
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRNA sequencing supports distinct reactive oxygen species-mediated pathways of apoptosis by high and low size mass fractions of Bay leaf (Lauris nobilis) in HT-29 cells
Annabelle L.
Rodd
ab,
Katherine
Ververis
ab,
Dheeshana
Sayakkarage
a,
Abdul W.
Khan
c,
Haloom
Rafehi
abc,
Mark
Ziemann
c,
Shanon J.
Loveridge
a,
Ross
Lazarus
d,
Caroline
Kerr
e,
Trevor
Lockett
e,
Assam
El-Osta
bcfg,
Tom C.
Karagiannis
*ab and
Louise E.
Bennett
*h
aEpigenomic Medicine, Baker IDI Heart and Diabetes Institute, The Alfred Medical Research and Education Precinct, Melbourne, Victoria, Australia
bDepartment of Pathology, The University of Melbourne, Parkville, Victoria, Australia
cEpigenetics in Human Health and Disease, Baker IDI Heart and Diabetes Institute, The Alfred Medical Research and Education Precinct, Melbourne, Victoria, Australia
dMedical Bioinformatics, Baker IDI Heart and Diabetes Institute, The Alfred Medical Research and Education Precinct, Melbourne, Victoria, Australia
eCSIRO Food and Nutrition Flagship, North Ryde, New South Wales, Australia
fEpigenomic Profiling Facility, Baker IDI Heart and Diabetes Institute, The Alfred Medical Research and Education Precinct, Melbourne, Victoria, Australia
gDepartment of Medicine, Monash University, Melbourne, Victoria, Australia
hCSIRO Food and Nutrition Flagship, Werribee, Victoria, Australia. E-mail: louise.bennett@csiro.au; Fax: +616 9731 3201; Tel: +616 9731 3200
First published on 15th June 2015
Abstract
Anti-proliferative and pro-apoptotic effects of Bay leaf (Laurus nobilis) in mammalian cancer and HT-29 adenocarcinoma cells have been previously attributed to effects of polyphenolic and essential oil chemical species. Recently, we demonstrated differentiated growth-regulating effects of high (HFBL) versus low molecular mass (LFBL) aqueous fractions of bay leaf and now confirm by comparative effects on gene expression, that HFBL and LFBL suppress HT-29 growth by distinct mechanisms. Induction of intra-cellular lesions including DNA strand breakage by extra-cellular HFBL, invoked the hypothesis that iron-mediated reactive oxygen species with capacity to penetrate cell membrane, were responsible for HFBL-mediated effects, supported by equivalent effects of HFBL in combination with γ radiation. Activities of HFBL and LFBL were interpreted to reflect differentiated responses to iron-mediated reactive oxygen species (ROS), occurring either outside or inside cells. In the presence of LFBL, apoptotic death was relatively delayed compared with HFBL. ROS production by LFBL mediated p53-dependent apoptosis and recovery was suppressed by promoting G1/S phase arrest and failure of cellular tight junctions. In comparison, intra-cellular anti-oxidant protection exerted by LFBL was absent for extra-cellular HFBL (likely polysaccharide-rich), which potentiated more rapid apoptosis by producing DNA double strand breaks. Differentiated effects on expression of genes regulating ROS defense and chromatic condensation by LFBL versus HFBL, were observed. The results support ferrous iron in cell culture systems and potentially in vivo, can invoke different extra-cellular versus intra-cellular ROS-mediated chemistries, that may be regulated by exogenous, including dietary species.
Introduction
Colorectal cancer (CRC) is the third most prevalent cancer worldwide, representing 10% of all cancers in 2008 and affecting 1.24 million people.1 CRC is the third leading cause of cancer-related deaths in industrialized cities2 and the incidence of CRC is increasing in countries where Western dietary and lifestyle practices are increasing.Around one third of the variation in CRC sucsceptibility is attributable to heritable genetic variants. While Mendelian syndromes that are associated with a high risk of developing CRC for carriers, notably Lynch Syndrome and Familial Adenomatous Polyposis, account for around 5% of the total CRC burden, modern genomics has identified a further 30 or more low penetrance predisposing variants.3 Colorectal carcinogenesis is widely accepted to be a sequential and multi-step process involving accumulation of mutations affecting oncogenes, tumor-suppressor and DNA-repair genes that control cell proliferation, cell survival and oncogenesis.2
Low levels of physical activity, and some dietary practices such as high red meat consumption, low fruit and vegetable intake and deficiency of some micro-nutrients have been associated with increased risk of CRC.4 Alternately, pre-clinical studies have demonstrated the potential of extracts from dietary plant including carotenoids,5 cruciferous vegetables rich in glucosinolates6 and isothiocyanates7 and specific phytochemicals such as curcumin8 and resveratrol9 to be protective against CRC.
The HT-29 human colonic adenocarcinoma cell line has been used as a model of cellular growth processes underlying CRC and a screening tool for investigating effectiveness of chemopreventative factors.10 More recently, effects of interventions have also been studied by differential gene regulation using RNA sequencing methodologies,11 which has extended the understanding of important growth-regulating genes beyond the previously narrow foci of mucin12 and heat-shock13 genes. Recent gene profiling studies have shown that the flavonoid apigenin stimulated 2–4-fold upregulation of caspase-3 and caspase-8 mRNA while down-regulating rapamycin and cyclin D1 to 0.4 and 0.2-times the level prior to flavonoid treatment, respectively.11 In addition, diallyl disulfide from garlic, exhibiting dose and time-dependent growth inhibition of HT-29 cells, was shown to alter the level of expression of 49 genes involved in controlling transduction, cell proliferation, cell growth, apoptosis and the protein composition and structure of the extracellular matrix.14 Apple polyphenolics that inhibited HT-29 growth have also been shown to modulate expression of genes involved with xenobiotic metabolism.15 All of these reported effects of dietary factors on HT-29 cell gene expression were substantiated by correlation with transcriptional activity.
More recent gene expression studies support the numerous in vitro studies demonstrating anti-proliferative and pro-apoptotic effects of a range of plant and fungal extracts on HT-29 cells. In these studies, bioactivity was associated with different chemical classes mostly extracted using organic solvents including: acid methanol extracts from bay leaf and other Australian native herbs;16 Bowman-Birk protease inhibitor from pea;17 ethanol extract of Phellinus linteus fungus growing on ginseng;18 butanol extract of Cordyceps militaris fungus growing on soybean;19 diallyl sulphide from garlic;14 polysaccharide from the fungus, Pleurotus ostreatus;20 polyphenolic-rich raspberry extract;21 maslinic and oleanolic acids (pentacyclic triterpenes) from olives;22 sulforaphane (isothiocyanate) from broccoli23 and epigallocatechin gallate from green tea.24 By using solvent-based extraction methods, these studies have all either intentionally or inadvertently focused on hydrophobic metabolites including polyphenolics and essential oils. In all cases, both anti-proliferative and apoptotic effects measured by cell cycle analysis or caspase-specific pathways were reported. Extracts of bay leaf are also reported to regulate growth in human melanoma, kidney, prostate and breast cancer cell lines.25 We have recently described divergent extents of anti-proliferation and apoptosis associated with higher molecular mass versus lower mass (polyphenolic, essential oil-rich) fractions of bay leaf extract on HT-29 cells.26
The aim of this study was to extend our previous studies by conducting RNA sequencing of HT-29 cells following exposure to unfractionated (UFBL), low (LFBL) and high (HFBL) molecular mass fractions of bay leaf. The hypothesis that LFBL and HFBL differentially interacts with reactive oxygen species (ROS) was explored by comparing effects of bay leaf fractions in the presence of Fe-mediated ROS production in cell culture or by γ radiation-mediated ROS production. The results confirm previous functional differences and extend our previous understanding, suggesting that LFBL and HFBL mediate differentiated rates of apoptosis involving reactive oxygen species (ROS), eliciting different pathways of toxicity and cellular defense and repair responses.
Results
Differentiated anti-proliferative and apoptotic effects of HFBL and LFBL on HT-29 cells
LFBL and HFBL were previously shown to exhibit divergent effects on inhibiting proliferation and promoting apoptosis, respectively, of HT-29 cells in vitro. The low and high mass bay leaf fractions were compositionally distinct with expected enrichment of polyphenolics and essential oils in the low mass fraction and of polysaccharides and proteins in the high mass fraction. We have now sought to substantiate these effects at the gene expression level. The effects of UFBL, HFBL and LFBL samples on cell viability and induction of caspase 3/7 activity in HT-29 cells indicated that the LFBL fraction was responsible for a dose-dependent decrease in cell viability and proliferation, with more prominent effects seen at 48 versus 24 hours incubation (Fig. 1). The EC50 values were calculated to be 30, 10 and 61.5 mg mL−1 for UFBL, LFBL and HFBL, respectively (Fig. 1D). Conversely, although caspase activity data indicated both HFBL and LFBL induced a significant increase in apoptosis, the HFBL induced a greater response in caspase 3/7 activity compared to LFBL and UFBL (Fig. 1C), as reported previously.26 The effects of all bay leaf samples produced significant shifts in the cell cycle with an increase of the proportion of cells in the S phase and sub-G1 phase and decreases in the proportion of cells in G1 and G2 phase, after 24 hours. Compared with the proportion of sub-G1 untreated cells (0.65%), HFBL treatment significantly increased the sub-G1 proportion to 60.5% (Table 1). Cell cycle analysis indicated that observed changes were similar for UFBL and LFBL treatments at either 24 or 48 h with relatively faster effects of HFBL on sub-G1 phase (Fig. 2).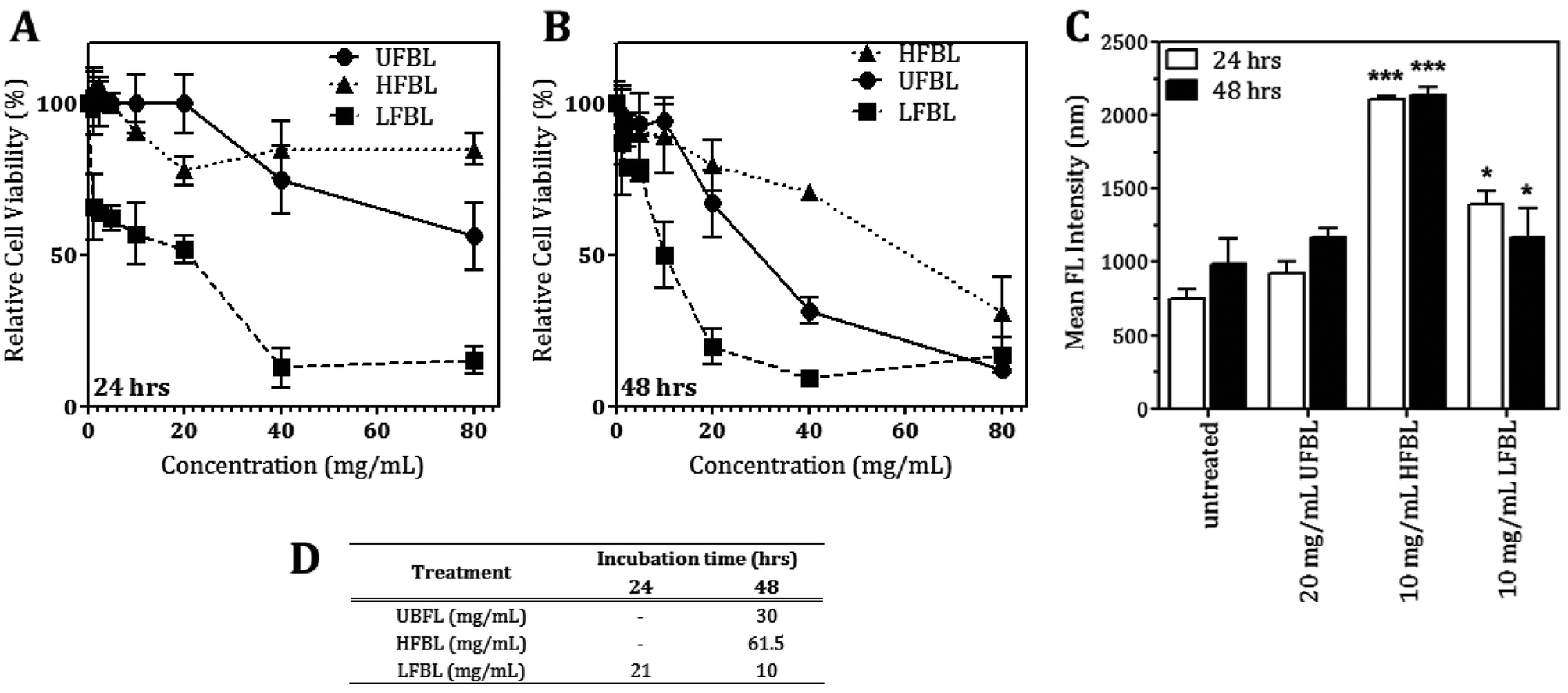 | ||
| Fig. 1 The effects of unfractionated (UFBL), high (HFBL) and low (LFBL) molecular weight fractions of bay leaf on HT-29 cell viability and apoptosis after treatment for 24 or 48 hours. Treatment with LFBL for 24 (A) and 48 (B) hours induces the greatest dose-dependent decrease in the cell viability of HT-29 cells. (C) HFBL and LFBL induce apoptosis in HT-29 cells after 48 hours, with HFBL exerting higher apoptotic activity than LFBL. (D) EC50 values of the cell viability for respective bay leaf samples. Shown is a representative experiment performed in triplicate, two independent experiments performed in total. Two-way ANOVA with Bonferroni post-tests were performed comparing each treatment to untreated cells, *P < 0.05, **P < 0.01, ***P < 0.001. | ||
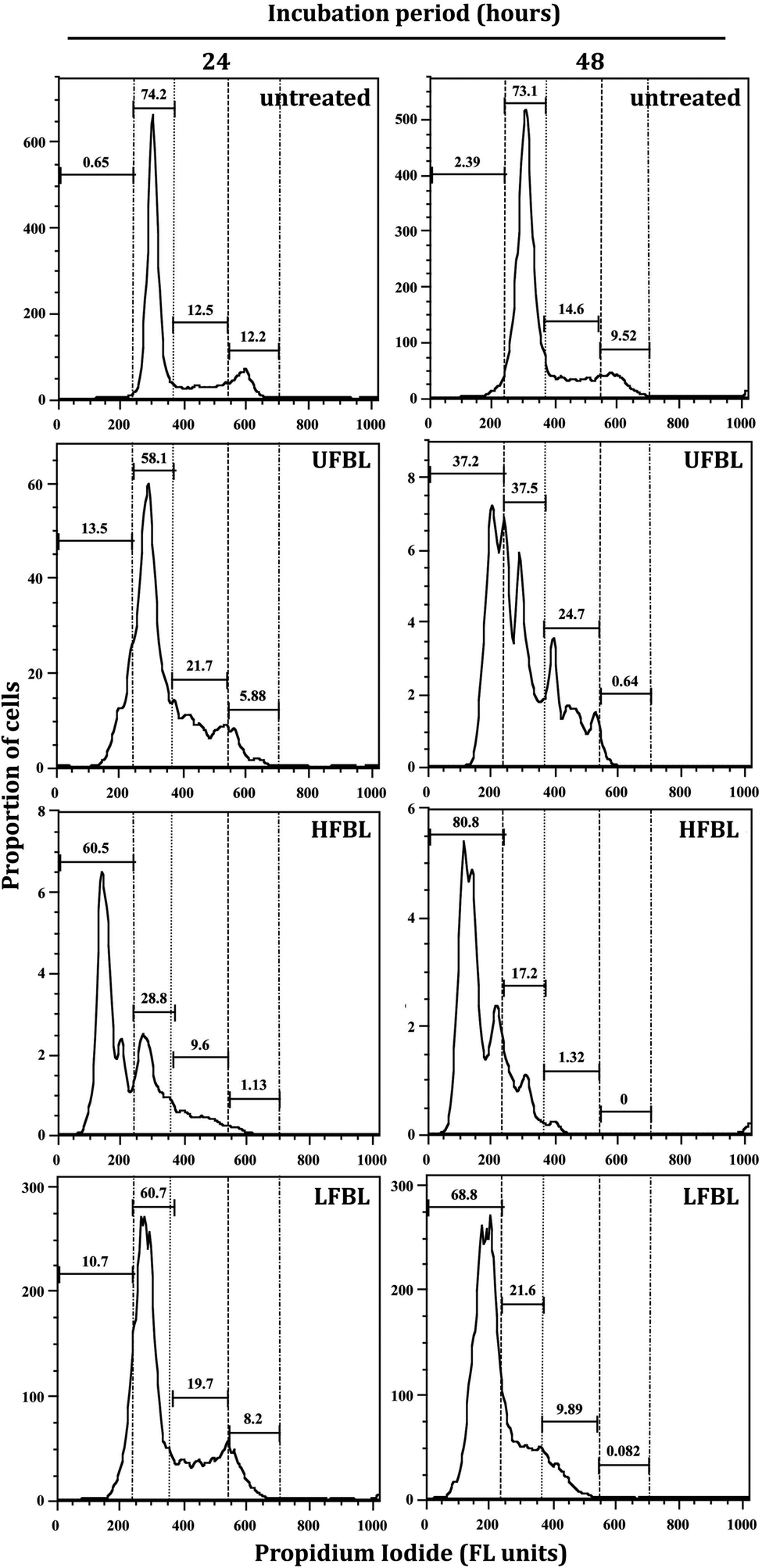 | ||
| Fig. 2 Cell cycle analysis by FACS of HT-29 cells treated with 20 mg mL−1 UFBL extract and 10 mg mL−1 of HFBL and LFBL fractions for 24 and 48 hours. Gatings from left show proportions of sub-G1, G0/G1, S, G2/M phase cells, respectively. Treatment with UFBL, HFBL and LFBL fractions produce a shift to sub-G1 phase comprising both apoptotic and necrotic cells. Shown are representative data from a single experiment performed in duplicates. | ||
| Treatment | Incubation time (h) | Sub-G1 | G1 | S | G2/M |
|---|---|---|---|---|---|
| Untreated | 0 | 1.5 ± 1.2 | 74.0 ± 0.8 | 10.9 ± 1.9 | 13.6 ± 1.5 |
| UFBL-20 mg ml−1 | 24 | 13.3 ± 0.4 | 56.8 ± 2.6 | 4.6 ± 1.8 | 25.3 ± 4.8 |
| UFBL-20 mg ml−1 | 48 | 47.4 ± 14.3 | 34.5 ± 4.0 | 0.4 ± 0.3 | 17.7 ± 10.0 |
| HFBL-10 mg ml−1 | 24 | 49.3 ± 8.7 | 36.5 ± 3.9 | 1.7 ± 0.8 | 12.4 ± 3.9 |
| HFBL-10 mg ml−1 | 48 | 73.6 ± 11.0 | 20.0 ± 3.9 | 0.1 ± 0.1 | 6.3 ± 7.0 |
| LFBL-10 mg ml−1 | 24 | 11.6 ± 1.2 | 59.0 ± 3.0 | 8.9 ± 0.9 | 20.5 ± 5.0 |
| LFBL-10 mg ml−1 | 48 | 62.5 ± 8.6 | 26.7 ± 7.4 | 0.3 ± 0.3 | 10.5 ± 0.9 |
HFBL but not LFBL induces DNA-double strand breaks
The comparative efficacy of the UFBL, HFBL and LFBL on the induction of DNA damage in HT-29 cells was measured using γH2AX, a marker for double-strand breaks (Fig. 3). Compared with non-significant production by UFBL and LFBL, induction of γH2AX foci per cell were significant for HFBL (Fig. 3A and 3B). HFBL-mediated γH2AX lesions, but not LFBL-mediated, increased as a function of time, and time-dependent production of γH2AX foci per cell were amplified by ionizing radiation (Fig. 3C and 3D). Treatment of HT-29 cells with HFBL but not LFBL, produced significant DNA damage with or without γ-radiation, suggesting that either γ-radiation-independent or γ-radiation-dependent DNA double-strand breaks mediated by HFBL were not repairable and could account for the pathway of cell apoptosis mediated by HFBL. These results also infer potentiation of γ-radiation-induced ROS free radical-mediated oxidative damage by HFBL, but not LFBL.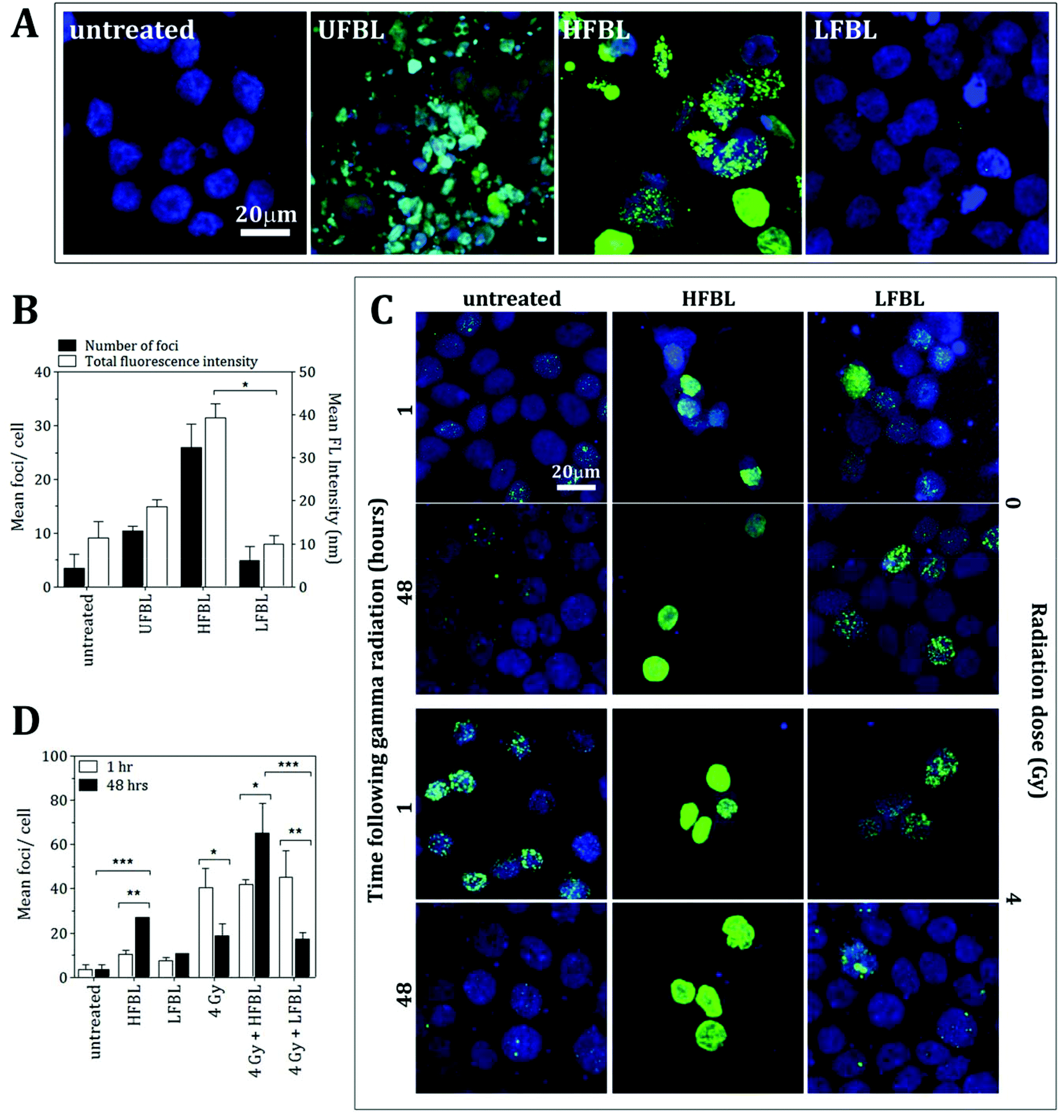 | ||
| Fig. 3 HFBL bayleaf fractions augment DNA double-strand breaks represented by γH2AX foci in human colon carcinoma HT-29 cells. (A) Representative photomicrographs of cells treated with 20 mg mL−1 UFBL, 10 mg mL−1 HFBL and 10 mg mL−1 LFBL samples for 48 hours prior to γH2AX staining. Merged image: nucleus stained with TO-PRO3 (blue), γH2AX foci (green). (B) HFBL significantly induced formation of γH2AX foci in HT-29. Images were analysed for the number of γH2AX foci per cell by image analysis and total fluorescence intensity of the γH2AX staining. Shown are the mean ± SD from two independent experiments each performed in duplicate. (C) Representative photomicrographs of cells treated with or without 4 Gy (137Cs) of γ-radiation following 48 hour incubation with 10 mg mL−1 HFBL and 10 mg mL−1 LFBL. Cells were stained for γH2AX foci at 1 hour post γ-radiation to depict initial DNA double-strand break formation and 48 hours post γ-radiation to measure the cells ability to repair. Merged image: nucleus stained with DAPI (blue), γH2AX foci (green). (D). Bayleaf samples did not induce radio-sensitivity in HT-29 cells but HFBL fractions prevented repair of γ-radiation-induced DNA double-strand breaks. Shown are the mean and SD from two independent experiments performed in duplicate; *p < 0.05, ***p < 0.001. | ||
mRNA-sequencing and pathway analysis
Global effects of UFBL, HFBL and LFBL on gene expression in HT-29 cells are shown in Fig. 4 and 5. Histograms of fold-change distribution (Fig. 4) demonstrated that treatment with all bay leaf samples primarily caused up-regulation of gene expression, with values mainly lying between 0 and 5. Furthermore, Venn diagram representation of up-regulated genes showed there was significant overlap between the genes up-regulated by all three samples of bay leaf, with approximately 50% of total up-regulated genes common across all bay leaf samples (Fig. 5A). These up-regulated genes were found to be mainly involved in cytoskeleton remodeling, cell cycle disruption, and induction of apoptotic pathways. LFBL treatment induced the majority of genes to be up-regulated, followed by the UFBL, and then HFBL. In contrast, however, a smaller percentage (∼17%) of the down-regulated genes was common across bay leaf samples (Fig. 5B). All three samples induced similar numbers of genes with decreased expression with HFBL > LFBL > UFBL. While a large proportion of genes were similarly affected by all three bay leaf samples, the genes that were differentially expressed highlight different biological pathways affected by respective fractions. Specific genes with altered expression were grouped by either cell cycle and apoptosis (Table 2), those regulating cell junctions and motility (Table 3) and defense against oxidative stress (Table 4).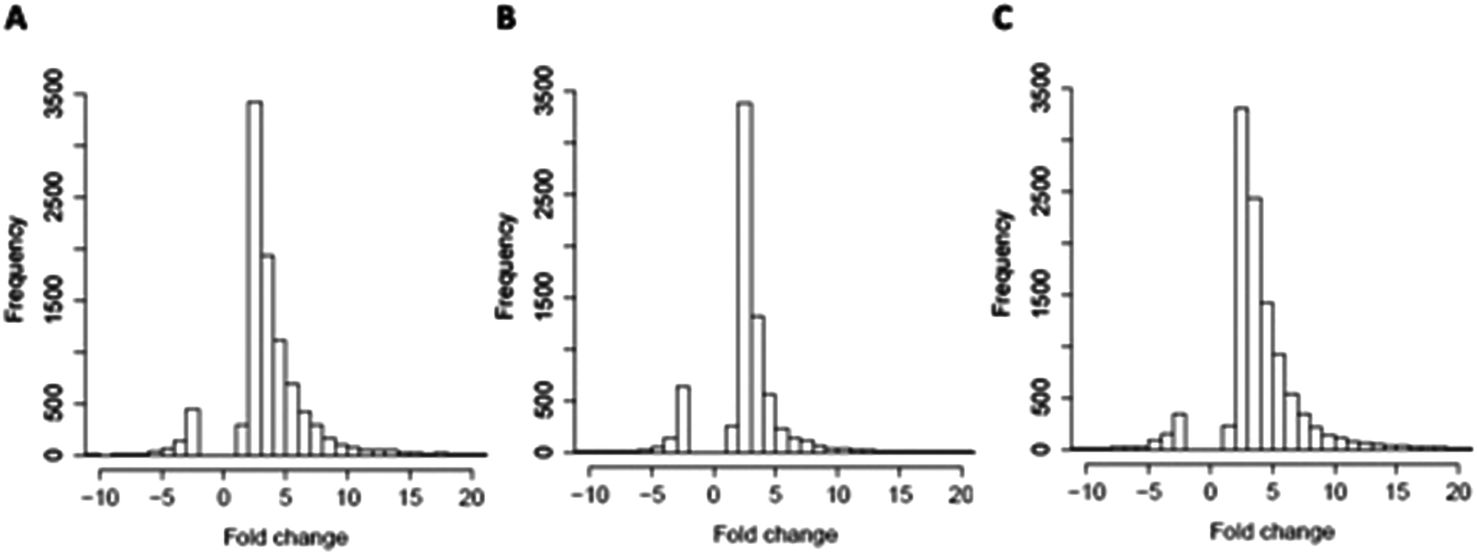 | ||
| Fig. 4 Effects on gene expression in HT-29 cells exposed to bay leaf size fractions for 48 h. Compared with the control, treatment with UFBL (A), HFBL (B) and LFBL (C) samples results primarily in up-regulation of genes, with fold-changes falling mainly between 0 and 5. | ||
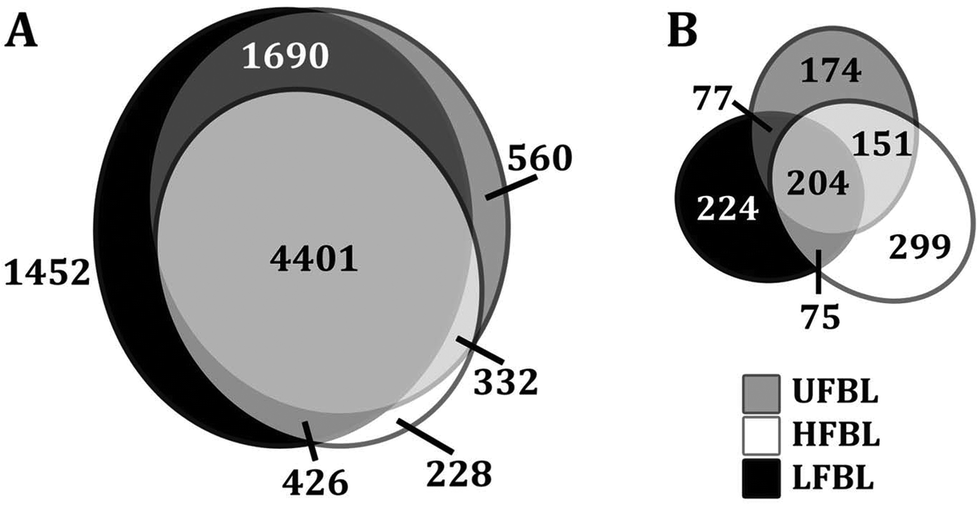 | ||
| Fig. 5 Majority of up-regulated gene expression changes are common for all three extracts of bay leaf. Venn diagrams demonstrating the overlap of changes in either up-regulation (A) and down-regulation (B) of gene expression for UFBL, HFBL and LFBL samples, compared with control. | ||

|
|
|
Expression of the tumor suppressor p53 was up-regulated post-treatment by all bay leaf samples, particularly LFBL. P53 activates both the cyclin-dependent kinase (CDK) inhibitors: p21 (up-regulated expression specifically by LFBL) and p27, resulting in their increased expression. These genes then inhibit the expression of CDK2. Moreover, treatment with LFBL resulted in a decrease in expression of both cyclin A and cyclin E. A decrease in expression of cyclin A/CDK2 and cyclin E/CDK complexes affected transition through the S and G1 phases respectively, possibly inducing cell cycle arrest during the G1/S transition, and subsequently leading to a reduction in cell proliferation (Fig. 6).
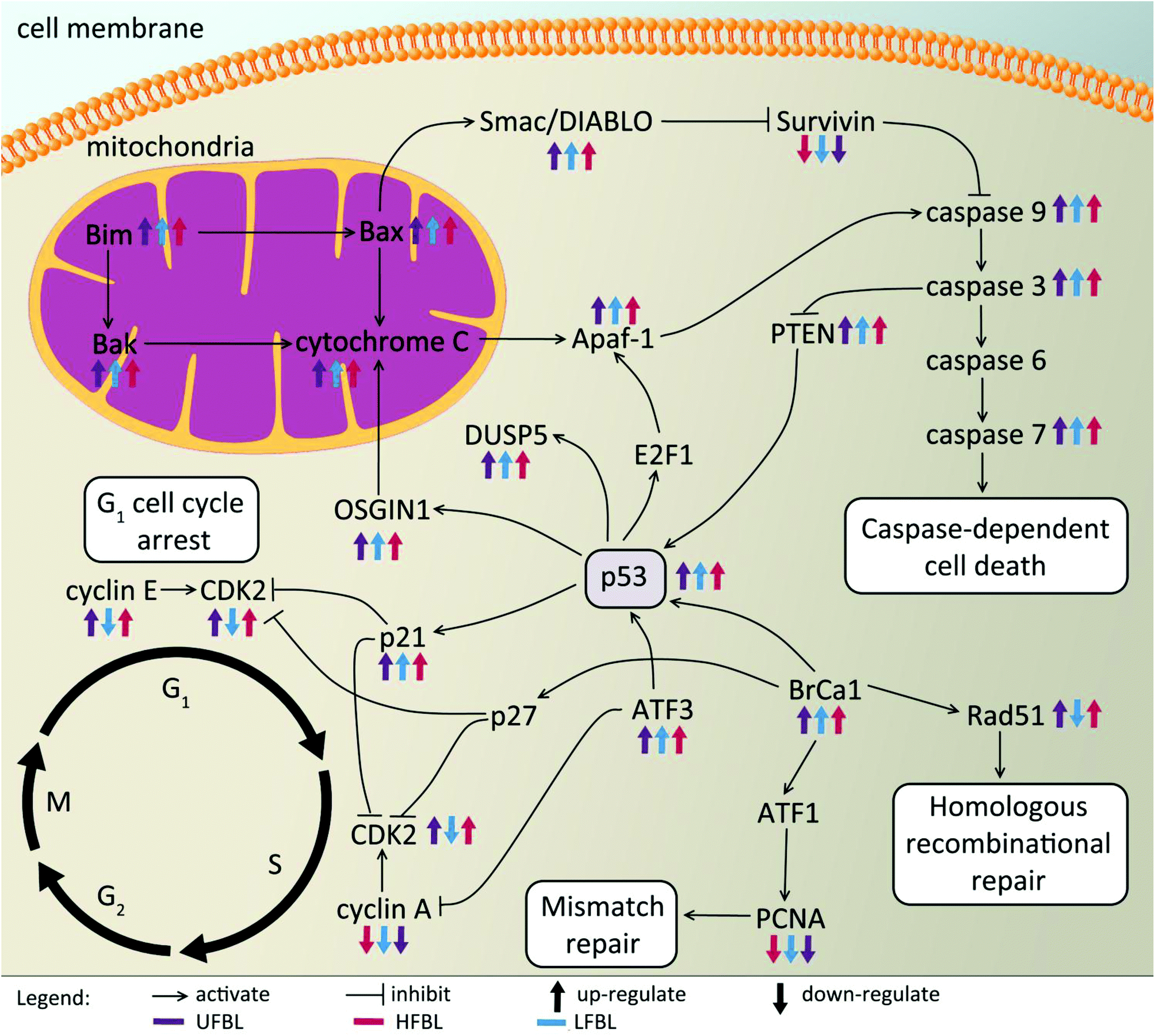 | ||
| Fig. 6 The expression of a number of genes involved in apoptosis and cell cycle regulation are affected by treatment with bay leaf extracts. Tumor suppressor p53 is primarily up-regulated by HFBL, which influences a number of genes involved in p53-dependent cell death, DNA repair, and initiation of cell cycle arrest. | ||
Treatment of HT-29 cells with bay leaf samples also resulted in distinct changes in expression of multiple genes involved in apoptosis (Table 2). The intrinsic pathway of cell death is expected to initiate release of a number of proteins, including cytochrome c from the inter-membrane space of the mitochondria into the cytoplasm. It is understood that the release of such proteins is a result of increased Bim expression (all three extracts, particularly LFBL, increase its expression), which directly mediates Bak and Bax (also increased expression post-treatment with bay leaf), leading to the cytochrome c release. Cytochrome c then binds to apaf-1 protein, in turn causing its activation. Apaf-1 subsequently binds ATP/dATP forming the apoptosome and mediating activation of caspase 9. This initiates the caspase cascade, leading to the activation of subsequent caspases, including caspase-3 and caspase-7, both which are up-regulated following treatment with bay leaf. Caspase 9 inhibits the expression of PTEN, which subsequently activates p53 thereby increasing its expression. Bax plays a pivotal role in inducing apoptosis by controlling the release of the IAP-binding mitochondrial protein, Smac/Diablo from the mitochondria. Smac/Diablo binds to the IAP protein, survivin, subsequently inhibiting it. Thus, post treatment with bay leaf samples, a decrease in expression of this protein is observed. Survivin blocks release of cytochrome c, hence a decrease of survivin leads to increased expression of cytochrome c, an observation seen following bay leaf treatment. P53 is also involved in initiation of apoptosis, activating E2F1, which then activates Apaf-1, eventually resulting in initiation of the caspase cascade. Further, p53 is responsible for the activation of both OSGIN1 and DUSP5, two proteins that have previously been implicated in malignant cell death. HFBL bay leaf caused a decrease in expression of DNA repair proteins: Rad51 and Proliferating Cell Nuclear Antigen (PCNA), both of which are mediated by Brca1 protein, which also displays elevated expression by bay leaf treatment (Fig. 6).
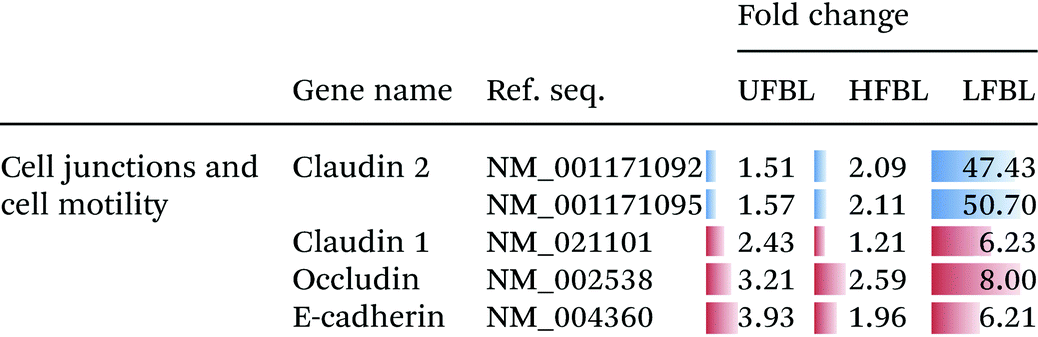
Tight junctions have been implicated in cell proliferation and cancer, where claudins and occludins are the major type of proteins involved. Claudin 2 is down-regulated following treatment with all three bay leaf samples, particularly the LFBL (Table 3). Conversely, expression of claudin 1 is increased, specifically by the LFBL sample. Occludin expression is also increased, consistent with it known activation by claudin 1, once again predominantly by the LFBL sample (Table 3). Increased expression in both claudin 1 and occludin results in tight junction disassembly. Furthermore, expression of E-cadherin (E-CD) is also increased, resulting in adherens junction disassembly (Fig. 7). Finally the expression of selected implicated genes were validated using immunofluorescence (Fig. 8) and qPCR analysis (Fig. 9) with similar fold changes of gene expression to observed, compared to mRNA-Seq analysis.
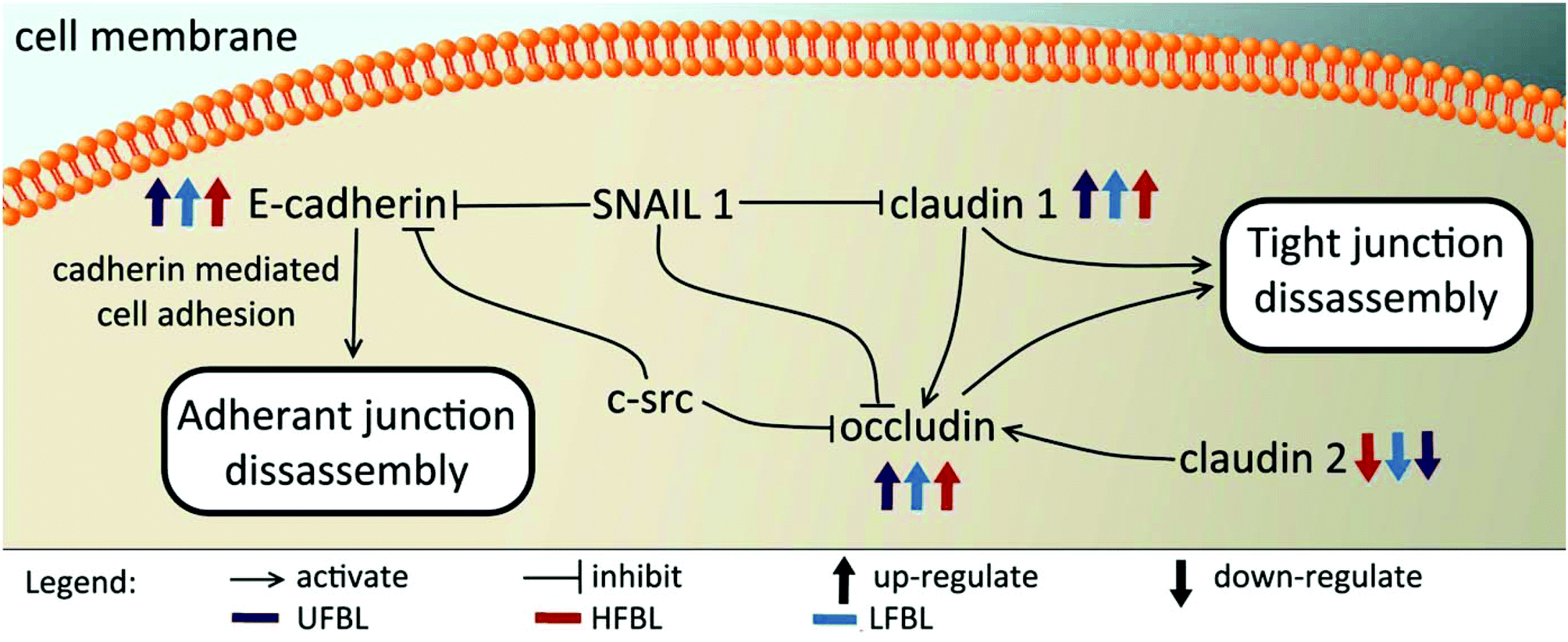 | ||
| Fig. 7 Treatment with bay leaf disrupts genes involved in both tight and adherin junctions, ultimately resulting in their disassembly. Post-treatment with bay leaf, a decrease in expression of Wnt, and simultaneous increase in expression of ID2, Axin 2, APC, β-catenin and E-cadherin is observed. Similarly, an increase of tight junction proteins claudin-1 and occludin is seen, while a decrease in claudin-2 is noticed. | ||
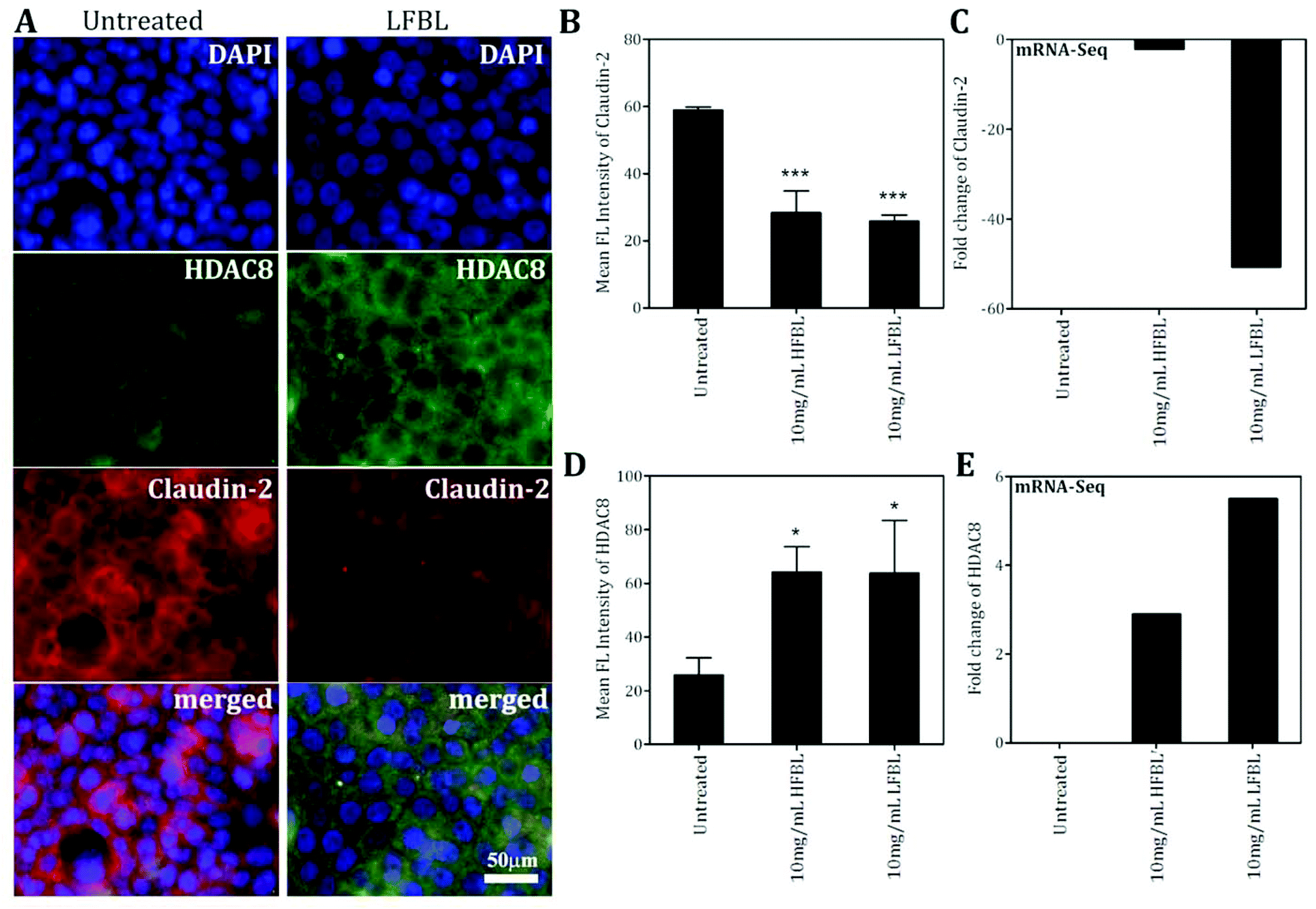 | ||
| Fig. 8 Validation of mRNA-Seq data was also employed by immunofluorescence assays. HT-29 cells pre-treated with HFBL and LFBL fractions were fixed and stained for anti-claudin-2 (red) and anti-HDAC8 (green). Shown are representative photomicrographs of HT-29 cells treated with HFBL or LFBL samples; bar = 50 μm (A). The mean ± SD of the total fluorescence intensity of claudin-2 (B) and HDAC8 (D) in a single cell are shown. Similar trends were observed in mRNA expression fold changes for genes claudin-2 (C) and HDAC8 (E). | ||
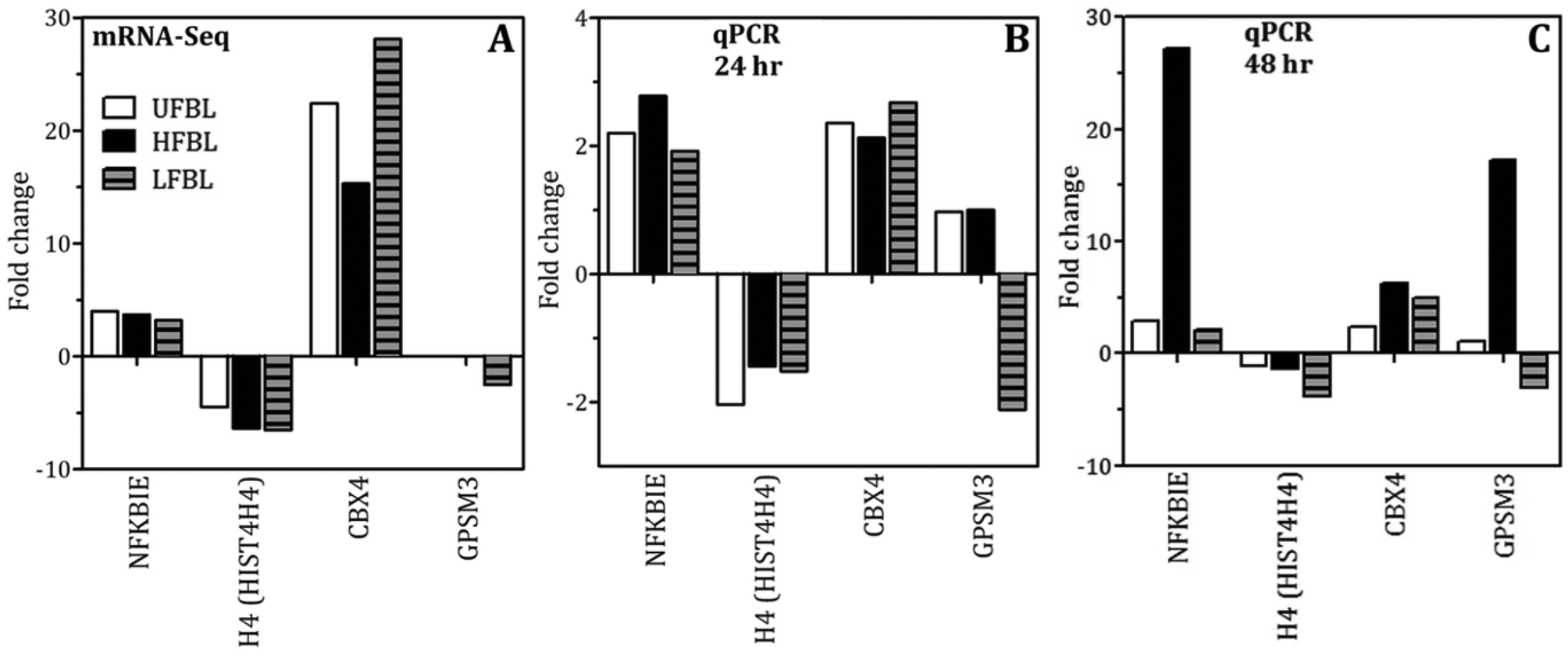 | ||
| Fig. 9 Trends in gene expression changes measured by qPCR were in agreement with those measured by mRNA-Seq. HT-29 cells were treated with UFBL, HFBL and LFBL samples prior to isolation of the RNA. The RNA of selected genes shown was amplified by RT-PCR and the results are expressed as the fold change in mRNA expression from the untreated cells. | ||
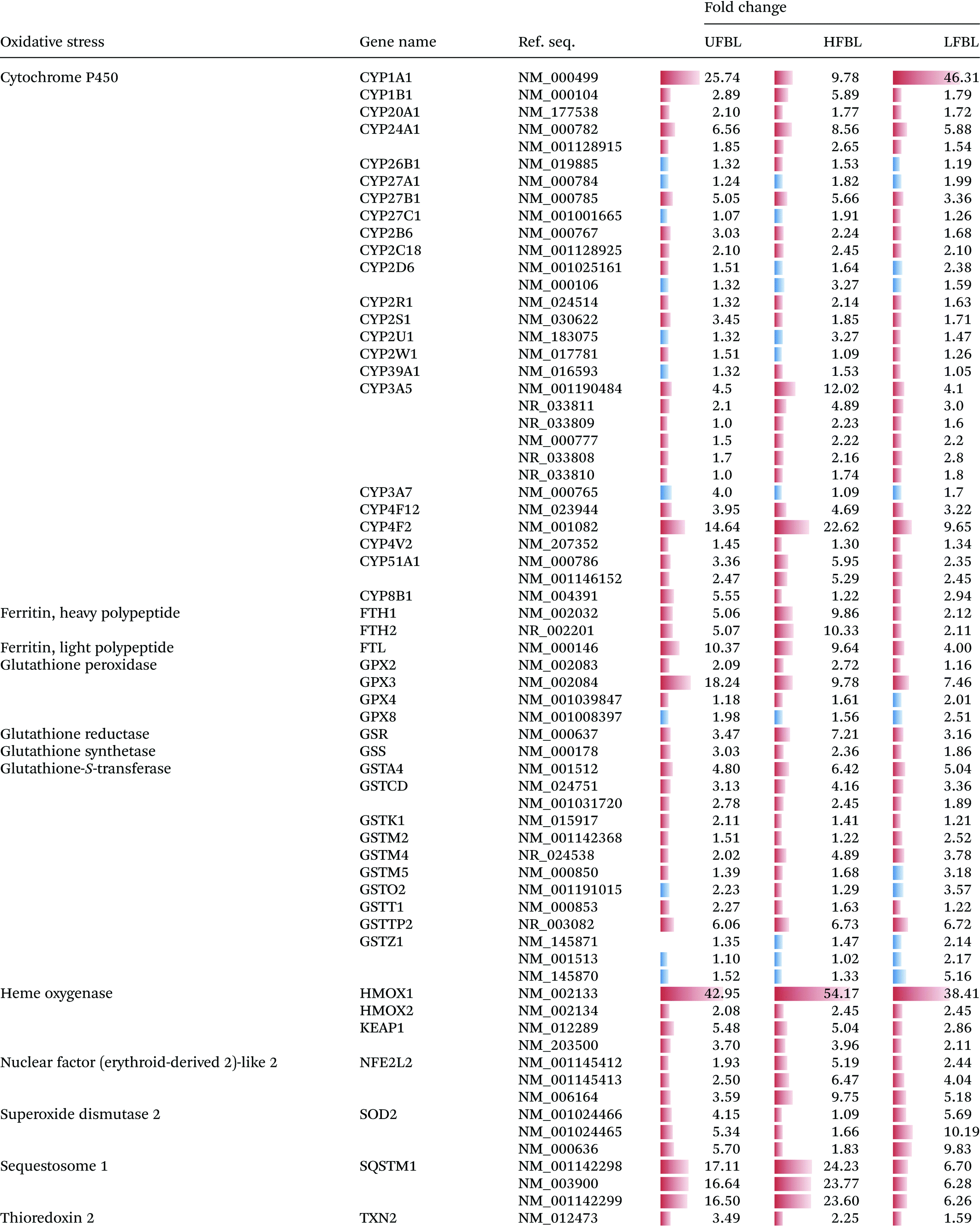
A substantial number gene families (13) and individual genes (67) related to management of oxidative stress were significantly up or down regulated by bay leaf fraction treatments (Table 4). Unlike for other gene classes, which responded most strongly to LFBL and UFBL, oxidative stress response pathways, except for SOD2, were more strongly influenced by HFBL (Table 4). Highest differential gene up-regulation by HFLB occurred for genes in cytochrome P450, ferritin, glutathione-S-transferase, heme oxygenase, nuclear factor (erythroid-derived 2) – like 2 and sequestome 1 families. These are all genes involved in direct management of ROS and electron exchanges involving oxygen. Conversely, genes differentially upregulated by LFBL which were also unregulated by UFBL, included cytochrome P450 (CYP1A1) and SOD2. Activation of these genes by LFBL and UFBL can account for P450-catalysed oxidation of LFBL substrates and production of hydrogen peroxide.
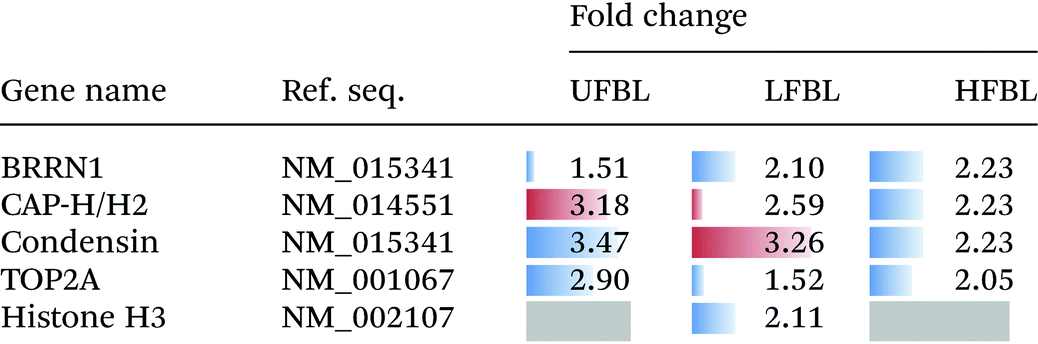
Chromatin condensation into chromosomes is an essential process in both malignant and normal cells and disruption of this pathway can also account for observed cell death (Fig. 2). Several proteins involved in this pathway were also affected by treatment with bay leaf extracts (Table 5). CDK1 phosphorylates the BRRN1 gene, therefore a decrease in CDK1 following treatment with bay leaf also results in a decrease in BRRN1 expression. BRRN1 activates both CAP-H/H2, which was down-regulated by LFBL but up-regulated by HFBL, and TOP2A, also involved with chromatin condensation, was down-regulated by UFBL, HFBL and LFBL. CAP-H/H2 activates condensin, which was down-regulated by both UFBL and LFBL. Condensin binds and activates histone H3 which was selectively down-regulated by HFBL but unaffected by UFLB or LFBL. Finally, BRRN1 binds and subsequently activates TOP2A and was down-regulated by UFBL, HLBL and LFBL.
|
Discussion
In our previous study, unfractionated bay leaf extract produced anti-proliferative effects on HT-29, SW-480 and HCT-116 human colon cancer cell lines.26 Furthermore, HFBL and LFBL were also shown to influence HT-29 cell death by complimentary apoptotic and glutathione-sensitive effects and it was proposed that redox-active polyphenol oxidase (PPO) could account for differentiated activity of HFBL versus LFBL26 The superior efficacy of HFBL versus LFBL on driving apoptotic cell death by caspase 3/7-dependent autophagic cell death via the mitochondria/cytochrome c-mediated activation pathways27 was confirmed in the present study (Fig. 1C). However, the demonstration that HFBL, but not LFBL, mediated DNA strand breaks necessitated the involvement of a toxic species that could penetrate cellular and nuclear membranes. Cell viability measures indicated that LFBL was more effective than HFBL and the potency of LFBL increased with incubation time (Fig. 1A and 1B). However, γH2AX lesions were exclusively mediated by HFBL, and amplified by γ-radiation, suggesting that unsuppressed free radical species were responsible for activity of HFBL. Subsequent gene expression profiles of HT-29 cells revealed that HFBL and LFBL induced differentiated adaptive cellular responses including in: cell cycle arrest and apoptosis, ROS defense and repair of DNA damage.Cell culturing and reactive oxygen species (ROS)
Oxidation of ferrous iron by oxygen under cell culturing conditions can produce ROS including superoxide, hydrogen peroxide and hydroxyl radicals.28 Likewise, selected phytochemicals with anti-oxidant properties (pAOX), can promote production of hydrogen peroxide in cell culture media such as DMEM containing ferrous iron.29–31 Cellular responses due to hydrogen peroxide toxicity can account for apparent anti-proliferative effects of pAOX species29 and explain up-regulation of similar genes for different pAOX species. For example, some of the p53-dependent apoptosis genes influenced by LFBL in this study were common to those up-regulated in HT-29 cells by rosemary extract also cultured in media containing iron.32 We hypothesize that although LFBL species effectively promoted ROS-mediated cell damage and apoptosis in the surrounding media including attacking cell junctions (Table 3), upon uptake of both ROS and LFBL into cells, LFBL mediated AOX protection, stimulated AOX cell defences, including upregulation of Cytochrome P450 and SOD2 (Table 4), and prevented DNA damage. The pattern of ROS/LFBL-mediated apoptosis of HT-29 cells was relatively delayed compared with HFBL (Fig. 2).Conversely, in the presence of HFBL, oxidation of polysaccharide species by hydroxyl radicals can produce hydroxyl-alkylperoxyl radicals, which in turn release superoxide,33 and could feasibly contribute to the iron-mediated ROS in the cell media. Transmission of ROS through cellular and nuclear membranes, but not extra-cellular HFBL, produced DNA damage and a distinct profile of oxidative stress gene responses compared with LFBL (Table 4). DNA strand breaks were also shown to be concentration-dependently mediated by superoxide in the presence of the large glycoprotein ricin.34 Likewise, superoxide-mediated damage to DNA was manifested as strand breakage, causing base modification and decrease in melting temperature.35 The gene expression responses associated with HFBL and LFBL can therefore be interpreted to reflect distinct ROS-mediated pathways of activity, and effects of intra-cellular protective effects that suppressed DNA damage, exerted by LFBL. The comparable pro-apoptotic effects of extracellular HFBL observed by γ radiation, a positive control for ROS production, support the interpretation that Fe-mediated ROS was also involved in HFBL-mediated apoptosis in cell culture experiments.
Unlike for other pathways (Tables 2 and 3), expression of genes regulating oxidative stress were mostly upregulated by HFBL and down regulated by LFBL (Table 4). The exception was SOD2 which was more strongly upregulated by LFBL versus HFBL, supporting the previous interpretation. Opposing directions of fold-changes between HFBL and LFBL, were observed for cytochrome P450, glutathione peroxidase (HFBL promoted defense against lipid peroxidation), glutathione-s-transferase (HFBL promoted glutathione-mediated reactivity with HFBL substrates). A key specific growth-suppressing bioactivity of HFBL appears to be the result of strong upregulation of sequestome 1 (p62) (>20-fold) which mediates autophagy-dependent tumour suppression through suppression of ROS, also involving KEAP1 (Table 4).36 LFBL displayed comparatively lower capacity to stimulate either sequestome 1 or KEAP1, instead expressing SOD2 (Table 4). Based on the distinct gene expression profile stimulated by HFBL versus LFBL (Table 4), it seems that ROS defense associated with LFBL were mediated by SOD2 and by sequestome 1 and KEAP1 for HFBL.
Effects on apoptosis
The intrinsic pathway of cell death was initiated following treatment of HT-29 cells with bay leaf, whereby similar levels of increased fold-change for caspase and other cell death-regulating genes was evident for either UFBL, HFBL or LFBL (Table 2). Cell death involves multiple proteins, beginning with release of cytochrome c from the inter-membrane space of the mitochondria into the cytoplasm. The release of such proteins is a result of increased Bim expression (elevated equally by UFBL, HFBL or LFBL), which directly mediates Bak and Bax (also increased expression post-treatment with bay leaf), leading to the cytochrome c release. Cytochrome c then binds to apaf-1 protein, in turn causing its activation. Apaf-1 subsequently binds ATP/dATP forming the apoptosome and mediating activation of caspase 9. This initiates the caspase cascade, leading to the activation of subsequent caspases, including caspase-3 and caspase-7, both up-regulated by bay leaf. Bax plays a pivotal role in inducing apoptosis by controlling the release of the IAP-binding mitochondrial protein, Smac/Diablo from the mitochondria. Smac/Diablo binds to the IAP protein, survivin, subsequently inhibiting it. Survivin expression is decreased by bay leaf extract, and leads to increased expression of cytochrome c, (Table 2). While the caspase 3/7 assay showed a significantly increased level of cell death post-treatment with the HFBL, this was not replicated in the gene expression levels determined by mRNA-Seq. The high dynamics and post-translational modifications of proteins can influence protein detectability, and may explain the differences observed in gene expression from RNA-Seq studies as compared to results obtained in the laboratory.37 Specifically, while mRNA expressions may remain constant between the cells treated with the three different bay leaf samples, the individual fractions may stimulate post-translational events of genes that will in turn affect their activation and subsequent action. Nevertheless, the increased expression of proteins driving intrinsic cell death, including by HFBL, together with the caspase 3/7 assay results, support the primary role of HFBL in initiation of cell death in HT-29 cells.Differential effects on DNA damage and repair by HFBL and LFBL
An early cellular response to DNA double-strand breaks (DSBs) is the phosphorylation of H2AX, a minor histone H2A variant.38,39 The rapid phosporylation of H2AX at the Ser-139 C-terminus to produce γH2AX can be measured as a biomarker of DNA damage and repair.40,41 DSBs are the most critical cell lesion with respect cell survival as they affect the linear continuity of the genome38 and can be produced by ionizing radiation.40 Following 48 hour incubation with HT-29 cells, bay leaf samples produced significant numbers of γH2AX foci, with highest effect by HFBL (>25 foci per cell) compared with UFBL and LFBL (both <15 foci per cell, Fig. 3). Effects of HFBL on the yield of DNA DSBs demonstrate the distinct nature of the cytotoxic lesion by HFBL alone, and not by LFBL, which was further amplified by ionizing radiation.CBX4 is an early DNA damage response protein that stimulates the accumulation of BMI1 at DNA damage sites.42,43 Furthermore, the removal of CBX4 results in decreased cellular resistance to ionizing radiation.42 Following treatment with UFBL, HFBL and LFBL, CBX4 gene expression was increased by 22.4, 15.3 and 28.1 fold change, respectively, suggesting that bay leaf samples would be differentially protective against ionizing radiation, with highest protection afforded by LFBL followed by UFBL. In addition, BMI1 was also increased 2.2, 0 and 4 fold for UFBL, HFBL and LFBL respectively (data not shown) which was validated by qPCR (Fig. 9). This can explain the lack of repair of HFBL-mediated DNA damage and significantly elevated γH2AX foci evident at 48 hours post irradiation (Fig. 3D).
Following DNA damage, cells undergo a series of complex processes including cell cycle arrest, activation of genes involved in repair of DNA, and in extreme cases the initiation of programmed cell death.44 In the case of malignant cells increasing their repair capacity, promotion of cellular resistance to the DNA-damaging agent will follow. As such, the genes and proteins involved with DNA repair may promote disease resistance to therapy. The Rad51 recombinase, a key player in eukaryotic DNA double-strand break repair, is an essential factor involved in homologous recombination repair.45,46 Tumor cells have increased levels of Rad5144,47 which results in increased resistance to DNA damage by chemotherapeutic drugs, which in turn, permits tumor progression, and may contribute to genomic instability.46 Relative to control, Rad51 expression was increased by HFBL (+1.26 fold), but decreased by LFBL (−2.1-fold, Table 2). In addition, PCNA augments DNA polymerase activity resulting in DNA replication and modulation of DNA repair processes including mismatch repair.48–50 As for Rad51, LFBL was also associated with a decrease in expression of PCNA, thus contributing to inhibition of repair of HFBL ROS-mediated DNA damage. Differential responses of chromatin condensation genes, condensin and CAP-H/H2 (Table 5), favoring their upregulation by HFBL but down-regualtion by LFBL, further supported that different apoptotic pathways were mediated by respective fractions.
Effects on cell cycle
Bay leaf extracts delayed cell cycling, reduced viability and increased apoptosis (as measured by caspase 3/7 assays, Fig. 1) and necrosis in cultured HT-29 cells. This is reflected in the increased proportions of bay leaf-treated cells in sub G1 phase, with the effects of HFBL being observed early (24 h) while the effects of LFBL were more delayed. (Fig. 2). The relative increase in the proportion of cells in S phase after 24 h suggested that both HFBL and LFBL elicited cell cycle arrest somewhere in the transition from G1 to S phase (Fig. 2).CDKs are a conserved family of protein kinases that control orderly progression through phases of the cell cycle, regulate the rate of proliferation and therefore represent promising targets for anti-cancer therapy.51,52 CDKs associate with various cyclin regulatory subunits, forming cyclin-CDK complexes which are critical regulators of cell-cycle progression in both malignant and normal cells.53 NGS analysis revealed altered expression of the CDKs by treatment with all bay leaf samples (Table 2). LFBL affected expression of a number of genes involved in the cell cycle progression by cyclin/CDK complex, and LFBL but not UFBL or HFBL, down-regulated CDK2 (Table 2). CDK2 associates with, and is regulated by both the regulatory cyclins E and A, both of which were also down-regulated by LFBL. While the CDK2/cyclin A complex is necessary for progression through the S phase of the cell cycle and studies have demonstrated that degradation of both CDK2 and cyclin A together causes massive tumor cell apoptosis both in vitro and in vivo,51 cyclin E is a requisite regulator of the G1–S transition. Cyclin E is often over-expressed in human malignancies, and supports the notion that deregulation of cyclin E/CDK2 may contribute to carcinogenesis.54
Specifically, p53 activates p21 leading to its subsequent increase, and mediation of p53-dependent growth arrest. CDK inhibitors (Cip/Kip) are well characterized for their role as negative regulators of cell cycle progression through interaction and subsequent inhibition of CDK molecules.55 In particular, p21cip1 and p27kip1 inhibit a broad-spectrum of cyclin-CDK complexes, including the cyclin A/CDK2 complex, and cyclin E/CDK2 complex. P21 is a cell cycle inhibitor, playing a significant role in halting cellular proliferation and having the ability to promote differentiation and cellular senescence via the formation of a quaternary complex with cyclins, CDKs and PCNA.56,57 P21 mediates its various biological activities mainly by inhibiting the kinase activity of the CDK1 and CDK2, leading to cell cycle arrest at specific stages. In the present study, levels of p21 were up-regulated following treatment with all three bay leaf samples, and a significant increase in p27 expression was also observed after treatment of cells with LFBL, but not UFBL or HFBL. Up-regulation of p21 is significant as inefficient expression of this gene equates to decreased survival rates in patients with CRC.58,59 Additionally, as CDK2 blocks p21 expression, the decreased levels of CDK2 lead to an up-regulation of p21. In this light, the suppression of cell proliferation and the cell cycle arrest at either the G1 or S phases induced by LFBL (i.e., peroxide) treatment most likely results from its ability to decrease expression of CDK2, cyclin A and cyclin E.
Alternatively, if DNA damage is severe, p53 promotes apoptosis. Activating transcription factor 3 (ATF3) is part of the ATF/CREB family of bZIP transcription factors, and is characterized as a stress-inducible or adaptive response gene.60,61 Effects of LFBL on cell cycle and apoptosis can also be accounted for by the significant up-regulation of ATF3 (214.3 fold, Table 2). ATF3 is described as a regulator of tumor suppressor functions in colorectal carcinogenesis62 that most likely stabilizes and increases transcriptional activity of p53 in the genotoxic response.63 Further, over-expression of ATF3 promotes induction of cell cycle arrest and inhibition of proliferation, through antagonising the expression of cyclin A,64 induction of apoptosis, and an overall retardation of tumor formation.65
Effects on tumor suppression
Previously described as the ‘guardian of the genome’,66 the development of more than 50% of human cancers is associated with the loss of, or mutations in the p53 gene,67 which can also cause resistance of therapy.68 By regulating an array of genes, p53 exerts its tumor suppressor effect as a molecular switch, inducing death of cells with irreversibly damaged DNA or other genomic errors.68,69 Over 75% of colorectal carcinomas involve a deletion at 17q, which is thought to affect the tumor suppressor gene, p53.70 Following DNA damage, p53 delays the G1 phase to permit DNA repair.71,72 All bay leaf samples caused up-regulation of p53 expression that can account for HT-29 growth arrest. However, p53 expression at 48 h was higher for LFBL than HFBL (Table 2) whereas apoptosis was more extensive for HFBL (Fig. 2), suggesting that DNA damage caused by HFBL was different and not assisted by this repair pathway to the same extent as LFBL.In addition to the p53 pathway, phosphatase and tensin homolog protein (PTEN) is a potent tumor suppressor gene that is also often mutated in human cancers.73 PTEN auto-regulates its expression through stabilization of p53, and has been shown to enhance p53 transactivation.74 Further, this gene down regulates the activity of the lipid second messenger phosphoinositol-3,4,5-triphosphate (PIP3) through dephosphorylation, thereby negatively regulating PIP3K-triggered signaling. The PI3 K/Akt pathway is an important driver of cell proliferation and survival. Through the negative regulation of this signaling pathway, PTEN modulates cell growth and survival causing a decrease in cell migration and invasion and additionally resulting in cell cycle inhibition.75,76 In this regard, PTEN is frequently deleted in advanced tumors, however post-treatment with HFBL, an increase in expression of this transcript was observed.
ATF3 and PTEN are among many known proteins that interact with p53.69 Following treatment with the HFBL, the expression of another p53-target gene, oxidative stress-induced growth inhibitor 1 (OSGIN1), was significantly increased (Table 2). OSGIN1 is ubiquitously expressed and a putative p53 binding site has been located at the OSGIN1 promoter, indicating that this gene is regulated by DNA damage in a p53-dependent manner.77 OSGIN1 regulates the differentiation and proliferation of cells, and its over-expression induces apoptosis and cell death, as shown in A498 (malignant kidney cells), buffalo rat liver, and MCF-7 (malignant breast cells).78–80 Loss of OSGIN1 is positively correlated with rapid tumor growth and metastasis.81 Conversely, its upregulation by bay leaf, particularly by LFBL (Table 2), likely contributed to induction of HT-29 apoptosis and cell death.
Another protein that also contains a p53-promoter region is the VH1-like enzyme, dual specificity protein 5 (DUSP5). Binding of p53 to the promoter region of DUSP5 has been shown to activate this gene in response to DNA damage in colon-cancer cells.82 Further, growth suppression in four distinct cancer cell lines was associated with elevation of DUSP5 expression, which may involve phosphatase activity toward mitogen-activated protein kinases, including MAPK3. As for OSGIN1, LFBL significantly up-regulated expression (∼271-fold), more effectively than either UFBL (∼1.6-fold) or HFBL (∼3-fold), and may also contribute to the growth-suppressing effects of bay leaf on HT-29 cells. Up-regulation of OSGIN1 and DUSP1 by a solvent extract of rosemary, likely enriched in low mass polyphenolics and under potential ROS-producing conditions, was recently reported.32
Effects on cell membrane integrity and tumor metastasis
Tight junctions are essential for the sealing of the cellular sheets, therefore controlling paracellular ion flux and maintaining tissue homeostasis. Tight junctions also play a crucial role in the maintenance of cell polarity by forming a barrier that acts to prevent lateral diffusion of membrane proteins and lipids, thereby maintaining the differential composition of the apical and basolateral domains. Tight junctions are composed of three major integral membrane proteins, occludins, claudins, and junctional adhesion molecules (JAMs). Loss of cellular organization due to impairment of apical and tight junctions has been implicated in cell proliferation and cancer.83 Silencing of occludin promotes cancer cell tumorigenesis and metastasis.84,85 Compared with other fractions, LFBL was associated with stronger increased expression of occludin (Table 3), also favouring more effective suppression of HT-29 proliferation.While decreased claudin expression is observed in a number of human malignancies, leading to compromised tight junction function, this does not seem to be the case for claudin-2, where high expression levels were seen in CRC.86 Treatment with the bay leaf, particularly LFBL, significantly decreased expression of the tight junction protein, claudin-2 Table 3). Claudin-2 is a unique member of the claudin family of transmembrane proteins, and its role in neoplastic transformation and growth was confirmed by up-regulation and correlation with cancer progression in tissues from 309 CRC patients.86 Moreover, expression of claudin-2 in breast cancer was correlated with liver metastasis.87 These data suggest that claudin-2 plays a role in cancer cell metastasis, possibly in the specific invasion of the liver, which is the most common site of metastasis and cause of death in CRC.48,88 It has been proposed that up-regulation or aberrant tissue expression of certain claudins may contribute to neoplasia by directly altering tight junction structure and function, and also by affecting cell signalling pathways.86 Effects of bay leaf treatment (both HFBL and LFBL, Table 3) on down-regulation of claudin-2 protein synthesis was confirmed by immunofluorescence (Fig. 7) supporting the potential for claudin-2-mediated tumor suppressing efficacy of bay leaf, particularly LFBL (−50.7-fold, Table 3), and another potentially protective pathway of CRC pathogenesis.
In contrast to the decreased expression of claudin-2, all bay leaf samples, particularly LFBL, increased expression of claudin-1 (Table 3). Claudin-1 expression is often down-regulated in human malignancies, thus an increase of this gene via treatment with LFBL offers a potential mechanism of action. When the integrity of the tight junction is lost, there are many events that may contribute towards the promotion of tumorigenesis. The ability for nutrients to diffuse into malignant cells encouraging cell growth and survival,89 decreased polarity and differentiation may influence the potential for metastasis90 and finally, the unrestrained growth of the cell all contribute to rapidly proliferating and cells.91
In addition to the claudins, calcium-dependent cell–cell adhesion transmembrane glycoprotein's (cadherins, E-CD), mediate homophilic adhesion between cells and are also linked with cancer metastasis.92,93 Treatment with bay leaf fractions increased expression of E-CD with higher fold-change for LFBL (+6.2-fold) and UFBL (+3.9-fold) versus HFBL (+2.0-fold, Table 3). In contrast, E-CD is typically under-expressed in cancer cell lines and down-regulation of E-CD in human carcinomas correlates positively with malignant cell invasion.94 Promotion of E-CD by LFBL and consequent suppression of cancer cell invasion94–96 might also be expected to protect against liver metastasis in CRC.97 Collectively, the significantly divergent effects of HFBL versus LFBL on cell membrane also support distinct bioactive species were responsible and that LFBL-associated hydrogen peroxide was particularly effective in undermining membrane integrity.
Experimental
Processing and size-fractionation of bay leaf
Dried bay leaf (Laurus nobilis, Hoyts Food Industries, Pty Ltd, Moorabbin, Victoria, Australia) was obtained from a local supermarket. Bay leaf was processed to maximize water solubility of components by dispersing in water, blending in a food processor, microwave heating, ultrasonicating and freeze drying (‘Stage 1’ product), as described previously.26 Briefly, ‘Stage 1’ bay leaf product was dispersed at 10% solids (w/w) with sonication (10 min, Unisonics Pty Ltd, Brookvale, New South Wales, Australia) before centrifuging (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 10 min at 20 °C, Model J2-MC, rotor JA-14, Beckmann Coulter Inc., Brea, CA, USA). The Stage 1 product was the same as that used and characterised in ref. 26 but it was the Stage 1 and not the Stage 2 product that was fractionated and used in the current study. Previously, the supernatant was fractionated using preparative gel filtration chromatography (PD-10 column, Amersham Biosciences, GE Healthcare, Uppsala, Sweden) with 6 successive 1.75 ml aliquots of water and finally 3.5 ml of methanol (fraction 7), under gravity-based elution. For the current study, bay leaf Stage 1 product supernatant fractions studied were: unfractionated (UFBL); high molecular mass fraction (HFBL, PD-10 fractions 1–3, ∼4–18 kDa) and low molecular mass fraction (LFBL, PD-10 fractions 4–7, <1.4 kDa) of Stage 1 bay leaf. Yields of HFBL and LFBL were 3.1% and 3.6% (w/w) of total Stage 1 product solids. The compositional characteristics of the original series of products using HPLC, LCMS and SDS-PAGE methods were described previously.26 Dried UFBL, HFBL or LFBL were prepared in 10% ethanol (v/v) at 1 g ml−1, filter sterilised (0.22 μm) and stored at −80 °C until use.
000g, 10 min at 20 °C, Model J2-MC, rotor JA-14, Beckmann Coulter Inc., Brea, CA, USA). The Stage 1 product was the same as that used and characterised in ref. 26 but it was the Stage 1 and not the Stage 2 product that was fractionated and used in the current study. Previously, the supernatant was fractionated using preparative gel filtration chromatography (PD-10 column, Amersham Biosciences, GE Healthcare, Uppsala, Sweden) with 6 successive 1.75 ml aliquots of water and finally 3.5 ml of methanol (fraction 7), under gravity-based elution. For the current study, bay leaf Stage 1 product supernatant fractions studied were: unfractionated (UFBL); high molecular mass fraction (HFBL, PD-10 fractions 1–3, ∼4–18 kDa) and low molecular mass fraction (LFBL, PD-10 fractions 4–7, <1.4 kDa) of Stage 1 bay leaf. Yields of HFBL and LFBL were 3.1% and 3.6% (w/w) of total Stage 1 product solids. The compositional characteristics of the original series of products using HPLC, LCMS and SDS-PAGE methods were described previously.26 Dried UFBL, HFBL or LFBL were prepared in 10% ethanol (v/v) at 1 g ml−1, filter sterilised (0.22 μm) and stored at −80 °C until use.
Cell culture
HT-29 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% (v/v) FBS, 1 mM sodium pyruvate and 1% antibiotic-antimycotic. Cells were grown as monolayers in a humidified 5% CO2 environment at 37 °C. Single cell suspensions were prepared by trypsinization (0.05% (v/v) trypsin/EDTA solution (Invitrogen, Carlsbad, CA, USA) for 10 minutes at 37 °C, as required. For maintenance, cells were passaged once per week and seeded at ratios 1:10.
Cell viability and apoptosis
Cells were seeded at a density of 15000 cells per well in black, flat bottom, 96-well plates (Nalge Nunc, Penfield, NY, USA) and allowed to attach overnight. Bay leaf samples were diluted in culture medium to the required concentrations and incubated at 37 °C, 5% CO2. Cell viability was determined after 24 and 48 hours exposure to the bayleaf extracts and measured using the Cell-Titer Blue® Assay kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. Cell-Titer Blue reagent was added to each well and incubated for 4 hours at 37 °C, 5% CO2, before reading fluorescence intensity (550 nm excitation; 615 nm emission) using the Perkin Elmer Victor3 (Perkin Elmer, MA, USA) multi-label plate reader. Apoptosis was measured by the level of Caspase 3/7 activity following 48 hours exposure to the bay leaf extracts, using the Apo-ONE Homogeneous Caspase 3/7 assay kit (Promega, NY, USA) according to the manufacturer's instructions. Fluorescence intensity (485 nm excitation; 525 nm emission) was measured using the Perkin Elmer Victor3 multi-label plate reader 4 hours following incubation with Apo-ONE reagent at 22 °C, on a rotating platform.
Flow cytometric analysis
Flow cytometry was utilized for cell cycle analysis. HT-29 cells were treated 20 mg ml−1 of UFBL and 10 mg ml−1 of HFBL and LFMBL fractions for 24 and 48 hours before they were harvested with 0.05% trypsin and washed with ice-cold PBS containing 2% (v/v) FBS twice and fixed overnight with continuous rotation in 70% (v/v) ethanol at 4 °C. Cells where then stained in 100 μg mL−1 propidium iodide containing 1 μg mL−1 RNase A (Qiagen Inc., Valencia, CA), overnight whilst on rotation at 4 °C. Samples were then transferred to FACS tubes and analyzed by flow cytometry using a Facs Calibur cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Fluorescent debris was gated using forward scatter versus orthogonal side scatter with a secondary gate placed around the single cell population using a pulse area versus plate width dot plot. The percentage of cells in each phase of the cell cycle was analyzed using Cell Quest software (version 5.1, BD Biosciences, San Jose, CA, USA).γH2AX immunofluorescence
HT-29 cells were seeded at 15000 cells per well in 8-well chamber slides (Lab-tek, Nunc, New York, USA) and allowed to attached overnight. Cells were then treated with 20 mg mL−1 crude extract, 10 mg mL−1 HFBL and 10 mg mL−1 LFBL fractions for 48 hours, at 37 °C, 5% CO2, prior to γ-radiation (137Cs) at 0 or 4 Gy, and incubated for a further hour at 37 °C, 5% CO2. Cells were then stained and quantified for the number of γH2AX foci per cell nuclei as previously described.39 All analysis was performed using image analysis freeware (FIJI Image J, version 1.46d). A minimum of 2 independent experiments were completed in duplicate and results are expressed as the mean ± standard deviation.
Library construction, mRNA sequencing and pathway analysis
Total RNA was isolated from cells pre-treated with either control (10% ethanol), UFBL (20 mg ml−1) or HFBL and LFBL (both 10 mg ml−1) for 48 hours using Trizol (Invitrogen), following manufacturer instructions. RNA extraction, library preparation, alignment to the genome and bioinformatics analysis to obtain fold changes in mRNA expression was performed as previously described.98 The online program MetaCore (GeneGo Inc. St. Joseph, MI, USA) was used for pathway analysis. A fold-change of >1.5 was considered statistically significant.qPCR validation of next generation gene sequencing data
Total RNA was purified from control and treated cells using TRIZOL following manufacturer instructions. RNA was quantified with a Nanodrop spectrophotometer (Thermo Fisher) and Qubit fluorometer (Life Technologies). The integrity of the RNA was assessed using a MultiNA Microchip Electrophoresis System (Shimadzu, Kyoto, Japan). High-capacity cDNA reverse transcription kit from Applied Biosystems (P/N 4375222) was used to reverse transcribe RNA following the manufacturer instructions. qPCR reactions were performed using Sybr fast (Roche, Penzberg, Germany) on cDNA prepared from RNA. Certain oligonucleotides were designed using Primer blast online tool on published sequences for human RNA (NCBI, Drumcondra, Dublin). Primers were synthesized by Geneworks (Gene Works Pty Ltd, Hindmarsh, SA, AUS): CBX4 5′-GGTGGTCGGGCCTTTTGAGCT-3′ and 5′-GCGCCCGGCCTCAGAGTTCTA-3′; NFKBIE 5′-ACCCACTGCAACTTGGAAAGGCA-3′ and 5′-GGCCCTGCTAGGTCACCCCT-3′; HIST4H4 5′-GTCTTGCGCTTGGCGTGCTC-3′ and 5′-ACGAGGAGACCCGGGGAGTC-3′; GPSM3 5′-CAGGTGGAGGCCTGAGCCCT-3′ and 5′-GGGGGATCCCAGGCACCCAT-3′ (Sigma-Aldrich, St Louis, MI, USA). PCR was performed on ABI Fast 7500 (Applied Biosystems, Carlsbad, CA, USA). The housekeeping gene used as an endogenous control for the experiment was β-actin. Key genes were validated by RT-PCR and immunofluorescence.Immunofluorescence staining
Immunofluorescence staining was employed for validation of gene expression changes as seen by mRNAseq analysis using expression of proteins: claudin-2 and HDAC8. HT-29 cells were seeded at 40000 cell per well in glass LabTek™ II 8-well chamber slides (Thermo scientific, Rochester, NY, USA) and allowed to attach overnight prior to 48 hour incubation with UFBL, HFBL and LFBL samples at a concentration of 10 mg ml−1. The cells were washed twice with PBS for one min on rotation and fixed with 4% PFA (Sigma-Aldrich) for 10 min at 22 °C. Following a brief wash with PBS, the cells were permeabilized using 0.1% Triton X-100 (Sigma-Aldrich) for 10 min at 22 °C, washed three times with PBS and blocked with 1% bovine serum albumin (BSA, Sigma-Aldrich) whilst on rotation. Cells were then incubated with rabbit monoclonal anti-claudin-2 (Epitomics, Burlingame, CA) and mouse monoclonal anti-HDAC8 (Sigma-Aldrich) primary antibodies diluted in 1% BSA (1:100; Sigma-Aldrich) for one hour at 22 °C in a humid chamber. Following three consecutive washes in PBS, cells were incubated with a secondary goat anti-rabbit antibody (1:100 in 1% BSA) conjugated with Alexa-546 (Molecular Probes, Oregon, USA) and goat anti- mouse antibody (1:100 in 1% BSA) conjugated with Alexa-488 (Molecular Probes) incubated for one hour in a dark humidified environment on a rotating platform at 22 °C. Following three consecutive washes, cells were mounted with prolong Gold anti-fade solution with DAPI (Molecular Probes), cover-slipped (Biolab, VIC, Australia) and sealed with nail polish. Images were acquired using an Olympus BX61 motorised upright fluorescence microscope automated with FVII Camera using a 40× Uplan FL oil objective with fluorescence filter cubes: DAPI (Ex: 350/50, Em: 460/50 nm) and FITC (Ex: 470/40, Em: 525/50 nm). Fluorescence intensity of each protein was analyzed using image analysis freeware FIJI Image J (version 1.46d, Image J software).
Statistical analysis
A minimum of 2 independent experiments were completed for cell-based assays. Results are expressed as the mean ± standard deviation (SD). Statistical analysis was performed using a Student's t-test in GraphPad Prism (version 5, GraphPad Software Inc, CA, USA), and results for significances P, designated by *P < 0.05, **P < 0.01, ***P < 0.001.Conclusion
We propose that HFBL and LFBL extracts each promoted HT-29 apoptosis by mechanisms involving ROS. The pro-apoptotic effects of Fe-mediated ROS production bay leaf fractions in cell culturing conditions were capitulated by ionizing radiation-mediated ROS production, supporting that bay leaf fractions modulated cellular responses involving ROS. Different mechanisms of apoptosis are inferred from different rates of apoptosis, gene responses and DNA-specific damage associated with LFBL versus HFBL. Patterns of gene expression reported for HT-29 challenge by rosemary extract, also cultured in DMEM containing ferrous iron,32 were comparable to that reported for LFBL, supporting a common mechanism. Similarly, the pattern of HFBL toxicity was comparable to superoxide-mediated toxicity observed in the presence of the glycoprotein ricin.34 Gene expression analysis provided a comprehensive mechanistic framework for understanding the distinct effects of HFBL versus LFBL on modulating ROS-mediated toxicity.HFBL-mediated ROS production initiated more rapid apoptosis by non-repairable DNA DSBs. In comparison, LFBL promoted extra-cellular toxicity of ROS but protected against DNA damage. LFBL promoted selective stimulation of genes driving G1 cell cycle arrest, failure of cell assembly at tight junctions and inducing p53-dependent apoptosis and attenuated cellular recovery by suppressing DNA repair. These distinct biological effects were not apparent for UFBL, and gene expression patterns associated with UFBL were more similar to LFBL than HFBL. HFBL-mediated ROS defense was associated with changes in cytochrome P450 (CYP3A5 and others), glutathione peroxidase, glutathione-s-transferase, heme oxygenase and sequestome 1 whereas LFBL-mediated ROS defense involved cytochrome P450 (CYP1A1 and others) and SOD2. This study offers information about signatures of gene expression to identify if ROS-mediated or direct effects are causing cell death, particularly to inform interpretation of effects of other phytochemical extracts.
Compliance with ethical guidelines
Annabelle Rodd, Katherine Ververis, Dheeshana Sayakkarage, Abdul Khan, Haloom Rafehi,1 Mark Ziemann, Shanon Loveridge, Ross Lazarus, Caroline Kerr, Trevor Lockett, Assam El-Osta, Tom Karagiannis and Louise Bennett declare that they have no conflict of interest.This article does not contain any studies with human or animal subjects performed by any of the authors.
Acknowledgements
The support of the Australian Institute of Nuclear Science and Engineering is acknowledged. TCK was the recipient of AINSE awards and ALR is the recipient of an AINSE PGRA. Epigenomic Medicine Laboratory is supported by an Australian Research Council Future Fellowship and McCord Research (the parental company of Pinnaclife, Inc). Supported in part by the Victorian Government's Operational Infrastructure Support Program. The authors would like to acknowledge the use of the facilities provided by Monash Micro Imaging @ AMREP and particularly, the expert assistance from Drs Stephen Cody and Iśka Carmichael and to thank Katherine Robinson and Hema Jegasothy for preparing processed and size-fractionated bay leaf products.References
- J. Ferlay, H. R. Shin, F. Bray, D. Forman, C. Mathers and D. M. Parkin, International Agency for Research on Cancer, Lyon, France, 2010 Search PubMed.
- C. N. Arnold, A. Goel, H. E. Blum and C. R. Boland, Cancer, 2005, 104, 2035–2047 CrossRef CAS PubMed.
- C. Esteban-Jurado, M. Garre, P. Vila, J. J. lozano, A. Pristoupilova, S. Beltran, A. Abuli, J. Munoz, F. Balaguer, T. Ocana, A. Castells, J. M. Pique, A. Carracedo, C. Ruiz-Ponte, X. Bessa, M. Andreu, L. Bujanda, T. Caldes and S. Castellvi-Bel, World J. Gastroenterol., 2014, 20, 1961–1971 CrossRef CAS PubMed.
- K. M. Emmons, C. M. McBride, E. Puleo, K. I. Pollak, B. H. Marcus, M. Napolitano, E. Clipp, J. Onken, F. A. Farraye and R. Fletcher, Prev. Med., 2005, 40, 527–534 CrossRef PubMed.
- T. Tanaka, Y. Matsuo and I. Kouno, Int. J. Mol. Sci., 2010, 11, 14–40 CrossRef CAS PubMed.
- I. Herr and M. W. Buchler, Cancer Treat. Rev., 2010, 36, 377–383 CrossRef CAS PubMed.
- C. Fimognari and P. Hrelia, Mutat. Res., Rev. Mutat. Res., 2007, 635, 90–104 CrossRef CAS PubMed.
- I. Villegas, S. Sanchez-Fidalgo and C. A. de la Lastra, Mol. Nutr. Food Res., 2008, 52, 1040–1061 CAS.
- A. Bishayee, Cancer Prev. Res., 2009, 2, 409–418 CrossRef CAS PubMed.
- H. T. Saied and A. M. Shamsuddin, Anticancer Res., 1998, 18, 1479–1484 Search PubMed.
- M. Turktekin, E. Konac, H. I. Onen, E. Alp, A. Yilmaz and S. Menevse, J. Med. Food, 2011, 14, 1107–1117 CrossRef CAS PubMed.
- H. Kitamura, M. Cho, B. H. Lee, J. R. Gum, B. B. Siddiki, S. B. Ho, N. W. Toribara, T. Lesuffleur, A. Zweibaum, Y. Kitamura, S. Yonezawa and Y. S. Kim, Eur. J. Cancer, 1996, 32A, 1788–1796 CrossRef CAS.
- Y. J. Lee, G. Erdos, Z. Z. Hou, S. H. Kim, J. H. Kim, J. M. Cho and P. M. Corry, Mol. Cell. Biochem., 1994, 137, 141–154 CrossRef CAS.
- Y. S. Huang, N. Xie, Q. Su, J. Su, C. Huang and Q. J. Liao, Mol. Med. Rep., 2011, 4, 553–559 CAS.
- S. Veeriah, T. Kautenburger, N. Habermann, J. Sauer, H. Dietrich, F. Will and B. L. Pool-Zobel, Mol. Carcinog., 2006, 45, 164–174 CrossRef CAS PubMed.
- K. Sakulnarmrat, M. Fenech, P. Thomas and I. Konczak, Food Chem., 2013, 136, 9–17 CrossRef CAS PubMed.
- A. Clemente, M. C. Marin-Manzano, E. Jimenez, M. C. Arques and C. Domoney, Br. J. Nutr., 2012, 108, S135–S144 CrossRef CAS PubMed.
- D. K. Park and H. J. Park, Food Sci. Biotechnol., 2012, 21, 683–690 CrossRef.
- M. L. Mollah, D. K. Park and H. J. Park, Evidence-Based Complementary Altern. Med., 2012, 249217, DOI:10.1155/2012/249217.
- I. Lavi, D. Friesem, S. Geresh, Y. Hadar and B. Schwartz, Cancer Lett., 2006, 244, 61–70 CrossRef CAS PubMed.
- E. M. Coates, G. Popa, C. I. Gill, M. J. McCann, G. J. McDougall, D. Stewart and I. Rowland, J. Carcinog., 2007, 6, 4, DOI:10.1186/1477-3163-6-4.
- M. E. Juan, U. Wenzel, V. Ruiz-Gutierrez, H. Daniel and J. M. Planas, J. Nutr., 2006, 136, 2553–2557 CAS.
- G. X. Shen, C. J. Xu, C. Chen, V. Hebbar and A. N. T. Kong, Cancer Chemother. Pharmacol., 2006, 57, 317–327 CrossRef CAS PubMed.
- C. Chen, G. X. Shen, V. Hebbar, R. Hu, E. D. Owuor and A. N. T. Kong, Carcinogenesis, 2003, 24, 1369–1378 CrossRef CAS PubMed.
- M. R. Loizzo, R. Tundis, F. Menichini, A. M. Saab and G. A. Stati, Anticancer Res., 2007, 27, 3293–3299 CAS.
- L. E. Bennett, M. Y. Abeywardena, S. Burnard, S. Forsyth, R. Head, K. King, G. S. Patten, P. Watkins, R. Williams, D. Zabaras and T. Lockett, Nutr. Cancer: Int. J., 2013, 65, 746–764 CrossRef CAS PubMed.
- H. M. Prince, M. J. Bishton and R. W. Johnstone, Future Oncol., 2009, 5, 601–612 CrossRef CAS PubMed.
- S. D. Aust, L. A. Morehouse and C. E. Thomas, J. Free Radicals Biol. Med., 1985, 1, 3–25 CrossRef CAS.
- L. H. Long, A. Hoi and B. Halliwell, Arch. Biochem. Biophys., 2010, 501, 162–169 CrossRef CAS PubMed.
- P. Bellion, M. Olk, F. Will, H. Dietrich, M. Baum, G. Eisenbrand and C. Janzowski, Mol. Nutr. Food Res., 2009, 53, 1226–1236 CAS.
- S. C. Roques, N. Landrault, P. L. Teissedre, C. Laurent, P. Besancon, J. M. Rouant and B. Caporiccio, Free Radical Res., 2002, 36, 593–599 CrossRef CAS PubMed.
- C. Ibanez, A. Valdes, V. Garcia-Canas, C. Simo, M. Celebier, L. Rocamora-Reverte, A. Gomez-Martinez, M. Herrero, M. Castro-Puyana, A. Segura-Carretero, E. Ibanez, J. A. Ferragut and A. Cifuentes, J. Chromatogr. A, 2012, 1248, 139–153 CrossRef CAS PubMed.
- E. Choe and D. B. Min, Crit. Rev. Food Sci. Nutr., 2006, 46, 1–22 CrossRef CAS PubMed.
- E. A. Hassoun and X. Wang, J. Biochem. Mol. Toxicol., 1999, 13, 179–185 CrossRef CAS.
- G. Waris, M. A. Khan, S. Khan and K. Alam, J. Exp. Clin. Cancer Res., 2007, 26, 499–504 CAS.
- J. Moscat and M. T. Diaz-Meco, Trends Biochem. Sci., 2012, 37, 230–236 CrossRef CAS PubMed.
- B. E. Bernstein, A. Meissner and E. S. Lander, Cell, 2007, 128, 669–681 CrossRef CAS PubMed.
- L. J. Mah, A. El-Osta and T. C. Karagiannis, Leukemia, 2010, 24, 679–686 CrossRef CAS PubMed.
- L. J. Mah, R. S. Vasireddy, M. M. Tang, G. T. Georgiadis, A. El-Osta and T. C. Karagiannis, J. Vis. Exp., 2010 DOI:10.3791/1957.
- T. C. Karagiannis and A. El-Osta, Cell. Mol. Life Sci., 2004, 61, 2137–2147 CrossRef CAS PubMed.
- E. P. Rogakou, W. Nieves-Neira, C. Boon, Y. Pommier and W. M. Bonner, J. Biol. Chem., 2000, 275, 9390–9395 CrossRef CAS PubMed.
- I. H. Ismail, J. P. Gagne, M. C. Caron, D. McDonald, Z. Xu, J. Y. Masson, G. G. Poirier and M. J. Hendzel, Nucleic Acids Res., 2012, 40, 5497–5510 CrossRef CAS PubMed.
- V. Ginjala, K. Nacerddine, A. Kulkarni, J. Oza, S. J. Hill, M. Yao, E. Citterio, M. van Lohuizen and S. Ganesan, Mol. Cell. Biol., 2011, 31, 1972–1982 CrossRef CAS PubMed.
- W. Henning and H. W. Sturzbecher, Toxicology, 2003, 193, 91–109 CrossRef CAS.
- H. Koehn, N. Magan, R. J. Isaacs and K. M. Stowell, Anti-Cancer Drugs, 2007, 18, 419–425 CrossRef CAS PubMed.
- H. L. Klein, DNA Repair, 2008, 7, 686–693 CrossRef CAS PubMed.
- E. Raderschall, K. Stout, S. Freier, V. Suckow, S. Schweiger and T. Haaf, Cancer Res., 2002, 62, 219–225 CAS.
- M. J. Gray, N. A. Dallas, G. Van Buren, L. Xia, A. D. Yang, R. J. Somcio, P. Gaur, L. S. Mangala, P. E. Vivas-Mejia, F. Fan, A. M. Sanguino, G. E. Gallick, G. Lopez-Berestein, A. K. Sood and L. M. Ellis, Oncogene, 2008, 27, 7192–7200 CrossRef CAS PubMed.
- M. L. Smith, I. T. Chen, Q. Zhan, I. Bae, C. Y. Chen, T. M. Gilmer, M. B. Kastan, P. M. O'Connor and A. J. Fornace Jr., Science, 1994, 266, 1376–1380 CAS.
- A. Umar, A. B. Buermeyer, J. A. Simon, D. C. Thomas, A. B. Clark, R. M. Liskay and T. A. Kunkel, Cell, 1996, 87, 65–73 CrossRef CAS.
- W. Chen, J. Lee, S. Y. Cho and H. A. Fine, Cancer Res., 2004, 64, 3949–3957 CrossRef CAS PubMed.
- A. Goga, D. Yang, A. D. Tward, D. O. Morgan and J. M. Bishop, Nat. Med., 2007, 13, 820–827 CrossRef CAS PubMed.
- P. Hauser, L. Ma, D. Agrawal, E. Haura, W. D. Cress and W. J. Pledger, Mol. Cancer Res., 2004, 2, 96–104 CAS.
- A. B. Gladden and J. A. Diehl, Cancer Cell, 2003, 4, 160–162 CrossRef CAS.
- C. Denicourt and S. F. Dowdy, Genes Dev., 2004, 18, 851–855 CrossRef CAS PubMed.
- Y. Xiong, H. Zhang and D. Beach, Genes Dev., 1993, 7, 1572–1583 CrossRef CAS.
- K. F. Macleod, N. Sherry, G. Hannon, D. Beach, T. Tokino, K. Kinzler, B. Vogelstein and T. Jacks, Genes Dev., 1995, 9, 935–944 CrossRef CAS.
- J. Roman-Gomez, J. A. Castillejo, A. Jimenez, M. G. Gonzalez, F. Moreno, C. Rodriguez Mdel, M. Barrios, J. Maldonado and A. Torres, Blood, 2002, 99, 2291–2296 CrossRef CAS PubMed.
- T. K. Zirbes, S. E. Baldus, S. P. Moenig, S. Nolden, D. Kunze, S. T. Shafizadeh, P. M. Schneider, J. Thiele, A. H. Hoelscher and H. P. Dienes, Int. J. Cancer, 2000, 89, 14–18 CrossRef CAS.
- D. Lu, J. Chen and T. Hai, Biochem. J., 2007, 401, 559–567 CrossRef CAS PubMed.
- C. Yan and D. D. Boyd, Cell Cycle, 2006, 5, 926–929 CrossRef CAS.
- N. C. Whitlock, J. H. Bahn, S. H. Lee, T. E. Eling and S. J. Baek, Cancer Prev. Res., 2011, 4, 116–127 CrossRef CAS PubMed.
- C. Yan, D. Lu, T. Hai and D. D. Boyd, EMBO J., 2005, 24, 2425–2435 CrossRef CAS PubMed.
- C. G. James, A. Woods, T. M. Underhill and F. Beier, BMC Mol. Biol., 2006, 7, 30 CrossRef PubMed.
- F. Fan, S. Jin, S. A. Amundson, T. Tong, W. Fan, H. Zhao, X. Zhu, L. Mazzacurati, X. Li, K. L. Petrik, A. J. Fornace Jr., B. Rajasekaran and Q. Zhan, Oncogene, 2002, 21, 7488–7496 CrossRef CAS PubMed.
- D. P. Lane, Nature, 1992, 358, 15–16 CrossRef CAS PubMed.
- L. Roger, G. Gadea and P. Roux, Biology of the cell/under the auspices of the European Cell Biology Organization, 2006, vol. 98, pp. 141–152 Search PubMed.
- A. J. Levine, Cell, 1997, 88, 323–331 CrossRef CAS.
- L. J. Ko and C. Prives, Genes Dev., 1996, 10, 1054–1072 CrossRef CAS.
- N. S. Fearnhead, J. L. Wilding and W. F. Bodmer, Br. Med. Bull., 2002, 64, 27–43 CrossRef CAS.
- M. B. Kastan, O. Onyekwere, D. Sidransky, B. Vogelstein and R. W. Craig, Cancer Res., 1991, 51, 6304–6311 CAS.
- S. J. Kuerbitz, B. S. Plunkett, W. V. Walsh and M. B. Kastan, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7491–7495 CrossRef CAS.
- H. Sawai, A. Yasuda, N. Ochi, J. Ma, Y. Matsuo, T. Wakasugi, H. Takahashi, H. Funahashi, M. Sato and H. Takeyama, BMC Gastroenterol., 2008, 8, 56 CrossRef PubMed.
- Y. Tang and C. Eng, Cancer Res., 2006, 66, 736–742 CrossRef CAS PubMed.
- H. Sun, R. Lesche, D. M. Li, J. Liliental, H. Zhang, J. Gao, N. Gavrilova, B. Mueller, X. Liu and H. Wu, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6199–6204 CrossRef CAS.
- S. Kim, C. Domon-Dell, J. Kang, D. H. Chung, J. N. Freund and B. M. Evers, J. Biol. Chem., 2004, 279, 4285–4291 CrossRef CAS PubMed.
- H. Yao, P. Li, B. J. Venters, S. Zheng, P. R. Thompson, B. F. Pugh and Y. Wang, J. Biol. Chem., 2008, 283, 20060–20068 CrossRef CAS PubMed.
- T. Wang, D. Xia, N. Li, C. Wang, T. Chen, T. Wan, G. Chen and X. Cao, J. Biol. Chem., 2005, 280, 4374–4382 CrossRef CAS PubMed.
- C. K. Ong, C. Y. Ng, C. Leong, C. P. Ng, K. T. Foo, P. H. Tan and H. Huynh, J. Biol. Chem., 2004, 279, 743–754 CrossRef CAS PubMed.
- C. K. Ong, C. Y. Ng, C. Leong, C. P. Ng, C. S. Ong, T. T. Nguyen and H. Huynh, Endocrinology, 2004, 145, 4763–4774 CrossRef CAS PubMed.
- R. Li, W. Chen, R. Yanes, S. Lee and J. A. Berliner, J. Lipid Res., 2007, 48, 709–715 CrossRef CAS PubMed.
- K. Ueda, H. Arakawa and Y. Nakamura, Oncogene, 2003, 22, 5586–5591 CrossRef CAS PubMed.
- Z. Nemeth, A. M. Szasz, A. Somoracz, P. Tatrai, J. Nemeth, H. Gyorffy, A. Szijarto, P. Kupcsulik, A. Kiss and Z. Schaff, Pathol. Oncol. Res., 2009, 15, 533–539 CrossRef CAS PubMed.
- M. Osanai, M. Murata, N. Nishikiori, H. Chiba, T. Kojima and N. Sawada, Cancer Res., 2006, 66, 9125–9133 CrossRef CAS PubMed.
- T. A. Martin, R. E. Mansel and W. G. Jiang, Int. J. Mol. Med., 2010, 26, 723–734 CAS.
- P. Dhawan, R. Ahmad, R. Chaturvedi, J. J. Smith, R. Midha, M. K. Mittal, M. Krishnan, X. Chen, S. Eschrich, T. J. Yeatman, R. C. Harris, M. K. Washington, K. T. Wilson, R. D. Beauchamp and A. B. Singh, Oncogene, 2011, 30, 3234–3247 CrossRef CAS PubMed.
- S. Tabaries, Z. Dong, M. G. Annis, A. Omeroglu, F. Pepin, V. Ouellet, C. Russo, M. Hassanain, P. Metrakos, Z. Diaz, M. Basik, N. Bertos, M. Park, C. Guettier, R. Adam, M. Hallett and P. M. Siegel, Oncogene, 2011, 30, 1318–1328 CrossRef CAS PubMed.
- D. E. Stange, F. Engel, T. Longerich, B. K. Koo, M. Koch, N. Delhomme, M. Aigner, G. Toedt, P. Schirmacher, P. Lichter, J. Weitz and B. Radlwimmer, GUT, 2010, 59, 1236–1244 CrossRef CAS PubMed.
- J. M. Mullin, J. Exp. Zool., 1997, 279, 484–489 CrossRef CAS.
- T. A. Martin and W. G. Jiang, Histol. Histopathol., 2001, 16, 1183–1195 CAS.
- P. J. Morin, Cancer Res., 2005, 65, 9603–9606 CrossRef CAS PubMed.
- A. Pizarro, N. Benito, P. Navarro, J. Palacios, A. Cano, M. Quintanilla, F. Contreras and C. Gamallo, Br. J. Cancer, 1994, 69, 157–162 CrossRef CAS PubMed.
- M. Takeichi, Science, 1991, 251, 1451–1455 CAS.
- U. H. Frixen, J. Behrens, M. Sachs, G. Eberle, B. Voss, A. Warda, D. Löchner and W. Birchmeier, J. Cell Biol., 1991, 113, 173–185 CrossRef CAS.
- J. Behrens, M. M. Mareel, F. M. Vanroy and W. Birchmeier, J. Cell Biol., 1989, 108, 2435–2447 CrossRef CAS.
- K. Vleminckx, L. Vakaet, M. Mareel, W. Fiers and F. Vanroy, Cell, 1991, 66, 107–119 CrossRef CAS.
- M. C. Uzquiano, V. G. Prieto, J. W. Nash, D. S. Ivan, Y. Gong, A. J. Lazar and A. H. Diwan, Modern Pathol., 2008, 21, 540–543 CrossRef CAS PubMed.
- H. Rafehi, A. J. Smith, A. Balcerczyk, M. Ziemann, J. Ooi, S. J. Loveridge, E. K. Baker, A. El-Osta and T. C. Karagiannis, Genes Nutr., 2012, 7, 343–355 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |