
Chemo-enzymatic synthesis of key intermediates (S)-γ-hydroxymethyl-α,β-butenolide and (S)-γ-hydroxymethyl-γ-butyrolactone via lipase-mediated Baeyer–Villiger oxidation of levoglucosenone†
A. L.
Flourat‡
abc,
A. A. M.
Peru‡
abc,
A. R. S.
Teixeira
ade,
F.
Brunissen
abc and
F.
Allais
*afg
aAgroParisTech, Chaire Agro-Biotechnologies Industrielles (ABI), 247 rue Paul Vaillant-Couturier F-51100, Reims, France. E-mail: florent.allais@agroparistech.fr
bAgroParisTech, UMR 1318 IJPB, Route de Saint-Cyr F-78026, Versailles, France
cINRA, UMR 1318 IJPB, Route de Saint-Cyr F-78026, Versailles, France
dAgroParisTech, UMR 1145 GENIAL, 1 avenue des Olympiades, F-91744 Massy, France
eINRA, UMR 1145 GENIAL, 1 avenue des Olympiades, F-91744 Massy, France
fAgroParisTech, UMR 782 GMPA, Site de Grignon, F-78850 Thiverval-Grignon, France
gINRA, UMR 782 GMPA, Site de Grignon, F-78850 Thiverval-Grignon, France
First published on 5th September 2014
Abstract
Levoglucosenone (LGO), a valuable chiral platform chemical that can be efficiently produced from catalytic fast pyrolysis of cellulose, has been efficiently converted into optically pure (S)-γ-hydroxymethyl-α,β-butenolide (HBO) using a two-step sequence involving a lipase-mediated Baeyer–Villiger oxidation and an acid hydrolysis. In the same fashion, (S)-γ-hydroxymethyl-γ-butyrolactone (2H-HBO) was successfully obtained through a three-step sequence (Baeyer–Villiger, palladium-catalysed hydrogenation and acid hydrolysis). The use of solid buffers in the lipase-mediated Baeyer–Villiger oxidation has proved beneficial in two ways: not only the reaction time and the enzymatic load were both reduced four-fold (from 8 to 2 hours and 464 to 113 U mmol−1) to reach conversions ≥83%, but solid buffers also prevented lipase from denaturation, thus preserving its enzymatic activity and allowing its use for further oxidation cycles.
Introduction
Faced with the inevitable decline of fossil fuels, and also more and more drastic regulations such as REACH, there is growing interest in the use of renewable sources as substitutes for petro-based resources. In this context, over the last few decades, much effort has been made to develop efficient methods to produce valuable chemicals from biomass, thus offering novel sustainable procedures as alternative routes to traditional synthetic pathways.1 In this context, lignocellulose, one of the most sustainable carbon sources for producing valuable synthons, is particularly of interest since its catalytic fast pyrolysis (CFP) has been deeply studied for the production of target products in high yields.2 Levoglucosenone (LGO), one of the products that can be produced from CFP, is recognized as a valuable product for the pharmaceutical industry for two primary reasons: (i) the preservation of one of the natural chiral centers in cellulose that can be transferred into chiral therapeutic agents able to inhibit enzymatic reactions in viruses and other pathogens, (ii) the α,β-unsaturated ketone and protected aldehyde functionality in the levoglucosenone molecule offers the organic chemist a wide range of synthetic opportunities. Following these considerations, LGO has been intensively used in the organic synthesis of several chemical compounds, such as natural products, nucleosides, anticancer drugs, and building blocks.3The phosphoric acid-catalysed pyrolysis process has proved to increase the selectivity towards LGO up to 30% when used on glucose, cellulose or birch and pine wood.4 Recently, the use of zeolites as acid catalysts improved LGO selectivity up to 40% of the produced oxygenated products, while increasing the production of other highly-valuable platform chemicals, such as furfural.5 Interestingly, a new and feasible procedure has been recently reported for recovering LGO from pyrolytic liquids by distillation.6
LGO is a synthon of choice for the production of several valuable products (Fig. 1).3,7 Among the different chemical intermediates that have been synthesized from LGO, the unsaturated chiral γ-lactone (S)-γ-hydroxymethyl-α,β-butenolide (aka HBO, Fig. 1) is certainly the most interesting since many drugs (such as Burseran or Isostegane),8 flavors9 and antiviral agents against HIV or hepatitis B virus10 can be efficiently synthesized from HBO.
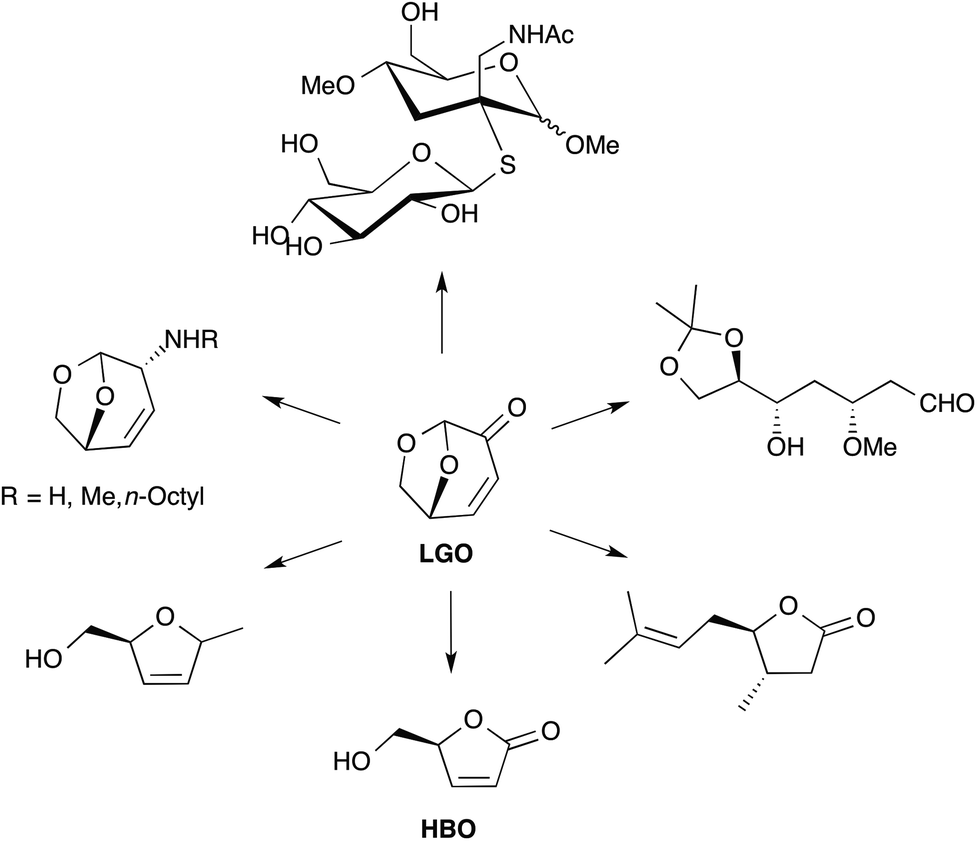 | ||
| Fig. 1 Some chemical intermediates obtainable from LGO. | ||
The most efficient methods described to date for the synthesis of HBO are the ones reported by Koseki11 and Paris.12 In both cases, HBO is obtained from LGO through a two-step procedure involving a Baeyer–Villiger oxidation of LGO into a mixture of formate lactone (FBO) and HBO, followed by an acid hydrolysis of the latter mixture to convert FBO into HBO (Scheme 1). Though the two procedures provide HBO in similar overall yields (ca. 80–90%), the one developed by Paris, that uses metal-based zeolites as a catalyst for the Baeyer–Villiger oxidation and Amberlyst-15 as acid-resins, allows the obtention of HBO in only 4 hours when it took more than 48 hours with Koseki's procedure using peracids (such as peracetic acid or m-chloroperbenzoic acid) and HCl–MeOH. Even though the zeolite-mediated procedure provides the desired target in a timely fashion, it must be noted that it has some drawbacks. Indeed, the synthesis of these relatively costly catalysts requires harsh conditions (e.g., aqueous HF solution, autoclave at 140 °C for 14 days, calcination at 580 °C). In addition, these tin- and aluminium-based catalysts may have a potential toxicity that may prevent their use for the synthesis of HBO as a precursor for drugs or food additives.
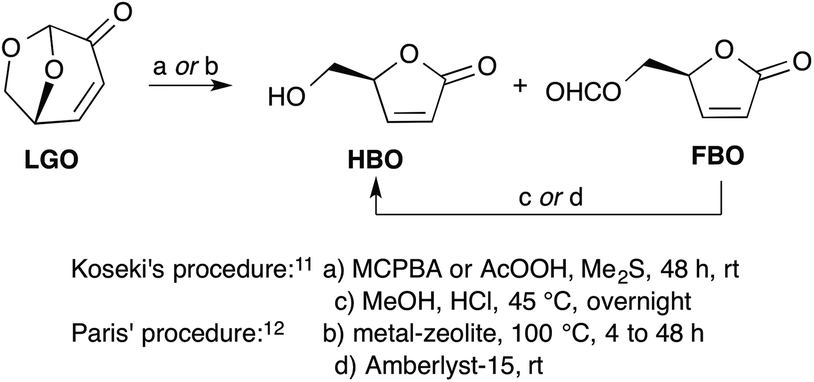 | ||
| Scheme 1 Baeyer–Villiger oxidation of LGO into HBO and FBO. | ||
In order to offer a cost-effective, low toxicity and greener transformation of LGO into HBO, the use of lipase as a biocatalyst seemed to be a promising alternative to the metal zeolites. Indeed, recent studies,13 particularly those of Kotlewska13d and Chavez13f have proved that the use of Candida antarctica B lipase (aka CAL-B) in the presence of both hydrogen peroxide and an acyl donor allows the efficient synthesis of lactones from cyclic ketones via a Baeyer–Villiger reaction (Scheme 2).
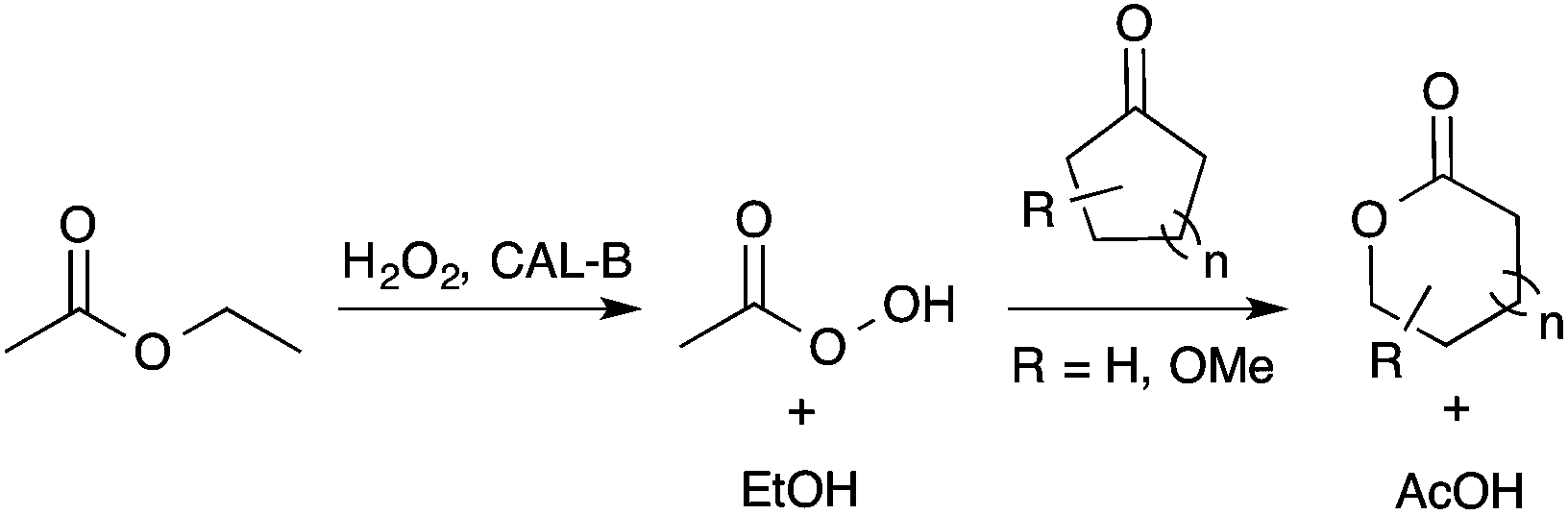 | ||
| Scheme 2 Lipase-mediated Baeyer–Villiger oxidation of cyclic ketones using AcOEt as an acyl donor and H2O2 as an oxidant. | ||
Having recently used CAL-B for the efficient synthesis of renewable bisphenols from ferulic acid and bio-based diols,14 we dedicated ourselves to its use towards the study and optimization of a sustainable lipase-mediated preparation of HBO (and its dehydro-derivative, (S)-γ-hydroxymethyl-γ-butyrolactone aka 2H-HBO) from LGO in a timely fashion and under mild conditions.
Results and discussion
The efficiency of the lipase-mediated Baeyer–Villiger oxidation described in this paper has been evaluated either by following the conversion of LGO into HBO and its formate (FBO)15 by HPLC (see the Experimental section) or by calculating yields of pure HBO after acid hydrolysis and purification on silica gel.The first stage of this study consisted of verifying the feasibility of the Baeyer–Villiger oxidation of levoglucosenone in the presence of Candida antarctica type B (CAL-B) and an acyl donor. To this aim, we first applied the reaction conditions reported by Chavez13f and used H2O2 as an oxidizing agent and ethyl acetate as both a solvent and an acyl donor (Scheme 3). Indeed, in their study, among all the acyl donors tested, ethyl acetate, that is a green solvent,16 was the one that gave the best conversions and yields. We were pleased to observe that under similar conditions (40 °C, [LGO] = 0.5 M in ethyl acetate, enzyme/LGO ratio 507 PLU mmol−1, 1.2 equiv. H2O2), 80% of LGO was converted (into a mixture of HBO and FBO) in only 8 hours while it took more than 48 hours to convert the same amount of cyclic ketones13f such as cyclopentanone, cyclohexanone or cyclooctanone. Such an increase in reactivity can be explained by the higher migratory ability of the acetal-substituent relative to the alkyl ones. Indeed, the two oxygens of the acetal greatly stabilize the transient carbocation through a +M mesomeric effect. To further explore the impact of the stability of such carbocation on the outcome of the lipase-mediated Baeyer–Villiger oxidation, LGO underwent palladium-catalysed hydrogenation to provide 2H-LGO that was then subsequently submitted to lipase-mediated oxidation under the above-mentioned conditions followed by acid hydrolysis (HCl, MeOH) (Scheme 3). Under such conditions, as previously described in Koseki's work17 on the peracetic acid-mediated oxidation of 2H-LGO, Baeyer–Villiger reaction on 2H-LGO in the presence of CAL-B also proved highly regioselective and led, after acid hydrolysis, to the corresponding dehydro-HBO (2H-HBO) exclusively and in high yield (78%). It is noteworthy that the latter γ-lactone can also be readily obtained from HBO through palladium-catalysed hydrogenation.
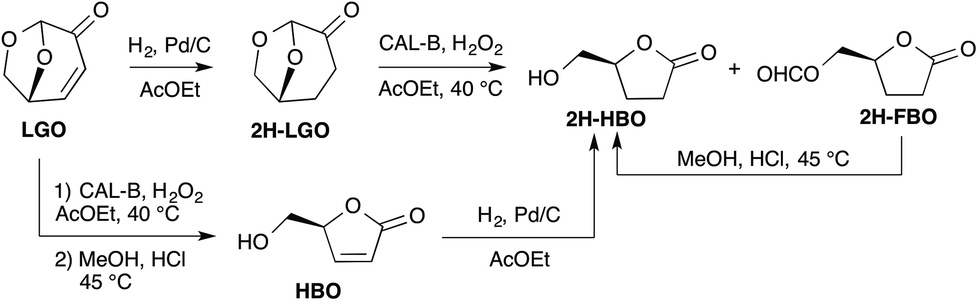 | ||
| Scheme 3 Chemo-enzymatic syntheses of 2H-HBO from LGO. | ||
Acid hydrolysis of the HBO and its formate
The oxidation of LGO leading to a mixture of HBO and FBO, it is essential to achieve an acid hydrolysis step to convert the latter into HBO. Two different procedures have been tested.The first one consisted in recovering the reaction mixture after oxidation and concentrating it under vacuum before diluting the resulting crude product in a methanol–hydrochloric acid solution and heating the solution to 45 °C until complete conversion of FBO (ca. 6–8 hours). The second method that used acid resin Amberlyst-15 and 1,4-dioxane instead of HCl and methanol, respectively,12 completely converted FBO in 6–8 hours at room temperature. It is noteworthy that replacing 1,4-dioxane by methanol or ethyl acetate resulted in longer reaction times. From the perspective of an industrial application, the procedure involving Amberlyst-15, performed at low temperature and allowing an easy recycling of the solid acid catalyst12b–e will be preferred over the hydrochloric acid method that requires the use of costly acid-resistant reactors and equipment.
Optimization of the lipase-mediated Baeyer–Villiger oxidation of levoglucosenone
In view of these promising early trials, we then optimized the reaction conditions by studying, successively, the effect of temperature, LGO concentration, enzyme load, as well as the nature, quantity and addition mode of the oxidizing agent.The effect of reaction temperature on the optimal conversion and reaction kinetics was first studied by running LGO oxidation at 20, 40 and 60 °C under the following conditions: [LGO] = 0.5 M in ethyl acetate, enzyme/LGO ratio 464 PLU mmol−1, 1.2 equiv. H2O2 (Fig. 2). As expected, the reaction proceeded very slowly when conducted at room temperature. Warming the reaction mixture up to 40 or 60 °C significantly increased the initial reaction rate. Nevertheless, after 8 hours, conversions were quite similar irrespective of the temperature applied. It is noteworthy that, as shown in Fig. 2, running the reaction for a longer time resulted in higher conversion up to 95% at 40 °C for 24 hours. In other words, an increase in yield of only 12% required a reaction time three times longer and thus an energy consumption three times higher, not to mention the greater risk of loss of enzyme activity. Furthermore, due to its acetal moiety, LGO degrades in slightly acidic aqueous solutions, therefore the shorter the residence time of LGO in the reaction medium, the better. Following these considerations, optimization experiments were conducted for reaction times of 8 hours in order to (i) prevent LGO degradation, (ii) preserve the activity of enzyme for potential further oxidation cycles, and (iii) reduce the energy and environmental impact of this lipase-mediated Baeyer–Villiger oxidation while maintaining high conversions of LGO. Furthermore, as warming the reaction at 60 °C compromises the enzyme stability, increases the risk of explosion linked to peracetic acid, and is more energy consuming than at 40 °C, the latter temperature was chosen for further experiments.
 | ||
| Fig. 2 Effect of temperature on LGO oxidation kinetics. | ||
The effect of LGO was studied for concentrations ranging from 0.25 to 1 M with a constant LGO–H2O2 ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2) at 40 °C, the latter being a critical factor in the rate of biocatalysed reactions.18 As shown in Table 1, increasing the concentration of LGO in the reaction resulted in an increase of the yields from 65 to 83% (entries 1–4). However, no increase was obtained for LGO concentration beyond 0.75 M (entries 3 and 4).
:1.2) at 40 °C, the latter being a critical factor in the rate of biocatalysed reactions.18 As shown in Table 1, increasing the concentration of LGO in the reaction resulted in an increase of the yields from 65 to 83% (entries 1–4). However, no increase was obtained for LGO concentration beyond 0.75 M (entries 3 and 4).
| Entry | [LGO] (mol L−1) | [CAL-B] (PLU mmol−1) | H2O2 (equiv.) | Yield HBOd (%) |
|---|---|---|---|---|
| a Reaction time: 8 hours. b H2O2 50% w/w. c H2O2–urea. d After acid hydrolysis and purification on silica gel. | ||||
| 1 | 0.25 | 464 | 1.2b | 65 |
| 2 | 0.5 | 464 | 1.2b | 74 |
| 3 | 0.75 | 464 | 1.2b | 83 |
| 4 | 1 | 464 | 1.2b | 83 |
| 5 | 0.5 | 695 | 1.2b | 72 |
| 6 | 0.5 | 927 | 1.2b | 75 |
| 7 | 0.5 | 2 × 464 | 1.2b | 75 |
| 8 | 0.5 | 464 | 1.5b | 70 |
| 9 | 0.75 | 139 | 1.2b | 72 |
| 10 | 0.75 | 232 | 1.2b | 72 |
| 11 | 0.75 | 676 | 1.2c | 70 |
The third reactant to be studied was the oxidizing agent. As shown in Table 1 (entries 2 and 8), the use of 1.2 equivalent of H2O2 was sufficient to observe an optimal yield (ca. 74%). The use of more equivalents of the oxidizing agent (1.5 equivalent and above) resulted in lower yields due to enzyme deactivation (entry 8). Indeed, previous studies demonstrated that high concentration of hydrogen peroxide slowly oxidizes sensitive amino acids and disrupts disulfide bridges, thus resulting in loss of structure and activity.19 H2O2–urea, considered as a milder oxidant,13b,c was also tested but did not improve the yield (entry 11).
Finally, the effect of enzyme loading was studied by varying the quantity of CAL-B from 139 to 464 PLU mmol−1 under the following conditions: 40 °C, [LGO] = 0.75 M in ethyl acetate, 1.2 equiv. H2O2. While optimal yield (83%) was obtained with the highest catalyst loading (Table 1, entry 3), a loading three times less nonetheless achieved a respectable yield of 72% (Table 1, entry 9). The mode of addition of the enzyme (in one portion or in two portions at t = 0 and t = 4 hours) did not have any impact on the yield (Table 1, entries 6 and 7).
Influence of acetic and formic acid by-product on residual enzymatic activity
Under the following reaction conditions (40 °C, [LGO] = 0.75 M in ethyl acetate, enzyme/LGO ratio 219 PLU mmol−1, 1.2 equiv. H2O2), determination of the residual enzyme activity of CAL-B during the course of oxidation revealed a relatively fast loss of bioactivity (e.g., 85% at 4 hours, 74% at 8 hours, Fig. 3), which jeopardizes its recycling. This loss of CAL-B enzymatic activity was attributed to the acetic acid and formic acid co-product generated during the reaction. Indeed, it has been previously proven that an acid environment dramatically modifies protein conformation and cleaves disulfide bridges thus impacting lipase bioactivity.13f,20 The enzyme thus loses its ability to form peracid and hence the quantity of HBO produced decreases. To overcome this problem, two solutions can be proposed: (i) reduce the reaction time by improving oxidation kinetics, or (ii) use buffer solutions/buffering salts in the reaction medium to neutralize acetic and formic acids formed.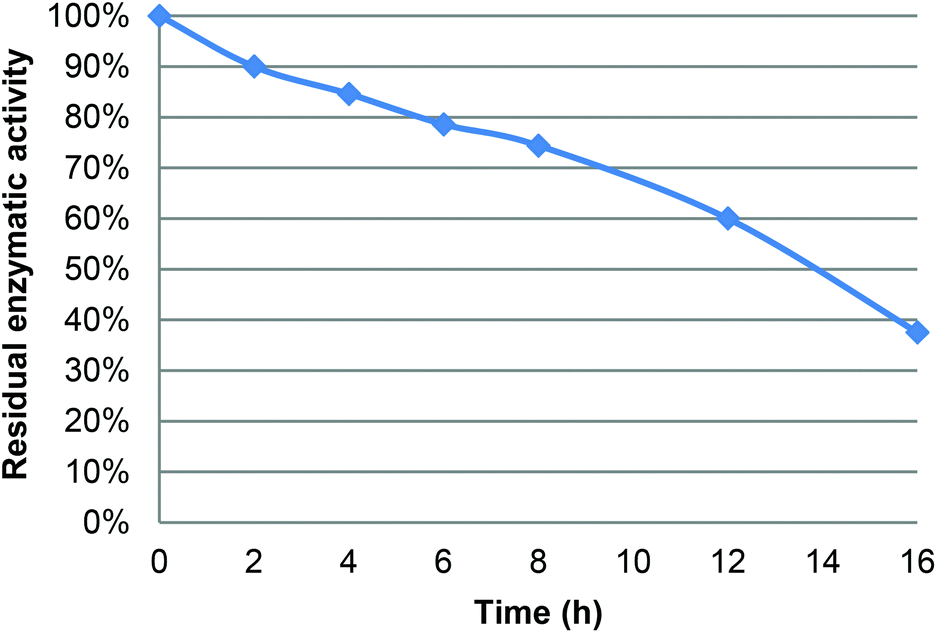 | ||
| Fig. 3 Residual activity of CAL-B during the lipase-mediated oxidation of LGO (measured using the lauric acid method – see the Experimental section). | ||
Influence of liquid and solid buffers on the lipase-mediated Baeyer–Villiger oxidation of LGO
CAL-B activity being optimal in a pH range of 7–10,21 oxidation of LGO was performed in the presence of a pH 7.2 aqueous phosphate buffer solution and resulted in only 47% yield after acid hydrolysis and purification on silica gel. Such a low yield can be attributed to the relatively high quantity of water in the reaction and its deleterious effect on enzyme activity.22 Indeed, though water is generally considered as the best solvent for enzymatic reactions, it can also affect the enzyme through the hydrolysis of peptide bonds, deamidation of amino acid chains or destruction of cysteine residues, thus causing its inactivation. Furthermore, the higher flexibility of the enzymes due to the presence of water (which results in their characteristic higher activity in aqueous environments) promotes these undesired reactions.22b Although organic solvents are able to stabilize enzymes by preventing such side reactions, enzymes still require a certain level of water in their structures in order to maintain their natural conformation, allowing them to deliver their full functionality.22b,c It is found that regardless of the type of reaction, the functionality of enzyme itself is maximum at an optimum level of water (aka water activity), beyond which the enzyme performance is decreased due to lower enzyme stability.22a Furthermore, up to a certain level, water can modify the solvent polarity/polarizability as well as the solubility of the reactants and the products that can cause mass transfer limitations and thus affect the enzyme performance at higher water levels. In addition, water can be a substrate or a product of the enzymatic reaction, influencing the enzyme turnover in different ways.To avoid any extra water in the reaction media than that coming from the aqueous hydrogen peroxide solution, we then turned to the use of solid buffers. For this study, three commercially available buffers (acid form and its corresponding sodium salt) were first tested: MOPS ((4-morpholinepropanesulfonic acid), pKa = 7.2), TAPS ((N-[tris(hydroxymethyl)methyl]-3-aminopropanesulfonic acid), pKa = 8.4), CAPSO ((3-(cyclohexylamino)-2-hydroxy-1-propanesulfonic acid), pKa = 9.6). Three oxidations have been performed under similar conditions (solid buffer and sodium salt 20 mg mL−1 each; 40 °C, [LGO] = 0.75 M in ethyl acetate, enzyme/LGO ratio 169 PLU mmol−1, 1.2 equiv. H2O2) and followed by HPLC to determine LGO conversion and kinetics. As shown in Fig. 4, in the presence of TAPS and CAPSO, the oxidation proved very efficient and led to conversion up to 87% in only 2 hours, while it took 8 hours to reach 72% without solid buffer (Table 1, entry 9) or 83% with an enzyme loading ca. 3 times higher (Table 1, entry 3).23 For comparison, conversion of 95% was achieved in 4 hours in the presence of metal-containing zeolites at 100 °C.12 With MOPS, the initial reaction rate was similar but optimal yield remained lower (72%). This decrease can be attributed to the fact that a gel formed in the reaction media and trapped CAL-B beads, thus limiting its access to hydrogen peroxide and LGO. It is noteworthy that the use of a solid buffer was also tested in the oxidation of H-LGO. In the presence of CAPSO, H-LGO led to H-HBO exclusively (75% yield; 40 °C, [LGO] = 0.75 M in ethyl acetate, enzyme/LGO ratio 113 PLU mmol−1, 1.2 equiv. H2O2, CAPSO, 8 hours), demonstrating that the use of solid buffer did not impact the regioselectivity of the lipase-mediated Baeyer–Villiger oxidation. Now that solid buffers have proved to reduce oxidation time substantially, their impact on the enzyme recyclability has been investigated.
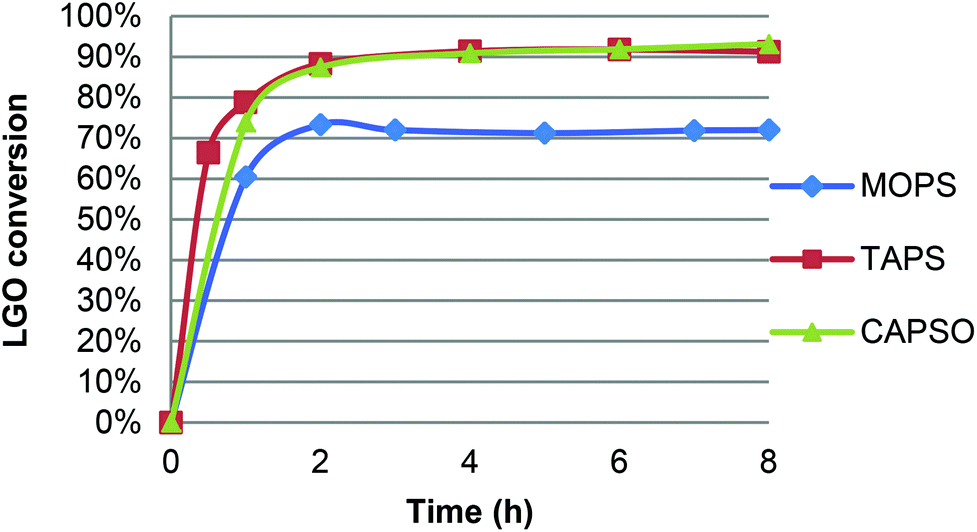 | ||
| Fig. 4 Effect of solid buffer on the lipase-mediated oxidation of LGO. | ||
Effect of solid buffer on CAL-B recyclability
The enzyme recyclability was evaluated by following the conversion of LGO after one, two, three and four 2- or 8-hour oxidation cycles at constant LGO concentration ([LGO] = 0.75 M in ethyl acetate, 40 °C, 1.2 equiv. H2O2) using the same CAL-B and solid buffer after adequate washing between each cycle (Fig. 5 and Table 2). This procedure was selected to mimic how this process would be implemented at the industrial scale.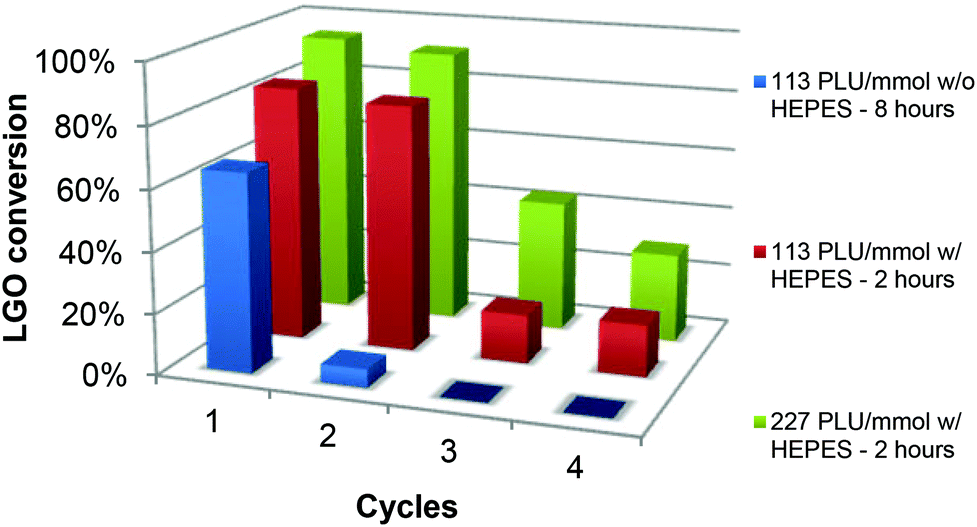 | ||
| Fig. 5 LGO conversion after repeated use of CAL-B with or without HEPES. | ||
| Entry | Solid buffer | [CAL-B] (PLU mmol−1) | Oxidation cycle | Reaction time (h) | LGO conversion (%) | Relative enzymatic activityb (%) |
|---|---|---|---|---|---|---|
| a [LGO] = 0.75 M in ethyl acetate, 40 °C, 1.2 equiv. H2O2. b Calculated percentage relatively to enzymatic activity at 1st cycle (see the Experimental section). | ||||||
| 1 | — | 113 | 1st | 8 | 65 | 100 |
| 2 | — | 113 | 2nd | 8 | 6 | 9 |
| 3 | HEPES | 113 | 1st | 2 | 84 | 100 |
| 4 | HEPES | 113 | 2nd | 2 | 81 | 96 |
| 5 | HEPES | 113 | 3rd | 2 | 16 | 19 |
| 6 | HEPES | 113 | 4th | 2 | 17 | 19 |
| 7 | HEPES | 227 | 1st | 2 | 94 | 100 |
| 8 | HEPES | 227 | 2nd | 2 | 91 | 97 |
| 9 | HEPES | 227 | 3rd | 2 | 43 | 46 |
| 10 | HEPES | 227 | 4th | 2 | 29 | 31 |
These 2- or 8-hour oxidation cycles have been performed with two different enzyme loadings (113 and 227 PLU mmol−1), with or without solid buffer. Since solid buffers with pKa between 8 and 10 resulted in conversions as high as 90% with an enzyme loading at 169 PLU mmol−1, and MOPS could not be used, we had to search for another solid buffer with a pKa close to CAL-B optimal pH (ca. 7.6). Considering the above conditions, we turned to HEPES ((N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)), pKa 7.5) that, in contrast to MOPS, did not form a gel.
As depicted in Fig. 5 and Table 2 – entries 1 & 2, in the presence of only 113 PLU mmol−1 of CAL-B, without any buffer, a 8-hour oxidation cycle was required to convert 65% of LGO. Upon a second 8-hour oxidation cycle, conversion dropped to 6%, thus proving that relative enzymatic activity in the second cycle was very low (9%) (see the Experimental section for relative enzyme activity definition and calculations). In the presence of HEPES (Table 2, entries 3–10), conversions up to 84 and 81% for the first and second cycles, respectively, were obtained in only 2 hours and dropped to 16–17% for the third and fourth ones. In the second, third and fourth oxidation cycles, relative enzymatic activities were thus 96%, 19% and 19%, respectively. It is noteworthy that in the presence of a higher amount of enzyme (227 PLU mmol−1), conversions and relative enzyme activities in the first, second, third and fourth cycles were even higher. In summary, the use of solid buffer not only improved the conversion rate, but also kept the enzyme from deactivation, thus allowing its recycling for further oxidation cycles.
Scale up process
The last important point that has been evaluated is the feasibility of scaling up the process described in Table 2 – entries 3 & 4 (Baeyer–Villiger oxidation of LGO in the presence of CAL-B and HEPES, recycling of CAL-B/HEPES through filtration, second Baeyer–Villiger oxidation of LGO using recycled enzyme/buffer, acid hydrolysis of the combined reactions mixture with HCl–MeOH, and finally purification on silica gel). To do so, scale up of this process was performed with 10 g of LGO per oxidation cycle. Under such conditions, conversions of LGO were comparable to those observed at the 500 mg scale (1st cycle: 83% and 81% respectively). The two crude reaction mixtures were then combined and subjected to the acid hydrolysis. Finally, after concentration to dryness, the crude mixture was easily purified by column chromatography and provided pure HBO in 67% yield (overall yield for the two 2-hours oxidation cycles).Conclusions
Lipase-mediated Baeyer–Villiger oxidation has been successfully applied to convert LGO into chiral lactones (S)-γ-hydroxymethyl-α,β-butenolide (HBO) and (S)-γ-hydroxymethyl-γ-butyrolactone (2H-HBO), two high-valuable chemical intermediates for the preparation of drugs or flavors. This enzymatic Baeyer–Villiger oxidation performed better on this peculiar cyclic ketone (+M mesomeric stabilization of the α-carbon) than on regular cyclic ones, giving optimal yields after only 8 hours (vs. 48 hours). Furthermore, the use of solid buffers in the reaction mixture not only shortens the reaction time from 8 to 2 hours and cuts the enzymatic loading by four, but it also prevents the deactivation of enzyme thus allowing its use for a second cycle without significant loss of activity. Compared to the previously reported preparations of HBO from LGO, this lipase-mediated Baeyer–Villiger oxidation procedure offers a cost-effective, low toxicity and greener alternative through the use of resin supported enzymes, green oxidants, and bio-based and low toxicity solvents/reactants under mild conditions.Experimental section
(–)-Levoglucosenone was kindly donated by the CIRCA Group (Knoxfield Victoria, Australia). Novozym 435® (aka CAL-B, lot no. SLBF9301 V, 9600 PLU g−1), hydrogen peroxide (50% w/w), hydrogen peroxide–urea, palladium on carbon (10%), MOPS, MOPS sodium salt, TAPS, TAPS sodium salt, CAPSO, CAPSO sodium salt, HEPES, HEPES sodium salt, lauric acid, 1-propanol and 12 N aqueous hydrochloric acid (36% w/w) were purchased from Sigma-Aldrich. Ethyl acetate, methanol, ethanol, hexane and acetonitrile (HPLC grade) were purchased from Thermofisher Scientific and used as received. Ultra-pure laboratory grade water (MilliQ, 18.2 megaOhms, 25 °C) was employed for HPLC analysis. TLC analyses were performed on an aluminium strip coated with Silica Gel 60 F254 from Merck, eluted with 20/80 cyclohexane–ethyl acetate, and revealed under UV-light (254 nm), then in the presence of a 20% w/w ethanolic solution of phosphomolybdic acid; LGO: Rf = 0.65 (20/80 cyclohexane–ethyl acetate), black spot. All the reactants were used without further purification. FT-IR and UV-visible spectra were recorded on Agilent Cary 630 and Cary 60, respectively. 1H and 13C NMR spectra were recorded in CDCl3 on a Bruker Fourier 300 MHz, reference in CDCl3 was: 7.26 ppm in 1H and 77.06 ppm in 13C; δ are given in ppm and J in Hz. Optical rotation values were measured using a 241 Perkin Elmer polarimeter. MS and HRMS were recorded on a Micromass GC-TOF. All reported yields are uncorrected and refer to the purified products.(S)-5-(Hydroxymethyl)furan-2(5H)-one (HBO)
Lipase-mediated Baeyer–Villiger reactions have been successfully performed with 250 mg to 10 g (–)-levoglucosenone.(–)-Levoglucosenone (LGO, 1 equiv.) was dissolved in ethyl acetate (C = 0.5–1 M). When specified, solid buffers, acid and salt forms (20 mg mL−1 each) were added. A catalytic amount of CAL-B was added to the reaction mixture, and then hydrogen peroxide (1.2 equiv. 50% w/w) was added at once. The mixture was incubated (20–60 °C) for 2 to 24 hours under shaking (250 rpm). CAL-B was removed by filtration and washed with ethyl acetate (10 mL). The filtrate and the latter ethyl acetate layer were combined and concentrated to dryness.
The crude mixture was evaporated to dryness with silica gel and purified by silica gel chromatography (elution with 75 to 100% ethyl acetate in cyclohexane) to yield pure HBO.
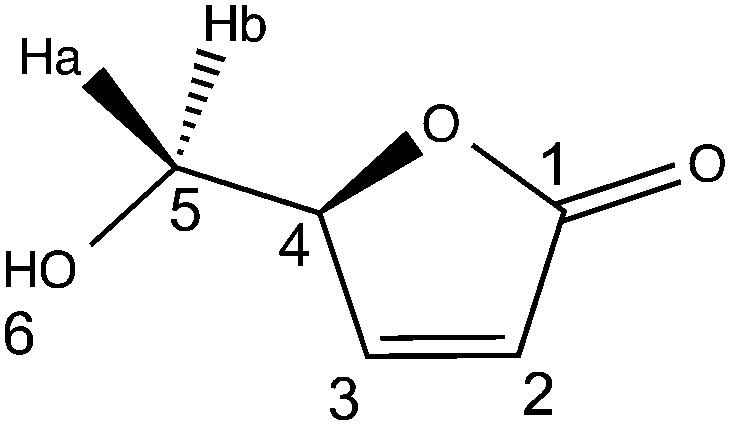
FT-IR (neat, cm−1): 3423 (OH), 1728 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1329, 1162, 1050.
O), 1329, 1162, 1050.
UV (EtOH, nm): 221.
[α]20D −112.0 (c 0.01, CHCl3). [(Lit.12 −114.5 (c 0.1, CHCl3).]
1H NMR (CDCl3, 300 MHz): δH 3.25 (s, 1H, H6), 3.79 (dd, 1H, J = 12 and 3.6 Hz, H5a), 3.99 (d, 1H, J = 12 Hz, H5b), 5.17 (m, 1H, H4), 6.20 (dd, 1H, J = 5.7 and 1.8 Hz, H2), 7.53 (dd, 1H, J = 5.7 and 1.5 Hz, H3).
13C NMR (CDCl3, 75 MHz): δC 62.2 (t, C1), 84.3 (d, C4), 122.8 (d, C2), 154.0 (d, C3), 173.5 (s, C1).
HRMS: m/z [M + H]+ calcd for C5H6O3: 115.0395, found: 115.0396.
(5R)-6,8-Dioxabicyclo[3.2.1]octan-4-one (2H-LGO)
10% Pd/C (10% w/w, 500 mg) was added to a solution of (–)-levoglucosenone LGO (5 g, 39.7 mmol) in ethyl acetate (50 mL, C = 0.8 M) at rt. The stirred suspension was degassed 3 times and kept under nitrogen. The suspension was then hydrogenated under a hydrogen atmosphere at rt until TLC showed complete consumption of the starting material. The crude mixture was filtered over a pad of Celite® and the filtrate was concentrated to dryness with silica gel. The crude product was purified by silica gel chromatography (elution with 10 to 60% ethyl acetate in cyclohexane) to yield pure 2H-LGO (colorless oil, 4.4 g, 87%).
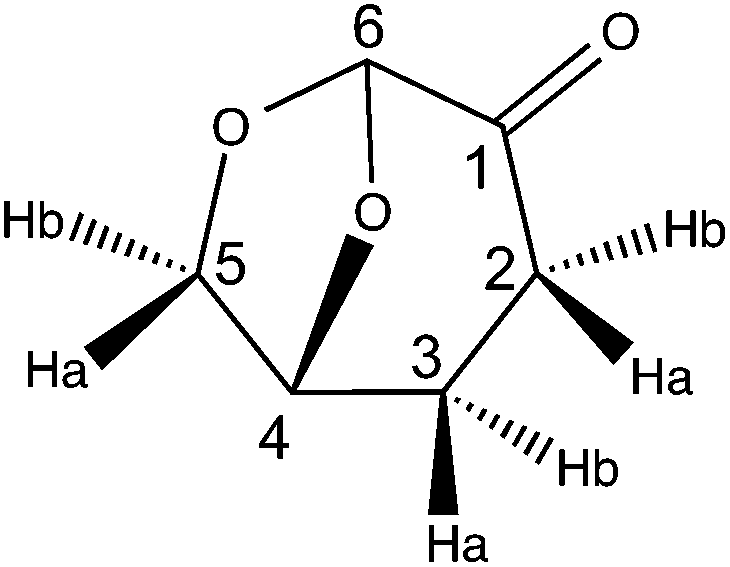
FT-IR (neat, cm−1): 2965, 1739 (CO), 1418, 1285, 1108.
UV (EtOH, nm): 308, 211.
[α]20D −253.6 (c 0.01, CHCl3).
1H NMR (CDCl3, 300 MHz): δH 2.02 (m, 1H, H3b), 2.34 (m, 2H, H2b,3a), 2.62 (m, 1H, H2a), 4.00 (m, 2H, H5a,4), 4.70 (m, 1H, H5b), 5.10 (s, 1H, H6).
13C NMR (CDCl3, 75 MHz): δC 29.9 (t, C3), 31.1 (t, C2), 67.5 (t, C5), 73.1 (d, C4), 101.5 (d, C6), 200.3 (s, C1).
HRMS: m/z [M + H]+ calcd for C6H9O3: 129.0552, found: 129.0553.
(S)-5-(Hydroxymethyl)dihydrofuran-2(3H)-one (2H-HBO)
2H-LGO (1 equiv.) was dissolved in ethyl acetate (C = 0.75 M). When specified, CAPSO, acid and salt forms (20 mg mL−1 each) were added. A catalytic amount of CAL-B was added to the reaction mixture, and then hydrogen peroxide (1.2 equiv. 50% w/w) was added at once. The mixture was incubated and shaken (250 rpm) at 40 °C until TLC showed complete consumption of the starting material. At the end of the reaction, CAL-B was removed by filtration and washed with ethyl acetate (10 mL). The filtrate and the latter ethyl acetate layer were combined and concentrated to dryness.
The crude mixture was evaporated to dryness with silica gel and purified by silica gel chromatography (elution with 75 to 100% ethyl acetate in cyclohexane) to yield pure 2H-HBO (CAL-B 507 PLU mmol−1, no solid buffer, 8 hours, 78% yield; CAL-B 113 PLU mmol−1, CAPSO, 4 hours, 75% yield).
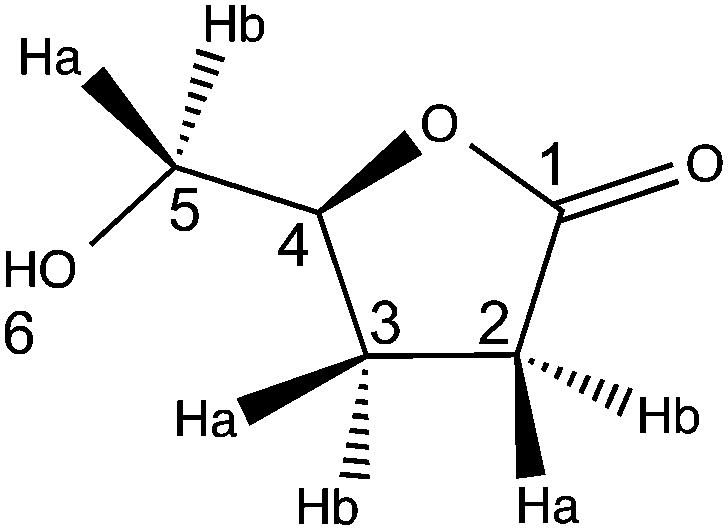
FT-IR (neat, cm−1): 3420 (OH), 2938, 1752 (CO), 1353, 1181.
UV (EtOH, nm): 207.
[α]20D +52.9 (c 0.01, CHCl3). [(Lit.24 +55.2 (c 0.1, CHCl3).]
1H NMR (CDCl3, 300 MHz): δH 2.20 (m, 2H, H3), 2.61 (m, 3H, H2,6), 3.66 (dd, 1H, J = 12.6 and 4.5 Hz, H5a), 3.92 (dd, 1H, J = 12.6 and 2.7 Hz, H5b), 4.64 (m, 1H, H4).
13C NMR (CDCl3, 75 MHz): δC 23.1 (t, C2), 28.7 (t, C3), 64.1 (t, C5), 80.8 (d, C4), 177.7 (s, C1).
HRMS: m/z [M + Na]+ calcd for C5H8NaO3: 139.0371, found: 139.0379.
Determination of residual enzyme activity – the Lauric acid method
A solution of lauric acid (74.65% w/w) in 1-propanol (22.37% w/w) and water (2.98% w/w) was prepared and heated at 60 °C. CAL-B (ca. 14 mg) was weighed in a vial and kept at 60 °C. After one hour, 5.0 g (ca. 6 mL) of the above solution was added to the enzyme and the mixture was stirred at 60 °C for 15 min. Samples (2 μL) were collected, weighed in a GC-vial and hexane (1 mL) was added. The vial was sealed and weighed again. This procedure was repeated three times for every single enzyme tested. Lauric acid conversion was determined by the following GC-MS method.Unit definition: 1 PLU unit = 1 μmol of 1-propyl laurate formed per gram of enzyme per minute at 60 °C (reaction time: 15 minutes).
It is noteworthy that the mere fact of putting CAL-B suspended in ethyl acetate resulted in a loss of ca. 45% of the enzymatic activity reported in the specification sheet of CAL-B lot no. SLBF9301 V. To determine the weight of CAL-B needed in the synthetic procedures, one must first determine the enzymatic activity after suspension in ethyl acetate to faithfully reproduce the CAL-B-mediated Baeyer–Villiger oxidations reported herein.
Relative enzymatic activities, which represent the overall enzyme activity of a given oxidation cycle compared to the first one, have been calculated using the following equations:
Relative enzymatic activity for the 1st cycle = 100
Relative enzymatic activity for the 2nd cycle = (LGO conversion (2nd cycle) × 100)/(LGO conversion (1st cycle))
Relative enzymatic activity for the 3rd cycle = (LGO conversion (3rd cycle) × 100)/(LGO conversion (1st cycle)) and so on.
GC-MS method
The GC-MS system consisted of an Agilent GC 5975 coupled with MS 7890 in electron impact mode with electron energy set at 70 eV and a mass range at m/z (30–350 amu), and equipped with a HP5-MS (30 m × 0.25 mm i.d., 0.25 μm film thickness) capillary column. The injector and transfer line temperatures were both at 280 °C. GC was performed in split mode (40:1). The temperature program was as follows: from 60 °C held for 1 min, then to 325 °C at 20 °C min−1, held at 325 °C for 5 min. The flow rate of hydrogen was set at 1.2 mL min−1 and the injected volume was 1 μL. The mass detector was set as follows: source temperature = 230 °C, quad temperature = 150 °C. A calibration curve was performed each time, with pure lauric acid (0.2–4.5 mg mL−1) in hexane. tr(lauric acid) = 7.22 min, tr(1-propyl laurate) = 7.92 min.
HPLC method
HPLC analyses were performed on a Thermofisher Ultimate 3000 equipped with a DAD detector (220 nm) and a Thermoscientific Syncronis aQ (250 × 4.6 mm, 5 μm) column. Samples were prepared by diluting 10 μL of the reaction mixture in 1.5 mL of acetonitrile. The following conditions were applied: injection 10 μL, oven temperature 30 °C, flow 0.8 mL min−1, elution method (water–acetonitrile): 0–5 min isocratic 85/15, 5–10 min from 85/15 to 90/10, 10–15 min isocratic 90/10, 15–20 min 90/10 to 85/15. Retention times: tr(LGO) = 8.40 min, tr(HBO) = 3.73 min, tr(FBO) = 3.87 min, tr(2H-LGO) = 8.40 min and tr(2H-HBO) = 3.61 min.Acknowledgements
The authors are grateful to the Circa Group for providing them with industrial grade levoglucosenone, and to Région Champagne-Ardenne, Conseil Général de la Marne and Reims Métropole for financial support.Notes and references
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed.
- (a) M. S. Miftakhov, F. A. Valeev and I. N. Gaisina, Russ. Chem. Rev., 1994, 63, 869 CrossRef PubMed; (b) A. V. Bridgwater, D. Meier and D. Radlein, Org. Geochem., 1999, 30, 1479 CrossRef CAS; (c) Q. Lu, W. M. Xiong, W. Z. Li, Q. X. Guo and X. F. Zhu, Bioresour. Technol., 2009, 100, 4871 CrossRef CAS PubMed.
- (a) F. Shafizadeh, R. H. Furneaux, T. T. Stevenson and T. G. Cochran, Carbohydr. Res., 1978, 61, 519 CrossRef CAS; (b) G. Dobele, T. Dizhbite, G. Rossinskaja, G. Telysheva, D. Mier, S. Radtke and O. Faix, J. Anal. Appl. Pyrolysis, 2003, 68–69, 197 CrossRef CAS; (c) J. Zandersons, A. Zhurinsh, G. Dobele, V. Jurkjane, J. Rizhikovs, B. Spince and A. Pazhe, J. Anal. Appl. Pyrolysis, 2013, 103, 222 CrossRef CAS PubMed.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257 CrossRef CAS PubMed.
- G. R. Court, C. H. Lawrence, W. D. Raverty and A. J. Duncan, WO2011/000030, 2011.
- (a) V. L. Budarin, P. S. Shuttleworth, J. R. Dodson, A. J. Hunt, B. Lanigan, R. Marriott, K. J. Milkowski, A. J. Wilson, S. W. Breeden, J. Fan, E. H. K. Sin and J. H. Clark, Energy Environ. Sci., 2011, 4, 471 RSC; (b) X. Hu, L. Wu, Y. Wang, D. Mourant, C. Lievens, R. Gunawan and C. Z. Li, Green Chem., 2012, 14, 3087 RSC.
- (a) K. Tomioka, T. Ishiguro and K. Koga, J. Chem. Soc., Chem. Commun., 1979, 652 RSC; (b) D. Enders, V. Lausberg, G. Del Signore and O. M. Berner, Synthesis, 2002, 515 CrossRef CAS.
- E. Takashi, M. Katsuya, Y. Hajime, K. Koseki, H. Kawakami and H. Matsushita, Heterocycles, 1990, 31, 1585 CrossRef PubMed.
- (a) H. Hawakami, T. Ebata, K. Koseki, K. Statsumoto, H. Matsushita, Y. Naoi and K. Itoh, Heterocycles, 1990, 31, 2041 CrossRef; (b) R. Flores, A. Rustullet, R. Alibes, A. Alvarez- Larena, P. de March, M. Figueredo and J. Font, J. Org. Chem., 2011, 76, 5369 CrossRef CAS PubMed; (c) A. Diaz-Rodriguez, Y. S. Sanghvi, S. Fernandez, R. F. Schinazi, E. A. Theodorakis, M. Ferreroa and V. Gotor, Org. Biomol. Chem., 2009, 7, 1415 RSC.
- K. Koseki, T. Ebata, H. Kawakawmi, H. Matsushita, K. Itoh and Y. Naoi, US 4,9947,585, Feb. 19th, 1991.
- (a) C. Paris, M. Moliner and A. Corma, Green Chem., 2013, 15, 2101 RSC; (b) P. Rammohan, S. Taradas and K. Shampa, ARKIVOC, 2012, 570 Search PubMed; (c) K. Tanabe and W. F. Hölderich, Appl. Catal. A., 1999, 181, 399 CrossRef CAS; (d) J. H. Clark, Acc. Chem. Res., 2002, 35, 791 CrossRef CAS PubMed; (e) Y. C. Sharma, B. Singh and J. Korstad, Biofuels, Bioprod. Bioref., 2011, 5, 69 CrossRef CAS.
- (a) P. Carlqvist, R. Eklund, K. Hult and T. Brinck, J. Mol. Model., 2003, 9(3), 164 CrossRef PubMed; (b) M. Y. Rios, E. Salazar and H. F. Olivo, Green Chem., 2007, 9, 459 RSC; (c) M. Y. Rios, E. Salazar and H. F. Olivo, J. Mol. Catal. B: Enzym., 2008, 54, 61 CrossRef CAS PubMed; (d) A. J. Kotlewska, F. van Rantwijk, R. A. Sheldon and I. W. C. E. Arends, Green Chem., 2011, 13, 2154 RSC; (e) A. Drozdz, A. Chrobok, S. Baj, K. Szymanska, J. Mrowiec-Bialon and A. B. Jarzebski, Appl. Catal., A, 2013, 467, 163 CrossRef CAS PubMed; (f) G. Chavez, R. Hatti-Kaul, R. A. Sheldon and G. Mamo, J. Mol. Catal. B: Enzym., 2013, 89, 67 CrossRef CAS PubMed.
- (a) F. Pion, F. A. Reano, P.-H. Ducrot and F. Allais, RSC Adv., 2013, 3, 8988 RSC; (b) F. Pion, P.-H. Ducrot and F. Allais, Macromol. Chem. Phys., 2014, 5, 431 CrossRef.
- Preliminary studies demonstrated that LGO was stable in the reaction medium (no degradation product) and that the only products obtained through its oxidation were HBO and FBO.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. Johnson, P. Kleine, C. Knight, M. A. Nagy, D. Perry and M. Stefaniakc, Green Chem., 2007, 10, 31–36 RSC.
- K. Koseki, T. Ebata, H. Kawakawmi, H. Matsushita, K. Itoh and Y. Naoi, US 5,112,994, 1992.
- (a) C. Orellana, S. Camocho, D. Adlercreutz, B. Mattiason and R. Hatti-Kaul, Eur. J. Lipid Sci. Technol., 2005, 107, 864 CrossRef; (b) S. Bhattachayra, A. Drews, E. Lyagin, M. Kraume and M. B. Ansorge-Schumacher, Chem. Eng. Technol., 2012, 35, 1 Search PubMed.
- (a) U. Törnvall, C. Melin, R. Hatti-Kaul and M. Hedström, Rapid Commun. Mass Spectrom., 2009, 2959 CrossRef PubMed; (b) U. Törnvall, M. Hedström, K. Schilléen and R. Hatti-Kaul, Biochimie, 2010, 92, 1867 CrossRef PubMed.
- A. R. Katrizky, N. G. Akhmedov and O. V. Denisko, Magn. Reson. Chem., 2003, 41, 37 CrossRef.
- (a) J. Sun, Y. Jiang, L. Zhou and J. Gao, New Biotechnol., 2010, 24, 53 CrossRef PubMed; (b) M. Adamczak, Pol. J. Food Nutr. Sci., 2003, 4, 3 Search PubMed.
- (a) K. Rezaei, Crit. Rev. Biotechnol., 2007, 27(4), 183 CrossRef CAS PubMed; (b) P. Adlercreutz, in Organic Synthesis with Enzymes in Non-Aqueous Media, ed. G. Carrea and S. Riva, Wiley-VCH Verlag Gmbh & Co KGaA, Weinheim, Germany, 2008, pp. 3–24 Search PubMed; (c) A. M. Klibanov, Nature, 2001, 409, 241 CrossRef CAS PubMed.
- Polarimetry analysis of the HBO obtained through solid buffer-based oxidation showed that the stereochemistry of HBO was retained (no racemization or inversion of configuration).
- (a) I. Bonnaventure and A. B. Charette, J. Org. Chem., 2008, 16, 6330 CrossRef PubMed; (b) In such a pH range, His224, Asp187 and Ser105 residues (the catalytic triad of the active site) are at the required protonation state to allow perhydrolysis of ethyl acetate.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H & 13C NMR spectra, and FT-IR spectra of LGO, HBO, 2-LGO and 2-HBO. See DOI: 10.1039/c4gc01231c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |