
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of the efficacy of dietary polyphenols in experimental models of inflammatory bowel diseases
Derek A.
Martin
and
Bradley W.
Bolling
*
Department of Food Science, University of Wisconsin-Madison, 1605 Linden Dr, Madison, WI 53706, USA. E-mail: bwbolling@wisc.edu; Fax: +011-608-262-6872; Tel: +011-890-0212
First published on 13th May 2015
Abstract
Crohn's disease and ulcerative colitis presently have no cure and are treated with anti-inflammatory drugs or monoclonal antibodies targeting pro-inflammatory cytokines. A variety of rodent models have been used to model chronic and acute colitis. Dietary polyphenols in foods and botanicals are of considerable interest for prevention and treatment of colitis. Many dietary polyphenols have been utilized for prevention of colitis in rodent models. Berries, green tea polyphenols, curcumin, and stilbenes have been the most extensively tested polyphenols in rodent models of colitis. The majority of polyphenols tested have inhibited colitis in rodents, but increasing doses of EGCG and green tea, isoflavones, flaxseed, and α-mangostin have exacerbated colitis. Few studies have examined combination of polyphenols or other bioactives for inhibition of colitis. Translating polyphenol doses used in rodent models of colitis to human equivalent doses reveals that supplemental doses are most likely required to inhibit colitis from a single polyphenol treatment. The ability to translate polyphenol treatments in rodent models is likely to be limited by species differences in xenobiotic metabolism and microbiota. Given these limitations, data from polyphenols in rodent models suggests merit for pursuing additional clinical studies for prevention of colitis.
 Derek A. Martin | Derek Martin is a PhD student at the University of Wisconsin-Madison Department of Food Science, and a Registered Dietitian. He earned a B.S. in Foods and Nutrition at Appalachian State University in Boone, North Carolina, and his Dietetic Internship was at Winthrop University in Rock Hill, South Carolina. Derek's graduate research is on the use of berry polyphenols for prevention of inflammation and development of Inflammatory Bowel Diseases. |
 Bradley W. Bolling | Bradley Bolling is an Assistant Professor of Food Chemistry at the University of Wisconsin-Madison Department of Food Science. His research program is focused on prevention of chronic diseases by consumption of polyphenols and other dietary bioactives in foods. His research approach includes human intervention studies, animal models of chronic disease, and cellular studies. Further, his group is working on characterizing the polyphenol content of foods and identifying the impact of polyphenol metabolism and bioavailability on prevention of chronic disease. |
1. Introduction
Inflammatory bowel diseases (IBD) include both ulcerative colitis (UC) and Crohn's disease (CD). UC may affect the rectum and entire colon in an uninterrupted pattern of inflammation that is generally confined to the mucosa.1 In CD, inflammation is typically located at the ileum and the colon, but it can affect any region of the intestine and often occurs in an interrupted pattern. CD may also involve strictures and fistulas, but these are not typical for UC.1 Pharmacotherapy for IBD includes anti-inflammatory drugs and anti-tumor necrosis factor-α (TNF-α) monoclonal antibodies, but treatment does not cure the disease.2 Thus, colectomy may be needed when pharmacotherapy is inadequate to control inflammation. A significant number of studies have investigated the role of diet on IBD prevention and treatment, as previously reviewed.3 The objective of this paper is to review recent data from rodent models about polyphenols for the prevention and treatment of IBD.Polyphenols and other phenolic compounds are secondary plant metabolites characterized by an aromatic or phenol ring structure. Polyphenols can be further divided into classes according to the number of phenolic rings and the bonds that join the rings. These classes include stilbenes such as resveratrol, flavonoids such as epigallocatechin gallate (EGCG), and phenolic acids such as caffeic acid, among others (Fig. 1). Polyphenols have antioxidant activity, modulate cell signaling pathways, and have anti-inflammatory properties.4 Dietary sources of polyphenols include berries, grapes and wine, tea, chocolate, coffee, and a variety of fruits and vegetables, and their content is indexed in nutrient databanks.5 Polyphenol intake can reach gram amounts, especially for individuals that consume multiple servings of fruits, vegetables, tea, or coffee each day.5 For example, habitual coffee consumption can supply 500–800 mg hydroxycinnamic acids.5
 | ||
| Fig. 1 Structures of polyphenols used singularly as treatment in rodent models of colitis. | ||
Human intervention trials for polyphenol-based IBD treatment are limited. However, pilot studies of curcumin for UC or CD,6–8 bilberries for UC,9 or ECGC for UC10 have had promising results. Other dietary and botanical interventions for colitis have been recently reviewed elsewhere.3,11,12 While polyphenol intake and IBD risk have not been directly studied by epidemiological approaches, IBD risk has been inversely associated with childhood fruit and vegetable consumption.13,14 These studies have increased interest in the therapeutic and prophylactic use of polyphenols for IBD. Pre-clinical evidence is needed to design effective clinical studies of polyphenols for IBD. Therefore, the purpose of this review is to summarize and analyze the use of polyphenols in animal models of IBD.
2. Experimental models of IBD
A number of rodent models have been used for investigating dietary treatments for IBD.15 Inflammation in rodent-based IBD models can be induced by chemicals, immune cell transfer, or target gene manipulations that reflect defects in epithelial integrity, innate immunity, or adaptive immunity. Inflammation characteristic of IBD can be chemically-induced by administration of dextran sulfate sodium (DSS), 2,4,6-trinitrobenzenesulfonic acid (TNBS), or acetic acid. Typically, colitis is induced in rodents by administration of DSS in drinking water with various cycles and doses, leading highly reproducible acute or chronic colitis.15 DSS disrupts epithelial barrier integrity and leads to an inflammatory immune response to gut microbiota. Consequently, DSS treatment induces inflammation through T helper (Th)1, Th2, and Th17 responses.16,17 TNBS induces colitis in susceptible rodent strains and alters colonic or microbiota proteins, rendering them immunogenic to the host.15 TNBS colitis induction has been useful in studying Th cell immune responses (primarily Th1), cytokine secretion patterns, cell adhesion and immunotherapy.15 Transfer of CD45RBHI T cells isolated from donor mice and transferred to severe combined immunodeficiency (SCID) or Recombinase Activating Gene (RAG) 1/2−/− mice causes intestinal inflammation.18 Lack of functional proteins in mice can also lead to colitis. These include interleukin (IL)-10 and IL-10 receptor knockouts, T-cell receptor (TCR)-α chain knockouts, and manipulations causing the overexpression of signal transducer and activator of transcription (STAT)4.15 To our knowledge the IL-10 receptor−/−, TCRα chain knockouts, and STAT4 overexpression models have not been used to evaluate the effects of polyphenols or other plant metabolites in colitis development. In contrast, most studies of polyphenols in IBD models have used chemically-induced colonic inflammation or IL-10−/− mice that spontaneously develop colitis (Table 1).| IBD Model | Polyphenol class | Intervention (reference) |
|---|---|---|
| DSS-induced colitis | Anthocyanin | Bilberries and bilberry anthocyanin extract,24 black raspberry powder26 |
| Flavan-3-ol | EGCG,31 green tea polyphenols33,39 | |
| Isoflavone | Genistein, daidzein and equol,49 daidzein rich isoflavones extract48 | |
| Proanthocyanidin | Oligonol,43 apple proanthocyanidins,41 cocoa extract,44 cocoa45 | |
| Flavanone | Hesperidin,52 naringenin53,54 | |
| Flavonol | Quercetin,55,57 rutin58,59,104 kaempferol,62 myricitrin64 | |
| Stilbene | Resveratrol,71–74,111 piceatannol73,76 | |
| Hydrolysable tannin | Ellagic acid,95 corilagin,98 pomegranate extract and urolithin A97 | |
| Curcuminoid | Curcumin,79,80Curcuma longa extract,81 curcumin solid lipid microparticles,87 curcumin with hydroxypropyl-β-cyclodextrin88 | |
| Hydroxycinnamic acid | Caffeic acid and chlorogenic acid,96 hydrocaffeic acid100 | |
| TNBS/DNBS-induced colitis | Anthocyanin | Grape juice concentrate,21,22 blueberry extract23 |
| Flavan-3-ol | EGCG30 | |
| Isoflavone | Genistein and daidzein50,51 | |
| Proanthocyanidin | Grape seed proanthocyanidins42 | |
| Curcuminoid | Curcumin83 | |
| Gingerol | Ginger extract, zingerone92 | |
| Hydrolysable tannin | Pomegranate extract96 | |
| IL-10−/− spontaneous colitis | Anthocyanin | Black raspberry25 |
| Stilbene | Resveratrol72 | |
| Mdr1a−/− or HLA-B27 transgenic rats, spontaneous colitis | Flavan-3-ol | Green tea polyphenols40 |
| Curcuminoids | Curcumin78 | |
| Acetic acid-induced colitis | Anthocyanins | Hawthorn berry extract27 |
| Flavonol | Quercetin56 | |
| Curcuminoid | Curcumin82,89 | |
| Gingerol | Ginger extract91 | |
| Hydroxycinnamic acid | Ferulate101 | |
| Adoptive transfer | Flavonol | Rutin61 |
3. Polyphenols and polyphenol-rich foods and extracts used in animal models of IBD
Polyphenol-rich fruits, vegetables, and other sources are composed of multiple polyphenol classes and other bioactives. For example, anthocyanin-rich berries also contain significant quantities of proanthocyanidins and dietary fiber.19,20 For the purposes of this review, preparations or unpurified sources of polyphenols are grouped with the most-abundant class of polyphenol identified in its composition. A number of polyphenols have been administered in animal models of colitis (Fig. 1, Table 1).3.1 Anthocyanins
Berries, grapes, and other pigmented plants can be rich sources of anthocyanins and have been applied to experimental models of IBD. The cost of isolation and purification of anthocyanins has precluded treatment with individual compounds.Dried bilberry (20% w/w) and anthocyanin-rich bilberry extract (10 or 1% w/w) were added to chow diet and fed to mice 2 week prior to and concurrent with acute (7 d) and chronic (4 × 7 d cycles) DSS treatment.24 Bilberry extract, but not whole bilberry, reduced acute-DSS colonic histological inflammation score, but both extracts and whole bilberry prevented acute decrease in colon length. Bilberries, but not the 10% anthocyanin extract, decreased mesenteric lymph node (MLN) TNF-α excretion, while both forms reduced MLN IFN-γ excretion. After chronic DSS-induced colitis, bilberry and bilberry extract reduced colon shortening, but only bilberry extract reduced colonic histological inflammation scores and excretion of IFN-γ and IL-6 from MLNs.
A 5% black raspberry diet was fed to spontaneously-colitic IL-10−/− mice for 8 week.25 Black raspberry feeding reduced colonic ulceration in the mucosa and submucosa in mice.25 Black raspberry powder was also administered at 5 or 10% of the diet to C57BL6/J mice treated with 3% DSS for 7 d.26 Both black raspberry doses prevented DSS-induced weight loss, colon shortening, and mucosal ulceration.26 The 10% black raspberry-treated mice had less colonic IL-1β and TNF-α mRNA, and decreased COX-2 and prostaglandin E2.26
Wistar rats were given 100 mg kg−1 hawthorn berry extract by oral gavage 3 d before and 7 d after induction of colitis by intrarectal 4% acetic acid.27 Hawthorn berry extract prevented colitis-induced body weight loss and inhibited increases in colonic MPO activity and nitric oxide.27 Hawthorn berry extract treatment prevented colonic lipid peroxidation and macroscopic and histopathological inflammatory damage scores.27
3.2 Flavan-3-ols and green tea
Flavan-3-ols, particularly (+)-catechin and (−)-epicatechin are widely distributed in plants, including nuts, berries, chocolate, wine.28 Green tea and green tea extract are rich in flavan-3-ols, including EGCG, epigallocatechin, epicatechin gallate, and epicatechin.29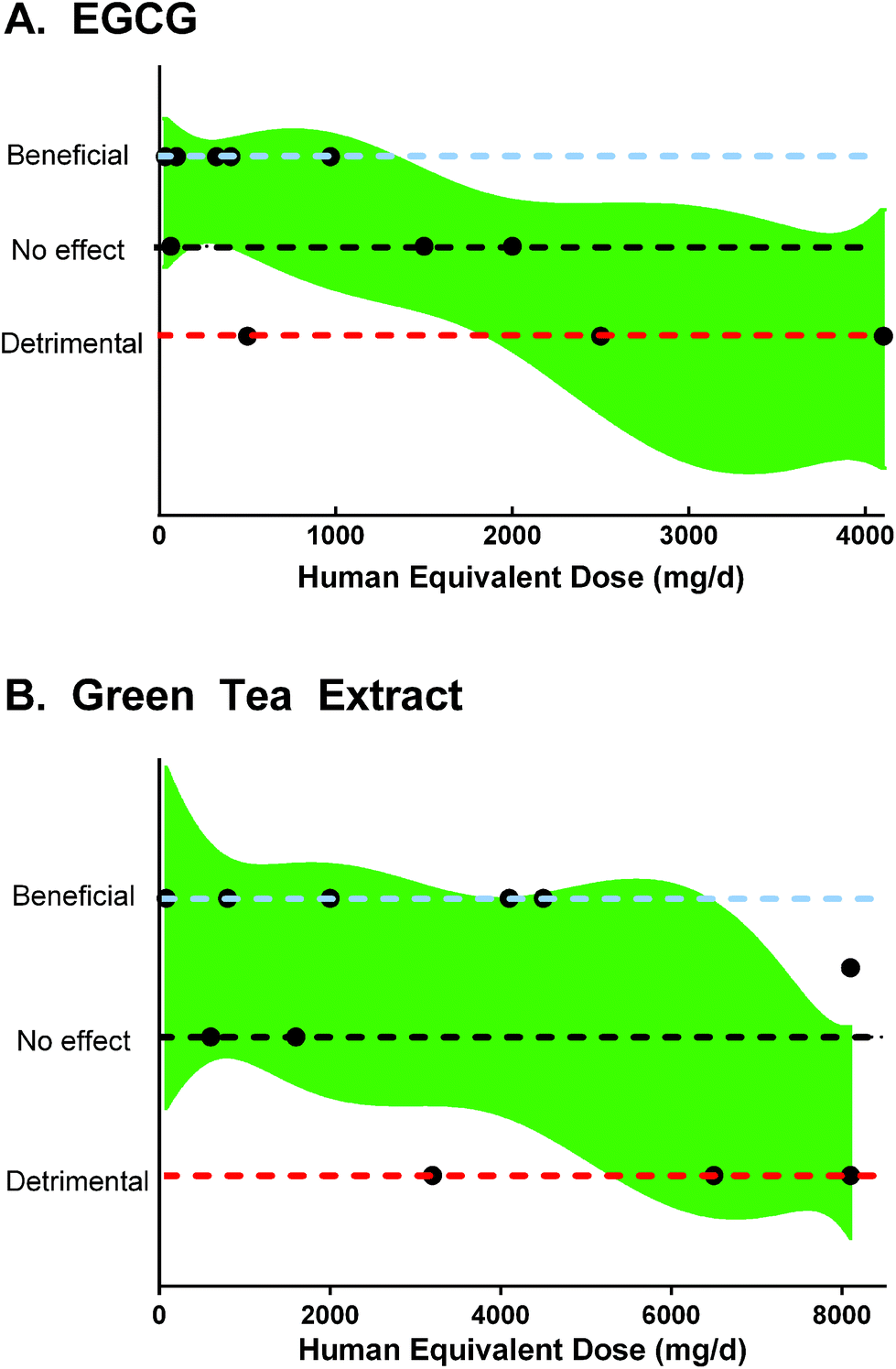 | ||
| Fig. 2 Results and human equivalent doses of A. EGCG and B. green tea polyphenols in rodent models of colitis, based on data from Table 2. A 90% confidence band of a best-fit quadratic non-linear regression is plotted for reference. | ||
3.3 Proanthocyanidins and cocoa
Proanthocyanidins are polymeric flavan-3-ols with 2 to >100 constituent units. Proanthocyanidin polymers are predominately “B-type”, having a single interflavan bond, although “A-type” proanthocyanidins are most abundant in cranberries. While cocoa, grapeseed extract, and berries are rich in proanthocyanidins, it should be noted that crude extracts are likely to contain other bioactive components, such as catechins, anthocyanins, and other flavonoids.Isolated apple proanthocyanidins, having degree of polymerization of 2–15, inhibited oxazolone and DSS-induced colitis.41 C57BL6 mice were provided 0.1, 0.3, or 1.0% apple proanthocyanidins in the drinking water for 14 d prior to a 4 d, 2.5% DSS treatment.41 DSS-treated mice consuming 1% apple proanthocyanidins lost significantly less weight, had less colon shortening and all survived to 20 d, compared to 40% survival in the control group.41 DSS-treated mice showed multifocal inflammatory cell infiltration, erosion of the epithelium and gland destruction, while consumption of 1% apple proanthocyanidins yielded almost normal histologic morphology.41
Intragastric administration of a grape-seed proanthocyanidin extract (95% proanthocyanidins) reduced recurrent TNBS-induced colitis in Wistar rats.42 Rats were given 100, 200, or 400 mg proanthocyanidins kg−1 24 h after a second TNBS administration and continued for 7 d.42 All doses of grape seed proanthocyanidins inhibited weight loss and macroscopic damage scores.42 The medium and high doses of proanthocyanidins preserved colonic antioxidant function by inhibiting reductions in superoxide dismutase and glutathione peroxidase activities.42 All proanthocyanidin doses decreased colonic TNF-α, the ratio of phosphorylated to unphosphorylated inhibitor of κB kinase, and NF-κB levels.42
Oligonol, an enzymatically-prepared low-molecular weight proanthocyanidin mixture also inhibited DSS-induced colitis in ICR mice.43 Mice consumed 0.5 or 5 mg kg−1 d−1 oligonol in water for 7 d prior to and during a 7 d, 3% DSS exposure.43 Oligonol attenuated body weight loss, rectal bleeding, colon shortening, and diarrhea during the DSS treatment.43 Oligonol dose-dependently decreased the colonic inflammatory histological grade.43 Furthermore, oligonol inhibited DSS-induced inhibitor of κB (IκB)α degradation, NF-κB p65 phosphorylation, and STAT3 phosphorylation in the colon.43
Cocoa and cocoa-extract consumption inhibited DSS-induced colitis in mice.44,45 A catechin and proanthocyanidin cocoa extract was administered at 500 mg kg−1 to mice on d 1 and d 4 during a 7 d 5% DSS administration.44 Mice treated with cocoa polyphenols were protected from colon shortening and weight loss.44 Cocoa polyphenols improved stool consistency, reduced colonic histological scores and MPO activity, which was accompanied by reduction in colonic phosphorylated STAT3 and phosphorylated STAT1α.44 In another study, consumption of 50 g cocoa kg−1 diet for 14 d before and during DSS treatment prevented colonic mononuclear cell infiltration and goblet cell loss and reduced serum TNF-α in Wistar rats.45
3.4 Isoflavones and soy
Dietary isoflavones are present in soy foods, including soymilk, tofu, miso, and soy protein. Dietary isoflavones are predominately malonyl-glycosides, but heat treatment and fermentation liberate genistein and daidzein, isoflavone aglycones.46 Equol and O-desmethylangolensin are unique microbial metabolites of daidzein.47Mixed soy isoflavones enriched in daidzein inhibited DSS-induced colitis in C57BL/6 female mice.48 Oral gavage of 80 mg isoflavones kg−1 d−1 (daidzein, genistein, and glycitein, 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2) was provided to mice for one week prior to and concurrent with a 4 d 2% DSS dose.48 Isoflavone-treated mice were protected from weight loss, had decreased severity and extent of inflammation and crypt damage.48 MLN cells from isoflavone-treated mice produced less IFN-γ, IL-12p40, and IL-6 and produced more IL-10 when stimulated ex vivo. The percentage of CD80+- and CD86+-CD11b+ cells in the MLN was also decreased by isoflavone treatment, indicating decreased activation in antigen presenting cells.48 In contrast, lower doses of individual isoflavones exacerbated a 4% DSS-induced colitis in female BALB/c mice.49 Gavage of 20 mg per kg bw of genistein, daidzein, or equol for a week prior to 4 d of 4% DSS treatment reduced survival relative to the non-treated DSS-control group.49 In contrast, lower equol concentrations (2 and 10 mg per kg bw) did not increase weight loss.49 MLN cells from 20 mg kg−1 equol-treated mice stimulated ex vivo with CD3 monoclonal antibody produced less IL-10 than control mice, but did not differ in TNF-α, IFN-γ, or IL-4.49 Exacerbation of colitis by genistein appears model-dependent, as 100 mg genistein per kg bw for 14 d prior to TNBS treatment in male Wistar rats inhibited colonic MPO activity and COX-2 protein.50 Exposure to ∼0.05% mg genistein and daidzein in prenatal and post-natal diets increased TNBS-induced colitis in Wistar rats, evidenced by colonic weights and MPO activity.50
:1:2) was provided to mice for one week prior to and concurrent with a 4 d 2% DSS dose.48 Isoflavone-treated mice were protected from weight loss, had decreased severity and extent of inflammation and crypt damage.48 MLN cells from isoflavone-treated mice produced less IFN-γ, IL-12p40, and IL-6 and produced more IL-10 when stimulated ex vivo. The percentage of CD80+- and CD86+-CD11b+ cells in the MLN was also decreased by isoflavone treatment, indicating decreased activation in antigen presenting cells.48 In contrast, lower doses of individual isoflavones exacerbated a 4% DSS-induced colitis in female BALB/c mice.49 Gavage of 20 mg per kg bw of genistein, daidzein, or equol for a week prior to 4 d of 4% DSS treatment reduced survival relative to the non-treated DSS-control group.49 In contrast, lower equol concentrations (2 and 10 mg per kg bw) did not increase weight loss.49 MLN cells from 20 mg kg−1 equol-treated mice stimulated ex vivo with CD3 monoclonal antibody produced less IL-10 than control mice, but did not differ in TNF-α, IFN-γ, or IL-4.49 Exacerbation of colitis by genistein appears model-dependent, as 100 mg genistein per kg bw for 14 d prior to TNBS treatment in male Wistar rats inhibited colonic MPO activity and COX-2 protein.50 Exposure to ∼0.05% mg genistein and daidzein in prenatal and post-natal diets increased TNBS-induced colitis in Wistar rats, evidenced by colonic weights and MPO activity.50
3.5 Flavanones
Flavanones include compounds such hesperetin, naringenin, and eriodictyol. Citrus polyphenols are predominately flavanone glycosides. Citrus juices contain hesperidin (hesperetin 7-O-rutinoside) from ∼1 to 50 mg/100 mL.28Hesperidin inhibited DSS-induced colitis in BALB/c mice.52 An 80 mg hesperidin kg−1 oral dose inhibited a 7 d, 5% DSS-induced colitis to a similar extent as a 500 mg sulfasalazine kg−1 treatment, based on an observed disease activity index.52 Similarly, the aglycone naringenin inhibited a 9 d, 2% DSS-induced colitis.53 Consumption of diets containing 0.3% naringenin protected mice from DSS-induced body weight loss, colon shortening, and an increase in the disease activity index score.53 Naringenin fed mice had improved intestinal barrier function determined by reduced colonic permeability and plasma lipopolysaccharide-binding protein, and higher concentrations of occludin, junctional adhesion molecule-A, and claudin-3 compared to the DSS treatment alone.53 Additionally, naringenin feeding inhibited the colonic cytokines IFN-γ, IL-6, and IL-17A.53 Another study of naringenin in a DSS-induced model of colitis in C57BL/6 mice reported that a 50 mg per kg bw oral dose reduced colitis and decreased colonic toll-like receptor 4, phospho-NFκB p65, TNF-α, and IL-6 protein.54
3.6 Flavonols
Quercetin,55–58 quercetin 3-O-rutinoside (rutin),59–61 kaempferol,62 morin,63 and myricitrin64 have been tested in rodent models of colitis. Delivery and slow-release of quercetin to the colon, through pectin and casein-based microcapsules, improved the efficacy of quercetin to inhibit acetic acid-induced colitis in Swiss mice.55 Doses of 100 mg quercetin kg−1 free or in microcapsules were provided 2 h prior and 10 h after administration of acetic acid. At 18 h, quercetin microcapsules, but not free quercetin reduced macroscopic colonic damage scores, MPO activity, IL-1β, and IL-33, and increased colonic IL-10.55 Consumption of a 2% rutin-supplemented diet by BALB/c mice inhibited markers of inflammation in 5% DSS-induced colitis models.58,60 Rutin decreased colonic IL-1β, IL-6, and TNF-α, and reduced colon shortening, but did not prevent colitis-induced weight loss.60 A lower dose of rutin, ∼0.05% in diets of C3H/HeOuJ mice for 7 d prior and during a 6 d 1.25% DSS treatment improved body weight, reduced colonic MPO activity, and inhibited colonic IL-17 and iNOS mRNA.59 However, this dose of rutin did not significantly reduce colon histopathology scores or protect from colon shortening.59The effect of pre-feeding kaempferol prior to DSS-treatment relative to concurrent treatment was examined by Park et al.62 C57BL/6J mice were fed diets enriched with 0.1% or 0.3% kaempferol starting 2 weeks before or concurrent to a 4 d, 2% DSS exposure.62 Kaempferol consumption continued until 1 week after the start of DSS exposure.62 The experimental diets significantly lowered disease activity index scores, with the greatest inhibition occurring in the mice fed 0.3% kaempferol prior to DSS exposure.62 All kaempferol treatments improved histological scores of colitis with the greatest effect in 0.3% kaempferol pre-fed group.62 Thus, kaempferol prefeeding appears to confer greater protection in the DSS-induced colitis than treatment post-initiation.
3.7 Other purified flavonoids
Gavage of luteolin protected against colitis in 2% DSS-treated C57BL6/6CrSlc mice.65 IP injection of astilbin to C57BL/6 mice prevented colonic inflammation and increased IL-10 and TGF-β in splenic dendritic cells after treatment of 2.5% DSS for 7 d.66 Consumption of 5 mg kg−1 apigenin reduced acetic-acid induced colitis in mice.67 Intragastric administration of 2 g baicalin kg−1 d−1 to Sprague Dawley rats prevented TNBS-induced colitic damage evaluated histologically.683.8 Stilbenes and resveratrol
Stilbenes are present in microgram to milligram quantities in grapes, wine, peanuts, tree nuts, and berries.28 Dietary stilbenes exist as glycosylated forms, e.g. resveratrol 3-O-glucoside, or as aglycones, e.g. resveratrol, and more than 40 types of stilbenes have been identified in foods.69The anti-colitic properties of supplemental resveratrol have been examined in DSS-treated mice70–74 and spontaneously-colitic IL-10−/− mice.75 Supplemental piceatannol has also been administered to DSS-treated mice.73,76 Supplemental resveratrol consumption (20 mg kg−1 diet) prevented 5% DSS-induced weight loss and colitis-induced mortality.70 In the same study, resveratrol consumption increased colonic IL-10 protein and inhibited colonic TNF-α protein after DSS treatment.70 Gavage of 100 mg resveratrol per kg bw to C57BL/6 mice during a 7 d, 3% DSS exposure decreased serum IFN-γ, TNF-α, IL-6, and IL-1β and reduced lamina propria CD4+ T cells expressing IFN-γ and TNF-α.72 Lamina propria cells from resveratrol-treated mice also had increased sirtuin 1 expression and decreased p-IκBα expression following DSS treatment.72 In IL-10−/− mice, 10, 50, or 100 mg resveratrol per kg bw was administered every other day after development of colitis.75 Treatment of 100 mg resveratrol per kg bw reduced colitic symptoms and weight loss, while lower doses did not.75 Immunosuppressive CD11b+Gr-1+ myeloid derived suppressor cells were increased in the spleen and lamina propria of mice given the highest dose of resveratrol.75 Resveratrol treatment also decreased colonic excretion of IFN-γ, TNF-α, IL-6, IL-12, and IL-1β.75
3.9 Curcuminoids and turmeric
Turmeric contains the curcuminoids curcumin, demethoxycurcumin, and bis-demethoxycurcumin.77 Curcumin treatment has been evaluated in Mdr1a−/− spontaneously colitic mice,78 DSS-treated BALB/c and C57BL/6 mice,79–81 Wistar rats with acetic acid-induced colitis,82 and Sprague-Dawley rats with TNBS-induced colitis.83 In these models, consumption of 0.2% to 0.6% curcumin-supplemented chow, or oral gavage of 50 to 200 mg curcumin per kg bw per d inhibited colitis. In a 52 d chronic DSS model of colitis, a 200 mg turmeric extract per kg bw dose reduced histological scores of inflammation and lowered the disease activity index.79 Consumption of 0.6% curcumin reduced DSS-induced colonic TNF-α, IFN-γ, COX-2, and iNOS proteins.80Notably, curcumin has poor solubility and oral bioavailability, so a number of studies have successfully improved delivery through emulsions, liposomes, and nanotechnology-based approaches.84–86 Strategies to increase curcumin solubility and delivery to the colon have increased the therapeutic efficacy of curcumin in rodent colitis models. Equivalent doses of curcumin loaded in solid lipid microparticles or hydroxypropyl-β-cyclodextrin increased recovery from DSS-induced colitis than curcumin alone (100 mg curcumin per kg bw).87,88 Similarly, curcumin-Zn(II) complexation improved curcumin water solubility and a 20 mg per kg bw dose improved histopathological scores moreso than curcumin alone in mice with acetic-acid induced colitis.89 Thus, solubility and bioavailability should be considered in formulating curcumin interventions for colitis.
3.10 Gingerols and ginger
Ginger contains the polyphenolic gingerols, shogaols, paradols, and zingerone.90 Consumption of ginger extract inhibited acetic acid-induced colitis in Wistar rats.91 Oral gavage of 200 or 400 mg ginger extract per kg bw inhibited colonic lesions to a similar extent as 500 mg sulfasalazine per kg bw. Ginger extract treatment improved colonic antioxidant function, and reduced colonic MPO activity and TNF-α.91 Ginger extract or zingerone was co-administered with TNBS to BALB/c mice.92 Treatment with ginger extract from 0.1 to 100 mg per kg bw or zingerone from 1 to 100 mg per kg bw dose-dependently decreased macroscopic colonic inflammation in TNBS-treated mice.92 Microarray analysis of colonic genes indicated IL-17, IL-1β, IL-6, IFN-γ, and TNF-α pathways were modulated by ginger extract and zingerone.923.11. Ellagitannins and pomegranate
Ellagitannins are hydrolysable tannins containing a hexahydroxydiphenoyl moiety which is metabolized to ellagic acid upon digestion. Ellagic acid is extensively metabolized by gut microbiota, and exists as urolithins or urolithin conjugates in circulation.93 A wide variety of ellagitannins have been identified in fruits such as pomegranates (punicalagin, corilagin), raspberries (sanguiin H-6, lambertianin C), and walnuts (glansrin A, B, and C, casuarictin).94 Ellagic acid,95,96 pomegranate extract,96,97 corilagin,98 and urolithin A97 have been administered in rodent models of colitis.Ellagic acid alone (0.5% diet) prevented chronic 1% DSS-induced disease activity index and histological markers of colitis in C57BL/6 mice.95 However, 2% ellagic acid supplemented diet did not prevent weight loss or disease activity index induced by a 7 d, 5% DSS treatment to BALB/c mice.95 Ellagic acid reduced DSS-induced colonic COX-2 and iNOS expression, p38 mitogen activated protein kinase (MAPK) phosphorylation, NF-κB IκBα phosphorylation, NF-κB p65 nuclear translocation, and STAT3 phosphorylation in mice.95 IP injection of corilagin at 7.5, 15, or 30 mg per kg bw for 7 d to C57BL/6 mice inhibited colitis induced by a 5 d, 2% DSS treatment.98 Corilagin treatment dose-dependently improved colon length, histological score and MPO activity.98 The colonic cytokines TNF-α, IL-1β, and IL-6, but not IL-10 were modulated by corilagin treatment.98 Since ellagitannins are extensively metabolized, it is likely that dietary intake of corilagin would lead to a different outcome than IP injection in a model of colitis. Urolithin A-supplemented diets, providing 2.2 mg per kg bw per d for 25 d concurrent and following a 5 d, 5% DSS exposure, inhibited colitis as graded histologically.97 Urolithin A consumption also increased fecal bifidobacteria and lactobacilli.97 Consumption of 250 or 500 mg pomegranate extract kg−1 chow by Wistar rats prior to and following TNBS-induced colitis prevented weight loss, and reduced colonic TNF-α and macroscopic and histological markers of colonic inflammation.96
3.12 Hydroxycinnamic acids
Chlorogenic acid,99 caffeic acid,59,99 hydrocaffeic acid,100 and sodium ferulate101 have been administered in rodent models of colitis. Chlorogenic acid is hydrolyzed to caffeic acid prior to absorption in the small intestine.102 Caffeic acid and chlorogenic acid were supplemented at 1 mM in the water of C57BL/6 mice 7 d prior to and concurrent with an 8 d, 3% DSS treatment.99 Chlorogenic acid, but not caffeic acid treatment prevented DSS-induced weight loss, although both caffeic and chlorogenic acids improved diarrhea scores and histological grading of colitis.99 Hydrocaffeic acid is a colonic catabolite of caffeic acid and other polyphenols. Hydrocaffeic acid prevented weight loss and reduced colonic TNF-α, IL-β, and IL-8 mRNA in Fischer 344 rats treated with 4% DSS.100 Thus, hydroxycinnamic acid catabolites appear to retain anti-inflammatory activity.3.13 Other purified phenolics
A number of other polyphenols have been solely administered in rodent models of colitis. These include apocynin (acetovanillone),58 canolol (4-vinyl-2,6-dimethoxyphenol),103 oleuropein, a phenolic secoiridoid from olive,60,104 chrysin, a flavone,105,106 silymarin,107 icariin (prenylated kaempferol diglucoside),108 esculetin and 4-methylesculetin (coumarin derivatives),109 hydroxytyrosol and tyrosol,110,111 and α-mangostin (xanthone).112 Notably, C57BL/6 mice consuming 112 mg α-mangostin per kg bw with DSS-induced colitis had worsened symptoms and greater colonic injury.1123.14 Other dietary sources of polyphenols
Other extracts or polyphenol-rich foods and ingredients have been used for treatment in rodent models of colitis. These extracts have a mixed composition that do not have a dominant polyphenol class or bioactive class. Foods that have been tested include cranberry,113 propolis,114 olive oil,110,111 almond skin powder,115 apples and apple polyphenols,116,117 flaxseed and its hulls and kernels (containing lignans),118 lemon verbena,119 and oat bran and blueberry fiber.120 While other polyphenol diets conferred protection against colitis in rodents, it should be noted that 10% flaxseed diets increased disease activity index in 2% DSS treated C57BL/6 mice.1184. Dose relevance of polyphenols in rodent models of IBD
It is critical to consider the dose of polyphenols when interpreting results of published studies or designing a new experiment. It may be possible to demonstrate a polyphenol or extract inhibits colitis in rodents, but if the dose is not relevant to human consumption the findings may not be translatable. Allometric scaling or body surface area approaches can be used to predict a “human equivalent dose” from rodent models (Table 2).121–123| Treatment and citation | Human equivalent doseb | Experimental dose | Time course | Outcome |
|---|---|---|---|---|
| a Abbreviations: EGCG: epigallocatechin gallate; GTE: green tea extract; DSS: dextran sodium sulfate. b Human equivalent dose based on 60 kg adult using body-surface area calculation.126 c Assuming 20 g bw for female C57BL/6 mice. d Assuming 30 g bw male ICR mice, consuming 4 g diet/d. e Based on 3 g diet/18 g mouse. | ||||
| Resveratrol71 | 8.6 mg d−1 | 1 mg kg−1 d−1, F344 rats | 20 d prior and concurrent with a 5 d 5% DSS treatment | Inhibition of colitis, modified microbiota |
| Resveratrol70 | 30 mg d−1 | 20 mg kg−1 diet, C57BL/6 mice | Concurrent with a 5 d, 3% DSS treatment, and 21 d post-DSS. | Improve mortality, inhibition of colitis |
| Resveratrol73 | 48.6 mg d−1 | 10 mg per kg bw, ICR mice | Concurrent with a 7 d, 2.5% DSS treatment | Inhibition of colitis |
| Resveratrol72 | 48.6, 243, 486 mg d−1 | 10, 50, 100 mg per kg bw, C57BL/6 mice | Concurrent with a 7 d, 3% DSS treatment, and for 7 d following DSS | Highest dose protective against colitis |
| Resveratrol74 | 58, 116, 232 mg d−1 | 75, 150, 300 mg kg−1 diet, C57BL/6 mice | 7 d prior, concurrent with a 7 d, 1% DSS treatment, and 7 d following DSS | Inhibition of colitis at two highest doses |
| Curcumin79 | 243 mg d−1 | 50 mg per kg bw, BALB/c mice | 7 d after a 7 d, 5% DSS treatment | Inhibition of colitis |
| Curcumin extract81 | 973 mg d−1 | 200 mg kg−1 d−1, BALB/c mice | 7 to 21 d following induction of colitis by 52 d, 2.5% DSS; For 7 d after a 7 d, 5% DSS treatment | Inhibition of chronic colitis at d 20, inhibition of acute colitis |
| Curcumin78 | ∼1.6–2.0 g d−1 | 0.2% diet, Mdr1a−/− mice | From 6 to 24 week | Inhibition of spontaneous colitis |
| Curcumin82 | 1.9 g d−1 | 100 mg d−1, Wistar albino rats | 10 d prior and 2 d following acetic acid treatment | Inhibition of colitis |
| Curcumin83 | 1.9 g d−1 | 100 mg per kg bw, Sprague-Dawley rats | 7 d after induction of colitis by TNBS | Inhibition of colitis |
| Curcumin80 | ∼4.4 g d−1c |
0.6% diet, 18 mg d−1 C57BL/6 mice | 2 week prior and concurrent with 0.7% DSS treatment for 15 cycles of 7 d DSS, 10 d no DSS (total 37 week) | Inhibition of colitis at cycle 11–15 |
| EGCG33 | 34 mg d−1 | 6.9 mg per kg bw, C57BL/6 mice | 1 week prior and concurrent with 60 d, 2% DSS alternating treatment | Inhibition of weight loss, some inhibition of colitis |
| EGCG31 | 65, 324 mg d−1d |
0.01, 0.05% diet, ICR mice | 7 d prior and concurrent with 7 d, 1.5% DSS treatment | Some inhibition of colitis at highest dose |
| EGCG30 | 97 mg d−1 (IP) | 20 mg per kg bw (IP), C57BL/6 mice | 1 to 7 d after induction of colitis by TNBS | Inhibition of colitis |
| EGCG32 | 405 mg d−1 | 50 mg per kg bw, Sprague-Dawley rats | 7 d after induction of colitis by acetic acid | Inhibition of colitis |
| EGCG34 | 0.5, 1.5, 2.5 g/2000 kcal32 | 0.1, 0.3, 0.5% EGCG diet, C57BL/6, CD-1 mice | 1 week prior, concurrent with 1.5% DSS treatment, and 3 d after DSS treatment | Highest dose increased rectal bleeding, lowest and highest doses increased weight loss |
| EGCG39 | 0.97, 2, 4.1 g d−1e |
0.12%, 0.25%, 0.5% diet, BALB/c | 10 d, concurrent and after 7 d, 3% DSS | Highest dose increased weight loss, lowest dose reduced colitis |
| GTE37 | 0.08, 0.8, 8.1 g d−1e |
0.01, 0.1, 1% GTE diet, ICR mice | Concurrent with 6 d, 5% DSS treatment | Highest dose induced mortality, lower doses inhibited colitis |
| GTE38 | 0.6, 1.6, 3.2, 6.5 g d−1d |
0.1, 0.25, 0.5, 1% GTE in diet, ICR mice | Concurrent with 6 d, 25% DSS treatment | Increased colitis at two highest doses |
| GTE39 | 2, 4.1, 8.1 g d−1e |
0.25, 0.5, 1% GTE diet, IL-10−/− mice; 1% GTE diet, BALB/c mice | IL-10−/−: from 4 week age; BALB/c: for 10 d, concurrent and after 7 d, 3% DSS | IL-10−/−: highest dose exacerbated colitis, lower doses improved colitis; BALB/c: prevented weight loss, improved colitis somewhat |
| GTE40 | 4.5 g d−1 | 0.6% GTE diet, Mdr1a−/− mice | 10 week | Inhibition of colitis |
| GTE36 | 8.1 g d−1e |
1% GTE in diet, ICR mice | Concurrent with 6 d, 5% DSS treatment | Disrupted kidney function |
As an example, treatment with a human equivalent of 30 mg resveratrol d−1 inhibited DSS-induced colitis in mice.70 Wine has a maximum of ∼14.3 mg resveratrol L−1, while most red wines have 2–5 mg resveratrol L−1.124 Other dietary sources of resveratrol have mg to sub-mg quantities.28 While consumption of 30 mg of resveratrol from the diet is not feasible, gram-quantities of supplemental resveratrol could be used to provide this dose.125 In contrast, another study treated colitic mice with a human dose equivalent to 8.4 g apple polyphenols d−1, which may not be possible to consume.116 Another study used a human equivalent of 121.6 mg morin d−1, but to our knowledge, supplemental morin is not on the market, and only sub-milligram quantities are present in food.28,63 Thus, to optimize translation of research using rodent models of colitis, doses and treatment regimens should be carefully selected. This is particularly necessary as over-supplementation of polyphenols could exacerbate colitis.36,37,112,118
5. Experimental limitations of studying molecular targets with respect to polyphenol metabolism
Upon consumption, polyphenols can be extensively catabolized by gut microbiota, subjected to biotransformation or be poorly absorbed. This complicates in vitro and animal models investigating the molecular targets of polyphenols as they may not reflect the metabolite(s) presented to cells or tissue in humans. Although many polyphenol metabolites are bioactive, they may not be active in the same manner as the parent compounds and generally have reduced function relative to parent compounds.126Unabsorbed and catabolized polyphenols may be active in the gastrointestinal system.126 This makes polyphenols an attractive target in the treatment of inflammatory bowel diseases. Phytochemicals must still be bioaccessible to be bioactive in the gut. Thus, they must be liberated from the food matrix to exert effects on the cells in the gut. Indeed, both polyphenols and other phenolic compounds exist in the fecal water and have high interindividual variation.127 Unabsorbed polyphenols may interact directly with microbiota, mucosal cells, and dendritic cell projections in the intestinal lumen. There is a greater need to understand how targeted polyphenol delivery systems could be used to optimize treatment for colitis.
For example, chlorogenic acid undergoes extensive transformation before absorption. Chlorogenic acids are hydroxycinnamic acids such as caffeic acid or ferulic acid linked to quinic acid through an ester bond.128 Before reaching the colon, chlorogenic acid can be cleaved to its phenolic acid and quinic acid portions, glucuronidated, sulfated, and methylated.102 It may enter circulation and re-enter the intestinal lumen via enterohepatic recirculation. Therefore, the compounds reaching the colon tissue and gut microbiota could be the parent compounds or one of the many metabolites which arise from its biotransformation of hydroxycinnamic acids. The bioactivity would then be dependent upon the metabolites’ interactions with particular signaling cascades or cellular proteins and membranes.
It should also be noted some aspects of rodent microbiota and xenobiotic metabolism are dissimilar to humans.129,130 Furthermore, colitis is associated with changes in gut microbiota and xenobiotic metabolism.131–133 A number of studies in rodents and humans have demonstrated that polyphenol consumption modifies microbiota composition.134–136 The extent that these species differences affect the ability to translate polyphenol treatment rodent models to humans is presently unknown.
6. Conclusions and future directions
A significant number of isolated polyphenols and polyphenol-rich extracts reduce colitis in rodent models. The benefits of polyphenols in rodent IBD models include reduction of bleeding, improvement in stool consistency, improved histological appearance, decreased weight loss, and protection from colon shortening. The doses of polyphenols applied in rodent IBD models vary widely, and should be given careful consideration in experimental design. Using infeasible human equivalent doses may not be warranted. Future consideration of targeted delivery of polyphenols may improve their ability to inhibit colitis.Polyphenols modulate a large number of targets relevant to inflammation and colitis. Polyphenols consistently modulate NF-κB pathway and a number of polyphenols inhibit the MAPK cascade and induce Nrf2. These targets enable polyphenols to inhibit inflammatory responses in a variety of ways. Polyphenols reduce proinflammatory cytokines, induce anti-inflammatory cytokines, inhibit NO production, induce immunosuppressive properties in immune cells, and inhibit COX-2 activity. IBD is characterized by a pronounced infiltration of innate and adaptive immune cells into the intestinal lamina propria with increased Th1 and Th17 cells in CD and increased Th2 and Th17 cells in UC.1 Therefore, it is necessary to establish the effects of polyphenols on the activities of antigen presenting cells, the regulation and differentiation of T cells into Th1, Th2, and Th17 cells. Substantial evidence implicates gut barrier dysfunction in the development of inflammatory bowel disease.137 A number of in vitro and some in vivo investigations have shown that polyphenols modulate intestinal barrier function.53,138–140 Modulation of intestinal barrier function is worthy of further investigation as a mechanism for the anti-colitic effects of polyphenols. In contrast to drugs, which generally have well-defined and specific targets, the broad actions of polyphenols on inflammation mechanisms could be an advantage to IBD therapy. However, caution should be exercised when designing adjuvant polyphenol treatments. Polyphenols can exert pharmacokinetic and pharmacodynamic interactions, leading to enhanced or antagonistic actions. Polyphenols can inhibit or induce Cyp3A4 expression, thus could potentially modulate pharmacokinetics of Cyp3A4-metabolized drugs such as prednisolone, budesonide, clarithromycin, cyclosporine used for IBD.141–143 Also, polyphenols inhibit TNF-α in rodent models of colitis, and biologic drugs targeting TNF-α and its signaling pathway are used for IBD treatment. More work is needed to test the safety of using polyphenols as adjuvants to current IBD drugs.
Resveratrol, curcumin, quercetin and anthocyanin-rich foods appear to have the most evidence for efficacy and safety in rodent colitis models. Despite the considerable number of positive studies on polyphenols for IBD treatment in rodents, few polyphenols have been examined in human intervention studies of colitis. Although evidence for the effectiveness of polyphenols for IBD in humans remains very limited, results from these pre-clinical studies indicate an opportunity for developing anti-colitic polyphenol treatments.
Acknowledgements
This work was supported by USDA National Institute of Food and Agriculture Hatch project 1005391. BB and DM conceived and designed this study, drafted the manuscript and approved publication.Notes and references
- C. Abraham and J. H. Cho, N. Engl. J. Med., 2009, 361, 2066–2078 CrossRef CAS PubMed.
- M. Fakhoury, R. Negrulj, A. Mooranian and H. Al-Salami, J. Inflammation Res., 2014, 7, 113–120 CrossRef PubMed.
- D. Lee, L. Albenberg, C. Compher, R. Baldassano, D. Piccoli, J. D. Lewis and G. D. Wu, Gastroenterology, 2015, 148, 1087–1106 CrossRef CAS PubMed.
- N. Hounsome, B. Hounsome, D. Tomos and G. Edwards-Jones, J. Food Sci., 2008, 73, R48–R65 CrossRef CAS PubMed.
- C. Manach, A. Scalbert, C. Morand, C. Rémésy and L. Jiménez, Am. J. Clin. Nutr., 2004, 79, 727–747 CAS.
- H. Hanai, T. Iida, K. Takeuchi, F. Watanabe, Y. Maruyama, A. Andoh, T. Tsujikawa, Y. Fujiyama, K. Mitsuyama, M. Sata, M. Yamada, Y. Iwaoka, K. Kanke, H. Hiraishi, K. Hirayama, H. Arai, S. Yoshii, M. Uchijima, T. Nagata and Y. Koide, Clin. Gastroenterol. Hepatol., 2006, 4, 1502–1506 CrossRef CAS PubMed.
- D. L. Suskind, G. Wahbeh, T. Burpee, M. Cohen, D. Christie and W. Weber, J. Pediatr. Gastroenterol. Nutr., 2013, 56, 277–279 CrossRef CAS PubMed.
- P. R. Holt, S. Katz and R. Kirshoff, Dig. Dis. Sci., 2005, 50, 2191–2193 CrossRef PubMed.
- L. Biedermann, J. Mwinyi, M. Scharl, P. Frei, J. Zeitz, G. A. Kullak-Ublick, S. R. Vavricka, M. Fried, A. Weber, H. U. Humpf, S. Peschke, A. Jetter, G. Krammer and G. Rogler, J. Crohn's Colitis, 2013, 7, 271–279 CrossRef PubMed.
- G. W. Dryden, A. Lam, K. Beatty, H. H. Qazzaz and C. J. McClain, Inflammatory Bowel Dis., 2013, 19, 1904–1912 Search PubMed.
- A. Charlebois, G. Rosenfeld and B. Bressler, CRC Crit. Rev. Food Sci. Nutr., 2015 DOI:10.1080/10408398.2012.760515.
- J. Langhorst, H. Wulfert, R. Lauche, P. Klose, H. Cramer, G. J. Dobos and J. Korzenik, J. Crohn's Colitis, 2015, 9, 86–106 CrossRef CAS PubMed.
- D. K. Amre, S. D'Souza, K. Morgan, G. Seidman, P. Lambrette, G. Grimard, D. Israel, D. Mack, P. Ghadirian, C. Deslandres, V. Chotard, B. Budai, L. Law, E. Levy and E. G. Seidman, Am. J. Gastroenterol., 2007, 102, 2016–2025 CrossRef CAS PubMed.
- J. K. Hou, B. Abraham and H. El-Serag, Am. J. Gastroenterol., 2011, 106, 563–573 CrossRef CAS PubMed.
- S. Wirtz and M. F. Neurath, Adv. Drug Delivery Rev., 2007, 59, 1073–1083 CrossRef CAS PubMed.
- M. Perse and A. Cerar, J. Biomed. Biotechnol., 2012, 2012, 718617 Search PubMed.
- J. Wen, P. Yang, X. Chen, Y. Fang, Q. Chang, C. Li and C. Zhang, Eur. J. Inflammation, 2015, 1–8, DOI:10.1177/1721727X15580902 , (in press).
- R. Eri, M. A. McGuckin and R. Wadley, Methods Mol. Biol., 2012, 844, 261–275 CAS.
- H. Hilz, E. J. Bakx, H. A. Schols and A. G. J. Voragen, Carbohydr. Polym., 2005, 59, 477–488 CrossRef CAS PubMed.
- R. L. Prior and L. Gu, Phytochemistry, 2005, 66, 2264–2280 CrossRef CAS PubMed.
- A. P. R. Paiotti, R. A. Neto, P. Marchi, R. M. Silva, V. L. Pazine, J. Noguti, M. M. Pastrelo, A. P. B. Gollucke, S. J. Miszputen and D. A. Ribeiro, Br. J. Nutr., 2013, 110, 973–980 CrossRef CAS PubMed.
- P. Marchi, A. P. Paiotti, R. Artigiani Neto, C. T. Oshima and D. A. Ribeiro, Environ. Toxicol. Pharmacol., 2014, 37, 819–827 CrossRef CAS PubMed.
- L. H. Wu, Z. L. Xu, D. Dong, S. A. He and H. Yu, J. Evidence–Based Complementary Altern. Med., 2011, 2011, 525462 Search PubMed.
- H. Piberger, A. Oehme, C. Hofmann, A. Dreiseitel, P. G. Sand, F. Obermeier, J. Schoelmerich, P. Schreier, G. Krammer and G. Rogler, Mol. Nutr. Food Res., 2011, 55, 1724–1729 CAS.
- L.-S. Wang, C.-T. Kuo, T. H.-M. Huang, M. Yearsley, K. Oshima, G. D. Stoner, J. Yu, J. F. Lechner and Y.-W. Huang, Cancer Prev. Res., 2013, 6, 1317–1327 CrossRef PubMed.
- D. C. Montrose, N. A. Horelik, J. P. Madigan, G. D. Stoner, L. S. Wang, R. S. Bruno, H. J. Park, C. Giardina and D. W. Rosenberg, Carcinogenesis, 2011, 32, 343–350 CrossRef CAS PubMed.
- H. Malekinejad, V. Shafie-Irannejad, R. Hobbenaghi, S. H. Tabatabaie and S. M. Moshtaghion, J. Med. Food, 2013, 16, 593–601 CrossRef CAS PubMed.
- J. A. Rothwell, J. Pérez-Jiménez, V. Neveu, A. Medina-Ramon, N. M'Hiri, L. P. Garcia, C. Manach, K. Knox, R. Eisner, D. Wishart and A. Scalbert, Database, 2013 DOI:10.1093/database/bat070.
- J. Blumberg, B. Bolling, C. Chen and H. Xiao, Eur. J. Nutr. Food Saf., 2015, 5, 1–31 CrossRef PubMed.
- P. A. Abboud, P. W. Hake, T. J. Burroughs, K. Odoms, M. O'Connor, P. Mangeshkar, H. R. Wong and B. Zingarelli, Eur. J. Pharmacol., 2008, 579, 411–417 CrossRef CAS PubMed.
- Y. S. Chiou, N. J. Ma, S. Sang, C. T. Ho, Y. J. Wang and M. H. Pan, J. Agric. Food Chem., 2012, 60, 3441–3451 CrossRef CAS PubMed.
- Z. H. Ran, C. Chen and S. D. Xiao, Biomed. Pharmacother., 2008, 62, 189–196 CrossRef CAS PubMed.
- M. Brückner, S. Westphal, W. Domschke, T. Kucharzik and A. Lügering, J. Crohn's Colitis, 2012, 6, 226–235 CrossRef PubMed.
- F. Guan, A. B. Liu, G. Li, Z. Yang, Y. Sun, C. S. Yang and J. Ju, Nutr. Cancer, 2012, 64, 847–855 CrossRef CAS PubMed.
- D. Komes, D. Horzic, A. Belscak, K. K. Ganic and I. Vulic, Food Res. Int., 2010, 43, 167–176 CrossRef CAS PubMed.
- H. Inoue, S. Akiyama, M. Maeda-Yamamoto, A. Nesumi, T. Tanaka and A. Murakami, Cell Stress Chaperones, 2011, 16, 653–662 CrossRef CAS PubMed.
- H. Inoue, M. Maeda-Yamamoto, A. Nesumi, T. Tanaka and A. Murakami, Biosci., Biotechnol., Biochem., 2013, 77, 1223–1228 CrossRef CAS PubMed.
- M. Kim, A. Murakami, S. Miyamoto, T. Tanaka and H. Ohigashi, BioFactors, 2010, 36, 43–51 CAS.
- H. S. Oz, T. Chen and W. J. de Villiers, Front. Immunol., 2013, 4, 132 Search PubMed.
- M. P. Barnett, J. M. Cooney, Y. E. Dommels, K. Nones, D. T. Brewster, Z. Park, C. A. Butts, W. C. McNabb, W. A. Laing and N. C. Roy, J. Nutr. Biochem., 2013, 24, 1678–1690 CrossRef CAS PubMed.
- Y. Yoshioka, H. Akiyama, M. Nakano, T. Shoji, T. Kanda, Y. Ohtake, T. Takita, R. Matsuda and T. Maitani, Int. Immunopharmacol., 2008, 8, 1802–1807 CrossRef CAS PubMed.
- Y. H. Wang, B. Ge, X. L. Yang, J. Zhai, L. N. Yang, X. X. Wang, X. Liu, J. C. Shi and Y. J. Wu, Int. Immunopharmacol., 2011, 11, 1620–1627 CrossRef CAS PubMed.
- H. W. Yum, X. Zhong, J. Park, H. K. Na, N. Kim, H. S. Lee and Y. J. Surh, Antioxid. Redox Signaling, 2013, 19, 102–114 CrossRef CAS PubMed.
- I. Andújar, M. C. Recio, R. M. Giner, E. Cienfuegos-Jovellanos, S. Laghi, B. a. Muguerza and J. L. Ríos, J. Agric. Food Chem., 2011, 59, 6474–6483 CrossRef PubMed.
- T. Perez-Berezo, C. Ramirez-Santana, A. Franch, S. Ramos-Romero, C. Castellote, F. J. Perez-Cano and M. Castell, Exp. Biol. Med., 2012, 237, 1181–1188 CrossRef CAS PubMed.
- H.-j. Wang and P. A. Murphy, J. Agric. Food Chem., 1994, 42, 1666–1673 CrossRef CAS.
- C. Atkinson, C. L. Frankenfeld and J. W. Lampe, Exp. Biol. Med., 2005, 230, 155–170 CAS.
- M. Morimoto, T. Watanabe, M. Yamori, M. Takebe and Y. Wakatsuki, J. Gastroenterol. Hepatol., 2009, 24, 1123–1129 CrossRef CAS PubMed.
- T. Sakai, S. Furoku, M. Nakamoto, E. Shuto, T. Hosaka, Y. Nishioka and S. Sone, Biosci., Biotechnol., Biochem., 2011, 75, 593–595 CrossRef CAS PubMed.
- J. Seibel, A. F. Molzberger, T. Hertrampf, U. Laudenbach-Leschowski and P. Diel, Eur. J. Nutr., 2009, 48, 213–220 CrossRef CAS PubMed.
- J. Seibel, A. F. Molzberger, T. Hertrampf, U. Laudenbach-Leschowski, G. H. Degen and P. Diel, Arch. Toxicol., 2008, 82, 941–950 CrossRef CAS PubMed.
- L. Xu, Z. L. Yang, P. Li and Y. Q. Zhou, Phytomedicine, 2009, 16, 989–995 CrossRef CAS PubMed.
- T. Azuma, M. Shigeshiro, M. Kodama, S. Tanabe and T. Suzuki, J. Nutr., 2013, 143, 827–834 CrossRef CAS PubMed.
- W. Dou, J. Zhang, A. Sun, E. Zhang, L. Ding, S. Mukherjee, X. Wei, G. Chou, Z. T. Wang and S. Mani, Br. J. Nutr., 2013, 110, 599–608 CrossRef CAS PubMed.
- C. F. S. Guazelli, V. Fattori, B. B. Colombo, S. R. Georgetti, F. T. M. C. Vicentini, R. Casagrande, M. M. Baracat and W. A. Verri, J. Nat. Prod., 2013, 76, 200–208 CrossRef CAS PubMed.
- R. Sotnikova, V. Nosalova and J. Navarova, Interdiscip. Toxicol., 2013, 6, 9–12 Search PubMed.
- E. Cavalcanti, E. Vadrucci, F. R. Delvecchio, F. Addabbo, S. Bettini, R. Liou, V. Monsurro, A. Y. Huang, T. T. Pizarro, A. Santino and M. Chieppa, PLoS One, 2014, 9, e88898 Search PubMed.
- M. Marin, R. M. Giner, J. L. Rios and C. Recio Mdel, Planta Med., 2013, 79, 1392–1400 CrossRef CAS PubMed.
- Z. Ye, Z. Liu, A. Henderson, K. Lee, J. Hostetter, M. Wannemuehler and S. Hendrich, Exp. Biol. Med., 2009, 234, 605–616 CrossRef CAS PubMed.
- E. Giner, I. Andujar, M. C. Recio, J. L. Rios, J. M. Cerda-Nicolas and R. M. Giner, J. Agric. Food Chem., 2011, 59, 12882–12892 CrossRef CAS PubMed.
- C. Mascaraque, C. Aranda, B. Ocon, M. J. Monte, M. D. Suarez, A. Zarzuelo, J. J. Marin, O. Martinez-Augustin and F. S. de Medina, Pharmacol. Res., 2014, 90, 48–57 CrossRef CAS PubMed.
- M. Y. Park, G. E. Ji and M. K. Sung, Dig. Dis. Sci., 2012, 57, 355–363 CrossRef CAS PubMed.
- J. Gálvez, G. Coelho, M. E. Crespo, T. Cruz, M. E. Rodríguez-Cabezas, A. Concha, M. Gonzalez and A. Zarzuelo, Aliment. Pharmcol. Ther., 2001, 15, 2027–2039 CrossRef.
- R. C. Schwanke, R. Marcon, F. C. Meotti, A. F. Bento, R. C. Dutra, M. G. Pizzollatti and J. B. Calixto, Mol. Nutr. Food Res., 2013, 57, 1938–1949 CAS.
- Y. Nishitani, K. Yamamoto, M. Yoshida, T. Azuma, K. Kanazawa, T. Hashimoto and M. Mizuno, BioFactors, 2013, 39, 522–533 CrossRef CAS PubMed.
- Y. Ding, Y. Liang, B. Deng, A. Qiao, K. Wu, W. Xiao and W. Gong, Biochem. Biophys. Res. Commun., 2014, 446, 529–534 CrossRef CAS PubMed.
- A. B. Ganjare, S. A. Nirmal and A. N. Patil, Fitoterapia, 2011, 82, 1052–1056 CrossRef CAS PubMed.
- S. X. Dai, Y. Zou, Y. L. Feng, H. B. Liu and X. B. Zheng, Phytother. Res., 2012, 26, 498–504 CrossRef CAS PubMed.
- R. Moss, Q. Mao, D. Taylor and C. Saucier, Rapid Commun. Mass Spectrom., 2013, 27, 1815–1827 CrossRef CAS PubMed.
- S. Sánchez-Fidalgo, A. Cárdeno, I. Villegas, E. Talero and C. A. de la Lastra, Eur. J. Pharmacol., 2010, 633, 78–84 CrossRef PubMed.
- M. Larrosa, M. J. Yanez-Gascon, M. V. Selma, A. Gonzalez-Sarrias, S. Toti, J. J. Ceron, F. Tomas-Barberan, P. Dolara and J. C. Espin, J. Agric. Food Chem., 2009, 57, 2211–2220 CrossRef CAS PubMed.
- U. P. Singh, N. P. Singh, B. Singh, L. J. Hofseth, R. L. Price, M. Nagarkatti and P. S. Nagarkatti, J. Pharmacol. Exp. Ther., 2010, 332, 829–839 CrossRef CAS PubMed.
- J. Youn, J. S. Lee, H. K. Na, J. K. Kundu and Y. J. Surh, Nutr. Cancer, 2009, 61, 847–854 CrossRef CAS PubMed.
- X. Cui, Y. Jin, A. B. Hofseth, E. Pena, J. Habiger, A. Chumanevich, D. Poudyal, M. Nagarkatti, P. S. Nagarkatti, U. P. Singh and L. J. Hofseth, Cancer Prev. Res., 2010, 3, 549–559 CrossRef CAS PubMed.
- U. P. Singh, N. P. Singh, B. Singh, L. J. Hofseth, D. D. Taub, R. L. Price, M. Nagarkatti and P. S. Nagarkatti, Brain, Behav., Immun., 2012, 26, 72–82 CrossRef CAS PubMed.
- Y. H. Kim, H. S. Kwon, D. H. Kim, H. J. Cho, H. S. Lee, J. G. Jun, J. H. Park and J. K. Kim, Int. Immunopharmacol., 2008, 8, 1695–1702 CrossRef CAS PubMed.
- C. E. Green, S. L. Hibbert, Y. A. Bailey-Shaw, L. A. D. Williams, S. Mitchell and E. Garraway, J. Agric. Food Chem., 2008, 2008 Search PubMed.
- K. Nones, Y. E. Dommels, S. Martell, C. Butts, W. C. McNabb, Z. A. Park, S. Zhu, D. Hedderley, M. P. Barnett and N. C. Roy, Br. J. Nutr., 2009, 101, 169–181 CrossRef CAS PubMed.
- L. Liu, Y. L. Liu, G. X. Liu, X. Chen, K. Yang, Y. X. Yang, Q. Xie, H. K. Gan, X. L. Huang and H. T. Gan, Int. Immunopharmacol., 2013, 17, 314–320 CrossRef CAS PubMed.
- I. Villegas, S. Sanchez-Fidalgo and C. A. de la Lastra, Mol. Nutr. Food Res., 2011, 55, 259–267 CAS.
- R. Aldini, R. Budriesi, G. Roda, M. Micucci, P. Ioan, A. D'Errico-Grigioni, A. Sartini, E. Guidetti, M. Marocchi, M. Cevenini, F. Rosini, M. Montagnani, A. Chiarini and G. Mazzella, PLoS One, 2012, 7, e44650 Search PubMed.
- Y. Topcu-Tarladacalisir, M. Akpolat, Y. H. Uz, G. Kizilay, M. Sapmaz-Metin, A. Cerkezkayabekir and I. K. Omurlu, J. Med. Food, 2013, 16, 296–305 CrossRef CAS PubMed.
- Z. Zeng, L. Zhan, H. Liao, L. Chen and X. Lv, Planta Med., 2013, 79, 102–109 CAS.
- P. Anand, A. B. Kunnumakkara, R. A. Newman and B. B. Aggarwal, Mol. Pharm., 2007, 4, 807–818 CrossRef CAS PubMed.
- J. Shaikh, D. D. Ankola, V. Beniwal, D. Singh and M. N. Kumar, Eur. J. Pharm. Sci., 2009, 37, 223–230 CrossRef CAS PubMed.
- L. Zhongfa, M. Chiu, J. Wang, W. Chen, W. Yen, P. Fan-Havard, L. D. Yee and K. K. Chan, Cancer Chemother. Pharmacol., 2012, 69, 679–689 CrossRef PubMed.
- V. Yadav, S. Suresh, K. Devi and S. Yadav, J. Pharm. Pharmacol., 2009, 61, 311–321 CrossRef CAS.
- V. Yadav, S. Suresh, K. Devi and S. Yadav, AAPS PharmSciTech, 2009, 10, 752–762 CrossRef CAS PubMed.
- R. Sareen, N. Jain and K. L. Dhar, Pharm. Dev. Technol., 2015, 1–6, DOI:10.3109/10837450.2015.1041042 (in press).
- Y. Tao, W. Li, W. Liang and R. B. Van Breemen, J. Agric. Food Chem., 2009, 57, 10014–10021 CrossRef CAS PubMed.
- H. S. El-Abhar, L. N. Hammad and H. S. Gawad, J. Ethnopharmacol., 2008, 118, 367–372 CrossRef CAS PubMed.
- C. Y. Hsiang, H. Y. Lo, H. C. Huang, C. C. Li, S. L. Wu and T. Y. Ho, Food Chem., 2013, 136, 170–177 CrossRef CAS PubMed.
- B. Cerda, F. A. Tomas-Barberan and J. Espin, J. Agric. Food Chem., 2005, 53, 227–235 CrossRef CAS PubMed.
- E. Bakkalbasi, O. Mentes and N. Artik, CRC Crit. Rev. Food Sci. Nutr., 2009, 49, 283–298 CrossRef CAS PubMed.
- M. Marin, R. Maria Giner, J. L. Rios and M. C. Recio, J. Ethnopharmacol., 2013, 150, 925–934 CrossRef CAS PubMed.
- M. A. Rosillo, M. Sanchez-Hidalgo, A. Cardeno, M. Aparicio-Soto, S. Sanchez-Fidalgo, I. Villegas and C. A. de la Lastra, Pharmacol. Res., 2012, 66, 235–242 CrossRef CAS PubMed.
- M. Larrosa, A. Gonzalez-Sarrias, M. J. Yanez-Gascon, M. V. Selma, M. Azorin-Ortuno, S. Toti, F. Tomas-Barberan, P. Dolara and J. C. Espin, J. Nutr. Biochem., 2010, 21, 717–725 CrossRef CAS PubMed.
- H. T. Xiao, C. Y. Lin, D. H. Ho, J. Peng, Y. Chen, S. W. Tsang, M. Wong, X. J. Zhang, M. Zhang and Z. X. Bian, J. Nat. Prod., 2013, 76, 2120–2125 CrossRef CAS PubMed.
- H. S. Shin, H. Satsu, M. J. Bae, Z. Zhao, H. Ogiwara, M. Totsuka and M. Shimizu, Food Chem., 2015, 168, 167–175 CrossRef CAS PubMed.
- M. Larrosa, C. Luceri, E. Vivoli, C. Pagliuca, M. Lodovici, G. Moneti and P. Dolara, Mol. Nutr. Food Res., 2009, 53, 1044–1054 CAS.
- W.-G. Dong, S.-P. Liu, B.-P. Yu, D.-F. Wu, H.-S. Luo and J.-P. Yu, World J. Gastroenterol., 2003, 9, 2533–2538 CAS.
- D. Del Rio, A. Stalmach, L. Calani and A. Crozier, Nutrients, 2010, 2, 820–833 CrossRef CAS PubMed.
- J. Fang, T. Seki, T. Tsukamoto, H. Qin, H. Yin, L. Liao, H. Nakamura and H. Maeda, Carcinogenesis, 2013, 34, 2833–2841 CrossRef CAS PubMed.
- E. Giner, M. C. Recio, J. L. Rios and R. M. Giner, J. Nat. Prod., 2013, 76, 1113–1120 CrossRef CAS PubMed.
- W. Dou, J. Zhang, E. Zhang, A. Sun, L. Ding, G. Chou, Z. Wang and S. Mani, J. Pharmacol. Exp. Ther., 2013, 345, 473–482 CrossRef CAS PubMed.
- E. K. Shin, H. S. Kwon, Y. H. Kim, H. K. Shin and J. K. Kim, Biochem. Biophys. Res. Commun., 2009, 381, 502–507 CrossRef CAS PubMed.
- A. E. Miroliaee, H. Esmaily, A. Vaziri-Bami, M. Baeeri, A. R. Shahverdi and M. Abdollahi, Toxicol. Mech. Methods, 2011, 21, 200–208 CrossRef CAS PubMed.
- F. Tao, C. Qian, W. Guo, Q. Luo, Q. Xu and Y. Sun, Biochem. Pharmacol., 2013, 85, 798–807 CrossRef CAS PubMed.
- A. Witaicenis, L. N. Seito and L. C. Di Stasi, Chem.-Biol. Interact., 2010, 186, 211–218 CrossRef CAS PubMed.
- T. Takashima, Y. Sakata, R. Iwakiri, R. Shiraishi, Y. Oda, N. Inoue, A. Nakayama, S. Toda and K. Fujimoto, J. Nutr. Biochem., 2014, 25, 186–192 CrossRef CAS PubMed.
- S. Sanchez-Fidalgo, A. Cardeno, M. Sanchez-Hidalgo, M. Aparicio-Soto and C. A. de la Lastra, J. Nutr. Biochem., 2013, 24, 1401–1413 CrossRef CAS PubMed.
- F. Gutierrez-Orozco, J. M. Thomas-Ahner, L. D. Berman-Booty, J. D. Galley, C. Chitchumroonchokchai, T. Mace, S. Suksamrarn, M. T. Bailey, S. K. Clinton, G. B. Lesinski and M. L. Failla, Mol. Nutr. Food Res., 2014, 58, 1226–1238 CAS.
- X. Xiao, J. Kim, Q. Sun, D. Kim, C. S. Park, T. S. Lu and Y. Park, Food Chem., 2015, 167, 438–446 CrossRef CAS PubMed.
- Y. Okamoto, T. Hara, T. Ebato, T. Fukui and T. Masuzawa, Int. Immunopharmacol., 2013, 16, 178–183 CrossRef CAS PubMed.
- G. Mandalari, C. Bisignano, T. Genovese, E. Mazzon, M. S. J. Wickham, I. Paterniti and S. Cuzzocrea, Int. Immunopharmacol., 2011, 11, 915–924 CrossRef CAS PubMed.
- J. A. Skyberg, A. Robison, S. Golden, M. F. Rollins, G. Callis, E. Huarte, I. Kochetkova, M. A. Jutila and D. W. Pascual, J. Leukocyte Biol., 2011, 90, 1043–1054 CrossRef CAS PubMed.
- C. Castagnini, C. Luceri, S. Toti, E. Bigagli, G. Caderni, A. P. Femia, L. Giovannelli, M. Lodovici, V. Pitozzi, M. Salvadori, L. Messerini, R. Martin, E. G. Zoetendal, S. Gaj, L. Eijssen, C. T. Evelo, C. M. Renard, A. Baron and P. Dolara, Br. J. Nutr., 2009, 102, 1620–1628 CrossRef CAS PubMed.
- L. Zarepoor, J. T. Lu, C. Zhang, W. Wu, D. Lepp, L. Robinson, J. Wanasundara, S. Cui, S. Villeneuve, B. Fofana, R. Tsao, G. A. Wood and K. A. Power, Am. J. Physiol.: Gastrointest. Liver Physiol., 2014, 306, G1042–G1055 CrossRef CAS PubMed.
- L. Lenoir, A. Rossary, J. Joubert-Zakeyh, J. Vergnaud-Gauduchon, M. C. Farges, D. Fraisse, O. Texier, J. L. Lamaison, M. P. Vasson and C. Felgines, Dig. Dis. Sci., 2011, 56, 3534–3545 CrossRef CAS PubMed.
- A. Hakansson, C. Branning, G. Molin, D. Adawi, M. L. Hagslatt, B. Jeppsson, M. Nyman and S. Ahrne, PLoS One, 2012, 7, e33510 Search PubMed.
- S. Reagan-Shaw, M. Nihal and N. Ahmad, FASEB J., 2008, 22, 659–661 CrossRef CAS PubMed.
- K. Schneider, J. Oltmanns and M. Hassauer, Regul. Toxicol. Pharmacol., 2004, 39, 334–347 CrossRef CAS PubMed.
- R. B. Rucker, J. Anim. Physiol. Anim. Nutr., 2007, 91, 148–156 CrossRef CAS PubMed.
- L. Mark, M. S. P. Nikfardjam, P. Avar and R. Ohmacht, J. Chromatogr. Sci., 2005, 43, 445–449 CAS.
- D. J. Boocock, G. E. S. Faust, K. R. Patel, A. M. Schinas, V. A. Brown, M. P. Ducharme, T. D. Booth, J. A. Crowell, M. Perloff, A. J. Gescher, W. P. Steward and D. E. Brenner, Cancer Epidemiol. Biomarkers, 2007, 16, 1246–1252 CrossRef CAS PubMed.
- B. Holst and G. Williamson, Curr. Opin. Biotechnol., 2008, 19, 73–82 CrossRef CAS PubMed.
- B. Halliwell, J. Rafter and A. Jenner, Am. J. Clin. Nutr., 2005, 81, 268S–276S CAS.
- M. N. Clifford, J. Sci. Food Agric., 1999, 79, 362–372 CrossRef CAS.
- P. I. Mackenzie, K. W. Bock, B. Burchell, C. Guillemette, S. Ikushiro, T. Iyanagi, J. O. Miners, I. Owens and D. W. Nebert, Pharmacogenet. Genomics., 2005, 15, 677–685 CrossRef CAS.
- T. L. Nguyen, S. Vieira-Silva, A. Liston and J. Raes, Dis. Models Mech., 2015, 8, 1–16 CrossRef PubMed.
- R. Hansen, R. K. Russell, C. Reiff, P. Louis, F. McIntosh, S. H. Berry, I. Mukhopadhya, W. M. Bisset, A. R. Barclay, J. Bishop, D. M. Flynn, P. McGrogan, S. Loganathan, G. Mahdi, H. J. Flint, E. M. El-Omar and G. L. Hold, Am. J. Gastroenterol., 2012, 107, 1913–1922 CrossRef CAS PubMed.
- S. Nell, S. Suerbaum and C. Josenhans, Nat. Rev. Microbiol., 2010, 8, 564–577 CrossRef CAS PubMed.
- T. Zhang, R. A. DeSimone, X. Jiao, F. J. Rohlf, W. Zhu, Q. Q. Gong, S. R. Hunt, T. Dassopoulos, R. D. Newberry, E. Sodergren, G. Weinstock, C. E. Robertson, D. N. Frank and E. Li, PLoS One, 2012, 7, e30044 CAS.
- T. E. Cowan, M. S. Palmnas, J. Yang, M. R. Bomhof, K. L. Ardell, R. A. Reimer, H. J. Vogel and J. Shearer, J. Nutr. Biochem., 2014, 25, 489–495 CrossRef CAS PubMed.
- R. V. Espley, C. A. Butts, W. A. Laing, S. Martell, H. Smith, T. K. McGhie, J. Zhang, G. Paturi, D. Hedderley, A. Bovy, H. J. Schouten, J. Putterill, A. C. Allan and R. P. Hellens, J. Nutr., 2014, 144, 146–154 CrossRef CAS PubMed.
- U. Etxeberria, A. Fernandez-Quintela, F. I. Milagro, L. Aguirre, J. A. Martinez and M. P. Portillo, J. Agric. Food Chem., 2013, 61, 9517–9533 CAS.
- J. R. Turner, Nat. Rev. Immunol., 2009, 9, 799–809 CrossRef CAS PubMed.
- T. Suzuki, Cell. Mol. Life Sci., 2013, 70, 631–659 CrossRef CAS PubMed.
- T. Suzuki and H. Hara, J. Nutr. Biochem., 2011, 22, 401–408 CrossRef CAS PubMed.
- M. Shieshiro, S. Tanabe and T. Suzuki, J. Funct. Foods, 2013, 5, 949–955 CrossRef PubMed.
- M. Schwab and U. Klotz, Clin. Pharmacokinet., 2001, 40, 723–751 CrossRef CAS PubMed.
- C. X. Liu, X. L. Yi, D. Y. Si, X. F. Xiao, X. He and Y. Z. Li, Curr. Drug Metab., 2011, 12, 835–849 CrossRef CAS.
- Y. Kimura, H. Ito, R. Ohnishi and T. Hatano, Food Chem. Toxicol., 2010, 48, 429–435 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |