent, compared with Nafion NR50, Amberlyst-15 resins, and sulfated ZrO2 catalyst, because of its good mass transfer on the one-dimensional channels of nanotubes (Fig. 3).
 | ||
| Fig. 3 TEM images of protonated TiO2 nanotubes with one-dimensional channels (reproduced with the permission of ref. 126. Copyright 2010 American Chemical Society). | ||
3.2 Hydrotalcite
Hydrotalcites (HT) are a class of layered materials with good surface areas, which have the trivalent and divalent cations contained within the octahedral sites of positively charged hydroxide sheets.127–135 In the conversion of biomass, the HT is not only used as a solid basic catalyst, but also combined with other active centers to catalyze the tandem reactions.136–142The Mg–Al based HTs were highly efficient for the production of biofuel derived chemicals such as aldol condensation of furfural with acetone136 and transesterification of tripalmitin with alcohols.137–139 For example, Hora et al.136 reported that HT gave furfural conversion at nearly 100% in the condensation of furfural with acetone.
Apart from these results, HT was also used to catalyze the tandem reactions. For example, Takagaki et al.140 found that the combination of HT and Amberlyst-15 resin could catalyze the conversion of glucose to HMF, where HT catalyzed the isomerization of glucose to fructose, while the Amberlyst-15 resin catalyzed the dehydration of fructose to HMF. Later, they supported Ru nanoparticles on HT for HMF oxidation.141 In this case, the combined catalysts of Ru/HT and Amberlyst-15, which are ternary catalysts with a base, an acid, and Ru sites, are efficient for one-pot conversion of glucose to 2,5-diformylfuran (Fig. 4).
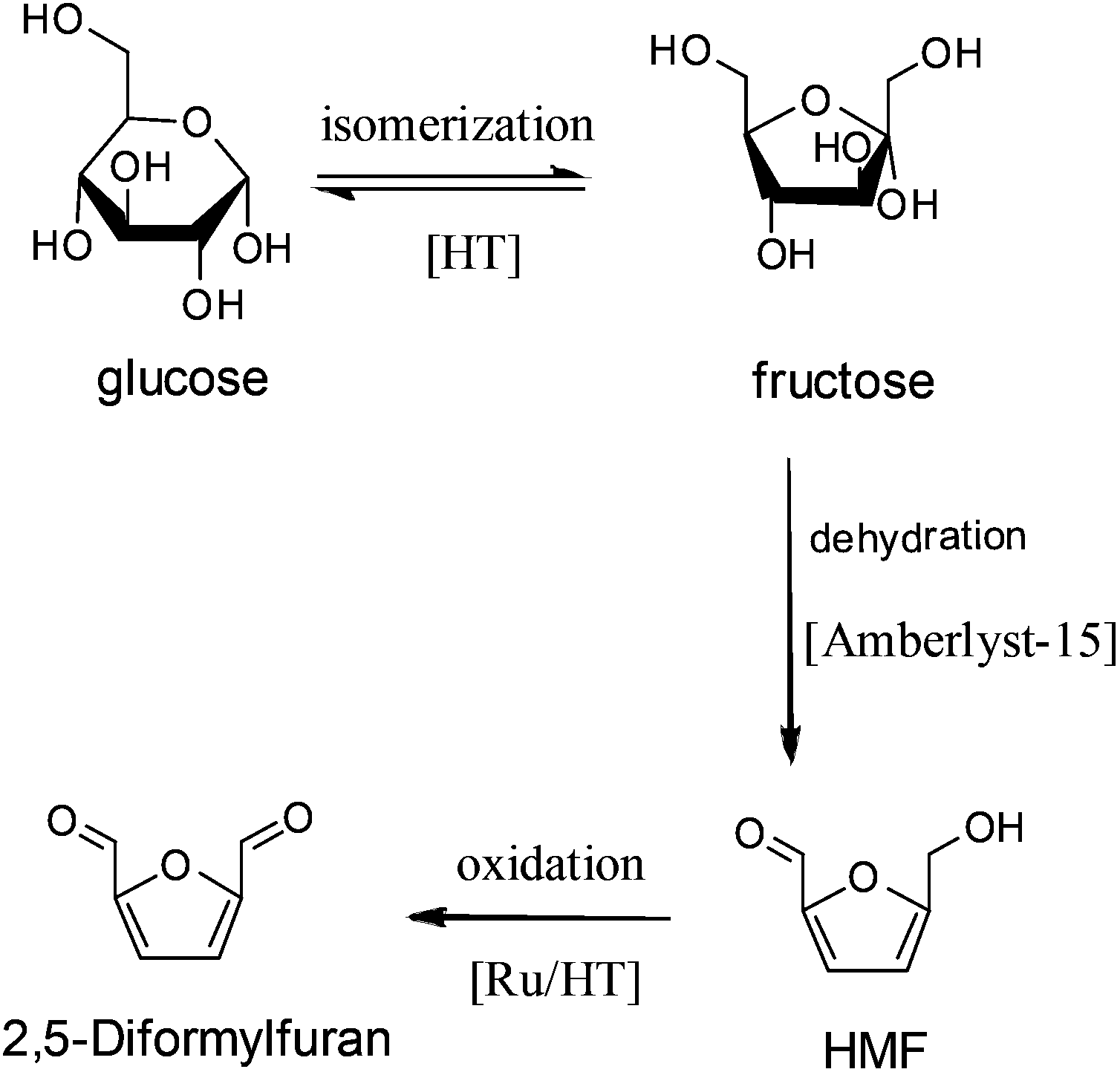 | ||
| Fig. 4 One-pot conversion of glucose to 2,5-diformylfuran over combined catalysts of Ru/HT and Amberlyst-15. | ||
In order to increase the sample stability, Matson et al.142 synthesized a porous metal oxide supported Cu catalyst (Cu20-PMO) by calcining a Cu-doped HT. It was found that the lignocellulose solids, including maple sawdust, pine flour, torrefied wood, and cellulose fibers, could be directly converted into alcohols, ethers, hydrogen, and C1 gases at 300–320 °C in supercritical methanol. It is worth emphasizing that nearly no coke is formed in the catalytic process, and the stable Cu20-PMO catalyst could be continuously used.143
4. Porous carbons
Considering the low thermal stability of sulfonated resins and poor porosity of sulfated metal oxides and HTs, nanoporous carbon-based catalysts have obvious advantages in the thermal stability and porosity. For example, the activated carbon, which is produced for nearly 1.5 million tons every year as an adsorbent, a capacitor, and a catalyst support, has very high surface area (∼1000 m2 g−1) and superior stability in acid/base media and/or under high temperature calcination at even 800 °C in an inert gas.144 Based on these excellent features, the nanoporous carbon supported acidic sites or metal nanoparticles are employed as efficient catalysts active for biomass conversion.4.1 Activated carbons and ordered mesoporous carbons (OMC)
Since the pioneering work of Hara et al. on sulfonated incompletely carbonized naphthalene as an efficient solid acid catalyst in 2004,145 sulfonated carbons have become a new type of solid acid. Recently, much attention has been paid to developing new sulfonated porous carbons for the conversion of biomass.146–149Followed by Hara's observation, the sulfonated activated carbons were synthesized from sulfonation of activated carbons with sulfuric acid or chlorosulfonic acid. However, the sulfonated activated carbons usually exhibited poor catalytic activities, which is due to the relatively weak interaction between sulphuric acid and the carbon surface.145 A similar phenomenon was also observed in the direct sulfonation of ordered mesoporous carbon (OMC) materials.150,151 Recently, to increase this interaction, Chang et al.150 have used H2O2 to pretreat the surface of OMC, obtaining the OMC with rich hydroxyl and carboxyl groups.152,153 After sulfonation with concentrated sulfuric acid, OMC-H2O2–SO3H gives an acid concentration of 2.09 mmol g−1, which is much higher than that (1.06 mmol g−1) of the directly sulfonated sample (OMC-SO3H). As a consequence, OMC-H2O2–SO3H exhibited a much higher catalytic activity than OMC-SO3H in the esterification of oleic acid with methanol, as shown in Fig. 5.152–154 Very importantly, even if recycled 5 times, there is no obvious activity loss compared with the fresh catalyst, which is reasonably attributed to the strong interaction between sulfonic groups and the carbon surface.
 | ||
| Fig. 5 Photographs of (A) OMC-SO3H and (B) OMC-H2O2-SO3H in methanol solvent. OMC-SO3H precipitates in a few minutes, while OMC-H2O2-SO3H forms a stable dispersion, because of its hydrophilic surface (reproduced with the permission of ref. 150. Copyright 2013 American Chemical Society). | ||
Except for using solid acids, porous carbons help to support metal nanoparticles for the oxidation and hydrogenation. Considering that these reactions are usually performed at relatively high temperature, a high stability and inert surface of porous carbons are suitable.155–157
One typical example is the activated carbon supported Pd nanoparticle catalyst (Pd/C). Since 1995, Pd/C has been widely applied in the conversion of biomass-based feedstocks.158 An important use is to catalyze the oxidation of glycerol, which is a major by-product for the production of biodiesel. Currently, about 1.2 million tons of glycerol were formed every year, mostly coming from the production of biodiesel. Therefore, the conversion of glycerol into valuable chemicals is of great importance. Garcia et al.158 reported a Pd/C catalyst used in the oxidation of glycerol by molecular oxygen, where the selectivity to glyceric acid could be as high as 70%. Interestingly, they found that adjusting the composition of metal species on activated carbons could effectively change the reaction route. For example, the deposition of Bi on Pt nanoparticles on activated carbon (Bi–Pt/C) orientated the selectivity towards the oxidation of the secondary hydroxyl group to yield dihydroxyacetone with a selectivity of 50%. Based on these observations, Bianchi et al. performed a systematic study on the glycerol oxidation over carbon-supported mono- and bimetallic catalysts based on Au, Pd, and Pt. It was found that the bimetallic Au-Pd/C and Au-Pt/C catalysts are more active than the monometallic catalysts.159
Besides the conversion of glycerol, the activated carbon supported metal nanoparticles are applied also for the conversion of sugars and cellulose.160 For example, Fuchigami et al. found that the Pd/C catalyst is highly active for the conversion of maleic anhydride (MAN) and succinic anhydride (SAN) in the presence of Cs2SO4, giving a γ-butyrolactone (GBL) yield of 97%. Ji et al.32 reported that activated carbon supported tungsten carbide with a small amount of Ni (Ni–W2C/C) can effectively catalyze cellulose conversion into ethylene glycol (yield of 61%). Very interestingly, this catalyst has a good recyclability, which is assigned to its high stability of the activated carbon support. Taking into consideration the huge amount (20 million tons in global 2011) of ethylene glycol produced from crude oil-based feedstocks, this route would be important for “green” synthesis of ethylene glycol from biomass.
The activated carbon supported metal catalysts are also good catalysts for upgrading of biofuels.161,162 Different from the conventional reaction systems, which generally produce alcohols and ketones from hydrogenation of phenols, Zhao et al.162 found that the Pd/C catalyst is efficient for the production of cyclohexane from hydrogenation of phenol, a model molecule of aromatic pyrolysis oil. In this case, a key factor is that the presence of homogeneous H3PO4 in the reaction system could catalyze the dehydration of cyclohexanol to cyclohexene at high reaction temperature (>170 °C). Furthermore, they also performed the hydrogenation of phenols (e.g. guaiacols and syringols) originating from lignin-based pyrolysis oil. In these cases, Pd/C catalysts exhibited high yield of C9 alkanes at 250 °C in the presence of H3PO4. As expected, the Pd/C catalysts with high stability exhibited excellent catalytic recyclability. However, it is still a challenge to recycle liquid H3PO4 in this system. The solution for this shortage will be discussed in the zeolite section.
The activated carbon supported Pt and Mo2C catalyst (Pt–Mo2C/C) is also effective for reforming CO, H2O, and cellulose to produce polyols.163 Here, the Pt–Mo2C/C was synthesized from impregnation of nitric acid-treated carbon with ammonium paramolybdate and chloroplatinic acid solution, followed by a temperature programmed reduction. As shown in Fig. 6, the small particle sizes of Pt and Mo2C at high weight loading should be attributed to the abundant nanoporosity of the activated carbon support, which has an extremely high surface area for the dispersion of Pt and Mo2C species. During this catalyst, the Pt–Mo2C catalyzed the water–gas shift reaction from CO and H2O to H2, while the Pt–C species catalyzed the cellulose conversion to polyols. This catalytic system exhibits much higher selectivity to polyols than the conventional system using pure molecular hydrogen, because the hydrogen species in situ produced on Pt–Mo2C in the nanopores of the activated carbon support immediately shifted to nearby Pt–C, thus taking the hydrogenation.
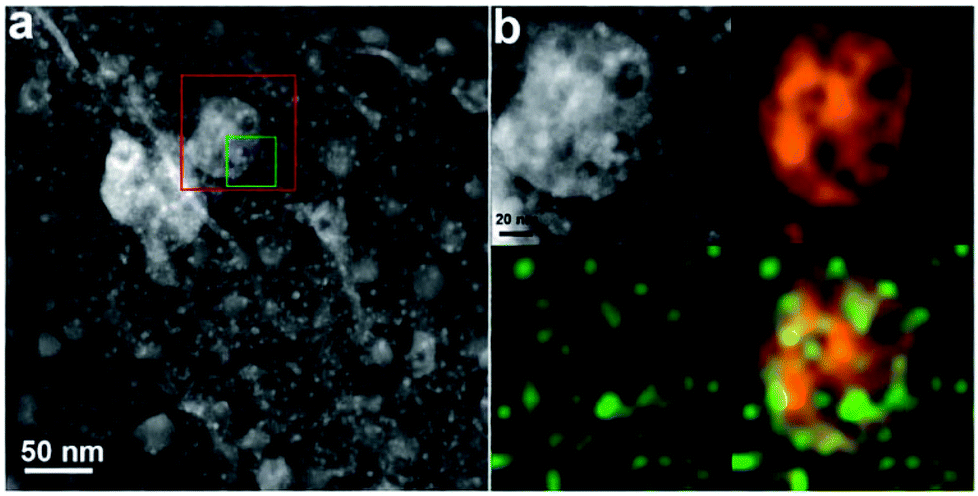 | ||
| Fig. 6 (a) A high-angle annular dark-field-STEM image of the Pt–Mo2C/C composite catalyst, showing two types of particles of different sizes; the large particles with a broad size distribution (20–50 nm) and the small particles with relatively uniform sizes (∼5 nm) correspond to Mo2C and Pt, respectively. (b) A magnified HAADF-STEM image (upper-left) of the red square region in (a) and the corresponding EDX elemental mappings of Mo (upper-right) and Pt (lower-left). The combined mapping image is shown in the lower right corner. These data confirmed that Pt-Mo2C/C have two types of Pt species on Mo2C and C, respectively (reproduced with the permission of ref. 163. Copyright 2013 Royal Society of Chemistry). | ||
4.2 Carbon nanotubes
Carbon nanotubes (CNTs) is a great discovery in carbon materials after fullerene.144 With a hollow structure, the CNTs show a large surface area of 200–300 m2 g−1 for multiwalled carbon nanotubes (MWCNTs) and 700–800 m2 g−1 for single-walled carbon nanotubes (SWCNTs). Although the surface areas of CNTs are lower than those of activated carbons (∼1000 m2 g−1), they have better mass transfer for the reactants/products than activated carbons.144In addition, the basic sites were easily functionalized on CNTs for catalyzing the biomass conversion. By carbometalation and C–C bond formation steps, Tessonnier et al.164 successfully synthesized a triethylamine-modified MWCNT catalyst (NEt3-MWCNT, Fig. 8), which is highly active and stable for transesterification of glyceryl tributyrate with methanol to produce biodiesel. In contrast, the NH2-MWCNT catalyst synthesized by the oxidation and amidation (Fig. 7) method gives lower activity and stability than NEt3-MWCNT, which might be due to the destruction of the CNT walls during the synthesis of NH2-MWCNT.165 These observations suggested the importance of a stable nanotube structure for the catalytic activity and stability of the CNTs-based catalysts.
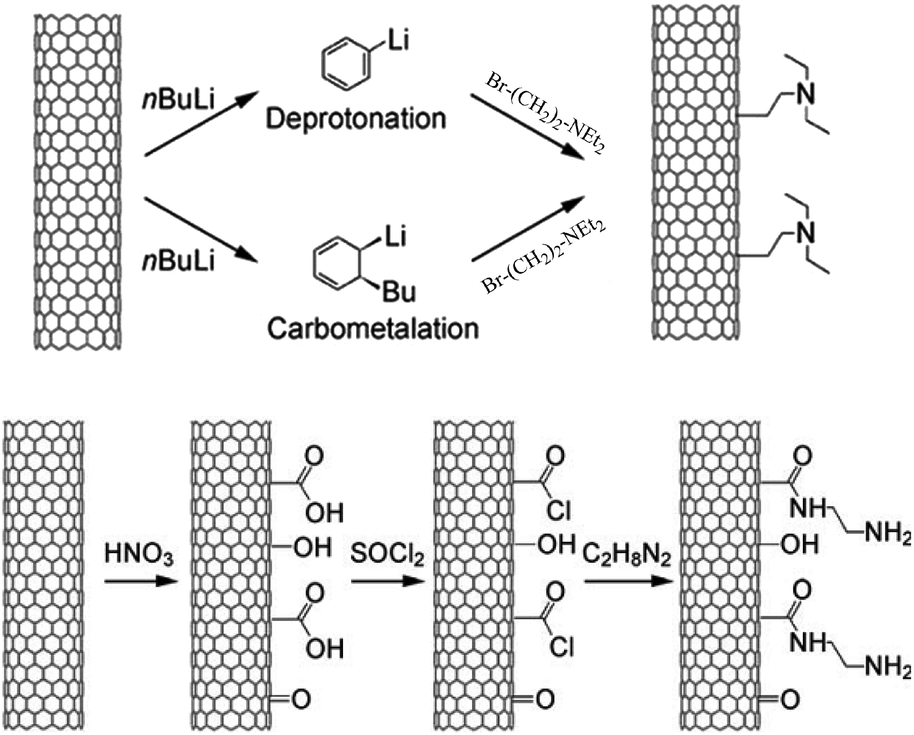 | ||
| Fig. 7 (Top) Synthesis of NEt3-MWCNT by the carbometalation and C–C bond formation method. (Bottom) Synthesis of NH2-MWCNT synthesized by the oxidation and amidation method (reproduced with the permission of ref. 164. Copyright 2009 Wiley-VCH). | ||
 | ||
| Fig. 8 TEM images of (a) SiO2@resin, (b) SiO2@HSC, and (c) HSC-SO3H; (d) hollow size distribution of HSC-SO3H. Clearly, the HSC-SO3H exhibits a hollow structure with a carbon shell thickness of 10–13 nm (reproduced with the permission of ref. 176. Copyright 2013 Royal Society of Chemistry). | ||
Besides modifications of basic sites, CNTs are also good supports to load metal nanoparticles for the conversion of biomass-based feedstocks. For example, Tan et al.166 reported the CNT supported Au nanoparticles as efficient catalysts for oxidation of cellobiose, giving a gluconic acid yield of 80% at 145 °C. Wang et al.167 found that CNT supported Ru nanoparticles are highly efficient for selective hydrogenolysis of glycerol to glycols. Later, Li et al.168 synthesized highly dispersed bimetallic Ru–Fe nanoparticles with a small size of 3 nm on CNTs, giving higher glycol yield than the mono-metallic catalysts in hydrogenolysis of glycerol. Recently, de Vlieger et al.169 extended the use of CNT supported Ru nanoparticles for reforming biomass-derived acetic acid to produce hydrogen.
4.3 Nitrogen-doped carbons
The activated carbon and carbon nanotubes have been widely used as catalyst supports. However, the shortage of strong interaction between the carbon surface and metals results in leaching of active metal sites.2,170–172 One solution to this problem is to dope nitrogen species in the carbons.173 As expected, N-doped porous carbon supported metal catalysts exhibited excellent catalytic performances in the biomass conversion. For example, Xu et al.174 synthesized N-doped porous carbons from the carbonization of dicyanamide-containing ion liquids of 3-methyl-1-butylpyridine dicyanamide using silica spheres as a hard template. After loading Pd nanoparticles, the obtained catalyst (Pd/CN0.132) is highly efficient for the hydrogenation of vanillin, a model reaction for biofuel upgrade, giving a product (2-methoxy-4-methylphenol) yield of 100%. In contrast, the conventional Pd/C showed the product yield of 72.5%.More recently, Wang et al.175 designed and synthesized a N-modified porous carbon and TiO2 composite supported Pd catalyst (Pd/TiO2@N-C). In this work, chitosan, a polysaccharide biomass with high nitrogen content, was used as a precursor for synthesizing TiO2@N-C by hydrothermal treatment with tetrabutyl titanate in acetic acid aqueous solution, followed by calcination. After loading Pd nanoparticles, the obtained Pd/TiO2@N-C is very active for catalytic hydrogenation of vanillin with formic acid as a hydrogen source, giving the product (2-methoxy-4-methylphenol) yield of 100%. The excellent catalytic performances over Pd/TiO2@N-C are reasonably attributed to the high activity of Pd nanoparticles on TiO2 and N-modified carbon for the tandem formic acid dehydrogenation and vanillin hydrogenation processes.
4.4 Carbon spheres
It is well known that an increase of mass transfer for heterogeneous catalysts is very helpful for enhancing catalytic performance, and an effective strategy is to introduce hierarchical porosity in the samples. Recently, Wang et al.176 synthesized a sulfonated hollow sphere carbon (HSC-SO3H) from carbonization of resin/SiO2 nanocomposites (SiO2@resin), removal of SiO2 (SiO2@HSC) by HF washing, and sulfonation with chlorosulfonic acid (Fig. 8). This unique sulfonated carbon catalyst with a hierarchical nanoporous structure shows comparable catalytic activities with a homogeneous sulphuric acid catalyst in acetalisation of glycerol with acetone to solketal. This phenomenon is assigned to its fast mass transfer due to the unique hollow sphere structure with rich micropores on the thin carbon shell (thickness of 10–13 nm), as shown in Fig. 8.4.5 Porous carbons synthesized from biomass feedstocks
The cellulose, starch, glucose, and sucrose are natural carbohydrates with rich hydrophilic groups. Hara and co-workers developed a series of sulfonated amorphous carbons from these natural materials.177–180 These results show a new route to synthesize carbon-based solid acids as highly efficient catalysts for transesterification of triolein, esterification of oleic acid, hydrolysis of cellobiose, and dehydration of fructose. It should be noted that the synthesized catalysts are non-porous with low surface area (<5 m2 g−1).177–180 Therefore, it is desirable to synthesize carbon materials with rich porosity from the natural feedstocks.181,182Starbon, a typical porous carbon derived from porous starch, leads to a hot topic focusing on synthesizing mesoporous porous carbons from the natural feedstocks in the absence of any template.183–192 Until now, the Starbon technique has been extended to synthesize mesoporous carbons from a series of biomass such as chitin, collagen, keratin, cellulose, lignin, furfural, and alginic acid, where the porosity, surface area, hydrophobicity/hydrophilicity, and functionalized groups could be easily controlled by adjusting the synthesis conditions.190–192 Interestingly, the sulfonated Starbon (Starbon-SO3H) is highly active for the esterification of diacids of succinic, fumaric, and itaconic acids, giving 5–10 times higher reaction rates than commercial solid acids of zeolites, Zr/SO42−, acidic clays, and resins.183,184,193
Notably, the mesopores in the carbons synthesized from the Starbon technique are disordered, because of the absence of any template during the synthesis process. More recently, Kubo et al.194 successfully synthesized ordered microporous and mesoporous carbons using fructose as feedstocks in the presence of the F127 triblock copolymer as a template (Fig. 9). Particularly, it is worth noting that although great success has been achieved in the synthesis of porous carbons from natural feedstocks, the study on their catalytic properties is just in the beginning stage. Considering the easy and low-cost synthesis process, rich porosity, and functionalized hydroxyl groups on these porous carbons, it is suggested that catalysts based on porous carbons from natural feedstocks would be potentially important for biomass conversion in the future.
 | ||
| Fig. 9 (A and B) TEM and (C and D) HR-TEM images of microporous and mesoporous carbons synthesized from fructose (reproduced with the permission of ref. 194. Copyright 2011 American Chemical Society). | ||
5. Ordered mesoporous silicas
Compared with nanoporous resins, metal oxides, and carbons, ordered mesoporous silicas (OMS) such as MCMs and SBAs materials have the obvious advantages of ordered open channels, controllable morphology and pore sizes, large surface areas, and rich functionalized groups. These features are attractive for functionalization of the OMS by active sites as efficient catalysts for biomass conversion.5.1 OMS with sulfonated groups
In 2003, Dufaud and Davis195 synthesized mesoporous silicas with various active sites such as sulfonated acid using organic–inorganic hybrid silane as a precursor. Recently, this method was applied to synthesize ordered mesoporous silicas with sulfonated groups for the biomass conversion.196–198 For example, Karaki et al.196 synthesized SBA-15 with a controlled local environment of the acid sites using different sulfonated silanes. They show high conversion for hydrolysis of cellobiose to glucose. Dhainaut et al.197 prepared hierarchical macroporous–mesoporous SBA-15 sulfonic acid catalysts in the presence of polystyrene in the starting gel. They are more active in the transesterification of tricaprylin and esterification of palmitic acid with methanol than the conventional sulfonated mesoporous SBA-15 because of the highly efficient mass transfer from the macroporous and mesoporous structure. Pirez et al.198 synthesized various sulfonated mesoporous KIT-6 catalysts, an ordered mesoporous silica with Ia3d architecture, by the post-grafting route (PrSO3H-KIT-6). They found that PrSO3H-KIT-6 is highly efficient for the esterification of free fatty acids (propanoic acid, hexanoic acid, lactic acid, and palmitic acid) with alcohols (methanol and ethanol), because of their interconnected pore network.5.2 OMS with metals
The metal species in OMS have Lewis acidic feature. Li et al.199 found that Zr, Hf, and Al in TUD-1 and MCM-41 catalysts are active for the acetalization of glycerol with acetone to produce solketal. However, due to a lack of Brønsted acid sites, their catalytic applications are severely limited. To create Brønsted acidic sites, Wang et al.200 synthesized extremely highly dispersed Sn species in the mesopores of SBA-15 (S–Sn–OH) by grafting dimethyldichlorostannane onto the silica surface, followed by calcination to transform the methyl groups into hydroxyl groups, where both Lewis and Brønsted acidic sites are present (the formation of Brønsted acidic sites is due to the presence of hydroxyl groups attached on the Sn species). This sample is designated as S–Sn–OH, giving comparable catalytic activity with that of sulphuric acid. Very importantly, no decrease of glycerol conversion and solketal selectivity was observed over a reaction period of 50 h in a continuous reaction system with a fixed-bed reactor, indicating its good stability.Interestingly, the S–Sn–OH catalyst exhibits unique product selectivities. For example, in the catalytic conversion of fructose into ethanol, ethyl lactate is the major product over SBA-15 with only Lewis sites, while ethyl levulinate is the major product over the S–Sn–OH catalyst. This phenomenon is also related to the presence of Brønsted acidity in the S–Sn–OH, which could catalyze the dehydration of fructose to HMF, followed by hydrolysis and esterification of HMF to ethyl levulinate. Possibly, this methodology for introducing Brønsted sites into the OMS samples with both Lewis and Brønsted acidity might be potentially suitable to extend other metals such as Zr and Nb.
Accordingly, the synthesis of OMS requires expensive surfactants as mesoporous templates, and removal of these templates by calcination always produces undesirable wastes. In addition, their relatively low hydrothermal stability and hydrophilic surface are not favourable for the biomass conversion. Therefore, synthesis of novel catalysts with abundant porosity, good stability, and controllable wettability is desirable.
6. Porous polydivinylbenzene
Nanoporous polydivinylbenzene (PDVB) synthesized from the solvothermal route201 has obvious advantages of abundant porosity, large pore volume, and superoleophilicity. Particularly, compared with conventional resins, the PDVB exhibits high stability and adsorption selectivity. For example, the PDVB almost does not adsorb either liquid or gaseous water and thus has a preferential selectivity for organic compounds. These features are helpful for the biomass conversion.202–208Motivated by the unique features of PDVB, a series of PDVB-based basic, acidic, and metal catalysts has been developed. For example, Liu et al.202 synthesized basic imidazole group modified nanoporous polydivinylbenzene by co-polymerization of divinylbenzene and 1-vinylimidazole (PDVB-VI), which showed high surface area (510–680 m2 g−1) and high alkaline density (0.953–2.822 mmol g−1). Interestingly, the PDVB-VI has a superhydrophobic surface, which is much more active in transesterification of tripalmitin with methanol than the conventional base catalysts such as Amberlyst-400 resin, hydrotalcite, CaO, NaOH, and 1-vinylimidazolate. Later, they functionalized the ionic liquid groups to the polydivinylbenzene to obtain nanoporous ionic liquid catalysts. These novel heterogeneous catalysts are even more active than the corresponding homogeneous ionic liquid catalysts.203 This phenomenon is due to the enrichment of reactants including tripalmitin and methanol in the nanopores of PDVB-based catalysts. Very interestingly, these catalysts have excellent catalytic recyclability. There is almost no activity loss over PDVB-[C1vim][SO3CF3] in the transesterification reaction after recycling 4 times. In contrast, the activity over Amberlyst-15 is remarkably reduced.
Both catalytic activity and product selectivity in the biomass conversion could be adjusted by the surface wettability of PDVB-based catalysts. It is well known that the key step in producing HMF from the biomass feedstocks is the dehydration of fructose, which is generally catalyzed by the acidic sites. Once HMF is formed, the hydration of HMF to levulinic acid (LA) and formic acid (FA) is also catalyzed in the same system, which results in a significant decrease of HMF yield.34–36,204–208 In order to investigate the reaction between water and HMF over acid catalysts, Wang et al.208 performed theoretical simulations on the hydration of HMF, and it was found that keeping the distance of protons on acidic sites to water molecules beyond the value of 2.05 Å could completely prevent the hydration of HMF to FA and LA. Prompted by this result, they designed the dehydration of fructose to HMF over a superhydrophobic PDVB-based solid acid catalyst (P-SO3H-154), which is synthesized from the co-polymerization of divinylbenzene and sodium p-styrene sulfonate, followed by ion-exchange in dilute sulphuric acid. As a result, the P-SO3H-154 catalyst shows a HMF yield of nearly 100% from the dehydration of fructose in a mixed solvent of THF–DMSO, because the hydrophobic surface of P-SO3H-154 could isolate the water molecules from the acid centers (Fig. 10), preventing the hydration of HMF. In contrast, the hydrophilic Amberlyst-15 shows a lower HMF yield of 45.7%, with LA and FA as by-products from the hydration of HMF.
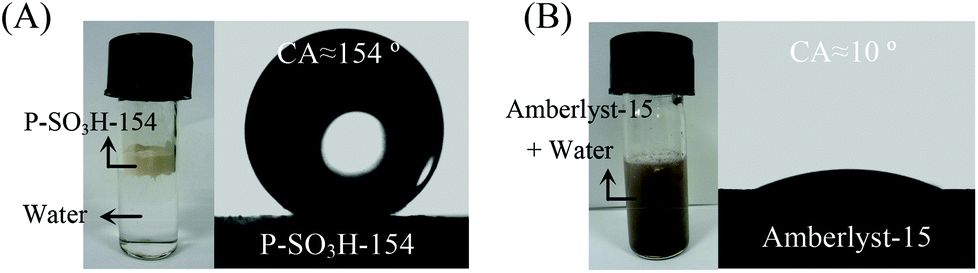 | ||
| Fig. 10 The photographs and water droplet contact angle tests of (A) P-SO3H-154 and (B) Amberlyst-15. The P-SO3H-154 sample is superhydrophobic with a water droplet contact angle (CA) of 154°, while Amberlyst-15 is superhydrophilic with CA ∼10° (reproduced with the permission of ref. 208. Copyright 2014 Wiley-VCH). | ||
Furthermore, the P-SO3H-154 catalyst was also extended to other biomass transforms such as dehydration of sorbitol to isosorbide, giving an isosorbide yield of 87.9%.209 In contrast, the sulfonated PDVB without any nanoporosity (nonporous-P-SO3H) which is also superhydrophobic gives a very low isosorbide yield of 3.4% due to the very low surface area of nonporous-P-SO3H (<5 m2 g−1). This result indicates the importance of the catalyst nanoporosity.
7. Zeolites
It is worth noting that there is always coke formed over the solid acid catalysts in the conversion of biomass, and calcination in air is regarded as an effective method to remove the coke. However, for the acid-functionalized resins, metal oxides, and PDVB catalysts, they could not bear the high temperature calcination.Zeolite crystals with intricate micropores, strong Brønsted and Lewis acidity have already been widely used as heterogeneous catalysts in the petrochemical and fine chemical industries.210–224 They have crystalline inorganic frameworks, offering a very high thermal stability.
7.1 Aluminosilicate zeolites
One of the best advantages of aluminosilicate zeolites is the product selectivity controlled by the micropore sizes. For example, Jae et al.225 reported that the ZSM-5, ZSM-11, TNU-9, ZSM-23, MCM-22, and IM-5 zeolites with small micropore sizes of 4.0–5.9 Å produced higher yield of aromatics in fast pyrolysis of glucose, while the SSZ-55, Beta, and Y zeolites with micropore sizes of 6.5–7.4 Å produced higher yield of coke, because of the large micropore sizes. In contrast, the ZK-5 zeolite with the smallest micropore size of 3.9 Å produced only oxygenated products without any aromatics, because the pore size (3.9 Å) is too small for the formation of aromatics. These results indicate the good feature of zeolites in influencing the product selectivity by micropore size limitation, which would be a renewable route to produce the desired chemicals from biomass by employing zeolites with suitable micropore sizes.Recently, Cheng et al.226 modified the conventional ZSM-5 zeolite by the chemical liquid deposition (CLD) method to obtain silylated ZSM-5, which has smaller micropore opening than the conventional ZSM-5. Interestingly, the silylated ZSM-5 exhibited a much higher p-xylene selectivity than conventional ZSM-5, because the decreased micropore size of silylated ZSM-5 is in good agreement with the kinetic diameter of p-xylene (Fig. 11). Later, Williams et al.227 reported the synthesis of p-xylene from dimethylfuran and ethylene in an aliphatic solvent at 300 °C over Y zeolite with a larger micropore size. After the cycloaddition of ethylene and 2,5-dimethylfuran and subsequent dehydration steps, the selectivity to p-xylene can reach as high as ∼75% with a dimethylfuran conversion of ∼95%. Furthermore, Do et al.228 proposed the reaction network of the side reactions in this reaction over Y zeolite by systematically identifying the structures of various by-products. Considering the importance of p-xylene in polyester fibre production in the industrial process (more than 30 million tons of p-xylene each year from crude-oil feedstock in the world), the green route for synthesizing p-xylene from the biomass should have a good future.
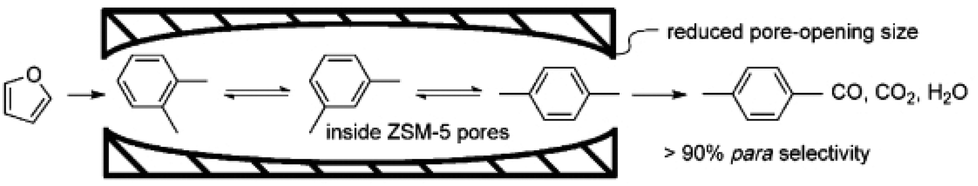 | ||
| Fig. 11 A scheme diagram showing the limitation of xylene isomers inside the decreased micropore of silylated ZSM-5 (reproduced with the permission of ref. 226. Copyright 2012 Wiley-VCH). | ||
As is known, upgrading of pyrolysis oil into alkanes goes through many steps including hydrogenation and isomerization, where the isomerization was conventionally performed over homogeneous acids.161 Based on the observations on pyrolysis oil upgrading over Pd/C and H3PO4 catalysts,161 Lercher and co-workers161,229–233 developed the upgrading of pyrolysis oil over aluminosilicate ZSM-5 and Beta zeolite supported Ni or Pd catalysts with high activity and good recyclability. In this case, liquid H3PO4 is not necessary. The avoidance of the liquid H3PO4 is greatly important for simplifying the process and saving energy.
Notably, upgrading of biofuels in a hot water for a long time is damaging for the zeolite framework owing to the destruction of Si–O–Si or Si–O–Al bonds.234,235 In order to improve the stability of Y zeolite, Zapata et al.236,237 functionalized the external surface of Y zeolite with organosilanes. The nanoporous structure of the hydrophobic Y zeolite obtained is much more stable in the water/oil emulsions (Fig. 12) than that of the untreated sample, giving excellent catalytic activity and recyclability in the biofuel upgrading performed at 200 °C.
 | ||
| Fig. 12 HRTEM of the Y zeolites: untreated zeolite (a) before and (c) after reaction; organosilane-functionalized zeolite (b) before and (d) after reaction. The crystalline structure of the functionalized zeolite remains unchanged after reaction, while that of the untreated zeolite is greatly affected, which indicates the high stability of organosilane-functionalized zeolite (reproduced with the permission of ref. 236. Copyright 2012 American Chemical Society). | ||
7.2 Sn-Beta, Ti-Beta, and Zr-Beta zeolites
Sn-, Ti-, and Zr-substituted Beta zeolites with Lewis acidic sites228,238–245 exhibit unique catalytic performance in biomass conversion. In 2010, Holm et al.246 found that Sn-, Ti-, and Zr-substituted microporous Beta zeolites could catalyze the one-pot conversion of glucose, fructose, and sucrose to lactic acid-derived products of methyl lactate and ethyl lactate at 160 °C. In contrast, the Al-Beta with Brønsted acid sites would catalyze the dehydration reaction to form HMF-derived products.In the same year, Davis et al.247–249 reported Sn-Beta zeolite as an efficient catalyst for isomerization of glucose into fructose in aqueous media with glucose feeds as high as 45 wt%. By 1H and 13C NMR spectroscopy on labelled glucose, they proposed the intramolecular hydride shift mechanism for glucose conversion to fructose, which is different from the proton transfer mechanism over base catalysts, as shown in Fig. 13. Later, they found that the glucose isomerization in methanol solvent in the Sn-Beta zeolite micropores could form mannose, while the reaction on the external surface of zeolite crystals over extraframework SnO2 nanoparticles could produce fructose.250 Based on these results, it could be easy to obtain desirable products from glucose isomerization inside and outside of the micropores of Beta zeolite by controlling the position of Sn species.
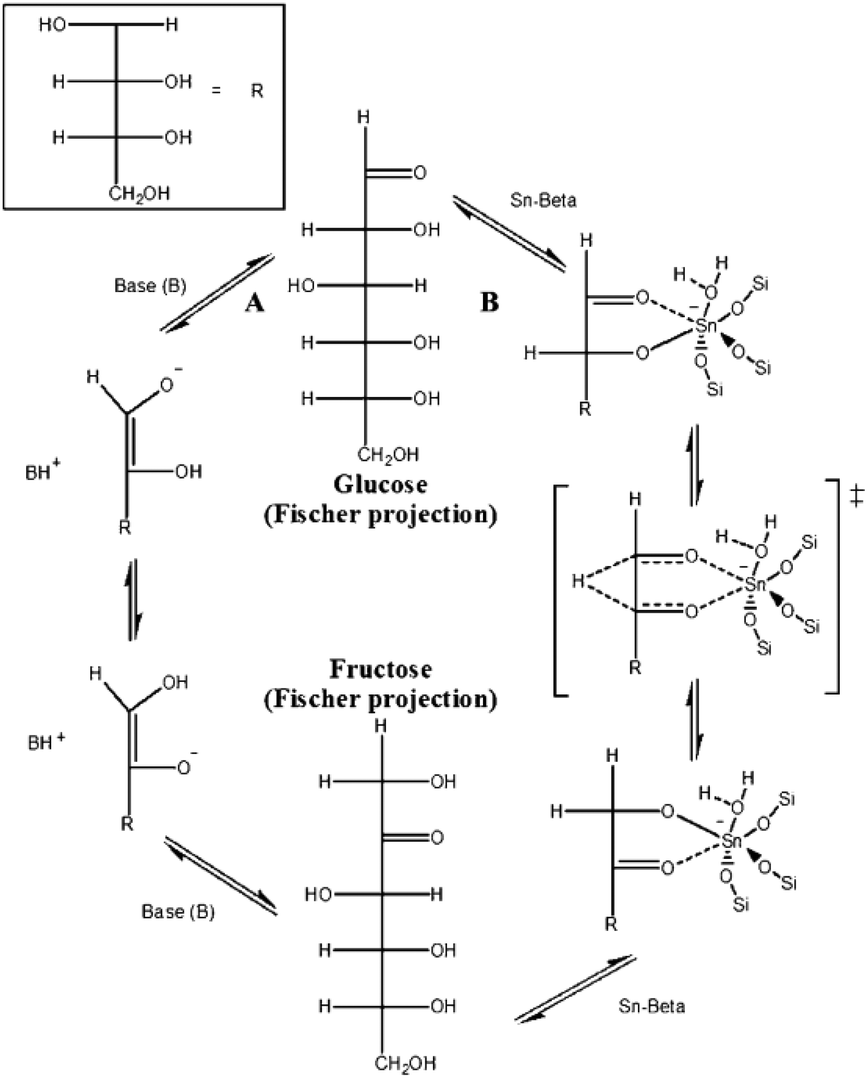 | ||
| Fig. 13 Glucose isomerization mechanisms by way of (A) proton transfer and (B) intramolecular hydride shift over base and Sn-Beta catalysts, respectively (reproduced with the permission of ref. 247. Copyright 2010 Wiley-VCH). | ||
To catalyze the one-pot production of HMF from glucose, cellobiose, and starch, a mixed catalyst of Sn-Beta and hydrochloric acid was used, where biphasic solvents were employed in order to immediately extract the HMF formed in the water phase to the organic phase, avoiding the side reaction of HMF hydration in water solvent.251 However, the use of homogeneous hydrochloric acid as a Brønsted acid makes it difficult to be regenerated, compared with the heterogeneous solid acids. Roman-Leshkov et al.252 performed the combination of Zr-Beta and aluminosilicate ZSM-5 nanosheet catalysts to one-pot production of γ-valerolactone from fructose. The whole process, which is designated as domino reaction, included dehydration and esterification catalyzed by the Brønsted acid sites on ZSM-5 nanosheets, and hydrogen transfer with 2-butanol as a hydrogen source catalyzed by Lewis acid of Zr-Beta. It is worth mentioning that they employed ZSM-5 nanosheets as Brønsted acid catalysts instead of the conventional ZSM-5 crystals, because the micropore size of ZSM-5 zeolite is too small for the relatively bulky molecule of fructose, while the ZSM-5 nanosheets have more exposed external acid sites.
The success of Davis and Roman-Leshkov's studies confirmed that adjusting metals and combining different types of zeolite catalysts could be used as a novel, simple, and efficient method to transform the biomass feedstocks by alternative routes.
8. Conclusions and perspectives
A brief overview was presented of recent studies that focused on the design, synthesis, and catalytic applications of nanoporous catalysts for the conversion of biomass-based feedstocks, including nanoporous resins, metal oxides, carbons, mesoporous silicas, polydivinylbenzene, and zeolites. For these catalysts, the nanoporous structures play an important role in dispersion of the active sites, access of reactants, mass transfer, and control of product selectivity. Notably, each type of nanoporous catalyst has its unique advantages. The sulfonated resins have high acidic density, but relatively low stability; the sulfated metal oxides have high stability, but relatively low surface areas; carbons and ordered mesoporous silicas have large surface areas, but the surface wettability of the nanopores is not easily controllable; the nanoporous polydivinylbenzenes have easily controllable wettability, but relatively low thermal stability; the zeolites have high thermal stability, but relatively low mass transfer for the sole micropores.Obviously, the advantages of each catalyst mentioned above are separate. Therefore, it is strongly expected that the combination of the advantages of various catalysts to synthesize novel nanoporous catalysts will result in more efficient catalytic performances. For example, introduction of mesoporosity into the microporous zeolite catalysts could form mesoporous zeolites with both micropores and mesopores, exhibiting excellent catalytic performance for the conversion of both small and bulky biomass molecules. Notably, the synthesis of a series of mesoporous zeolites such as MFI and BEA structures has been successful,253–257 but the catalytic conversion of biomass molecules is still under investigation currently. The synthesis of mesoporous zeolites offers a good opportunity for catalyzing the biomass to valuable fuels and chemicals.
Finally, it is emphasized that the discovery of novel catalysts is fundamental for developing efficient catalysts for the biomass conversion. This review briefly summarized recent studies on the nanoporous catalysts with various features, which might be valuable for the rational design of highly active catalysts in the future.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21273197 and U1162201) and the China Postdoctoral Science Foundation (2014M550318).Notes and references
- K. T. Wan and M. E. Davis, Nature, 1994, 370, 449 CrossRef CAS.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481 RSC.
- C. Perego and R. Millini, Chem. Soc. Rev., 2013, 42, 3956 RSC.
- S. C. Reyes and E. Iglesia, J. Catal., 1991, 129, 457 CrossRef CAS.
- L. Wang, J. Zhang, J. Sun, L. Zhu, H. Zhang, F. Liu, D. Zheng, X. Meng, X. Shi and F.-S. Xiao, ChemCatChem, 2013, 5, 1606 CrossRef CAS.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083 RSC.
- R. J. Davis and E. G. Derouane, Nature, 1991, 349, 313 CrossRef CAS.
- H. M. Hultman, M. de Lang, I. W. C. E. Arends, U. Hanefeld, R. A. Sheldon and T. Maschmeyer, J. Catal., 2003, 217, 275 CrossRef CAS.
- J. M. Thomas, R. Raja, G. Sankar and R. G. Bell, Nature, 1999, 398, 227 CrossRef CAS PubMed.
- G. Busca, Chem. Rev., 2007, 107, 5366 CrossRef CAS PubMed.
- L. Wang, X. Meng, B. Wang, W. Y. Chi and F.-S. Xiao, Chem. Commun., 2010, 46, 5003 RSC.
- W. Wang, C.-L. Chen, N.-P. Xu and C.-Y. Mou, Green Chem., 2002, 4, 257 RSC.
- G. Sartori and R. Maggi, Chem. Rev., 2006, 106, 1077 CrossRef CAS PubMed.
- M. Pera-Titus, Chem. Rev., 2014, 114, 1413 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed.
- T. J. Morgan and R. Kandiyoti, Chem. Rev., 2014, 114, 1547 CrossRef CAS PubMed.
- F. Su and Y. Guo, Green Chem., 2014, 16, 2934 RSC.
- M. E. Himmel, S. Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804 CrossRef CAS PubMed.
- A. T. W. M. Hendriks and G. Zeeman, Bioresour. Technol., 2009, 100, 10 CrossRef CAS PubMed.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982 CrossRef CAS PubMed.
- R. Luque and A. M. Balu, Producing Fuels and Fine Chemicals from Biomass Using Nanomaterials, Wiley, 2013 Search PubMed.
- H. B. Nisbet, J. Inst. Pet., 1946, 32, 162 Search PubMed.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed.
- H. de Lasa, E. Salaices, J. Mazumder and R. Lucky, Chem. Rev., 2011, 111, 5404 CrossRef CAS PubMed.
- M. Jarvis, Nature, 2003, 426, 611 CrossRef CAS PubMed.
- E. Bahcegul, S. Apaydin, N. I. Haykir, E. Tatlic and U. Bakir, Green Chem., 2012, 14, 1896 RSC.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787 CrossRef CAS PubMed.
- R. Rinaldi, R. Palkovits and F. Schuth, Angew. Chem., Int. Ed., 2008, 47, 8047 CrossRef CAS PubMed.
- J. B. Binder and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4516 CrossRef CAS PubMed.
- F. Liu, R. K. Kamat, I. Noshadi, D. Peck, R. S. Parnas, A. Zheng, C. Qi and Y. Lin, Chem. Commun., 2013, 49, 8456 RSC.
- N. Ji, T. Zhang, M. Y. Zheng, A. Q. Wang, H. Wang, X. D. Wang and J. G. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510 CrossRef CAS PubMed.
- M. Rose, Angew. Chem., Int. Ed., 2013, 52, 9613 CrossRef CAS.
- Y. Roman-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933 CrossRef CAS PubMed.
- H. Zhang, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597 CrossRef PubMed.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754 RSC.
- C. Moreau, A. Finiels and L. Vanoye, J. Mol. Catal. A: Chem., 2006, 253, 165 CrossRef CAS PubMed.
- C. Moreau, R. Durand, S. Razigade, J. Duhamet, O. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538 RSC.
- S. Hu, Z. Zhang, J. Song, Y. Zhou and B. X. Han, Green Chem., 2009, 11, 1746 RSC.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Green Chem., 2008, 10, 799 RSC.
- A. A. Kiss, A. C. Dimian and G. Rothenberg, Adv. Synth. Catal., 2006, 348, 75 CrossRef CAS.
- L. Bournay, D. Casanave, B. Delfort, G. Hillion and J. A. Chodorge, Catal. Today, 2005, 106, 190 CrossRef CAS PubMed.
- M. G. Kulkarni and A. K. Dalai, Ind. Eng. Chem. Res., 2006, 45, 2901 CrossRef CAS.
- M. L. Granados, M. D. Zafra Poves, D. M. Alonso, R. Mariscal, F. C. Galisteo, R. Moreno-Tost, J. Santamaría and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 317 CrossRef CAS PubMed.
- G. J. Suppes, M. A. Dasari, E. J. Doskocil, P. J. Mankidy and M. J. Goff, Appl. Catal., A, 2004, 257, 213 CrossRef CAS PubMed.
- H. Fukuda, A. Kondo and H. Noda, J. Biosci. Bioeng., 2001, 92, 405 CrossRef CAS.
- G. Arzamendi, I. Campo, E. Arguiñarena, M. Sánchez, M. Montes and L. M. Gandía, Chem. Eng. J., 2007, 134, 123 CrossRef CAS PubMed.
- W. L. Xie and X. M. Huang, Catal. Lett., 2006, 107, 53 CrossRef CAS.
- C. R. V. Reddy, R. Oshel and J. G. Verkade, Energy Fuels, 2006, 20, 1310 CrossRef CAS.
- W. Song, C. Zhao and J. A. Lercher, Chem. – Eur. J., 2013, 19, 9833 CrossRef CAS PubMed.
- B. Peng, X. Yuan, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 9400 CrossRef CAS PubMed.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. B. Li and J. A. Lercher, Chem. Commun., 2010, 46, 412 RSC.
- M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854 CrossRef CAS PubMed.
- C. Moreau, M. N. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11 CrossRef CAS.
- F. K. Kazi, A. D. Patel, J. C. Serrano, J. A. Dumesic and R. P. Anex, Chem. Eng. J., 2011, 169, 329 CrossRef CAS PubMed.
- Y. Nakagawa and K. Tomishige, Catal. Commun., 2010, 12, 154 CrossRef CAS PubMed.
- T. Buntara, S. Noel, P. H. Phua, I. Melian-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083 CrossRef CAS PubMed.
- J. A. Libra, K. S. Ro, C. Kammann, A. Funke, N. D. Berge, Y. Neubauer, M.-M. Titirici, C. Fuhner, O. Bens, J. Kern and K.-H. Emmerich, Biofuels, 2011, 2, 89 CrossRef.
- M. Chidambaram and A. T. Bell, Green Chem., 2010, 12, 1253 RSC.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2014, 16, 191 RSC.
- P. Neves, S. Lima, M. Pillinger, S. M. Rocha, J. Rocha and A. A. Valente, Catal. Today, 2013, 218, 76 CrossRef PubMed.
- L. Petrus and M. A. Noordermeer, Green Chem., 2006, 8, 861 RSC.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893 RSC.
- C. Zhao, T. Bruck and J. A. Lercher, Green Chem., 2013, 15, 1720 RSC.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516 RSC.
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548 RSC.
- J. Y. Park, Z. M. Wang, D. K. Kim and J. S. Lee, Renewable Energy, 2010, 35, 614 CrossRef CAS PubMed.
- T. A. Peters, N. E. Benes, A. Holmen and J. T. F. Keurentjes, Appl. Catal., A, 2006, 297, 182 CrossRef CAS PubMed.
- Y. J. Liu, E. Lotero and J. G. Goodwin Jr., J. Catal., 2006, 243, 221 CrossRef CAS PubMed.
- T. D. Courtney, V. Nikolakis, G. Mpourmpakis, J. Chen and D. G. Vlachos, Appl. Catal., A, 2012, 449, 59 CrossRef CAS PubMed.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, ChemSusChem, 2012, 5, 1812 CrossRef CAS PubMed.
- W. Long and C. W. Jones, ACS Catal., 2011, 1, 674 CrossRef CAS.
- A. M. Ruppert, A. N. Parvulescu, M. Arias, P. J. C. Hausoul, P. C. A. Bruijnincx, R. J. M. K. Gebbink and B. M. Weckhuysen, J. Catal., 2009, 268, 251 CrossRef CAS PubMed.
- B. M. E. Russbueldt and W. F. Hoelderich, Appl. Catal., A, 2009, 362, 47 CrossRef CAS PubMed.
- A. Takagaki and K. Ebitani, Chem. Lett., 2009, 38, 650 CrossRef CAS.
- R. Rinaldi and F. Schuth, ChemSusChem, 2009, 2, 1096 CrossRef CAS PubMed.
- R. Rinaldi, N. Meine, J. Vom Stein, R. Palkovits and F. Schuth, ChemSusChem, 2010, 3, 266 CrossRef CAS PubMed.
- K. Shimizu, R. Uozumi and A. Satsuma, Catal. Commun., 2009, 10, 1849 CrossRef CAS PubMed.
- M. Hara, Energy Environ. Sci., 2010, 3, 601 CAS.
- G. A. Olah, T. Keumi and D. Meidar, Synthesis, 1978, 929 CrossRef CAS.
- G. K. Chuah, S. H. Liu, S. Jaenicke and L. J. Harrison, J. Catal., 2001, 200, 352 CrossRef CAS.
- T. Kornon, M. E. Jamroz, P. Niewiadomski, M. Malanowski and J. Kijenski, Polimery, 2012, 57, 728 CrossRef.
- E. Lam, E. Majid, A. C. W. Leung, J. H. Chong, K. A. Mahmoud and J. H. T. Luong, ChemSusChem, 2011, 4, 535 CrossRef CAS PubMed.
- Y. J. Liu, E. Lotero and J. G. Goodwin Jr., J. Catal., 2006, 242, 278 CrossRef CAS PubMed.
- Z. Huang, W. Pan, H. Zhou, F. Qin, H. Xu and W. Shen, ChemSusChem, 2013, 6, 1063 CrossRef CAS PubMed.
- M. Brandle and J. Sauer, J. Am. Chem. Soc., 1998, 120, 1556 CrossRef.
- F. Liu, J. Sun, Q. Sun, L. Zhu, L. Wang, X. Meng, C. Qi and F.-S. Xiao, Catal. Today, 2012, 186, 115 CrossRef CAS PubMed.
- J.-P. Dacquin, H. E. Cross, D. R. Brown, T. Duren, J. J. Williams, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 1383 RSC.
- M. Kitano, E. Wada, K. Nakajima, S. Hayashi, S. Miyazaki, H. Kobayashi and M. Hara, Chem. Mater., 2013, 25, 385 CrossRef CAS.
- M. A. Harmer, W. E. Farneth and Q. Sun, J. Am. Chem. Soc., 1996, 118, 7708 CrossRef CAS.
- P. Kalita, N. M. Gupta and R. Kumar, J. Catal., 2007, 245, 338 CrossRef CAS PubMed.
- M. Selvaraj, B. R. Min, Y. G. Shul and T. G. Lee, Microporous Mesoporous Mater., 2004, 74, 143 CAS.
- X. Zhang, Y. Zhao, S. Xu, Y. Yang, Y. Wei and Q. Yang, Nat. Commun., 2014, 5, 3170 Search PubMed.
- H. Adkins, E. E. Burgoyne and H. J. Schneider, J. Am. Chem. Soc., 1950, 72, 2626 CrossRef CAS.
- R. V. Jagadeesh, A. E. Surkus, H. Junge, M. M. Pohl, J. Radnik, J. Rabeah, H. M. Huan, V. Schunemann, A. Bruckner and M. Beller, Science, 2013, 342, 1073 CrossRef CAS PubMed.
- R. Schaub, E. Wahlstrom, A. Ronnau, E. Laegsgaard, I. Stensgaard and F. Besenbacher, Science, 2003, 299, 377 CrossRef CAS PubMed.
- N. Feng, Q. Wang, A. Zheng, Z. Zhang, J. Fan, S.-B. Liu, J.-P. Amoureux and F. Deng, J. Am. Chem. Soc., 2013, 135, 1607 CrossRef CAS PubMed.
- N. Feng, A. Zheng, Q. Wang, P. Ren, X. Gao, S.-B. Liu, Z. Shen, T. Chen and F. Deng, J. Phys. Chem. C, 2011, 115, 2709 CAS.
- W. Li, F. Ma, F. Su, L. Ma, S. Zhang and Y. Guo, ChemSusChem, 2011, 4, 744 CrossRef CAS PubMed.
- A. M. Alsalme, P. V. Wiper, Y. Z. Khimyak, E. F. Kozhevnikova and I. V. Kozhevikov, J. Catal., 2010, 276, 181 CrossRef CAS PubMed.
- T. Jin, T. Yamaguichi and K. Tanabe, J. Phys. Chem., 1986, 90, 4794 CrossRef CAS.
- B. M. Reddy and M. K. Patil, Chem. Rev., 2009, 109, 2185 CrossRef CAS PubMed.
- X. Chen, Y. Ju and C. Mou, J. Phys. Chem. C, 2007, 111, 18731 CAS.
- K. Arata, Green Chem., 2009, 11, 1791 RSC.
- G. D. Yadav and J. J. Nair, Microporous Mesoporous Mater., 1999, 33, 1 CrossRef CAS.
- K. Arata, Appl. Catal., A, 1996, 146, 3 CrossRef CAS.
- R. A. Comelli, C. R. Vera and J. M. Parera, J. Catal., 1995, 151, 96 CrossRef CAS.
- B. M. Reddy, P. M. Sreekanth and P. Lakshmanan, J. Mol. Catal. A: Chem., 2005, 237, 93 CrossRef CAS PubMed.
- A. S. Khder, E. A. EI-Sharkawy and A. I. Ahmed, Catal. Commun., 2008, 9, 769 CrossRef CAS PubMed.
- A. Corma, A. Martinez and C. Martinez, Appl. Catal., A, 1996, 144, 249 CrossRef CAS.
- H. Matsuhashi, T. Tanaka and K. Arata, J. Phys. Chem. B, 2001, 105, 9669 CrossRef CAS.
- C. Gannoun, A. Turki, H. Kochkar, R. Delaigle, P. Eloy, A. Ghorbel and E. M. Gaigneaux, Appl. Catal., B, 2014, 147, 58 CrossRef CAS PubMed.
- Z. Wang, Q. Lu, X. Zhu and Y. Zhang, ChemSusChem, 2011, 4, 79 CrossRef CAS PubMed.
- V. Degirmenci, D. Uner, B. Cinlar, B. H. Shanks, A. Yilmaz, R. A. van Santen and E. J. M. Hensen, Catal. Lett., 2011, 141, 33 CrossRef CAS.
- C. Liu, S. Lee, D. Su, Z. Zhang, L. Pfefferle and G. L. Haller, J. Phys. Chem. C, 2012, 116, 21742 CAS.
- W. Li, F. Ma, F. Su, L. Ma, S. Zhang and Y. Guo, ChemCatChem, 2012, 4, 1798 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Catal. Commun., 2009, 10, 1771 CrossRef CAS PubMed.
- K. Suwannakarn, E. Lotero, J. G. Goodwin and C. Lu, J. Catal., 2008, 255, 279 CrossRef CAS PubMed.
- Y. Sun, S. Ma, Y. Du, L. Yuan, S. Wang, J. Yang, F. Deng and F.-S. Xiao, J. Phys. Chem. B, 2005, 109, 2567 CrossRef CAS PubMed.
- I. Jiménez-Morales, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2011, 103, 91 CrossRef PubMed.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160 CrossRef CAS.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Adv. Mater., 1999, 11, 1307 CrossRef CAS.
- D. V. Bavykin, J. M. Friedrich and F. C. Walsh, Adv. Mater., 2006, 18, 2807 CrossRef CAS.
- M. Kitano, K. Nakajima, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2010, 132, 6622 CrossRef CAS PubMed.
- L. Wang, H. Wang, W. Zhang, J. Zhang, J. P. Lewis, X. Meng and F.-S. Xiao, J. Catal., 2013, 298, 186 CrossRef CAS PubMed.
- L. Wang, W. Zhang, S. Zeng, D. Su, X. Meng and F.-S. Xiao, Chin. J. Chem., 2012, 30, 2189 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- A. Abad, P. Comcepcion, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066 CrossRef CAS PubMed.
- L. Wang, W. Zhang, D. S. Su, X. Meng and F.-S. Xiao, Chem. Commun., 2012, 49, 5476 RSC.
- B. M. Choudary, S. Madhi, N. S. Chowdari, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127 CrossRef CAS PubMed.
- L. Wang, X. Meng and F.-S. Xiao, Chin. J. Catal., 2010, 31, 943 CrossRef CAS.
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526 CrossRef CAS.
- K. K. Rao, M. Gravelle, J. S. Valente and F. Figueras, J. Catal., 1998, 173, 115 CrossRef CAS.
- L. Hora, V. Kelbichová, O. Kikhtyanin, O. Bortnovskiy and D. Kubička, Catal. Today, 2014, 223, 138 CrossRef CAS PubMed.
- M. D. Serio, M. Cozzolino, M. Giordano, R. Tesser, P. Patrono and E. Santacesaria, Ind. Eng. Chem. Res., 2007, 46, 6379 CrossRef.
- H. J. Kim, B. S. Kang, M. J. Kim, Y. M. Park, D. K. Kim, J. S. Lee and K. Y. Lee, Catal. Today, 2004, 93, 315 CrossRef PubMed.
- T. Ebiura, T. Echizen, A. Ishikawa, K. Murai and T. Baba, Appl. Catal., A, 2005, 283, 111 CrossRef CAS PubMed.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Commun., 2009, 6276 RSC.
- A. Takagaki, M. Takahashi, S. Nishimura and K. Ebitani, ACS Catal., 2011, 1, 1562 CrossRef CAS.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090 CrossRef CAS PubMed.
- J. Wang, J. Ren, X. Liu, J. Xi, Q. Xia, Y. Zu, G. Lu and Y. Wang, Green Chem., 2012, 14, 2506 RSC.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782 CrossRef CAS PubMed.
- M. Hara, T. Yoshida, A. Takagaki, T. Takata, J. N. Kondo, S. Hayashi and K. Domen, Angew. Chem., Int. Ed., 2004, 43, 2955 CrossRef CAS PubMed.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033 RSC.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588 RSC.
- X. Mo, E. Letero, C. Lu, Y. Liu and J. G. Goodwin, Catal. Lett., 2008, 123, 1 CrossRef CAS.
- A. Onda, T. Ochi and K. Yanagisawa, Top. Catal., 2009, 52, 801 CrossRef CAS.
- B. Chang, J. Fu, Y. Tian and X. Dong, J. Phys. Chem. C, 2013, 117, 6252 CAS.
- L. Geng, G. Yu, Y. Wang and Y. X. Zhu, Appl. Catal., A, 2012, 427, 137 CrossRef PubMed.
- G. Guo, Z. L. Xiu and Z. X. Liang, Appl. Energy, 2012, 98, 47 CrossRef PubMed.
- R. Buzzoni, S. Bordiga, G. Ricchiardi, G. Spoto and A. Zecchina, J. Phys. Chem., 1995, 99, 11937 CrossRef CAS.
- M. Hara, Energy Environ. Sci., 2010, 3, 601 CAS.
- Z. Liu, X. Ling, X. Su and J. Lee, J. Phys. Chem. B, 2004, 108, 8234 CrossRef CAS.
- E. Auer, A. Freund, J. Pietsch and T. Tacke, Appl. Catal., A, 1998, 173, 259 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, Appl. Catal., A, 1996, 142, 97 CrossRef CAS.
- R. Garcia, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 127, 165 CrossRef CAS.
- C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta and L. Prati, Catal. Today, 2005, 102, 203 CrossRef PubMed.
- C. Perego, A. Carati, P. Ingallina, M. A. Mantegazza and G. Bellussi, Appl. Catal., A, 2001, 221, 63 CrossRef CAS.
- J. He, C. Zhao and J. A. Lercher, J. Catal., 2014, 309, 362 CrossRef CAS PubMed.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987 CrossRef CAS PubMed.
- J. Li, L. Liu, M. Li, Y. Zhu, H. Liu, Y. Kou, J. Zhang, Y. Han and D. Ma, Energy Environ. Sci., 2014, 7, 393 CAS.
- J. P. Tessonnier, A. Villa, O. Majoulet, D. S. Su and R. Schlogl, Angew. Chem., Int. Ed., 2009, 48, 6543 CrossRef CAS PubMed.
- A. Villa, J. P. Tessonnier, O. Majoulet, D. S. Su and R. Schlogl, Chem. Commun., 2009, 4405 RSC.
- X. Tan, W. Deng, M. Liu, Q. Zhang and Y. Wang, Chem. Commun., 2009, 7179 RSC.
- J. Wang, S. Shen, B. Li, H. Lin and Y. Yuan, Chem. Lett., 2009, 38, 572 CrossRef CAS.
- B. Li, J. Wang, Y. Yuan, H. Ariga, S. Takakusagi and K. Asakura, ACS Catal., 2011, 1, 1521 CrossRef CAS.
- D. J. M. de Vlieger, L. Lefferts and K. Seshan, Green Chem., 2014, 16, 864 RSC.
- F. Clippel, M. Dusselier, R. Van Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089 CrossRef PubMed.
- S. Crossley, J. Faria, M. Shen, M. Resasco and D. E. Resasco, Science, 2010, 327, 68 CrossRef CAS PubMed.
- X. Xu, Y. Li, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987 CrossRef CAS PubMed.
- S. B. Yang, X. L. Feng, X. C. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339 CrossRef CAS PubMed.
- X. Xu, Y. Li, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987 CrossRef CAS PubMed.
- L. Wang, B. Zhang, X. Meng, D. S. Su and F.-S. Xiao, ChemSusChem, 2014, 7, 1537 CrossRef CAS PubMed.
- L. Wang, J. Zhang, S. Yang, Q. Sun, L. Zhu, Q. Wu, H. Zhang, X. Meng and F.-S. Xiao, J. Mater. Chem. A, 2013, 1, 9422 CAS.
- M. Toda, A. Takagaki, M. Okamura, J. N. Kondo, S. Hayashi, K. Domen and M. Hara, Nature, 2005, 438, 178 CrossRef CAS PubMed.
- M. Okamura, A. Takagaki, M. Toda, J. N. Kondo, T. Tatsumi, K. Domen, M. Hara and S. Hayashi, Chem. Mater., 2006, 18, 3039 CrossRef CAS.
- K. Nakajima, M. Okamura, J. N. Kondo, T. Tatsumi, K. Domen, S. Hayashi and M. Hara, Chem. Mater., 2009, 21, 186 CrossRef CAS.
- K. Nakajima and M. Hara, ACS Catal., 2012, 2, 1296 CrossRef CAS.
- M. M. Titirici, Hydrothermal Nanocarbons, in Advanced Hierarchical Nanostructured Material, Wiley, 2013 Search PubMed.
- M. M. Titirici, Green Nanostructured Carbons, in Producing Fuels and Fine Chemicals from Biomass using Nanomaterials, Taylor & Francis, 2013 Search PubMed.
- V. L. Budarin, J. H. Clark, R. Luque, D. J. Macquarrie, K. Milkowski and R. J. White, PCT Int. Patent WO 2007104798, A2 20070920 Search PubMed.
- V. L. Budarin, R. Luque, D. M. Macquarrie and J. H. Clark, Chem. – Eur. J., 2007, 13, 6914 CrossRef CAS PubMed.
- R. J. White, V. L. Budarin and J. H. Clark, ChemSusChem, 2008, 1, 408 CrossRef CAS PubMed.
- V. L. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., Int. Ed., 2006, 45, 3782 CrossRef CAS PubMed.
- M. M. Titirici, M. Antonietti and N. Baccile, Green Chem., 2008, 10, 1204 RSC.
- Y. Huang, H. Cai, D. Feng, D. Gu, Y. Deng, B. Tu, H. Wang, P. A. Webley and D. Zhao, Chem. Commun., 2008, 2641 RSC.
- S. Polarz, B. Smarsly and J. H. Schattka, Chem. Mater., 2002, 14, 2940 CrossRef CAS.
- A. Lua and T. Yang, J. Colloid Interface Sci., 2004, 274, 594 CrossRef CAS PubMed.
- J. R. Dodson, V. L. Budarin, A. J. Hunt, P. S. Shuttleworth and J. H. Clark, J. Mater. Chem. A, 2013, 1, 5203 CAS.
- R. J. White, V. Budarin, R. Luque, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 3401 RSC.
- V. Budarin, R. Luque, J. H. Clark and D. J. Macquarrie, Chem. Commun., 2007, 634 RSC.
- S. Kubo, R. J. White, N. Yoshizawa, M. Antonietti and M.-M. Titirici, Chem. Mater., 2011, 23, 4882 CrossRef CAS.
- V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2003, 125, 9403 CrossRef CAS PubMed.
- M. Karaki, A. Karout, J. Toufaily, F. Rataboul, N. Essayem and B. Lebeau, J. Catal., 2013, 305, 204 CrossRef CAS PubMed.
- J. Dhainaut, J. Dacquin, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 296 RSC.
- C. Pirez, J.-M. Caderon, J.-P. Dacquin, A. F. Lee and K. Wilson, ACS Catal., 2012, 2, 1607 CrossRef CAS.
- L. Li, T. I. Koranyi, B. F. Sels and P. P. Pescarmona, Green Chem., 2012, 14, 1611 RSC.
- L. Wang, J. Zhang, X. Wang, B. Zhang, W. Ji, X. Meng, J. Li, D. S. Su, X. Bao and F.-S. Xiao, J. Mater. Chem. A, 2014, 2, 3725 CAS.
- Y. Zhang, S. Wei, F. Liu, Y. Du, S. Liu, Y. Ji, T. Yokoi, T. Tatsumi and F.-S. Xiao, Nano Today, 2009, 4, 135 CrossRef CAS PubMed.
- F. Liu, W. Li, Q. Sun, L. Zhu, X. Meng, Y. Guo and F.-S. Xiao, ChemSusChem, 2011, 4, 1059 CrossRef CAS PubMed.
- F. Liu, L. Wang, Q. Sun, L. Zhu, X. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2012, 134, 16948 CrossRef CAS PubMed.
- E. I. Gurbuz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 125, 1308 CrossRef.
- C. Sievers, I. Musin, T. Marzialetti, M. B. V. Olarte, P. K. Agrawal and C. W. Jones, ChemSusChem, 2009, 22, 665 CrossRef PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827 CrossRef CAS PubMed.
- L. Wang, H. Wang, F. Liu, A. Zheng, J. Zhang, Q. Sun, J. P. Lewis, L. Zhu, X. Meng and F.-S. Xiao, ChemSusChem, 2014, 7, 402 CrossRef CAS PubMed.
- J. Zhang, L. Wang, F. Liu, X. Meng, J. Mao and F.-S. Xiao, Catal. Today DOI:10.1016/j.cattod.2014.04.017.
- W. E. Farneth and R. J. Gorte, Chem. Rev., 1995, 95, 615 CrossRef CAS.
- C. A. Fyfe, Y. Feng, H. Grondey, G. T. Kokotailo and H. Gies, Chem. Rev., 1991, 91, 1525 CrossRef CAS.
- K. Tarach, K. Góra-Marek, J. Tekla, K. Brylewska, J. Datka, K. Mlekodaj, W. Makowski, M. C. I. López, J. M. Triguero and F. Rey, J. Catal., 2014, 312, 46 CrossRef CAS PubMed.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS PubMed.
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249 CrossRef CAS.
- K. Tanabe and W. F. Holderich, Appl. Catal., A, 1999, 181, 399 CrossRef CAS.
- W. Holderich, M. Hesse and F. Naumann, Angew. Chem., Int. Ed., 1988, 27, 226 CrossRef.
- Y. J. Li and J. N. Armor, Appl. Catal., B, 1992, 1, L31 CrossRef CAS.
- H. Y. Chen and W. M. H. Sachtler, Catal. Today, 1998, 42, 73 CrossRef CAS.
- A. Corma, V. Fornes, S. B. Pergher, T. L. M. Maesen and J. G. Buglass, Nature, 1998, 396, 353 CrossRef CAS PubMed.
- J. C. Lavalley, Catal. Today, 1996, 27, 377 CrossRef CAS.
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D. H. Choi and R. Ryoo, Nat. Commun., 2006, 5, 718 CAS.
- J. R. Anderson, K. Foger, T. Mole, R. A. Rajadhyaksha and J. V. Sanders, J. Catal., 1979, 58, 114 CrossRef CAS.
- S. Sato, Y. Yoshihiro, H. Yahiro, N. Mizuno and M. Iwammoto, Appl. Catal., 1991, 70, L1 CrossRef CAS.
- R. M. West, M. S. Holm, S. Saravanamurugan, J. Xiong, Z. Beversdorf, E. Taarning and C. H. Christensen, J. Catal., 2010, 269, 122 CrossRef CAS PubMed.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257 CrossRef CAS PubMed.
- Y.-T. Cheng, Z. Wang, C. J. Gilbert, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 11097 CrossRef CAS PubMed.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935 CrossRef CAS.
- P. T. M. Do, J. R. McAtee, D. A. Watson and R. F. Lobo, ACS Catal., 2013, 3, 41 CrossRef CAS PubMed.
- C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 5935 CrossRef CAS PubMed.
- J. He, L. Lu, C. Zhao, D. Mei and J. A. Lercher, J. Catal., 2014, 311, 41 CrossRef CAS PubMed.
- J. He, C. Zhao, D. Mei and J. A. Lercher, J. Catal., 2014, 309, 280 CrossRef CAS PubMed.
- C. Zhao, D. M. Carmaioni and J. A. Lercher, J. Catal., 2012, 288, 92 CrossRef CAS PubMed.
- C. Zhao, S. Kasakov, J. He and J. A. Lercher, J. Catal., 2012, 296, 12 CrossRef CAS PubMed.
- R. M. Ravenelle, F. D. Schualer, A. Amico, N. Danilina, J. A. van Bokhoven, J. A. Lercher, C. W. Jones and C. Sievers, J. Phys. Chem. C, 2010, 114, 19582 CAS.
- T. Okuhara, Chem. Rev., 2002, 102, 3641 CrossRef CAS PubMed.
- P. A. Zapata, J. Faria, M. P. Ruiz, R. E. Jentoft and D. E. Resasco, J. Am. Chem. Soc., 2012, 134, 8570 CrossRef CAS PubMed.
- P. A. Zapata, Y. Huamg, M. A. Gonzalez-Borja and D. E. Resasco, J. Catal., 2013, 308, 82 CrossRef CAS PubMed.
- T. Blasco, M. A. Camblor, C. Corma and J. Perezpariente, J. Am. Chem. Soc., 1993, 115, 11806 CrossRef CAS.
- V. Choudhary, A. B. Pinar, R. F. Lobo, D. G. Vlachos and S. I. Sandler, ChemSusChem, 2013, 6, 2369 CrossRef CAS PubMed.
- T. Blasco, M. A. Camblor, P. Esteve, J. M. Guil, A. Martinez, J. A. Perdigon-Melon and S. Valencia, J. Phys. Chem. B, 1998, 102, 75 CrossRef CAS.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423 CrossRef CAS PubMed.
- A. Corma, M. E. Domine and S. Valencia, J. Catal., 2003, 215, 294 CrossRef CAS.
- A. Corma, M. E. Domine, L. Nemeth and S. Valencia, J. Am. Chem. Soc., 2002, 124, 3194 CrossRef CAS PubMed.
- A. Corma and M. Renz, Chem. Commun., 2004, 550 RSC.
- M. Boronat, A. Corma and M. Renz, J. Phys. Chem. B, 2006, 110, 21168 CrossRef CAS PubMed.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602 CrossRef CAS PubMed.
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954 CrossRef PubMed.
- M. Moliner, Y. Román-Leshkov, J. A. Labinger and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164 CrossRef CAS PubMed.
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Román-Leshkov, S. Hwang, A. Palsdottir, D. Silvermana, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727 CrossRef CAS PubMed.
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705 CrossRef CAS.
- E. Nikolla, T. Roman-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408 CrossRef CAS.
- L. Bui, H. Luo, W. R. Gunther and Y. Roman-Leshkov, Angew. Chem., Int. Ed., 2013, 52, 8022 CrossRef CAS PubMed.
- K. Na, M. Choi and R. Ryoo, Microporous Mesoporous Mater., 2013, 166, 3 CrossRef CAS PubMed.
- J. Zhu, Y. Zhu, L. Zhu, M. Rigutto, A. van der Made, C. Yang, S. Pan, L. Wang, L. Zhu, Y. Jin, Q. Sun, Q. Wu, X. Meng, D. Zhang, Y. Han, J. Li, Y. Chu, A. Zheng, S. Qiu, X. Zheng and F.-S. Xiao, J. Am. Chem. Soc., 2014, 136, 2503 CrossRef CAS PubMed.
- X. Y. Zhang, D. X. Liu, D. D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684 CrossRef CAS PubMed.
- F. Liu, T. Willhammar, L. Wang, L. Zhu, Q. Sun, X. Meng, W. Carrillo-Cabrera, X. Zou and F.-S. Xiao, J. Am. Chem. Soc., 2012, 134, 4557 CrossRef CAS PubMed.
- W. C. Yoo, S. Kumar, R. L. Penn, M. Tsapatsis and A. Stein, J. Am. Chem. Soc., 2009, 131, 12377 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |