
Efficient synthesis of quinoxalines from 2-nitroanilines and vicinal diols via a ruthenium-catalyzed hydrogen transfer strategy†
Feng
Xie
ab,
Min
Zhang
*a,
Huanfeng
Jiang
a,
Mengmeng
Chen
ab,
Wan
Lv
a,
Aibin
Zheng
a and
Xiujuan
Jian
a
aSchool of Chemistry & Chemical Engineering, South China University of Technology, Wushan Rd-381, Guangzhou 510641, People's Republic of China. E-mail: minzhang@scut.edu.cn; Fax: (+86)-020-39925999; Tel: (+86)-13424037838
bSchool of Chemical & Material Engineering, Jiangnan University, Wuxi 214122, People's Republic of China
First published on 21st August 2014
Abstract
Via a ruthenium-catalyzed hydrogen transfer strategy, we have demonstrated a one-pot method for efficient synthesis of quinoxalines from 2-nitroanilines and biomass-derived vicinal diols for the first time. In such a synthetic protocol, the diols and the nitro group serve as the hydrogen suppliers and acceptors, respectively. Hence, there is no need for the use of external reducing agents. Moreover, it has the advantages of operational simplicity, broad substrate scope and the use of renewable reactants, offering an important basis for accessing various quinoxaline derivatives.
Introduction
Concerning the gradual depletion of fossil resources, the utilization of renewable reactants for chemical production has become a key issue in sustainable chemistry. As a result, the biomass-derived alcohols have emerged as desirable candidates for various synthetic purposes: (1) condensation reactions with the use of in situ formed carbonyl intermediates via transition-metal catalysed alcohol dehydrogenation;1 (2) reduction reactions using alcohols as alternative hydrogen sources for reducing a number of organic chemicals;2 (3) in line with the principles of green chemistry, the application of alcohols as both hydrogen suppliers and coupling components for benign construction of carbon–carbon and carbon–heteroatom bonds is, in synthetic chemistry, of particular importance.3–5 In these processes, the screening of a suitable catalyst system that is compatible with both dehydrogenation of alcohols and hydrogen transfer processes is considered as the main challenging point.Quinoxaline derivatives constitute an important class of N-containing heterocycles that exhibit diverse biological activities such as antitumor,6 antiviral,7 antibacterial,8 anti-inflammatory,9 anti-HIV,10 and anticancer activities.11 Moreover, quinoxalines have been widely applied as building blocks for the preparation of dyes,12 cavitands,13 luminescent materials,14 semiconductors,15 chemically controllable switches,16 dehydroannulenes,17etc. Due to the interesting functions, the development of efficient methods for accessing quinoxalines has long been a subject of synthetic chemists. Conventionally, quinoxalines could be prepared via a double condensation of 1,2-phenylenediamines with 1,2-diketones (Scheme 1, method A).18 Other elegant contributions mainly involve the oxidative trapping of vicinal diols or α-hydroxy ketones with 1,2-diamines (method B),19,20 1,4-addition of 1,2-diamines to diazenylbutenes (method C),21 the coupling of epoxides with ene-1,2-diamines (method D),22 2-nitroanilines with phenethylamines (method E),23 alkynes or ketones with 1,2-diamines via a key oxidation process (method F),24 and the sequential reductive coupling and cyclization of polymer-linked 2-nitrophenyl carbamate with α-bromoketones (method G).25 Nevertheless, many of these methods require the addition of excessive additives, the use of special pre-functionalized or less environmentally benign halogenated reagents, which could constantly result in preparation difficulties and/or have a detrimental influence on the environment. In 2012, the Corma and Iborra group demonstrated an interesting synthesis of quinoxalines from glycols with 1,2-phenylenediamines or 1,2-dinitrobenzenes by employing heterogeneous catalysis.26 However, the use of 1,2-dinitrobenzenes need to undergo the nitro group reduction using an external high-pressure hydrogen source and the oxidative cyclization processes (method H). From the viewpoint of step-economy concern, the development of environmentally friendly shortcuts for the synthesis of quinoxalines from renewable reactants would be of important significance.
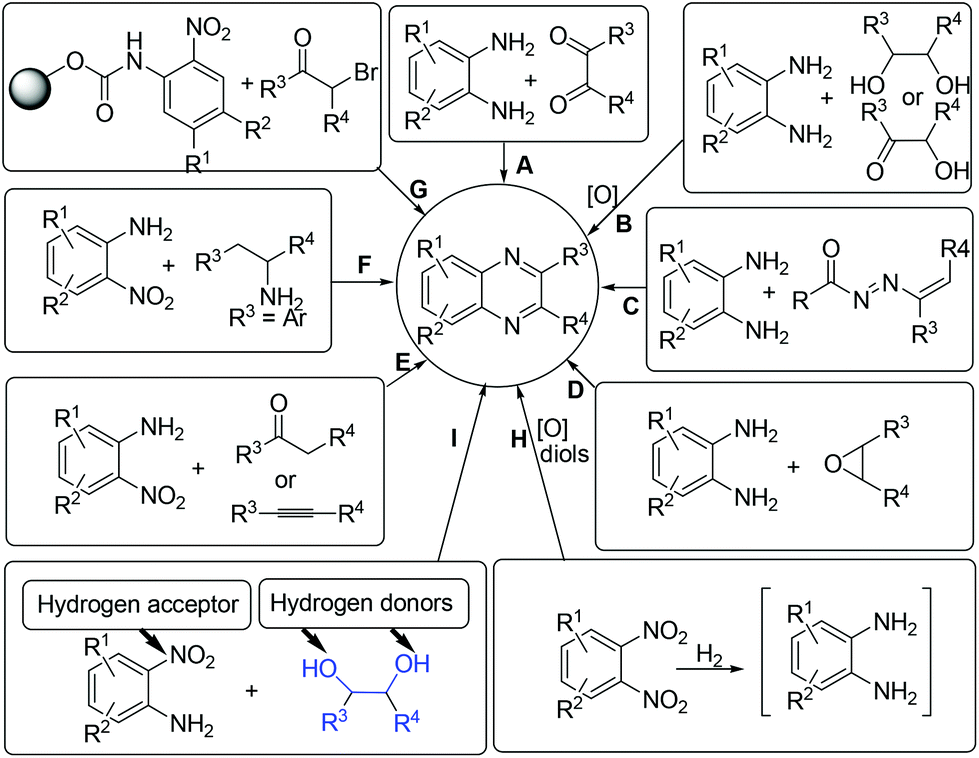 | ||
| Scheme 1 Various methods for accessing quinoxalines. | ||
Herein, via a ruthenium-catalyzed hydrogen transfer strategy, we report a straightforward method for efficient synthesis of quinoxalines from biomass-derived glycols27 and stable 2-nitroanilines for the first time. In such a synthetic protocol, the vicinal diols and the nitro group of 2-nitroanilines serve as the hydrogen donors and hydrogen acceptor, respectively. Hence, there is no need for the use of external reducing agents (Scheme 1, method I).
Results and discussion
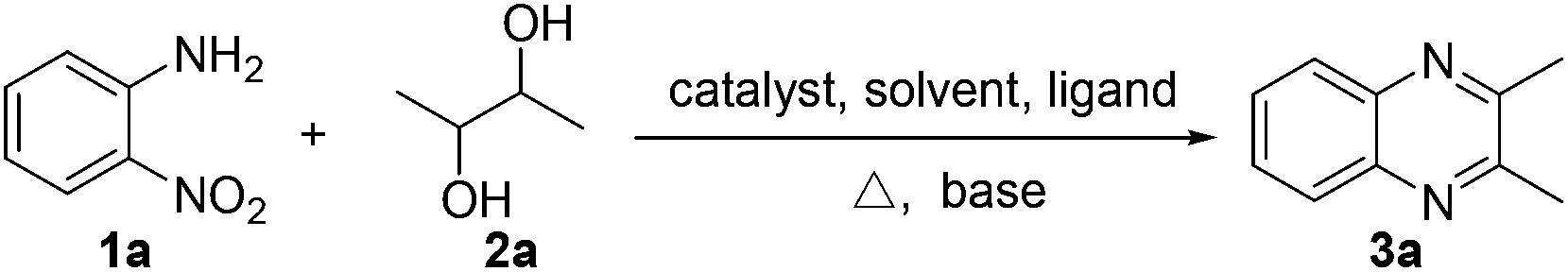
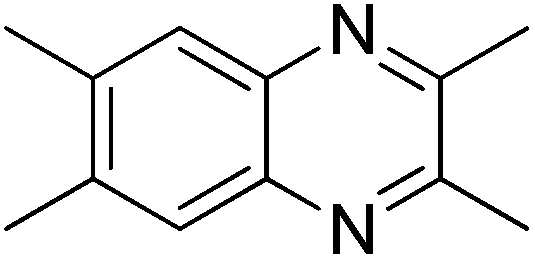
We initiated our investigations by choosing the synthesis of 2,3-dimethylquinoxaline 3a from 2-nitroaniline 1a and butane-2,3-diol 2a as a model reaction to determine different reaction parameters. First, 7 ruthenium catalysts (Scheme 2, see Cat 1–Cat 7) were tested by performing the reaction at 150 °C for 8 h using t-BuOK as the base and t-amyl alcohol as the solvent (Table 1, entries 1–7). Ru3(CO)12 (Cat 4) exhibited the highest activity in the formation of product 3a. The absence of ruthenium catalyst failed to give any desired product (Table 1, entry 8). Further investigation showed that the ligand played an important role in the reaction yield (Table 1, entry 9). By using Cat 4, the diphosphine L4 was proven to be the most effective one among various phosphine ligands tested (Scheme 2: L1–L7 and Table 1: entries 10–15). Subsequently, several inorganic and organic bases were examined, CsOH·H2O was the best choice (Table 1, entries 16–20). Other polar and less-polar solvents were proven to be inferior to t-amyl alcohol (Table 1, entry 21). Finally, we employed the combination of Cat 4, L4, CsOH·H2O and t-amyl alcohol, a decrease of reaction temperature led to a decreased product yield (Table 1, entry 22). And 50 mol% of the base was sufficient to obtain a desirable yield (Table 1, entry 23). An increase in the diol amount to 2 mmol resulted in the best product yield (Table 1, entry 24). Hence, the optimal reaction conditions can be as indicated in entry 24 of Table 1 (Table 1, entry 24).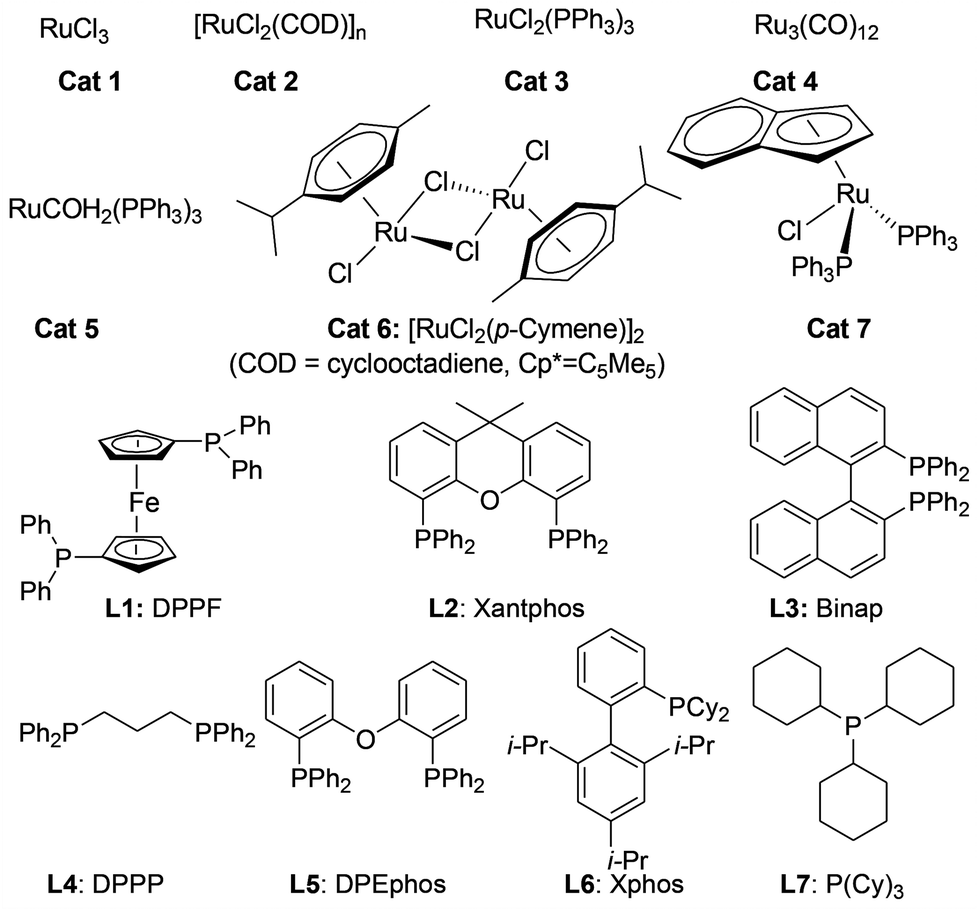 | ||
| Scheme 2 Catalysts and ligands employed for the optimization of reaction conditions. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | 3a, Yieldb % |
| a Reaction conditions: all reactions were carried out under a nitrogen atmosphere by using 1a (0.5 mmol), 2a (3 equiv.), catalyst (1 mol%), ligand (3 mol%), solvent (1.5 mL), temperature (150 °C), base (20 mol%), reaction time (8 h). b GC yield using hexadecane as an internal standard. c Yields are with respect to toluene, DMSO and diglyme used as the reaction solvents, respectively. d Reaction temperature (140 °C). e Base (50 mol%). f Base (70 mol%). g 2a: (4 equiv.), base (50 mol%). | ||||
| 1 | Cat 1 | L1 | t-BuOK | 12 |
| 2 | Cat 2 | L1 | t-BuOK | 16 |
| 3 | Cat 3 | L1 | t-BuOK | 25 |
| 4 | Cat 4 | L1 | t-BuOK | 62 |
| 5 | Cat 5 | L1 | t-BuOK | 45 |
| 6 | Cat 6 | L1 | t-BuOK | 31 |
| 7 | Cat 7 | L1 | t-BuOK | 14 |
| 8 | — | L1 | t-BuOK | — |
| 9 | Cat 4 | — | t-BuOK | 8 |
| 10 | Cat 4 | L2 | t-BuOK | 23 |
| 11 | Cat 4 | L3 | t-BuOK | 57 |
| 12 | Cat 4 | L4 | t-BuOK | 68 |
| 13 | Cat 4 | L5 | t-BuOK | 61 |
| 14 | Cat 4 | L6 | t-BuOK | 16 |
| 15 | Cat 4 | L7 | t-BuOK | <10 |
| 16 | Cat 4 | L4 | K2CO3 | 64 |
| 17 | Cat 4 | L4 | Cs2CO3 | 75 |
| 18 | Cat 4 | L4 | CsOH·H2O | 79 |
| 19 | Cat 4 | L4 | KOH | 63 |
| 20 | Cat 4 | L4 | NEt3 | 22 |
| 21 | Cat 4 | L4 | CsOH·H2O | [65, 45, 72]c |
| 22 | Cat 4 | L4 | CsOH·H2O | 62d |
| 23 | Cat 4 | L4 | CsOH·H2O | 83e, 83f |
| 24 | Cat 4 | L4 | CsOH·H2O | 87g |
With the availability of optimized reaction conditions, we then examined the generality of the synthetic protocol. First, we focused on the synthesis of quinoxaline 3 by testing a variety of 2-nitroanilines 1 with symmetrical vicinal diols 2. As shown in Table 2, both alkyl (i.e.2a, 2b) and aryl (i.e.2c) substituted vicinal diols underwent smooth cyclization to afford the 2,3-dialkyl and 2,3-diaryl quinoxalines in moderate to excellent yields upon isolation (Table 2, entries 1–16). The ortho-substituent of 2-nitroaniline 1d has little influence in affording the desired product 3d (Table 2, entry 4). Cyclohexane-1,2-diol 2b resulted in the tricyclic products efficiently (entries 8–12), these examples demonstrate the potential of the methodology for further construction of polycyclic products. Interestingly, ethylene glycol can also be applied for the preparation of 2,3-non-substituted products in reasonable yields (entries 17–19). Among all the examples examined, it was found that the electronic properties of the substituents on the aryl ring of substrate 1 influenced the product yields significantly. Specially, the electron-donating groups (i.e., –Me, –OMe) containing 2-nitroanilines (Table 2, entries 2–4, 9, 10, 14, 17 and 18) afforded the products in higher yield than the electron-deficient ones (i.e., –Cl, –F) (Table 2, entries 5, 6, 11, 15 and 19). This phenomenon can be rationalized as the electron-donating groups could enhance the nucleophilicity of the anilines 1, thus favoring the imination step of the annulation process. It is noteworthy that owing to the aryl groups are ortho to the nitrogen atom of the quinoxalines, the 2,3-diaryl quinoxalines could be applied as the C^N ligands for the preparation of organometallic complexes or materials28 (Table 2, entries 13–16).
|
|
||||
|---|---|---|---|---|
| Entry | 1 | 2 | Product 3 | Yieldb % |
| a Reaction conditions: all reactions were carried out under a nitrogen atmosphere by using 1a (0.5 mmol), 2a (4 equiv.), catalyst (1 mol%), ligand (3 mol%), solvent (1.5 mL), temperature (150 °C), base (50 mol%), reaction time (8 h). b Isolated yield. c Reaction time (12 h). | ||||
| 1 | 1a | 2a |

|
3a, 82 |
| 2 | 1b | 2a |

|
3b, 86 |
| 3 | 1c | 2a |
|
3c, 71 |
| 4 | 1d | 2a |
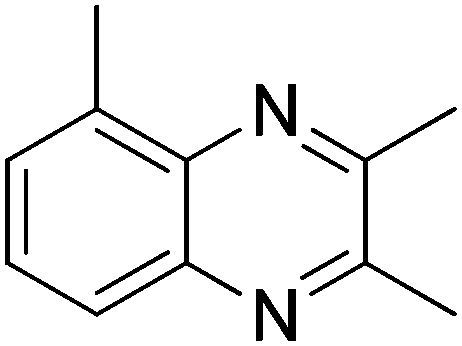
|
3d, 80 |
| 5 | 1e | 2a |
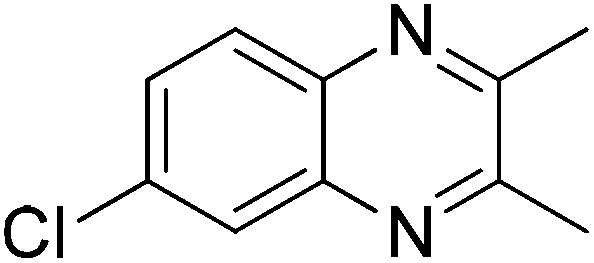
|
3e, 68 |
| 6 | 1f | 2a |
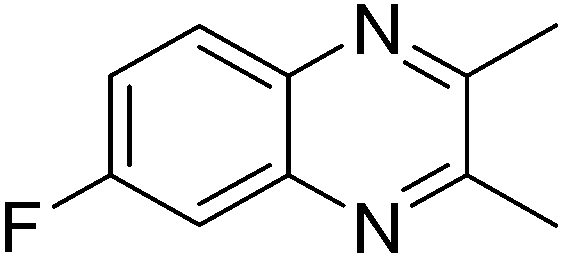
|
3f, 61 |
| 7 | 1g | 2a |

|
3g, 45c |
| 8 | 1a | 2b |

|
3h, 79 |
| 9 | 1b | 2b |
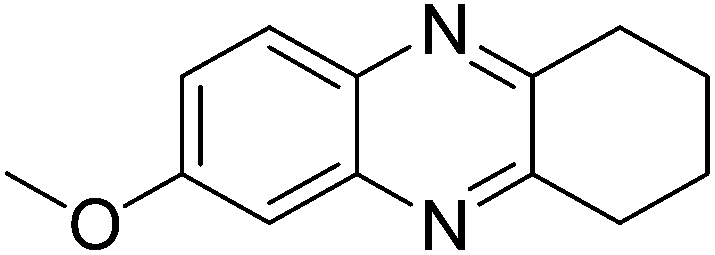
|
3i, 83 |
| 10 | 1c | 2b |

|
3j, 84 |
| 11 | 1e | 2b |
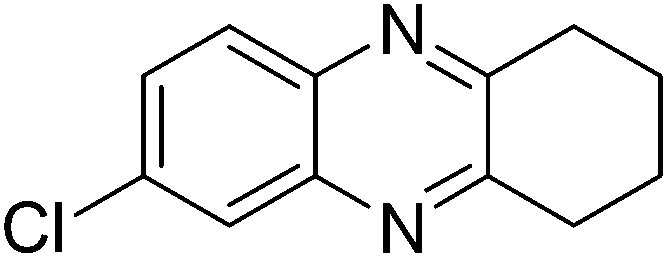
|
3k, 56 |
| 12 | 1g | 2b |
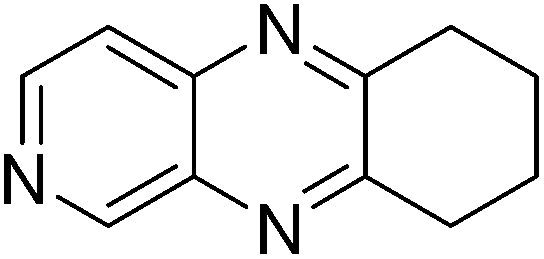
|
3l, 43c |
| 13 | 1a | 2c |

|
3m, 84 |
| 14 | 1b | 2c |

|
3n, 87 |
| 15 | 1e | 2c |
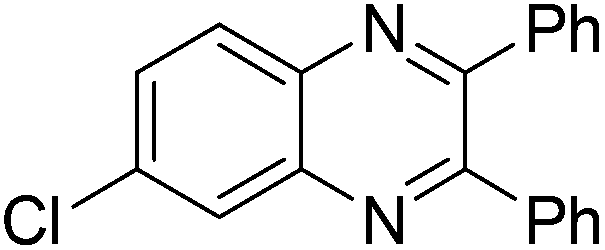
|
3o, 70 |
| 16 | 1g | 2c |

|
3p, 63c |
| 17 | 1b | 2d |

|
3q, 65 |
| 18 | 1c | 2d |
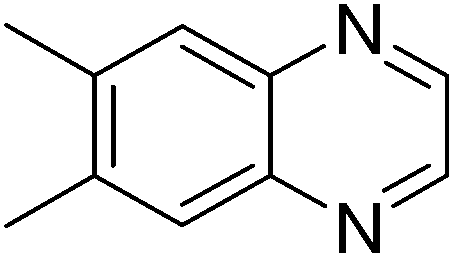
|
3r, 68 |
| 19 | 1e | 2d |
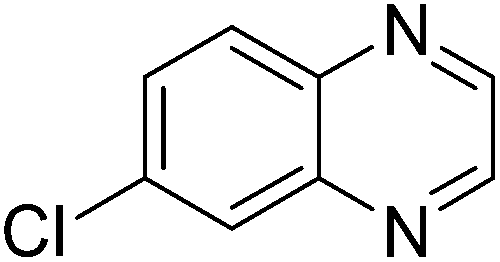
|
3s, 58 |
Subsequently, we turned our attention to employ unsymmetrical vicinal diols with our synthetic protocol. Representative substrates such as 1-phenylethane-1,2-diol 2e and propane-1,2-diol 2f in combination with various 2-nitroanilines were tested. All the reactions underwent efficient cyclization to afford the desired products in moderate to good isolated yields (Table 3, entries 1–9). Similar to the results described in Table 2, the electron-rich 2-nitroanilines could give the products in relatively higher yields (Table 3, entries 2, 4 and 7) than the electron-poor ones (Table 3, entries 3 and 5). Based on 1H-NMR analysis, the reactions of 4-methoxy-2-nitrobenzenamine 1b and 4-chloro-2-nitrobenzenamine 1e with 2e gave two regioisomers in ratios of 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :57 and 52:48, respectively (Table 3, entries 4 and 5). Interestingly, glycerol 2g could also be transformed in combination with 2-nitroanilines into the 2-methyl quinoxalines in reasonable yields (Table 3, entries 8 and 9), indicating that glycerol can be utilized as an alternative of propane-1,2-diol 2f.
:57 and 52:48, respectively (Table 3, entries 4 and 5). Interestingly, glycerol 2g could also be transformed in combination with 2-nitroanilines into the 2-methyl quinoxalines in reasonable yields (Table 3, entries 8 and 9), indicating that glycerol can be utilized as an alternative of propane-1,2-diol 2f.
|
|
||||
|---|---|---|---|---|
| Entry | 1 | 2 | Product 3 | Yieldb (%) |
| a Reaction conditions: all reactions were carried out under a nitrogen atmosphere by using 1a (0.5 mmol), 2a (4 equiv.), catalyst (1 mol%), ligand (3 mol%), solvent (1.5 mL), temperature (150 °C), base (50 mol%), reaction time (8 h). b Isolated yield. c Reaction time (12 h). | ||||
| 1 | 1a | 2e |
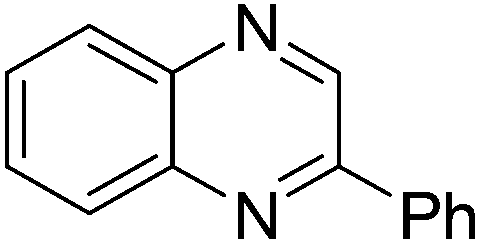
|
3t, 78 |
| 2 | 1c | 2e |
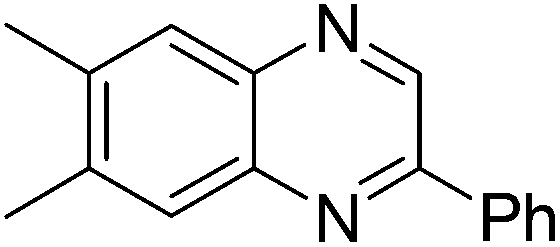
|
3u, 70 |
| 3 | 1h | 2e |
|
3v, 40c |
| 4 | 1b | 2e |
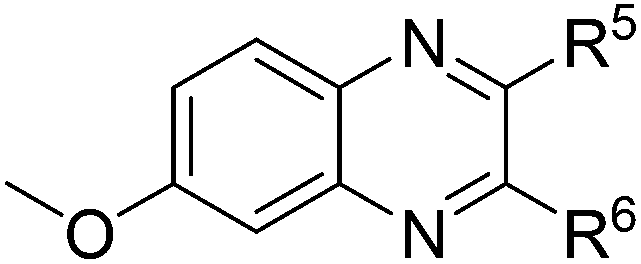
|
(3w:3w′ = 43:57), 75 |
| 5 | 1e | 2e |
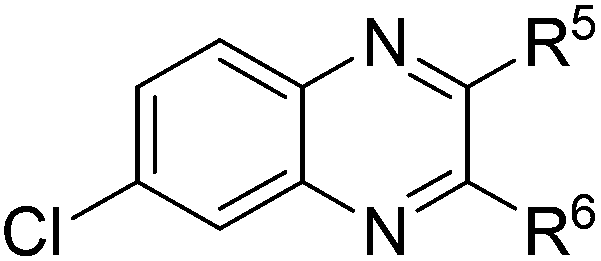
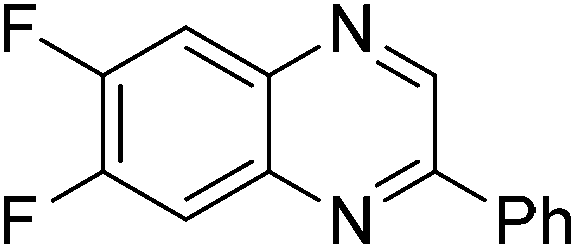
|
(3x:3x′ = 52:48), 61 |
| 6 | 1a | 2f |
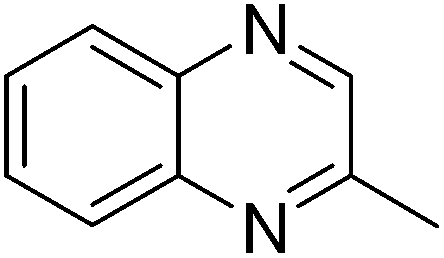
|
3y, 74 |
| 7 | 1c | 2f |
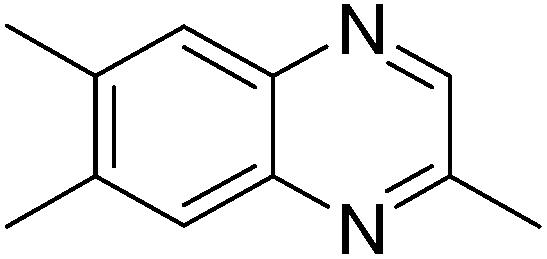
|
3z, 69 |
| 8 | 1a | 2g |

|
3y, 36c |
| 9 | 1c | 2g |
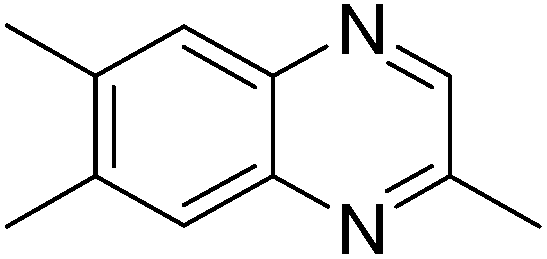
|
3z, 38c |

Upon the GC and GC-MS analyses, it was found that the vicinal diols undergo partial decomposition to form aldehydes under the standard reaction conditions. It is noteworthy that the aldehydes can be easily trapped by 1,2-phenylenediamine to form benzimidazoles.24 Interestingly, in all of our tested examples (Tables 2 and 3), we did not observe any benzimidazole by-products. Further, the reaction of 1a and 2a was interrupted after 3 h to analyze the reaction intermediates. We detected only the product 3a in 42% yield without observation of any 1,2-phenylenediamine (Scheme 3, eqn (1)). Moreover, the reaction of 4-chlorobenzene-1,2-diamine 1e′ with 2e gave products 3x and 3x′ in a ratio of 30:70 upon 1H-NMR analysis (Scheme 3, eqn (2)), which is inconsistent with the result of the reaction of 1e with 2e (Table 3, entry 5). These results suggest that the reactions involving 1,2-phenylenediamine intermediates are less likely, and the imination of the amino group of 2-nitroanilines should occur prior to the reduction of the nitro group.
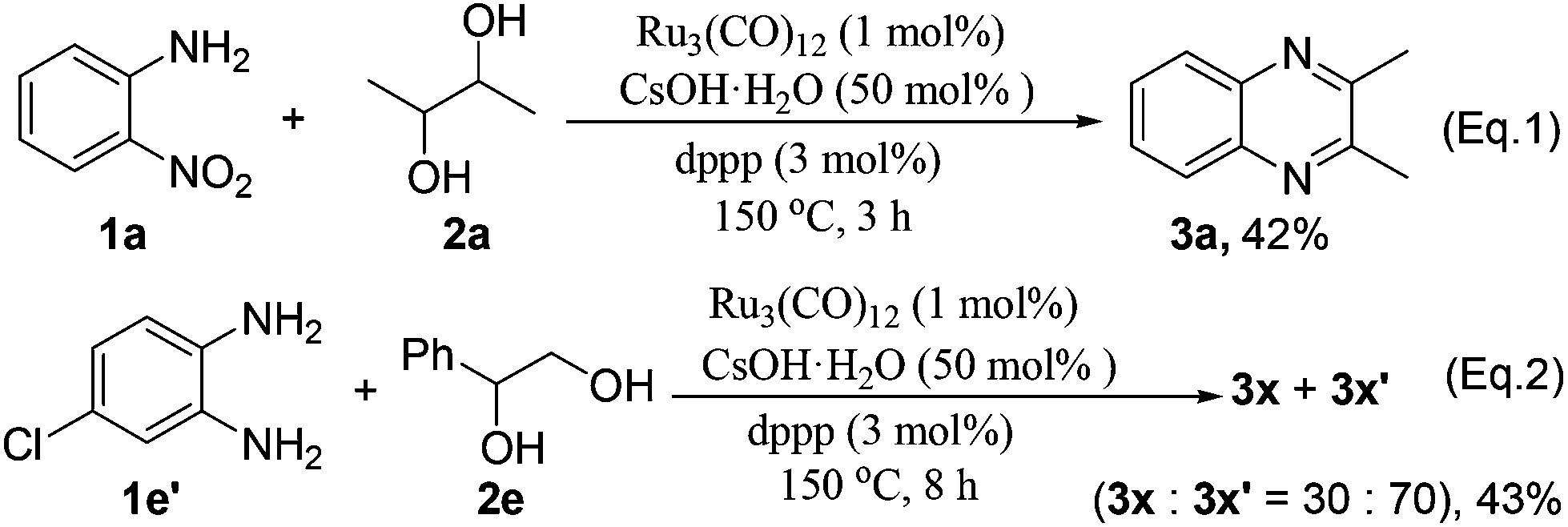 | ||
| Scheme 3 Verification experiments. | ||
On the basis of the above-described results as well as the related processes,19,20,24 a possible reaction pathway is depicted in Scheme 4, which comprises the following tandem sequences: (1) the reaction initiates with the dehydrogenation of vicinal diol 2via cooperative actions of the ruthenium catalyst and base;3 (2) then, the imination of 2-nitroaniline 1 gave α-hydroxy imine A1 or A2; (3) the transfer hydrogenation of the nitro group and tautomerization result in intermediate A3 or A4; (4) finally, the intramolecular condensation of A3 or A4 and dehydrogenative aromatization would afford desired product 3 or 3′ (Scheme 4).
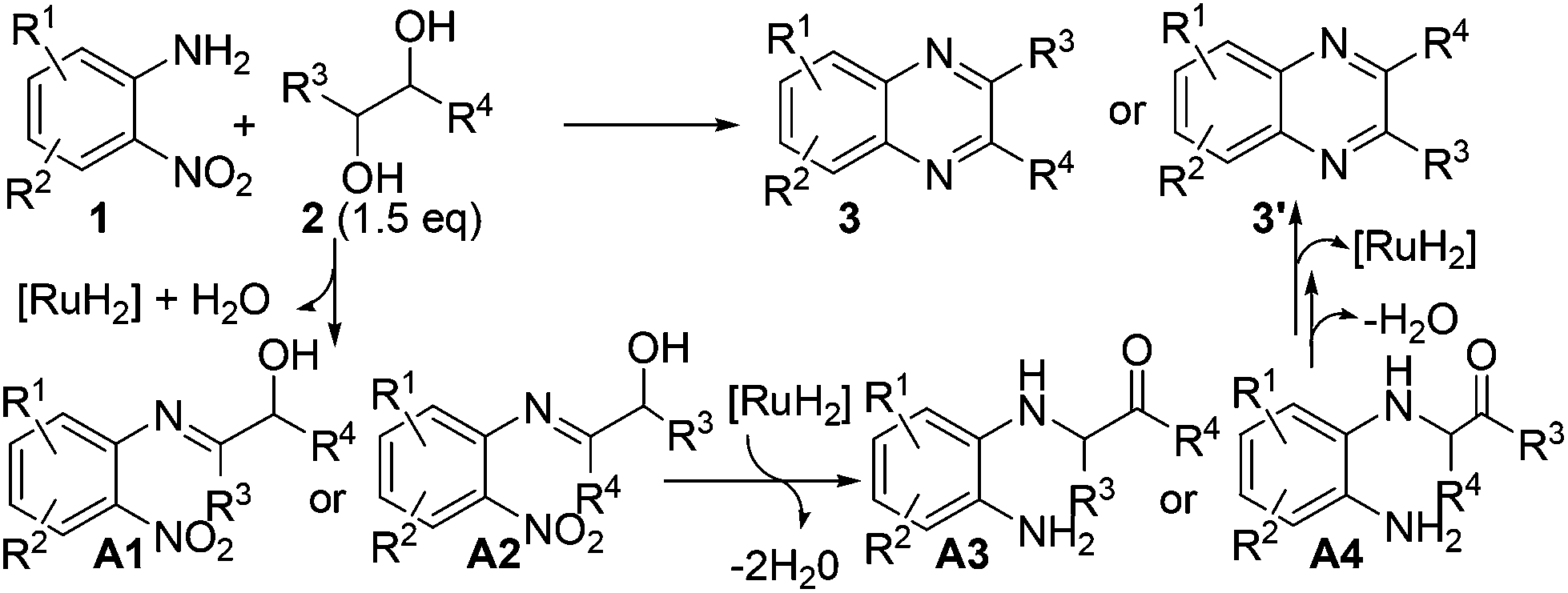 | ||
| Scheme 4 Possible pathway for the formation of quinoxalines. | ||
Experimental
General information
All the obtained products were characterized by melting points (m.p.), 1H-NMR, 13C-NMR, infrared spectra (IR), and mass spectra (MS), the NMR spectra of the known compounds were found to be identical with the ones reported in the literature. Melting points were measured on an Electrothermal SGW-X4 microscope digital melting point apparatus and are uncorrected; IR spectra were recorded on a FTLA2000 spectrometer; 1H-NMR and 13C-NMR spectra were obtained on Bruker-400; mass spectra were recorded on Trace DSQ GC/MS. Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns are described as singlet (s), doublet (d), triplet (t), multiplet (m); TLC was performed using commercially prepared 100–400 mesh silica gel plates (GF254), and visualization was effected at 254 nm; All the reagents were purchased from commercial sources (J&KChemic, TCI, Fluka, Acros, SCRC), and used without further purification.Typical procedure for synthesis of 2,3-dimethylquinoxaline 3a
[Ru3(CO)12] (3.2 mg, 0.005 mmol), dppp (6.2 mg, 0.015 mmol), CsOH·H2O (37 mg, 0.25 mmol), and 2-nitroaniline (1a; 69 mg, 0.5 mmol) were added successively to a Schlenk tube (50 mL) equipped with a magnetic stirrer bar, and the pressure tube was then purged. Under nitrogen atmosphere 2, 3-butanediol (2a; 180 mg, 2.0 mmol), and tert-amyl alcohol (1.5 mL) were added. The reaction mixture was heated at 150 °C for 8 h in a sealed tube under a nitrogen atmosphere. After cooling to room temperature, the solvent was removed under vacuum, and then it was directly purified by preparative TLC on silica, eluting with petroleum ether (60–90 °C) : ethyl acetate (12:1) to give 2,3-dimethylquinoxaline (3a) as a brown solid (62 mg, 82%).
Conclusions
In conclusion, we have demonstrated a new and straightforward method for efficient synthesis of quinoxalines via a ruthenium-catalyzed hydrogen-transfer strategy. By employing a commercially available catalyst system [Ru3(CO)12/dppp/CsOH·H2O], different 2-nitroanilines were efficiently converted in combination with a variety of biomass-derived glycols into various substituted products in moderate to excellent isolated yields. The synthetic protocol has the advantages that there is no need for the use of external reducing agents, and has operational simplicity, broad substrate scope and the utilization of renewable reactants, offering an important basis for accessing quinoxaline derivatives.Acknowledgements
The authors are grateful to the funds of the “National Natural Science Foundation of China (21472052 and 21101080)”, “Fundamental Research Funds for the Central Universities of China (2014ZZ0047)”, and “Distinguished talent program of SCUT”.Notes and references
- C. Gunanathan and D. Milstein, Science, 2013, 341, 249 CrossRef CAS PubMed.
- (a) W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS PubMed; (b) G. Wienhoefer, F. A. Westerhaus, K. Junge and M. Beller, J. Organomet. Chem., 2013, 744, 156–159 CrossRef CAS PubMed; (c) S. Werkmeister, C. Bornschein, K. Junge and M. Beller, Chem. – Eur. J., 2013, 19, 4437–4440 CrossRef CAS PubMed; (d) R. V. Jagadeesh, G. Wienhoefer, F. A. Westerhaus, A.-E. Surkus, H. Junge, K. Junge and M. Beller, Chem. – Eur. J., 2011, 17, 14375–14379 CrossRef CAS PubMed; (e) S. Horn and M. Albrecht, Chem. Commun., 2011, 47, 8802–8804 RSC; (f) R. L. Patman, M. R. Chaulagain, V. M. Williams and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 2066–2067 CrossRef CAS PubMed; (g) D. Gnanamgari, A. Moores, E. Rajaseelan and R. H. Crabtree, Organometallics, 2007, 26, 1226–1230 CrossRef CAS.
- For reviews on borrowing-hydrogen methodology, see: (a) R. H. Crabtree, Organometallics, 2011, 30, 17–19 CrossRef CAS; (b) G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed; (c) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed; (d) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC; (e) G. W. Lamb and J. M. J. Williams, Chim. Oggi, 2008, 26, 17–19 CAS; (f) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS PubMed; (g) G. Guillena, D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358–2364 CrossRef CAS PubMed.
- Selected examples on C–C or C–N bond formations via hydrogen transfer: (a) X. Cui, C. Zhang, F. Shi and Y. Deng, Chem. Commun., 2012, 48, 9391–9393 RSC; (b) A. Zanardi, J. A. Mata and E. Peris, Chem. – Eur. J., 2010, 16, 10502–10506 CrossRef CAS PubMed; (c) S. Michlik and R. Kempe, Chem. – Eur. J., 2010, 16, 13193–13198 CrossRef CAS PubMed; (d) B. Blank and R. Kempe, J. Am. Chem. Soc., 2010, 132, 924–925 CrossRef CAS PubMed; (e) B. Blank, S. Michlik and R. Kempe, Chem. – Eur. J., 2009, 15, 3790–3799 CrossRef CAS PubMed; (f) M. Zhang, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 597–601 CrossRef CAS PubMed; (g) M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384–11388 CrossRef CAS PubMed; (h) D. Srimani, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 4012–4015 CrossRef CAS PubMed; (i) C. Gunanathan and D. Milstein, Science, 2013, 341, 249 CrossRef CAS PubMed.
- Selected examples on sequential hydrogen transferring reduction and cyclization: (a) L. Tang, X. Guo, Y. Yang, Z. Zha and Z. Wang, Chem. Commun., 2014, 50, 6145–6148 RSC; (b) H. Wang, X. Cao, F. Xiao, S. Liu and G.-J. Deng, Org. Lett., 2013, 15, 4900–4903 CrossRef CAS PubMed; (c) F. Xiao, Y. Liu, C. Tang and G.-J. Deng, Org. Lett., 2012, 14, 984–987 CrossRef CAS PubMed; (d) M. Wu, X. Hu, J. Liu, Y. Liao and G.-J. Deng, Org. Lett., 2012, 14, 2722–2725 CrossRef CAS PubMed; (e) Y. Liu, W. Chen, C. Feng and G. Deng, Chem. – Asian J., 2011, 6, 1142–1146 CrossRef CAS PubMed; (f) S. Liu, R. Chen and G.-J. Deng, Chem. Lett., 2011, 40, 489–491 CrossRef CAS; (g) C. Feng, Y. Liu, S. Peng, Q. Shuai, G. Deng and C.-J. Li, Org. Lett., 2010, 12, 4888–4891 CrossRef CAS PubMed.
- S. T. Hazeldine, L. Polin, J. Kushner, J. Paluch, K. White, M. Edelstein, E. Palomino, T. H. Corbett and J. P. Horwitz, J. Med. Chem., 2001, 44, 1758–1776 CrossRef CAS PubMed.
- (a) N. S. Hari Narayana Moorthy, E. Manivannan, C. Karthikeyan and P. Trivedi, Mini-Rev. Med. Chem., 2013, 13, 1415–1420 CrossRef CAS; (b) F. Rong, S. Chow, S. Yan, G. Larson, Z. Hong and J. Wu, Bioorg. Med. Chem. Lett., 2007, 17, 1663–1666 CrossRef CAS PubMed.
- A. K. Parhi, Y. Zhang, K. W. Saionz, P. Pradhan, M. Kaul, K. Trivedi, D. S. Pilch and E. J. LaVoie, Bioorg. Med. Chem. Lett., 2013, 23, 4968–4974 CrossRef CAS PubMed.
- R. A. Smits, H. D. Lim, A. Hanzer, O. P. Zuiderveld, E. Guaita, M. Adami, G. Coruzzi, R. Leurs and I. J. P. de Esch, J. Med. Chem., 2008, 51, 2457–2467 CrossRef CAS PubMed.
- (a) X. Hui, J. Desrivot, C. Bories, P. M. Loiseau, X. Franck, R. Hocquemiller and B. Figadere, Bioorg. Med. Chem. Lett., 2006, 16, 815–820 CrossRef CAS PubMed; (b) Y. B. Kim, Y. H. Kim, J. Y. Park and S. K. Kim, Bioorg. Med. Chem. Lett., 2004, 14, 541–544 CrossRef CAS PubMed.
- F. A. R. Rodrigues, I. d. S. Bomfim, B. C. Cavalcanti, C. d. O. Pessoa, J. L. Wardell, S. M. S. V. Wardell, A. C. Pinheiro, C. R. Kaiser, T. C. M. Nogueira, J. N. Low, L. R. Gomes and M. V. N. de Souza, Bioorg. Med. Chem. Lett., 2014, 24, 934–939 CrossRef CAS PubMed.
- E. D. Brock, D. M. Lewis, T. I. Yousaf and H. H. Harper, The Procter and Gamble Company, USA, WO9951688, 1999.
- J. L. Sessler, H. Maeda, T. Mizuno, V. M. Lynch and H. Furuta, J. Am. Chem. Soc., 2002, 124, 13474–13479 CrossRef CAS PubMed.
- (a) B. D. Lindner, Y. Zhang, S. Hoefle, N. Berger, C. Teusch, M. Jesper, K. I. Hardcastle, X. Qian, U. Lemmer, A. Colsmann, U. H. F. Bunz and M. Hamburger, J. Mater. Chem. C, 2013, 1, 5718–5724 RSC; (b) K. R. J. Thomas, M. Velusamy, J. T. Lin, C.-H. Chuen and Y.-T. Tao, Chem. Mater., 2005, 17, 1860–1866 CrossRef CAS.
- S. Dailey, W. J. Feast, R. J. Peace, I. C. Sage, S. Till and E. L. Wood, J. Mater. Chem., 2001, 11, 2238–2243 RSC.
- M. J. Crossley and L. A. Johnston, Chem. Commun., 2002, 1122–1123 RSC.
- S. Ott and R. Faust, Synlett, 2004, 1509–1512 CAS.
- (a) M. Ayaz, Z. Xu and C. Hulme, Tetrahedron Lett., 2014, 55, 3406–3409 CrossRef CAS PubMed; (b) C. Srinivas, C. N. S. S. P. Kumar, V. J. Rao and S. Palaniappan, J. Mol. Catal. A: Chem., 2007, 265, 227–230 CrossRef CAS PubMed; (c) S. V. More, M. N. V. Sastry and C.-F. Yao, Green Chem., 2006, 8, 91–95 RSC; (d) R. S. Bhosale, S. R. Sarda, S. S. Ardhapure, W. N. Jadhav, S. R. Bhusare and R. P. Pawar, Tetrahedron Lett., 2005, 46, 7183–7186 CrossRef CAS PubMed; (e) Z. Zhao, D. D. Wisnoski, S. E. Wolkenberg, W. H. Leister, Y. Wang and C. W. Lindsley, Tetrahedron Lett., 2004, 45, 4873–4876 CrossRef CAS PubMed.
- (a) T. Hille, T. Irrgang and R. Kempe, Chem. – Eur. J., 2014, 20, 5569–5572 CrossRef CAS PubMed; (b) C. S. Cho and S. G. Oh, Tetrahedron Lett., 2006, 47, 5633–5636 CrossRef CAS PubMed.
- (a) V. Jeena and R. S. Robinson, Tetrahedron Lett., 2014, 55, 642–645 CrossRef CAS PubMed; (b) S. Sithambaram, Y. Ding, W. Li, X. Shen, F. Gaenzler and S. L. Suib, Green Chem., 2008, 10, 1029–1032 RSC; (c) R. S. Robinson and R. J. K. Taylor, Synlett, 2005, 1003–1005 CAS; (d) S. Y. Kim, K. H. Park and Y. K. Chung, Chem. Commun., 2005, 1321–1323 RSC; (e) S. A. Raw, C. D. Wilfred and R. J. K. Taylor, Org. Biomol. Chem., 2004, 2, 788–796 RSC.
- D. Aparicio, O. A. Attanasi, P. Filippone, R. Ignacio, S. Lillini, F. Mantellini, F. Palacios and J. M. de Santos, J. Org. Chem., 2006, 71, 5897–5905 CrossRef CAS PubMed.
- (a) M. M. Ibrahim, D. Grau, F. Hampel and S. B. Tsogoeva, Eur. J. Org. Chem., 2014, 1401–1405 CrossRef CAS PubMed; (b) S. Antoniotti and E. Dunach, Tetrahedron Lett., 2002, 43, 3971–3973 CrossRef CAS.
- T. B. Nguyen, P. Retailleau and A. Al-Mourabit, Org. Lett., 2013, 15, 5238–5241 CrossRef CAS PubMed.
- (a) Y. Xu and X. Wan, Tetrahedron Lett., 2013, 54, 642–645 CrossRef CAS PubMed; (b) S. Shi, T. Wang, W. Yang, M. Rudolph and A. S. K. Hashmi, Chem. – Eur. J., 2013, 19, 6576–6580 CrossRef CAS PubMed; (c) S. Okumura, Y. Takeda, K. Kiyokawa and S. Minakata, Chem. Commun., 2013, 49, 9266–9268 RSC; (d) C.-Y. Chen, W.-P. Hu, M.-C. Liu, P.-C. Yan, J.-J. Wang and M.-I. Chung, Tetrahedron, 2013, 69, 9735–9741 CrossRef CAS PubMed; (e) C. Zhang, Z. Xu, L. Zhang and N. Jiao, Tetrahedron, 2012, 68, 5258–5262 CrossRef CAS PubMed.
- S. K. Singh, P. Gupta, S. Duggineni and B. Kundu, Synlett, 2003, 2147–2150 CAS.
- M. J. Climent, A. Corma, J. C. Hernandez, A. B. Hungria, S. Iborra and S. Martinez-Silvestre, J. Catal., 2012, 292, 118–129 CrossRef CAS PubMed.
- (a) V. Lehr, M. Sarlea, L. Ott and H. Vogel, Catal. Today, 2007, 121, 121–129 CrossRef CAS PubMed; (b) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed; (c) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- (a) B. Pena, A. David, C. Pavani, M. S. Baptista, J.-P. Pellois, C. Turro and K. R. Dunbar, Organometallics, 2014, 33, 1100–1103 CrossRef CAS; (b) J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Chem. Sci., 2013, 4, 1234–1244 RSC; (c) Y. P. Dong, M. J. Shi, B. H. Tong and Q. F. Zhang, Luminescence, 2012, 27, 414–418 CrossRef CAS PubMed; (d) W. Yang, H. Fu, Q. Song, M. Zhang and Y. Ding, Organometallics, 2011, 30, 77–83 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Images of 1H and 13C NMR of all products. See DOI: 10.1039/c4gc01316f |
| This journal is © The Royal Society of Chemistry 2015 |