Spatio-temporal resolution of primary processes of photosynthesis
Wolfgang
Junge
*
Dept. Biology & Chemistry, University of Osnabrück, R. 35/E42 Barbarastrasse 11, 49076 Osnabrück, Germany. E-mail: junge@uos.de; Tel: +49-15112180743
First published on 31st March 2015
Abstract
Technical progress in laser-sources and detectors has allowed the temporal and spatial resolution of chemical reactions down to femtoseconds and Å-units. In photon-excitable systems the key to chemical kinetics, trajectories across the vibrational saddle landscape, are experimentally accessible. Simple and thus well-defined chemical compounds are preferred objects for calibrating new methodologies and carving out paradigms of chemical dynamics, as shown in several contributions to this Faraday Discussion. Aerobic life on earth is powered by solar energy, which is captured by microorganisms and plants. Oxygenic photosynthesis relies on a three billion year old molecular machinery which is as well defined as simpler chemical constructs. It has been analysed to a very high precision. The transfer of excitation between pigments in antennae proteins, of electrons between redox-cofactors in reaction centres, and the oxidation of water by a Mn4Ca-cluster are solid state reactions. ATP, the general energy currency of the cell, is synthesized by a most agile, rotary molecular machine. While the efficiency of photosynthesis competes well with photovoltaics at the time scale of nanoseconds, it is lower by an order of magnitude for crops and again lower for bio-fuels. The enormous energy demand of mankind calls for engineered (bio-mimetic or bio-inspired) solar-electric and solar-fuel devices.
1 Introduction
When the founders of atomic and molecular physics convened in 1927 at the 5th Solvay Conference on “Electrons and Photons” in Brussels (see photograph in ref. 1) they had established quantum mechanics, relativity and the interplay of stochastic and conservative forces in the nano-world. Despite their immense insight it was probably all but evident to them how molecular events could ever be resolved down to molecular scales of time (femto-second) and space (1 Å-unit = 0.1 nanometer). Neither was it evident that such a high resolution might be applicable not only to small molecules but also to complex systems such as oligomers, interfaces, proteins, membranes, cells and organisms. High resolution became feasible by dramatic innovations in physical instrumentation. In the 1960s pulse lasers opened up the time domain of nanoseconds, soon followed by pico- to femtoseconds (ps–fs) in the seventies. The whole spectral range from the infrared (IR) over the visible to the ultraviolet (UV) has been covered by pulsed photon sources. It culminated in the free electron “laser” (rather a coherent super-radiation source) in the seventies, which provides femtosecond X-ray or electron pulses. Pulse sources together with greatly improved detectors and data handling capacities have allowed the high resolution of structure and kinetics, particularly in those physical, chemical and biological systems that can be stimulated by light.Recent technical progress was covered in the Faraday Discussion on “Emerging Photon Technologies for Chemical Dynamics” (FD 171). The present Faraday Discussion (FD 177) expands this theme into “Temporally and Spatially Resolved Molecular Science”. It aims at a rigorous exploration of the energy landscape and the path of extremely rapid photophysical and photochemical reactions involving a manifold of vibronic and torsional states. Two- and more-dimensional photon-echo spectroscopies have been essential to overcome the lack of spectral resolution when using photon-pulses of fs-duration. Prominent topics of FD 177 have been quantum coherence, failure of the micro-canonical approximation, solvent effects on vibrational spectra and relaxations, and time-resolved structure analysis by fs-pulses of X-rays or electrons. A common quest in most contributions to FD 177 has been the perfection of data and the outmost rigor in their theoretical interpretation. It has called for simple and well defined systems, mostly small inorganic or organic molecules. The paradigms derived from the behaviour of simple systems – physical chemistry at its best – have provided firm grounds for bio-physical-chemists and biophysicists to address their more complex research objects. Although being complex in design some of nature's essential constructs, like the proteins that supply cells with energy, originated about three billions years ago and have been largely conserved during evolution. They are thus standardized and often as well defined as simple chemical compounds.
When dealing with these fundamental biological systems the canon of questions to be asked is expanded. The key questions of physical chemistry – what, where and how fast – are to be supplemented by what for and why. The former three address the object, its molecular structure and evolution in time. The question what for addresses the physiological role of a given construct and the question why whether a particular construct is exacted by laws of physics or merely reveals nature's fancy as a legacy of evolution. In the following the relation between pure and biologically oriented physical chemistry is illustrated by taking oxygenic photosynthesis as an example.
2 Oxygenic photosynthesis
Oxygenic photosynthesis by cyanobacteria and plants uses sunlight and produces oxygen and biomass. Biomass serves man as food, fuel, fibre and platform chemical. Early attempts to understand this process go as far back as the 18th century. Jan Ingen-Housz in his study on vegetables noticed their “Great Power of Purifying the Common Air in the Sunshine and of Injuring it in the Shade and at Night”.2 It was a first appreciation of the production and re-consumption – in the reaction cycle between photosynthesis and cell respiration – of the gases that were later coined oxygen and carbon dioxide. A rigorous spectroscopic analysis of photosynthesis started half a century ago using microsecond flashes of light for excitation. In 1961 Duysens,3 Kok,4 and Witt5 independently arrived at the conclusion that green plant photosynthesis is powered by two photosystems in series. They drive electrons from water to NADP+. Water is oxidized by photosystem II (PSII) to yield oxygen and protons. PSII, in turn, reduces photosystem I (PSI) which reduces NADP+ to NADPH, as illustrated in Fig. 1.6 In that same year Mitchell (Nobel Prize 1978) postulated that ATP is synthesized from ADP and Pi at the expense of a proton-motive force across the respective coupling membrane.7 The energy conserved in the chemical difference of the products NADPH/O2 (by PSII and PSI) and ATP/ADP·Pi (by ATP synthase (FOF1)) drives the reduction of CO2 to biomass. | ||
| Fig. 1 Electron and proton transfer in the coupling membrane of oxygenic photosynthesis. (a) Electron transfer (red arrows) and proton transfer (purple arrows) involving photosystems I and II (PSI and PSII), cytochrome b6f (cyt b6f), and FOF1. It produces O2, NADPH and ATP. Modified with permission from the Annual Review of Biochemistry, Volume 84. © 2015, Annual Reviews, http://www.annualreviews.org.6 | ||
A decade later the time resolution of the primary electron transfer was advanced into the picosecond range.8 But it took more than another decade until Deisenhofer, Huber and Michel (Nobel Prize 1988) published the first structural model of a photosynthetic reaction centre (resolution 3 Å).9 It was the first ever structural model of any membrane protein. Today, structural models are available for all the proteins central to photosynthesis. PSI of a cyanobacterium is resolved at a 2.5 Å resolution,10 and even the larger PSI of green plants with a molecular mass of 660![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 a.u. is resolved at 3.4 Å.11 The latter hosts almost 200 chlorophyll molecules, mostly with antennae function, plus the redox-active core with four chlorophyll- and two pheophytin molecules.12 Cyanobacterial PSII, the water–quinone oxidoreductase, has been resolved starting at 3.8 Å13 and now at a 1.9 Å resolution.14 The structure of ATP synthase (FOF1) has only partially been determined at the atomic resolution.15,16 However, its full structure is evident from low resolution data of the holo-enzyme (see ref. 6 and 17 and references therein). PSII and PSI are solid-state devices. FOF1 is a most agile rotary machine composed of two rotary motor generators. FO, the ion-driven motor18,19 is mechanically coupled20 to F1, the chemical generator.15,21
000 a.u. is resolved at 3.4 Å.11 The latter hosts almost 200 chlorophyll molecules, mostly with antennae function, plus the redox-active core with four chlorophyll- and two pheophytin molecules.12 Cyanobacterial PSII, the water–quinone oxidoreductase, has been resolved starting at 3.8 Å13 and now at a 1.9 Å resolution.14 The structure of ATP synthase (FOF1) has only partially been determined at the atomic resolution.15,16 However, its full structure is evident from low resolution data of the holo-enzyme (see ref. 6 and 17 and references therein). PSII and PSI are solid-state devices. FOF1 is a most agile rotary machine composed of two rotary motor generators. FO, the ion-driven motor18,19 is mechanically coupled20 to F1, the chemical generator.15,21
The key machinery of bioenergetics (photosynthesis and respiration) dates about three billion years ago. Its design has not changed much since then. It justifies casting into one coherent scheme the kinetic and structural insights which have been elaborated on enzymes from different organisms (microorganisms, plants and animals).
3 Excitation energy transfer
Photosynthetic reaction centres are surrounded by pigments with antenna function, in PSII between 200 and 300 chlorophyll-molecules. Being embedded in membrane proteins they enhance the absorption cross-section of the photo-reactive core. In chlorophyll-based reaction centres from all organisms the core hosts four plus two tetra-pyrroles, the former four being bacterio-chlorophyll and the latter two either bacterio-pheophytin or bacterio-chlorophyll (see Fig. 2). These pigments are very closely spaced and thus strongly coupled. The core was first isolated in the pure form from bacteria. Starting in the eighties femtosecond spectroscopy revealed that the first electron transfer step from bacterio-chlorophyll to bacterio-pheophytin relaxes in picoseconds.22,23 Soon thereafter vibrational coherence between these pigments became evident.24 As in several contributions to this Faraday Discussion femtosecond excitation and multidimensional photon-echo spectroscopy became the clue to investigate processes where the micro-canonical approximation fails. An impressive series of studies on photosynthesis has revealed quantum beats in antennae proteins (for reviews and references see ref. 25–27, for a recent survey of quantum biology see ref. 28). | ||
| Fig. 2 Structural model of the redox cofactors in the core of cyanobacterial PSII (modified with permission of Science).114 The electron transfer uses the D1-branch of the hetero-dimer. Chorophyll a molecules are given in green and blue, pheophytin a in blue and the primary (QA) and secondary (QB) quinones in purple. The catalytic CaMn4-cluster is linked via a tyrosine (TyrZ) to the chlorophyll (PD1). Centre-to-centre distances in Å-units. | ||
Taking up the above biophysical question of physical necessity versus legacy of evolution one wonders whether coherent excitation transfer is a necessary prerequisite for the high quantum yield of photosynthesis. In a theoretical study on a structurally well characterized bacterial antenna complex it is argued that it is rather unecessary.29 If the reorganisation time of the environment is not much faster than the one of the system coherence is prolonged. However, it does not increase the efficiency of energy transfer, except when excitation decay competes with trapping.29 An experimental study on the same antenna protein was presented at this meeting in a poster by Battacharyya and Sebastian. Mechanisms for the dephasing of coherence by the environment were discussed in posters by Kayal et al. and Roy et al., and the mechanism of photon-induced intra-molecular excimer formation and charge transfer was treated by Mishra et al.30
In photosynthetic antennae the excitation decay does not to compete with trapping. This is has been corroborated by the following recent experiments. Van Grondelle's group analysed the excitation transfer between six pigments (4 chlorophyll a, 2 pheophytin a) in the purified core of PSII.31 Quantum beats persisted for several 100 femtoseconds even at ambient temperature. Several coherent excimer-/charge-transfer states coexisted before the creation of a metastable radical pair. The authors suggested that quantum design of the core is pivotal for the high quantum yield.31 In these experiments the core of PSII is isolated from its antennae complement and it is excited by femtosecond pulses. In vivo the core is surrounded by antennae. Quanta trickle in at a rate of much less than 1000 per second. Holzwarth and his colleagues studied this situation in oxygen evolving PSII particles containing a complement of about 80 chlorophyll molecules.32,33 To avoid singlet–singlet annihilation they kept the photon density of the exciting pulse low. The total time for exciton trapping and charge separation was about 100 ps.32 The calculated intrinsic time of charge separation in the core, on the other hand, was much shorter, 2.7 ps.33 This situation is paralleled in purple photosynthetic bacteria. Excitation transfer in the B850-ring of the antenna LH2 as well as in the B875-ring of LH1 is very fast (about 100 fs) and it involves coherence. The transfer from LH2 to LH1 takes 3 ps, and further on from LH1 into the trap 35 ps.25 The long and trap-limited lifetime of excitation greatly exceeds the decay time of coherence. Independent of whether coherent transfer is involved or not, the only condition for high quantum yield is that the transfer beats the dissipative loss of excitation in the whole system. This physical constraint is rather mild, and it provides a leeway for various transfer mechanisms and structural designs. In fact, photosynthetic organisms use a wide palette of mechanisms for excitation transfer, ranging from hopping by Förster-resonance over Frenkel-excitons to more delocalized modes involving the coherent states of many pigment molecules. Accordingly, the antennae structure varies considerably between organisms. Chlorophyll molecules are either enwrapped in proteins as in the LHC2 of plants,34 and in the LH1 and LH2 of purple bacteria,35,36 or self-organized as in the chlorosomes of green bacteria.37 The energy transfer may be directed towards a deep trap (low energy trap), as in PSI, or undirected if the trap is shallow, as in PSII.33 In all cases the quantum yield of trapping is close to one.
4 Electron transfer
Excitation transfer into the very core of any reaction centre results in a very rapid charge separation. The first metastable product is a radical pair which is formed in picoseconds. In PSII this pair is pheophytinD1+–chlorophyllD1− (see Fig. 2). Herein the subscript D1 refers to the subunit to which the respective pigment is attached. While the positive charge is localized on this particular chlorophyll in PSII (PD1 in Fig. 2), it is delocalized over two bacterio-chlorophylls, the “special pair”, in the reaction centre of purple bacteria.38 The delocalization of the unpaired electron over this “special pair” hence is not pivotal for a high quantum yield, in contrast with previous beliefs.39The charge separation is directed across the membrane. The membrane is charged by 50 mV (field strength 107 V m−1) per single turnover of each photosystem. Light induced voltage transients were first detected in plant chloroplasts. Electrochromic absorption changes of intrinsic pigments served as a molecular voltmeter40,41 with a very high time-resolution.42 The vibrational Stark-effect as a molecular voltmeter was discussed in a poster of this Faraday Discussion by S. Bagchi et al.
Structural pseudo-C2-symmetry is a common feature of most reaction centres. Their hetero-dimeric structure is considered an evolutionary legacy of a homo-dimeric and truly C2-symmetrical common ancestor.43–46 Anaerobic green bacteria still host a homo-dimeric reaction centre.43
Independent of the type of reaction centre and organism, the positions and orientations of the six pigments in the core are very much the same. There are three pigments on each branch of the dimer. While only one branch is redox-active in type II centres (of purple bacteria and in PSII of plants and cyanobacteria), both are similarly active in a type I centre (PSI). The functional asymmetry in the former has been attributed (by Stark-effect spectroscopy) to different dielectric screenings in the two branches.47 The asymmetric activity of type II centres is linked to its function as a one-electron to two-electron gate, whereas PSI, with its longer line of secondary acceptors, functions mono-electronically (for a thorough discussion and references see ref. 48).
The mechanism of electron transfer between cofactors that are embedded in the protein is adequately described by the Marcus theory.49,50 It models tunnelling within the limits of the Born–Oppenheimer approximation. The rate is a function of the (edge-to-edge) distance between donor and acceptor (r), and it depends on three parameters: the standard free energy difference between the donor and the acceptor (ΔG0), the reorganization energy of their environment (λ) and the decay constant of wave-function-overlap (β):
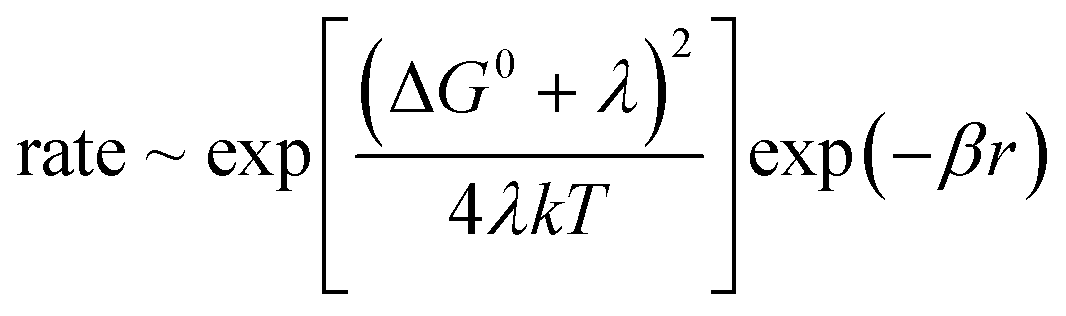 | (1) |
This simple relation in terms of only three parameters has been experimentally very well met. In synthetic donor–spacer–acceptor triads it holds over three orders of magnitude.51 As for the electron transfer between cofactors in a protein, various pathways were screened in cytochrome c. Its heme served as the electron acceptor for a photosensitizer which was covalently attached to various positions at the surface. The transfer rate decreased depending on whether the electron passed mainly along single bonds, hydrogen bonds or through a void volume in the protein.52,53 In a related study, Dutton and colleagues54 analysed the wealth of kinetic and structural data on the electron transfer in photosynthesis and cell respiration. They corrected the observed rates for the matching driving force (determined!) and reorganization energy (assumed!), namely −ΔG0 = λ. Over 13 orders of magnitude the normalized rates decline mono-exponentially with the edge-to-edge distance (r) between donor and acceptor. Deviations by one order of magnitude can be attributed to local variations of λ and β. The donor–acceptor distance is hence the major determinant of the rate of electron transfer.55 The reorganization energy (λ) and the decay constant of the electronic wave functions (β) in proteins do not vary too much. Nature's engineering of structures for rapid electron transport has thus relied mainly on the distance between redox-cofactors (r) and on the driving force (ΔG0).55
Once the core of the reaction centre is hit by an excitation the primary electron transfer is very fast. In PSII the radical pair (chlorophyll+–pheophytin−) appears in picoseconds. The electron is then transferred to a first quinone (QA) in about 400 picoseconds and from there on to a second quinone (QB) in some 100 microseconds.56,57 The electron hole, on the other hand, is rapidly transferred in some ten nanoseconds to a tyrosine58 and further in microseconds to the Mn4Ca-cluster. All of these reactions outrun by orders of magnitude the respective wasteful back-reactions of excited states and radical pairs. Rapid forward reactions and slow back-reactions guarantee the high quantum yield of photosynthesis. On the other hand they require losses of free energy. In other words, photosynthesis sacrifices energy efficiency for directionality.48
5 Water oxidation
PSII is a water–quinone oxidoreductase. The structure of cyanobacterial PSII is resolved at 3.8 Å,13 3.5 Å,59 and 1.9 Å.14 Water (-derivatives) are oxidized by the catalytic Mn4Ca-cluster and oxygen plus protons are liberated. When dark adapted PSII is excited by a series of short light flashes the Mn4Ca-cluster is clocked through sequentially higher and metastable oxidation states, until reaching the highest state (see ref. 60 and references therein). Only then the reaction with bound water proceeds (in one millisecond) to yield dioxygen (see ref. 61). The stepwise accumulation and pooling of four oxidizing equivalents, before initiating what still appears as the reaction of four-electrons-at-once, serves two purposes. It homogenizes the energy demand of successive electron transfer steps, and controls hazardous intermediates (e.g. hydroxyl radical and super-oxide) on the way from water to dioxygen. There is a wealth of detailed kinetic studies on water oxidase, addressing the chlorophyll-moiety, an intermediate tyrosine, the Mn4Ca-cluster, electrons, protons and oxygen. A wide range of optical (X-ray, UV, VIS and IR), magnetic and mass spectroscopic techniques have been utilized to resolve electron transfer, proton transfer, conformational changes of the protein, substrate binding and product release in the time-domain from nano- to milliseconds (for reviews and references see ref. 62–65).The precise structure of the manganese cluster is still under contention. During standard X-ray exposure of PSII-crystals Mn4 is the prime target of radiation damage66,67 (for radiation damage of metal markers for protein structural studies see Helliwell et al.).68 The reduction of Mn may influence its ligation state. The lack of unequivocal structural models of the Mn4Ca-cluster in its sequential oxidation states is a major obstacle for a thorough understanding of the final reaction cascade. For the time being, two non-invasive approaches, namely by magnetic resonance spectroscopy (Endor69) and by theoretical chemistry (density functional theory71), have converged towards one particular structural model of the CaMn4-cluster and its ligands including water (derivatives). X-ray crystal structural analysis will eventually take up, either challenging or corroborating the present structural concept by a probe-before-destroy-approach (for principles and perspectives of this technique see Miller et al.).70 PSII-crystals have been exposed to an ultra-short and intense X-ray pulse (100 fs) of a free-electron-laser.72,73 The feasibility of analysing the cluster-structure in its sequential oxidation states has been clearly demonstrated, although at a limited resolution so far (5–5.5 Å).73 Structural details on the Mn4Ca-moiety, bound water (derivatives), and amino acid ligands is the essential complement of kinetic data when aiming to understand the detailed reaction mechanism of this “holy grail” of photosynthesis.
6 ATP synthesis
Mitchell's daring hypothesis on proton-driven ATP synthesis7 roused the unprepared bioenergetics community. Soon thereafter essential elements of his hypothesis were detected in plant chloroplasts. Illumination generates transmembrane voltage40 and a pH-difference,74 and the pH-difference75 and/or the voltage76 can drive the synthesis of ATP. The molecular structure of ATP synthase was then still unknown. Only its bipartite construction with a membrane portion, FO, which conducts protons, and a peripheral portion, F1, which interacts with nucleotides, was apparent. Boyer (Nobel Prize 1996) and his colleagues found that F1 hosts two, if not three identical catalytic sites, that operate alternately,77 if not in a rotary mode.78 Conformational energy stored in the enzyme might drive the formation of ATP from ADP and P1.79 In 1994 Walker (Nobel Prize 1996) and his colleagues presented the first structural model of its chemical generator at a 2.3 Å resolution,15 now at 1.9 Å,80 which strongly favoured a rotary function of F1. F1 is a pseudo-hexagon of two types of subunits arranged as (αβ)3 with a cranked shaft (subunit γ) in its centre. It suggests that the rotation of the central shaft drives ATP synthesis when progressing from one catalytic site on β to the next. Soon thereafter the rotation of the central shaft was experimentally established, starting without time resolution by the biochemical crosslink technique,81 and then time resolved either by polarized photobleaching and recovery82,63 or fluorescence microscopy.83 The latter technique became the gold standard in the field (for original video recordings see ref. 119). Its principle is illustrated in Fig. 3a. The hexagonal body of the enzyme is fixed on a solid support so that the hydrolysis of ATP drives the shaft around. In a pioneering experiment, a fluorescent actin filament was attached to the foot of the central stalk of F1 and the rotation of the filament was video-recorded in a fluorescence microscope.83 In the author's lab the fluorescent probe was attached to the holo-enzyme, FOF1, as shown in Fig. 3a. Notably the procession of this enzyme over its reaction coordinate can be recorded in real-time. By certain tricks it has been feasible to correlate the dynamic jumps and transient dwells of the live enzyme with the still pictures of the inhibited enzyme as a crystal structure.84–86 Moreover, the rotor position has been externally manipulated by an attached hyper-paramagnetic bead.87,88 Ongoing studies in this line will likely provide more precise data on the energy landscape of F1, a prerequisite for thorough theoretical descriptions (see ref. 89–91 and references therein). The wealth of structural and kinetic data qualifies ATP synthase as a hydrogen atom of nano-motoring. The sophisticated, yet simple electro-mechano-chemical operation of ATP synthase has been described in several reviews.6,17–20,92,93 For animations of the enzyme and its two motors, FO and F1, see ref. 121. A few salient features shall be emphasized in the following. | ||
| Fig. 3 The micro-videographic assay for the rotary activity of FOF1.115,116 (a) The holo-enzyme is solubilized in detergent and the crown of F1 is attached by His-tags to a solid support. A fluorescent actin filament is attached to the rotor ring of FO (modified with permission from FEBS Lett.116). (b) When ATP hydrolysis drives the rotor ring round, the actin filament, subject to viscous drag on the filament, is curved. The elastic parameters of the filament are determined by fluctuation analysis (modified figures from ref. 115 with permission). For the original video-recordings see ref. 120. | ||
Both FO and F1 are stepping motors. When driven by ATP hydrolysis F1 rotates with a period of 120°, with substeps of 40° and 80°.94–96 This stepping reflects the presence of three equivalent reaction sites on F1 and torque production during substrate binding, catalysis and product release. On the other hand FO steps by 36°,97,98 reflecting the C10 symmetry of the particular bacterial FO. The mechanism and the structure of the rotary ion-motor, FO, is not discussed here. For the principle of operation see ref. 18, for the kinetic properties ref. 99, for the structure ref. 16 and for simulations of its reaction dynamics ref. 89,99,100. Here we focus on the cooperation of FO and F1 in the holo-enzyme.
If the proton-motive force is larger than the thermodynamic force of ATP hydrolysis the electrochemical motor of ATP synthase, FO, drives the chemical generator, and F1 synthesizes ATP. If the force-relation is reversed, motor and generator change roles. ATP hydrolysis by F1 drives FO to generate the proton-motive force. If the forces match the enzyme rests. Thermodynamic quasi-equilibrium between the two motors was used to determine the torque generated by ATP hydrolysis.101 As illustrated in Fig. 3a a long filament (∼3 μm) was attached to the rotary electromotor of the holo-enzyme. It slows the turnover rate of the enzyme by several orders of magnitude into a state of quasi-equilibrium. The torque was calculated from the elastic deformation of the filament (documented in Fig. 3b), which reflects the counteraction of the drive and the viscous drag. The mean torque, 56 pN nm, conformed with the calculated driving force of ATP-hydrolysis (70 kJ mol−1),101 which implies that the two motors do not slip against each other.
Although torque production by F1 was expected to progress in steps (under the given conditions), the torque output to the actin filament on FO was almost constant. This surprising observation has been explained by solving the Fokker–Planck equation of a stepping nanomotor when elastically coupled to a heavy load.101 Decreasing the stiffness of the elastic buffer between the stepping drive and the load flattens the torque profile at the output. It increases the turnover rate by orders of magnitude over the one with a stiff transmission (see Fig. 4a). In other words, an elastic force–torque transmission is pivotal for high kinetic efficiency of the stepping nanomotor that drives a heavy load.102 This benefit of a compliant transmission applies to all of nature's intrinsically stepping nanomotors. The distribution of elastically compliant and stiff domains over FOF1 was determined by fluctuation analysis.20,103,104 The result is illustrated in Fig. 4b. The most compliant domain (stiffness 70 pN nm) of the holo-enzyme is located on the central rotor and between the sites where the respective power strokes are generated (see ref. 6 and 20 for details and references). The rest of the enzyme, and in particular the stator, are stiffer by an order of magnitude. The elastic transmission decouples the two motors kinetically while keeping them strictly coupled, both thermodynamically and, in the time average, also under a steady turnover. It explains why this enzyme can run with different gears in different organisms. The gear ratio of FO:F1, and likewise the expected H+:ATP ratio, ranges from 8:3 in mammalian mitochondria105 to 10:3 in yeast mitochondria105 and 14:3 in chloroplasts,106 and it varies between 10:3 and 15:3 in different bacteria (see ref. 107 and references therein). Organisms (organelles) thriving at a large and constant ion-motive force, like mammalian mitochondria, run at low gear (speedsters), and those at a low and/or fluctuating force, like chloroplasts and in alkaliphilic bacteria, at high gear (tractors).6 Because the two motors are kinetically decoupled the enzyme can operate with a strictly proton specific ion motor in some organisms,99 and optionally on sodium or proton108,109 in others, depending on the ambient ion concentration. The elastically compliant transmission accounts for considerable freedom of design for both rotor and stator in the similarly bipartite relatives of FOF1, namely the archeal A-ATPase and the V-ATPase of eukaryotes.6,110 They all share with other nucleotide triphosphatases the (pseudo-)hexagonal design of the catalytic headpiece, namely with helicases and bacterial DNA-translocases which rotate and/or translocate proteins, RNA or DNA, in their central cavity and probably share a common ancestry.111
 | ||
| Fig. 4 On the elastic torque transmission between the ion-driven motor, FO, and the chemical generator, F1, of rotary ATP synthase. (a) The distribution of elastically compliant (red) and stiff domains (grey) over the enzyme. Numbers give the torsion stiffness in pN nm.6,117,118 (b) Calculated dependence of the turnover rate of a stepping and rotary nanomotor, which is coupled to a heavy load, on the torsion stiffness of the transmission to the load.115 With permission from the Annual Review of Biochemistry, Volume 84. © 2015, Annual Reviews, http://www.annualreviews.org. | ||
7 Outlook
Oxygenic photosynthesis in its present and fossil forms provides man with food, fiber and fuel. The quantum yield of photosynthesis is almost perfect. As to primary energy efficiency in relation to the solar spectrum (about 20%), oxygenic photosynthesis compares well with single-band-gap photovoltaic cells.112 For the yearly average of crops in the field however the efficiency drops down to 2%, and for bio-ethanol production to 0.2%, if not being energy negative (as were biofuels of the first generation). The total useful energy of biomass which is globally produced in one year (on land) is only about 5-times greater than man's yearly energy consumption. It is obvious that present day biomass, and even more so biofuels, cannot satisfy man's ever growing energy demand. Although bio-ethanol is produced and used on a large scale in Brazil, it is no option for countries with higher population densities and/or industrialization, namely China, the EU and India. The products of photosynthesis should thus rather be reserved for food, feed, fibre and platform chemicals. A sustainable energy supply calls for technical utilization of solar energy. Wind-power, photo-thermal and photo-voltaic devices are technically established, economically competitive, and increasingly installed worldwide. It is a major scientific challenge to develop new techniques for the production of solar fuels, artificial photosynthesis with greater efficiency, and based on low cost catalysts.113References
- http://en.wikipedia.org/wiki/Solvay_Conference accessed on the 25th of March 2015.
- J. Ingen-Housz, Experiments Upon Vegetables, P. Elmsly and H. Payne, London, 1779 Search PubMed.
- L. N. Duysens, J. Amesz and B. M. Kamp, Nature, 1961, 190, 510–511 CrossRef CAS.
- B. Kok, Biochim. Biophys. Acta, 1961, 48, 527–533 CrossRef CAS.
- H. T. Witt, A. Mueller and B. Rumberg, Nature, 1961, 192, 967–969 CrossRef CAS.
- W. Junge and N. Nelson, Annu. Rev. Biochem., 2015, 84, 631–657 Search PubMed.
- P. Mitchell, Nature, 1961, 191, 144–148 CrossRef CAS.
- T. L. Netzel, P. M. Rentzepis and J. Leigh, Science, 1973, 182, 238–241 CAS.
- J. Deisenhofer, O. Epp, K. Miki, R. Huber and H. Michel, J. Mol. Biol., 1984, 180, 385–398 CrossRef CAS.
- P. Jordan, P. Fromme, H. T. Witt, O. Klukas, W. Saenger and N. Krauss, Nature, 2001, 411, 909–917 CrossRef CAS PubMed.
- A. Amunts, O. Drory and N. Nelson, Nature, 2007, 447, 58–63 CrossRef CAS PubMed.
- N. Nelson and W. Junge, Annu. Rev. Biochem., 2015, 84, 659–683 CrossRef PubMed.
- A. Zouni, H. T. Witt, J. Kern, P. Fromme, N. Krauss, W. Saenger and P. Orth, Nature, 2001, 409, 739–743 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- J. P. Abrahams, A. G. W. Leslie, R. Lutter and J. E. Walker, Nature, 1994, 370, 621–628 CrossRef CAS PubMed.
- T. Meier, P. Polzer, K. Diederichs, W. Welte and P. Dimroth, Science, 2005, 308, 659–662 CrossRef CAS PubMed.
- J. E. Walker, Biochem. Soc. Trans., 2013, 41, 1–16 CrossRef CAS PubMed.
- W. Junge, H. Lill and S. Engelbrecht, Trends Biochem. Sci., 1997, 22, 420–423 CrossRef CAS.
- C. von Ballmoos, A. Wiedenmann and P. Dimroth, Annu. Rev. Biochem., 2009, 78, 649–672 CrossRef CAS PubMed.
- W. Junge, H. Sielaff and S. Engelbrecht, Nature, 2009, 459, 364–370 CrossRef CAS PubMed.
- P. D. Boyer, Annu. Rev. Biochem., 1997, 66, 717–749 CrossRef CAS PubMed.
- J. M. Breton, J. L. Martin, A. Migus, A. Antonetti and A. Orszag, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 957–961 CrossRef.
- W. Holzapfel, U. Finkele, W. Kaiser, D. Oesterhelt, H. Scheer, H. U. Stilz and W. Zinth, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5168–5172 CrossRef CAS.
- M. H. Vos, J. C. Lambry, S. J. Robles, D. C. Youvan, J. Breton and J. L. Martin, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 8885–8889 CrossRef CAS.
- G. R. Fleming and R. van Grondelle, Curr. Opin. Struct. Biol., 1997, 7, 738–748 CrossRef CAS.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- G. D. Scholes and C. Smyth, J. Chem. Phys., 2014, 140 Search PubMed.
- Quantum Effects in Biology, ed. M. Mohseni, Y. Omar and G. Engel and M. B. Plenio, Cambridge University Press, Cambridge, UK, 2014 Search PubMed.
- A. G. Dijkstra and Y. Tanimura, New J. Phys., 2012, 14 Search PubMed.
- A. K. Pati, S. J. Gharpure and A. K. Mishra, Faraday Discuss., 2015 10.1039/C4FD00170B.
- E. Romero, R. Augulis, V. I. Novoderezhkin, M. Ferretti, J. Thieme, D. Zigmantas and R. Van Grondelle, Nat. Phys., 2014, 10, 676–682 CrossRef CAS.
- G. H. Schatz, H. Brock and A. R. Holzwarth, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8414–8418 CrossRef CAS.
- G. H. Schatz, H. Brock and A. R. Holzwarth, Biophys. J., 1988, 54, 397–405 CrossRef CAS.
- W. Kuhlbrandt, D. N. Wang and Y. Fujiyoshi, Nature, 1994, 367, 614–621 CrossRef CAS PubMed.
- M. Z. Papiz, S. M. Prince, T. Howard, R. J. Cogdell and N. W. Isaacs, J. Mol. Biol., 2003, 326, 1523–1538 CrossRef CAS.
- R. J. Cogdell, N. W. Isaacs, A. A. Freer, J. Arrelano, T. D. Howard, M. Z. Papiz, A. M. Hawthornthwaite-Lawless and S. Prince, Prog. Biophys. Mol. Biol., 1997, 68, 1–27 CrossRef CAS.
- A. R. Holzwarth, K. Griebenow and K. Schaffner, J. Photochem. Photobiol., A, 1992, 65, 61–71 CrossRef CAS.
- J. J. Katz, J. R. Norris, L. L. Shipman, M. C. Thurnauer and M. R. Wasielewski, Annu. Rev. Biophys. Bioeng., 1978, 7, 393–434 CrossRef CAS PubMed.
- L. L. Shipman, T. M. Cotton, J. R. Norris and J. J. Katz, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1791–1794 CrossRef CAS.
- W. Junge and H. T. Witt, Zeitschrift für Naturforschung, 1968, 23b, 244–254 Search PubMed.
- W. Junge, Annu. Rev. Plant Physiol., 1977, 28, 503–536 CrossRef CAS.
- C. Wolff, H. E. Buchwald, H. Ruppel, H. Witt and H. T. Witt, Zeitschrift für Naturforschung, 1969, B 24, 1038–1042 Search PubMed.
- M. Buttner, D. L. Xie, H. Nelson, W. Pinther, G. Hauska and N. Nelson, Biochim. Biophys. Acta, Bioenerg., 1992, 1101, 154–156 CrossRef CAS.
- G. Hauska, T. Schoedl, H. Remigy and G. Tsiotis, Biochim. Biophys. Acta, Bioenerg., 2001, 1507, 260–277 CrossRef CAS.
- A. Ben-Shem, F. Frolow and N. Nelson, FEBS Lett., 2004, 564, 274–280 CrossRef CAS.
- N. Nelson and A. Ben-Shem, BioEssays, 2005, 27, 914–922 CrossRef CAS PubMed.
- M. A. Steffen, K. Q. Lao and S. G. Boxer, Science, 1994, 264, 810–816 CAS.
- A. W. Rutherford, A. Osyczka and F. Rappaport, FEBS Lett., 2012, 586, 603–616 CrossRef CAS PubMed.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS PubMed.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- G. L. Closs and J. R. Miller, Science, 1988, 240, 440–447 CAS.
- H. B. Gray and J. R. Winkler, Annu. Rev. Biochem., 1996, 65, 537–561 CrossRef CAS PubMed.
- J. N. Onuchic, D. B. Beratan, J. R. Winkler and H. B. Gray, Annu. Rev. Biophys. Biomol. Struct., 1992, 21, 349–377 CrossRef CAS PubMed.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS PubMed.
- D. Noy, C. C. Moser and P. L. Dutton, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 90–105 CrossRef CAS PubMed.
- F. J. E. van Mieghem, K. Brettel, B. Hillmann, A. Kamlowski, A. W. Rutherford and E. Schlodder, Biochemistry, 1995, 34, 4798–4813 CrossRef CAS.
- B. Hillmann, K. Brettel, F. J. E. van Mieghem, A. Kamlowski, A. W. Rutherford and E. Schlodder, Biochemistry, 1995, 34, 4814–4827 CrossRef CAS.
- M. Haumann, A. Mulkidjanian and W. Junge, Biochemistry, 1999, 38, 1258–1267 CrossRef CAS PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- P. Joliot and B. Kok, in Bioenergetics of photosynthesis, ed. R. A. Govindjee, Academic Press., New York, 1975, pp. 387–412 Search PubMed.
- A. Klauss, M. Haumann and H. Dau, J. Phys. Chem. B, 2015, 119, 2677–2689 CrossRef CAS PubMed.
- J. Messinger and G. Renger, in Primary processes of photosynthesis – part 2, ed. G. Renger, The Royal Society of Chemistry, Cambridge, 2008, vol. 1, pp. 291–349 Search PubMed.
- D. Sabbert, S. Engelbrecht and W. Junge, Proc. Natl. Acad. Sci, 1997, 94, 4401–4405 CrossRef CAS.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- W. Junge, M. Haumann, R. Ahlbrink, A. Mulkidjanian and J. Clausen, Philos. Trans. R. Soc. London, Ser. B, 2002, 357, 1407–1417 CrossRef CAS PubMed.
- J. Yano, J. Kern, K. D. Irrgang, M. J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12047–12052 CrossRef CAS PubMed.
- M. H. Grabolle, C. Müller, P. Liebisch and H. Dau, J. Biol. Chem., 2006, 281, 4580–4588 CrossRef CAS PubMed.
- J. R. Helliwell, A. Brink, S. Kaenket, V. L. Starkey and S. W. M. Turner, Faraday Discuss., 2015 10.1039/C4FD00166D.
- L. Rapatskiy, N. Cox, A. Savitsky, W. M. Ames, J. Sander, M. M. Nowaczyk, M. Rogner, A. Boussac, F. Neese, J. Messinger and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 16619–16634 CrossRef CAS PubMed.
- Stephanie Manz, Albert Casandruc, Dongfang Zhang, Yinpeng Zhong, Rolf A. Loch, Alexander Marx, Taisuke Hasegawa, Lai Chung Liu, Shima Bayesteh, Hossein Delsim-Hashemi, Matthias Hoffmann, Matthias Felber, Max Hachmann, Frank Mayet, Julian Hirscht, Sercan Keskin, Masaki Hada, Sascha W. Epp, Klaus Flöttmann and R. J. Dwayne Miller, Faraday Discuss., 2015 10.1039/C4FD00204K.
- P. E. Siegbahn, J. Photochem. Photobiol., B, 2011, 104, 94–99 CrossRef CAS PubMed.
- J. Kern, R. Alonso-Mori, R. Tran, J. Hattne, R. J. Gildea, N. Echols, C. Glockner, J. Hellmich, H. Laksmono, R. G. Sierra, B. Lassalle-Kaiser, S. Koroidov, A. Lampe, G. Han, S. Gul, D. Difiore, D. Milathianaki, A. R. Fry, A. Miahnahri, D. W. Schafer, M. Messerschmidt, M. M. Seibert, J. E. Koglin, D. Sokaras, T. C. Weng, J. Sellberg, M. J. Latimer, R. W. Grosse-Kunstleve, P. H. Zwart, W. E. White, P. Glatzel, P. D. Adams, M. J. Bogan, G. J. Williams, S. Boutet, J. Messinger, A. Zouni, N. K. Sauter, V. K. Yachandra, U. Bergmann and J. Yano, Science, 2013, 340, 491 CrossRef CAS PubMed.
- C. Kupitz, S. Basu, I. Grotjohann, R. Fromme, N. A. Zatsepin, K. N. Rendek, M. S. Hunter, R. L. Shoeman, T. A. White, D. Wang, D. James, J. H. Yang, D. E. Cobb, B. Reeder, R. G. Sierra, H. Liu, A. Barty, A. L. Aquila, D. Deponte, R. A. Kirian, S. Bari, J. J. Bergkamp, K. R. Beyerlein, M. J. Bogan, C. Caleman, T. C. Chao, C. E. Conrad, K. M. Davis, H. Fleckenstein, L. Galli, S. P. Hau-Riege, S. Kassemeyer, H. Laksmono, M. Liang, L. Lomb, S. Marchesini, A. V. Martin, M. Messerschmidt, D. Milathianaki, K. Nass, A. Ros, S. Roy-Chowdhury, K. Schmidt, M. Seibert, J. Steinbrener, F. Stellato, L. Yan, C. Yoon, T. A. Moore, A. L. Moore, Y. Pushkar, G. J. Williams, S. Boutet, R. B. Doak, U. Weierstall, M. Frank, H. N. Chapman, J. C. Spence and P. Fromme, Nature, 2014, 513, 261–265 CrossRef CAS PubMed.
- W. Schliephake, W. Junge and H. T. Witt, Zeitschrift für Naturforschung, 1968, 23, 1571–1578 CAS.
- A. T. Jagendorf and E. Uribe, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 170–177 CrossRef CAS.
- W. Junge, B. Rumberg and H. Schroeder, Eur. J. Biochem., 1970, 14, 575–581 CrossRef CAS PubMed.
- C. Kayalar, J. Rosing and P. D. Boyer, J. Biol. Chem., 1977, 252, 2486–2491 CAS.
- P. D. Boyer and W. E. Kohlbrenner, in Energy coupling in photosynthesis, eds. B. R. Selman and S. Selman-Reimer, Elsevier, Amsterdam, 1981, pp. 231–241 Search PubMed.
- P. D. Boyer, Trends Biochem. Sci., 1977, 2, 38–41 CrossRef CAS.
- M. W. Bowler, M. G. Montgomery, A. G. Leslie and J. E. Walker, J. Biol. Chem., 2007, 282, 14238–14242 CrossRef CAS PubMed.
- T. M. Duncan, V. V. Bulygin, Y. Zhou, M. L. Hutcheon and R. L. Cross, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 10964–10968 CrossRef CAS.
- D. Sabbert, S. Engelbrecht and W. Junge, Nature, 1996, 381, 623–625 CrossRef CAS PubMed.
- H. Noji, R. Yasuda, M. Yoshida and K. Kinosita, Nature, 1997, 386, 299–302 CrossRef CAS PubMed.
- H. Sielaff, H. Rennekamp, S. Engelbrecht and W. Junge, Biophys. J., 2008, 95, 4979–4987 CrossRef CAS PubMed.
- D. Okuno, R. Fujisawa, R. Iino, Y. Hirono-Hara, H. Imamura and H. Noji, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20722–20727 CrossRef CAS PubMed.
- T. Masaike, F. Koyama-Horibe, K. Oiwa, M. Yoshida and T. Nishizaka, Nat. Struct. Mol. Biol., 2008, 15, 1326–1333 CAS.
- H. Itoh, A. Takahashi, K. Adachi, H. Noji, R. Yasuda, M. Yoshida and K. Kinosita, Nature, 2004, 427, 465–468 CrossRef CAS PubMed.
- R. Watanabe and H. Noji, Sci. Rep., 2014, 4, 4962 CAS.
- S. Mukherjee and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14876–14881 CrossRef CAS PubMed.
- S. Mukherjee and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20550–20555 CrossRef CAS PubMed.
- K.-i. Okazaki and G. Hummer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16468–16473 CrossRef CAS PubMed.
- D. Stock, A. G. Leslie and J. E. Walker, Science, 1999, 286, 1700–1705 CrossRef CAS.
- K. Kinosita Jr, Adv. Exp. Med. Biol., 2012, 726, 5–16 CrossRef.
- R. Yasuda, H. Noji, M. Yoshida, K. Kinosita Jr and H. Itoh, Nature, 2001, 410, 898–904 CrossRef CAS PubMed.
- K. Shimabukuro, R. Yasuda, E. Muneyuki, K. Y. Hara, K. Kinosita Jr and M. Yoshida, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14731–14736 CrossRef CAS PubMed.
- H. Ueno, T. Suzuki, K. Kinosita Jr and M. Yoshida, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1333–1338 CrossRef CAS PubMed.
- M. G. Düser, N. Zarrabi, D. J. Cipriano, S. Ernst, G. D. Glick, S. D. Dunn and M. Börsch, EMBO J., 2009, 28, 2689–2696 CrossRef PubMed.
- R. Ishmukhametov, T. Hornung, D. Spetzler and W. D. Frasch, EMBO J., 2010, 29, 3911–3923 CrossRef CAS PubMed.
- B. A. Feniouk, M. A. Kozlova, D. A. Knorre, D. Cherepanov, A. Mulkidjanian and W. Junge, Biophys. J., 2004, 86, 4094–4109 CrossRef CAS PubMed.
- D. Pogoryelov, A. Krah, J. D. Langer, O. Yildiz, J. D. Faraldo-Gomez and T. Meier, Nat. Chem. Biol., 2010, 6, 891–899 CrossRef CAS PubMed.
- O. Pänke, D. A. Cherepanov, K. Gumbiowski, S. Engelbrecht and W. Junge, Biophys. J., 2001, 81, 1220–1233 CrossRef.
- W. Junge, O. Pänke, D. A. Cherepanov, K. Gumbiowski, M. Muller and S. Engelbrecht, FEBS Lett., 2001, 504, 152–160 CrossRef CAS.
- A. Wächter, Y. Bi, S. D. Dunn, B. D. Cain, H. Sielaff, F. Wintermann, S. Engelbrecht and W. Junge, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3924–3929 CrossRef PubMed.
- H. Sielaff, H. Rennekamp, A. Wächter, H. Xie, F. Hilbers, K. Feldbauer, S. D. Dunn, S. Engelbrecht and W. Junge, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17760–17765 CrossRef CAS PubMed.
- I. N. Watt, M. G. Montgomery, M. J. Runswick, A. G. Leslie and J. E. Walker, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16823–16827 CrossRef CAS PubMed.
- H. Seelert, A. Poetsch, N. A. Dencher, A. Engel, H. Stahlberg and D. J. Mueller, Nature, 2000, 405, 418–419 CrossRef CAS PubMed.
- D. Pogoryelov, A. L. Klyszejko, G. O. Krasnoselska, E. M. Heller, V. Leone, J. D. Langer, J. Vonck, D. J. Muller, J. D. Faraldo-Gomez and T. Meier, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1599–E1608 CrossRef CAS PubMed.
- W. Laubinger and P. Dimroth, Eur. J. Biochem., 1987, 168, 475–480 CrossRef CAS PubMed.
- A. Krah, D. Pogoryelov, J. D. Langer, P. J. Bond, T. Meier and J. D. Faraldo-Gomez, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 763–772 CrossRef CAS PubMed.
- N. Nelson, Biochim. Biophys. Acta, Bioenerg., 1992, 1100, 109–124 CrossRef CAS.
- A. Y. Mulkidjanian, M. Y. Galperin, K. S. Makarova, Y. I. Wolf and E. V. Koonin, Biol. Direct, 2008, 3, 13 CrossRef PubMed.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- T. A. Faunce, W. Lubitz, A. W. B. Rutherford, D. R. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 Search PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- O. Pänke, D. A. Cherepanov, K. Gumbiowski, S. Engelbrecht and W. Junge, Biophys. J., 2001, 81, 1220–1233 CrossRef.
- O. Pänke, K. Gumbiowski, W. Junge and S. Engelbrecht, FEBS Lett., 2000, 472, 34–38 CrossRef.
- A. Wächter, Y. Bi, S. D. Dunn, B. D. Cain, H. Sielaff, F. Wintermann, S. Engelbrecht and W. Junge, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3924–3929 CrossRef PubMed.
- H. Sielaff, H. Rennekamp, A. Wächter, H. Xie, F. Hilbers, K. Feldbauer, S. D. Dunn, S. Engelbrecht and W. Junge, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17760–17765 CrossRef CAS PubMed.
- Original video recordings of rotary motion of F1: http://www.k2.phys.waseda.ac.jp/Movies.html.
- Original video recordings of FOF1: http://www.home.uni-osnabrueck.de/wjunge/Media.html.
- Animations of the rotary activity of F1, FO, and FOF1: http://www.mrc-mbu.cam.ac.uk/research/atp-synthase/molecular-animations-atp-synthasehttp://www.home.uni-osnabrueck.de/wjunge/Media.html.
| This journal is © The Royal Society of Chemistry 2015 |