Spatial resolution in Raman spectroscopy
Volker
Deckert
*ab,
Tanja
Deckert-Gaudig
a,
Marco
Diegel
a,
Isabell
Götz
a,
Lucas
Langelüddecke
b,
Henrik
Schneidewind
a,
Gaurav
Sharma
b,
Prabha
Singh
b,
Pushkar
Singh
a,
Steffen
Trautmann
a,
Matthias
Zeisberger
a and
Zhenglong
Zhang
a
aLeibniz Institute of Photonic Technology, Albert-Einstein-Str. 9, 07745 Jena, Germany. E-mail: Volker.deckert@ipht-jena.de; Fax: +49 3641 206 139; Tel: +49 3641 206 113
bInstitute of Physical Chemistry and Abbe Center of Photonics, University of Jena, Helmholtzweg 4, 07743 Jena, Germany
First published on 31st March 2015
Abstract
This article is intended to set the scope of the meeting, in particular, the high spatial resolution section.
1 Introduction
A constant motivation to develop advanced technologies addressing temporal and spatial resolution is the investigation of structural changes of a single molecule while it is reacting. The dream is to follow a bond breakage or bond formation of a single selected molecule with a method that provides structural sensitivity, time resolution, and local specificity. Presently, such a combined technique is not yet available, however, regarding each single requirement of the mentioned list, tremendous progress has been made over the last few decades.This overview will specifically discuss the spatial resolution aspects that are required to address, if not a single molecule, at least a sample volume that is small enough to distinct the target structure from the background. The 2014 Chemistry Nobel prize awarded to Moerner, Hell, and Betzig, highlighted the importance of high resolution techniques specifically for biochemical applications. While the awarded fluorescence approach is certainly extremely useful whenever dye labelling is possible and no interference with the function of the target system is expected, in particular for smaller molecular systems such labelling is not practical or even possible. In such cases vibrational spectroscopy techniques can be used, often at the cost of lower sensitivity, however, as structural information is obtained directly no labelling is required and sample preparation becomes easier. In this article we will solely present the high lateral resolution aspects of Raman spectroscopy, many of those aspects can be easily applied to infrared absorption spectroscopy if the specific factors of the particular wavelength range are considered. As mentioned before, the sensitivity of fluorescence label techniques easily allows the investigation of single molecules. With normal Raman scattering this is not possible as the scattering cross sections are several orders of magnitude lower compared to fluorescence. Since the mid 1970s a method has existed to enhance the Raman signals considerably.1–3 By using roughened noble metal surfaces or noble metal nanoparticles an enhancement of several orders of magnitude can be achieved mainly due to plasmonic effects. This bridges the gap between fluorescence and Raman spectroscopy and allows the investigation of single molecules with so called surface-enhanced Raman scattering (SERS).4–6
At present the only possible way to overcome the diffraction limit7,8 with Raman spectroscopy requires the use of optical near-fields. Consequently, we will deal solely with the special properties of near-field Raman scattering,9–11 which luckily is in many ways directly associated with the aforementioned plasmonic effects. Accordingly a combination of near-field optics and SERS provides access to a high lateral resolution, low detection limit, and high structural sensitivity. Furthermore, many aspects regarding the investigation of small sample volumes, and thus restricted number of sampled molecules, can be regarded as general aspects whenever such small dimensions are probed.
2 Experimental
General setup
All investigations mentioned here were measured using a so-called tip-enhanced Raman scattering (TERS) setup. Fig. 1 shows the schematic sketch of our system. The main components are an atomic force microscope (AFM) and an inverted optical microscope equipped with an x, y, z piezo stage for automated sample scanning. The whole system is designed for a precise positioning of the near-field optical probe with respect to laser focus and sample. These parts are shown in dark grey. Additionally, coarse sample and tip alignment standard xy stages with ∼2 μm precision are also available, but not shown in detail. Further details about this particular setup can be found in the literature where a straightforward adaptation to a reflection setup giving access to opaque samples is also presented.12–14 The illumination through substrate and sample on to the plasmonic tip seems to be demanding and prone to losses, however, this setup allows for the best collection efficiency and implementing proper polarisation allows an optimal excitation of the nanoparticles. | ||
| Fig. 1 Schematic view of a TERS epi-illumination setup. Tip and sample can be scanned independently using precision piezo stages. Illumination and collection for the Raman system is carried out via a high N. A. objective. The detection of the Raman signal is done after appropriate filtering using a standard multichromator/CCD detector arrangement. | ||
TERS tips
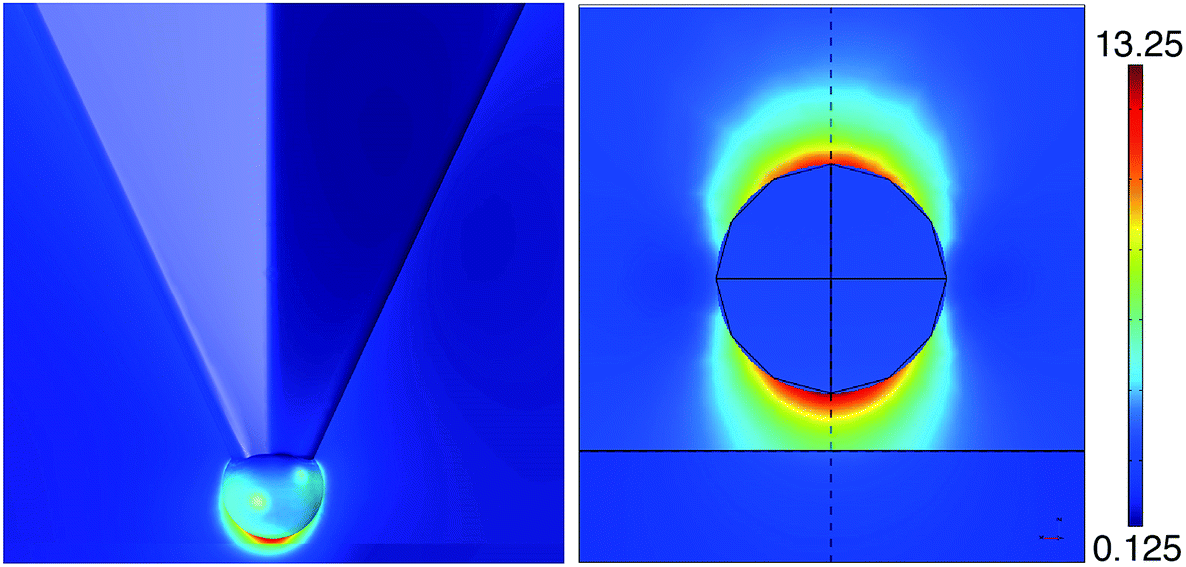 | ||
| Fig. 2 Left panel: field distribution of an ideal TERS probe with one single nanoparticle attached to the apex of an AFM tip. Right panel: detail of the field distribution E(x, z) obtained by FEM simulation for a silver sphere (similar to the dipol–mirror dipole model) of radius 10 nm and a distance from the surface of 5 nm, λ = 413 nm. Field distributions in both cases are comparable. | ||
 | ||
| Fig. 3 SEM images of the main manifestations of the silver nanoparticles on top of the TERS tips: a single nanoparticle at the outermost top (a), two nanoparticles at the top (b), or a single nanoparticle at the edge of the AFM tip (c), respectively. The scale bar applies to all images. The insets show the same tips using the material contrast. | ||
Application examples
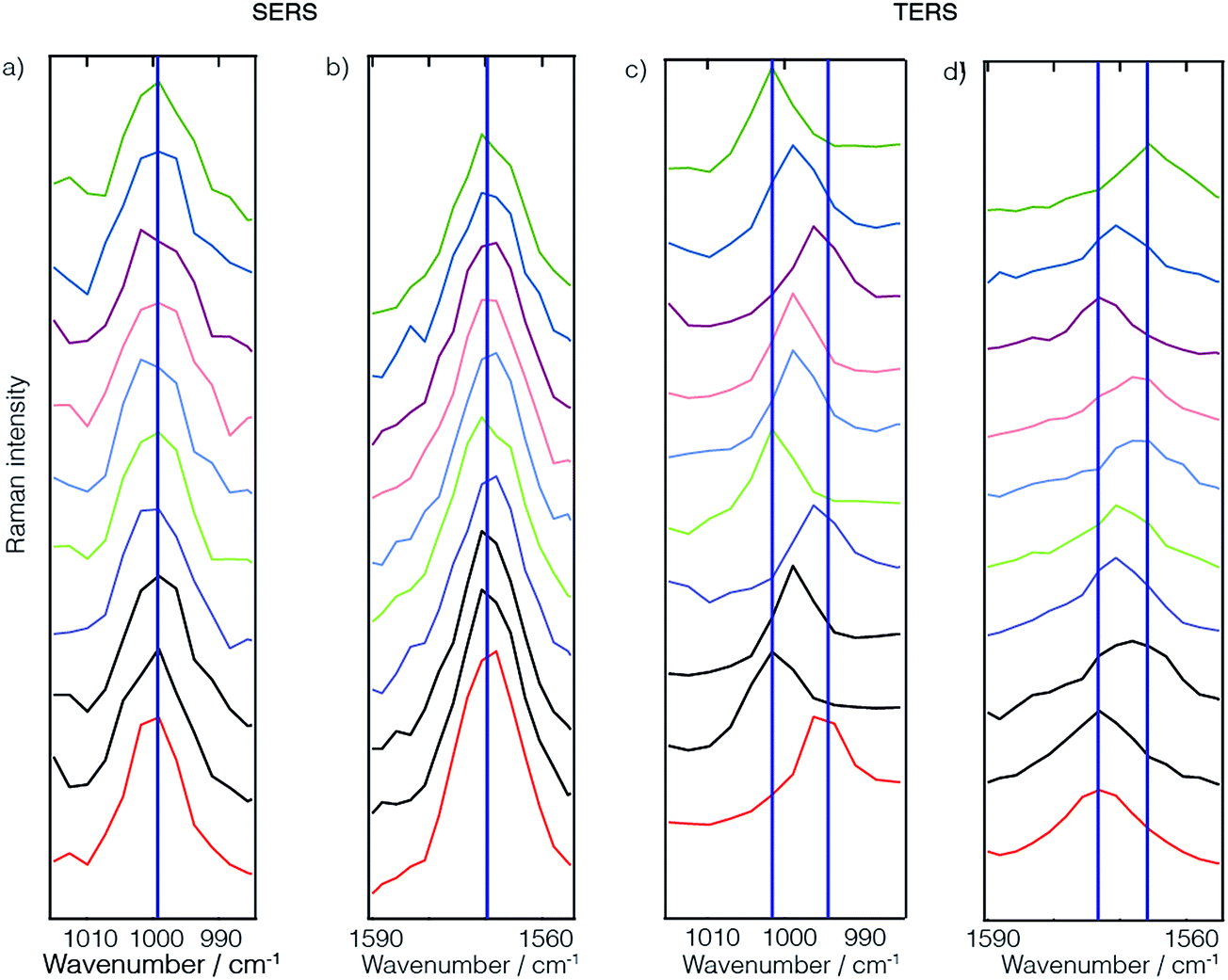 | ||
| Fig. 4 Comparison between SERS (a and b) and TERS (c and d) spectra of thiophenol monolayers. Due to the small number of molecules detected in the TERS case, averaging effects like in the case of the SERS spectra are absent or not yet pronounced. This leads to stronger fluctuations in the TERS case. | ||
Protein structures and spatial resolution
As a kind of by-product of our TERS investigations of protein structure surfaces, namely amyloid fibrils, the lateral resolution aspect emerged again.40–44 When investigating fibrils with TERS using a step size of 5 Å between subsequent positions, surprisingly pronounced spectral changes could be observed if a long enough acquisition time was chosen to allow a detailed band analysis. It should be mentioned here that Paulite et al. have presented TERS images of well-ordered amyloid nanotapes that appear homogeneous.45 Unfortunately the signal-to-noise ratio in those experiments was chosen such that apart from the phenylalanine “ring breathing” mode no other signal was detected and a prediction regarding structural changes was hardly possible. Hence, we consider the signal variations detected in our experiments to be significant and related to the actual sample composition. If one considers furthermore the findings of the previous experimental and theoretical section concerning the lateral resolution, the choice of step size to be 5 Å was set quite conservatively to avoid accidental undersampling of the dataset. When investigating the data we typically find results like those shown in Fig. 5. Here we investigated an insulin amyloid protofilament and clearly saw structural changes occurring on a length scale of a few Ångströms. For instance, in the first four spectra one can observe an amide I band (at 1604 cm−1) representing α-helix/unordered secondary structures. This is also observed, however slightly weaker, in the fifth spectrum, but then vanishes in the remaining two spectra. Similar patterns can be observed for the disappearance of the phenylalanine “ring breathing” mode and the appearance of a tyrosine marker band. The marker bands of cystine also change, however, as many different conformations around the disulfide bridge affect the exact spectral position and intensity ratios of the associated bands, a full assignment would be too complex here. Similarly, as detected in the self-assembled thiophenol monolayer case, one can observe similar spectral band position variations that are related to the immediate local environment of the sample. This can of course also involve the tip. These results can be simply understood as a way to investigate structural changes with high lateral resolution. But, in fact, this resolution is rather unexpected as we do not observe average peptide spectra, but rather locally dependent changes that reveal distinct amino acid contents.44 Looking at the data of Fig. 4 and considering previous data on fibrils41,42,44 and on DNA bases46,47 a lateral resolution of about 1 nm can be estimated which is actually at the limit of sampling in the case presented. Bearing in mind the TERS SAM experiments where a direct estimation of the resolution was not feasible, such a lateral resolution would relate to less than 10 molecules in the active region under the tip. For such a small number a “non-average” behavior becomes reasonable.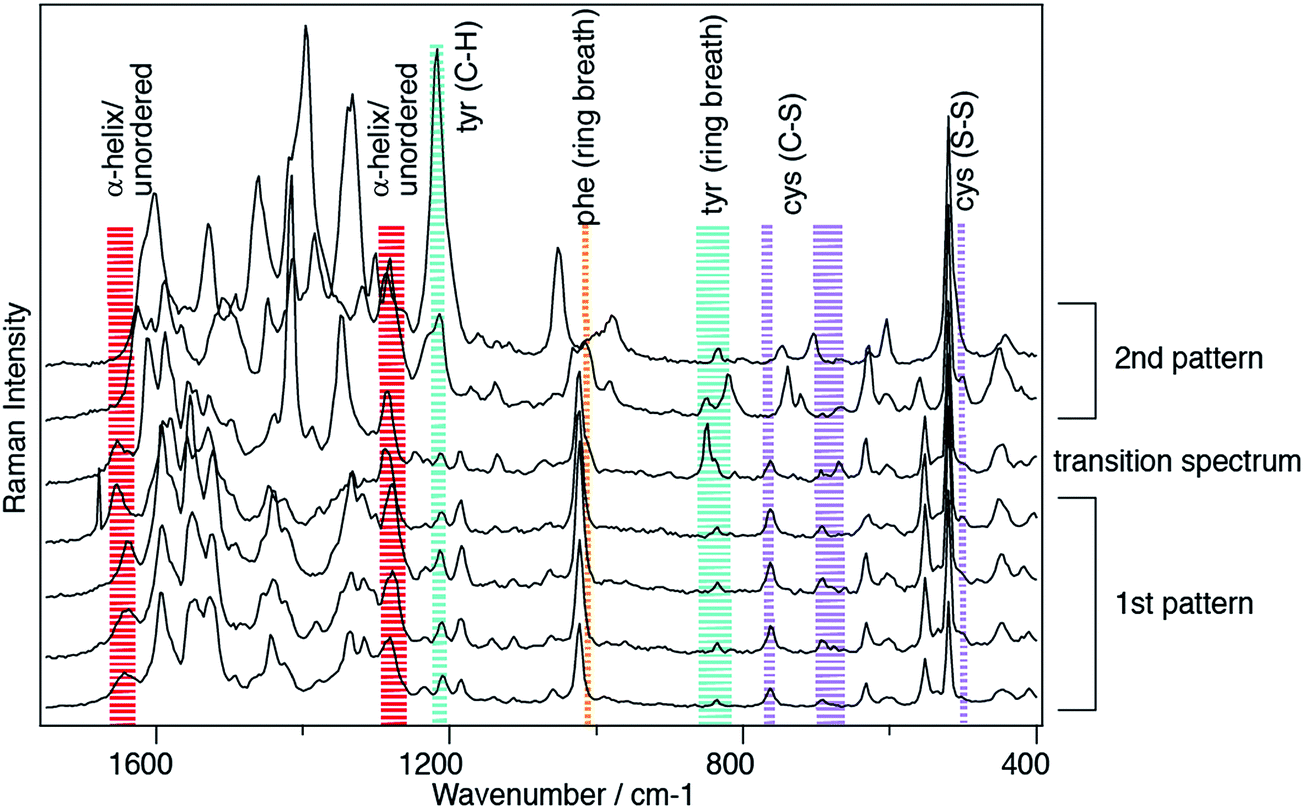 | ||
| Fig. 5 TERS spectra detected on an insulin amyloid protofilament generated at pH 1.5, laser excitation 532 nm, acquisition time 10 s/spectrum, distance between positions 0.5 nm. Two major patterns can be clearly distinguished: only in the first 5 spectra α-helix/unordered structures can be clearly identified by amide I bands at 1640 cm−1. A transition is also apparent in the synchronous appearance and disappearance of phenylalanine, tyrosine and cystine. | ||
There is an important caveat to the generalization implied here. At the moment we are not aware of a full theoretical description that explains the lateral resolution of the experiments. It seems that either the size and shape parameters of the plasmonic tips have to be modeled differently, or that direct interactions of the tip and the sample provide an additional contrast that leads to the enhanced resolution observed. The latter aspect is often termed “chemical enhancement”. While currently strong efforts are being made to unravel this issue, we think that any high resolution beyond 3–5 nm can be utilized to sequence virtually any material.
General method for sequencing a single strand
For such sequencing we will provide the outline of this method and an estimation of the precision that is required for a successful application.48 One can consider a tip as a simple point dipole, with the dipole in the z-direction, with a given field distribution that results in a TERS interaction volume as mentioned previously. This means for the following ideas we assume the theoretically predicted lateral resolution values and not even the values we experimentally determined. If, for instance a DNA single strand is aligned along the x-axis with bases at the positions xk (k = 1, 2, …, K) and z = 0, and the tip moves along x at height z, the signal at position xn for one particular Raman peak will bewhere bk equals either 1 or 0, depending on whether the monomer investigated (here a nucleobase) is present at xk (or not). P(x,z) is related to the influence of the tip and is proportional to the 4th power of the field. For N > K, the coefficients bk can be calculated using a least square fitting method that requires the inversion of the matrix (M).

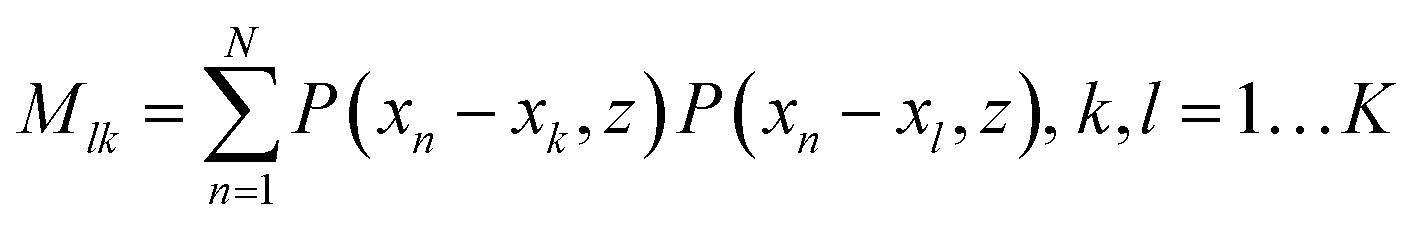
Such inverse problems are often ill conditioned, i.e. a small error in the input values (S(xn)) can result in huge errors in the reconstructed solution (bk). A useful method to evaluate this problem is the singular value decomposition of the matrix M which provides the condition number C. This number allows the estimation of the error of the reconstructed b values for a given error ΔS/S of the actually measured signal S.

Fig. 6 shows the condition number of our problem as a function of the distance between tip dipole and base size normalized with respect to the base-to-base distance. We assumed 5 samples per base, i.e. N = 5K, in other words, the polymer strand is scanned in such a way that each single base is sampled at five different subsequent TERS tip positions. The data show that a direct reconstruction of the bk values is only possible for extremely small distances between the bases in the DNA strand and the tip dipole which corresponds to the center of the Ag sphere in the experiment. If, for example, the error in the Raman signal is 1% this estimation results in a maximum distance z of two base-to-base distances corresponding to roughly 1 nm. In addition to this general estimation we performed simulations where we started with several arbitrary base arrangements and calculated the corresponding TERS signal. After adding a certain noise to the signal we recalculated the base positions. The simulations show that slightly higher distances are allowed compared with the estimation given above, but the general trend was confirmed. While this renders the sequencing a challenging problem, it is also clear that the method's quality will improve dramatically with improving lateral resolution. Taking into account the experimental resolution value estimations and further improvements in the above mentioned method (for details see ref. 48) a direct sequencing as proposed already in the first TERS paper on DNA strands49 is possible.
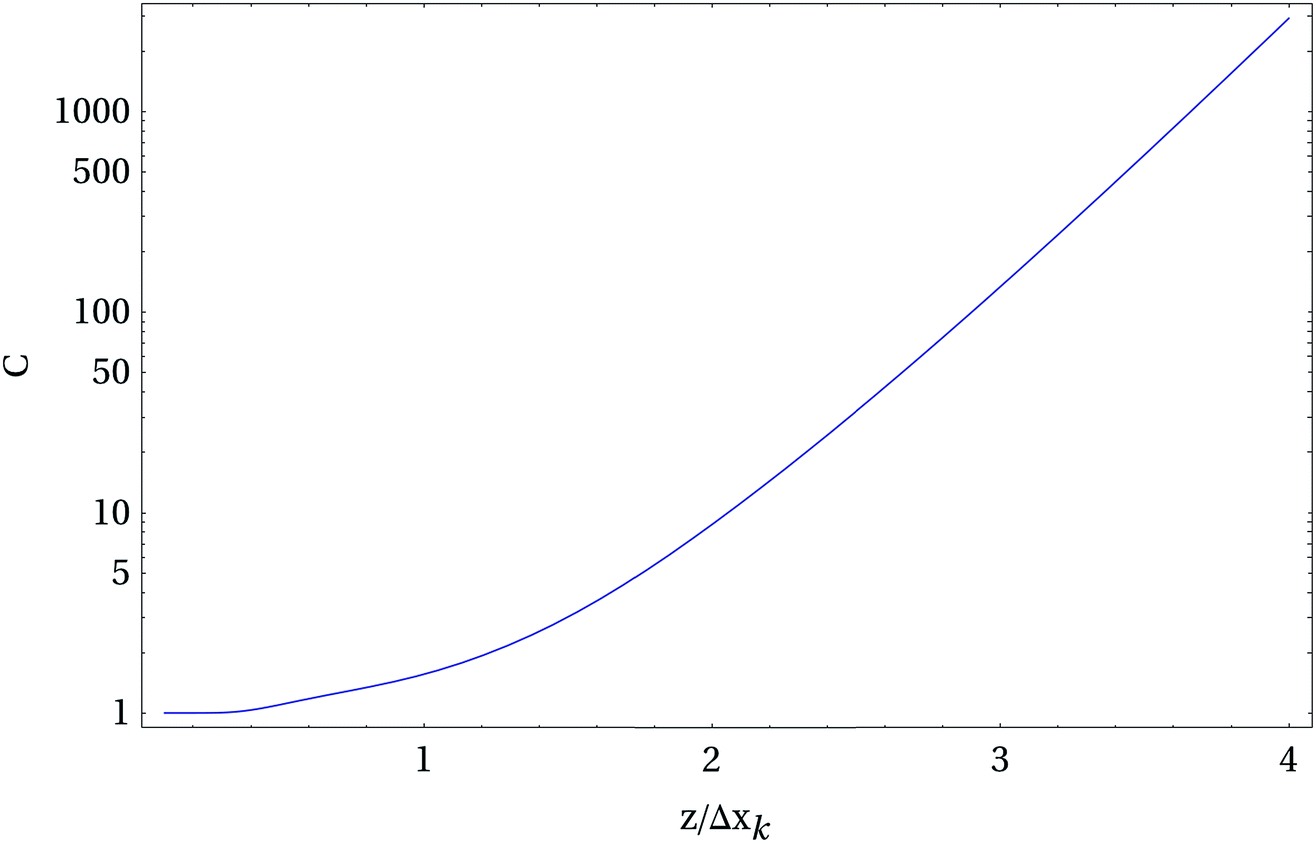 | ||
| Fig. 6 Condition number C of the inverse sequencing problem using the theoretically modeled enhancement profile of a TERS tip. The graph shows how quickly the method becomes unreliable if the distance z between tip and sample increases. The x-axis values represent normalized distances normalized to a single unit of interest Δxk (for instance a base-to-base distance in DNA). Interestingly an AFM can achieve the small distances required. | ||
3 Conclusion
We presented the high resolution aspects of Raman spectroscopy using different approaches. By studying homogeneous samples under controlled conditions, a comparison of signal position fluctuations indicates that TERS probes very small sample volumes where individual orientation effects are not averaged. Complementary studies on proteins and protein fibrils reveal a surprising capability to distinguish amino acid distributions locally on a length scale of at least 1 nm, also confirming the high spatial resolution of TERS. Interestingly, no consistent theory is at hand to explain this resolution with the usual approach using “macroscopic” tip parameters. Last but not least we presented a general method that allows a direct sequencing of single polymer stands with TERS that would already work with the current theoretically predicted resolution.The time resolution aspects of TERS have not been addressed here, but are currently being successfully investigated by the van Duyne group.50 In this way the goal to combine ultimate temporal, spatial and spectral resolution as a general tool should be reached in the not too distant future.
Acknowledgements
We gratefully acknowledge support from the Deutsche Forschungsgemeinschaft (FR 1348/19-1) the Alexander von Humboldt foundation, the Carl-Zeiss Foundation and the Thüringer Aufbaubank (FKZ: 2011 FE 9048; 2011 VF 0016) for their financial support.References
- M. Fleischmann, P. Hendra and A. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- D. Jeanmaire and R. van Duyne, J. Electroanal. Chem., 1977, 84, 1–20 CrossRef CAS.
- S. Nie and S. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- E. C. Le Ru, M. Meyer and P. G. Etchegoin, J. Phys. Chem. B, 2006, 110, 1944–1948 CrossRef CAS PubMed.
- E. Abbe, Arch. Mikrosk. Anat. Entwicklungsmech., 1873, 9, 413–418 CrossRef.
- Rayleigh, London, Edinburgh Dublin Philos. Mag. J. Sci., 1879, 8, 261–274 CrossRef.
- D. A. Smith, S. Webster, M. Ayad, S. D. Evans, D. Fogherty and D. Batchelder, Ultramicroscopy, 1995, 61, 247–252 CrossRef CAS.
- C. L. Jahncke, H. D. Hallen and M. A. Paesler, J. Raman Spectrosc., 1996, 27, 579–586 CrossRef CAS.
- D. Zeisel, B. Dutoit, V. Deckert, T. Roth and R. Zenobi, Anal. Chem., 1997, 69, 749–754 CrossRef CAS.
- A. Rasmussen and V. Deckert, J. Raman Spectrosc., 2006, 37, 311–317 CrossRef CAS.
- E. Bailo and V. Deckert, Chem. Soc. Rev., 2008, 37, 921–930 RSC.
- T. Deckert-Gaudig, M. Richter, D. Knebel, T. Jähnke, T. Jankowski, E. Stock and V. Deckert, Appl. Spectrosc., 2014, 68, 916–919 CrossRef CAS PubMed.
- L. Novotny and B. Hecht, Principles of Nano-Optics, Cambridge University Press, 2012 Search PubMed.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783–826 CrossRef CAS.
- M. Kerker, SPIE Milestone Series, 1990, 10, 696 Search PubMed.
- A. Otto, I. Mrozek, H. Grabhorn and W. Akemann, J. Phys.: Condens. Matter, 1992, 4, 1143–1212 CrossRef CAS.
- H. Kim, K. M. Kosuda, R. P. van Duyne and P. C. Stair, Chem. Soc. Rev., 2010, 39, 4820–4844 RSC.
- K. J. Savage, M. M. Hawkeye, R. Esteban, A. G. Borisov, J. Aizpurua and J. J. Baumberg, Nature, 2012, 491, 574–577 CrossRef CAS PubMed.
- R. Stockle, Y. Suh, V. Deckert and R. Zenobi, Chem. Phys. Lett., 2000, 318, 131–136 CrossRef CAS.
- R. Stockle, V. Deckert, C. Fokas and R. Zenobi, Appl. Spectrosc., 2000, 54, 1577–1583 CrossRef CAS.
- N. Hayazawa, Y. Inouye, Z. Sekkat and S. Kawata, Opt. Commun., 2000, 183, 333–336 CrossRef CAS.
- B. Ren, G. Picardi and B. Pettinger, Rev. Sci. Instrum., 2004, 75, 837–841 CrossRef CAS PubMed.
- C. Leiterer, T. Deckert-Gaudig, P. Singh, J. Wirth, V. Deckert and W. Fritzsche, Electrophoresis DOI:10.1002/elps.201400530.
- G. Xu, Z. Liu, K. Xu, Y. Zhang, H. Zhong, Y. Fan and Z. Huang, Rev. Sci. Instrum., 2012, 83, 103708 CrossRef PubMed.
- P. R. Brejna and P. R. Griffiths, Appl. Spectrosc., 2010, 64, 493–499 CrossRef CAS.
- T. Kim, K.-S. Jeon, K. Heo, H. M. Kim, J. Park, Y. D. Suh and S. Hong, Analyst, 2013, 138, 5588–5593 RSC.
- B. Pettinger, B. Ren, G. Picardi, R. Schuster and G. Ertl, Phys. Rev. Lett., 2004, 92, 096101 CrossRef.
- B. Ren, G. Picardi, B. Pettinger, R. Schuster and G. Ertl, Angew. Chem., Int. Ed., 2005, 44, 139–142 CrossRef CAS PubMed.
- R. D. Rodriguez, E. Sheremet, S. Müller, O. D. Gordan, A. Villabona, S. Schulze, M. Hietschold and D. R. T. Zahn, Rev. Sci. Instrum., 2012, 83, 123708 CrossRef CAS PubMed.
- T. Deckert-Gaudig and V. Deckert, Small, 2009, 5, 432–436 CrossRef CAS PubMed.
- T. Deckert-Gaudig and V. Deckert, J. Raman Spectrosc., 2009, 40, 1446–1451 CrossRef CAS.
- T. Deckert-Gaudig, E. Bailo and V. Deckert, Phys. Chem. Chem. Phys., 2009, 11, 7360–7362 RSC.
- J.-S. Huang, V. Callegari, P. Geisler, C. Brüning, J. Kern, J. C. Prangsma, X. Wu, T. Feichtner, J. Ziegler, P. Weinmann, M. Kamp, A. Forchel, P. Biagioni, U. Sennhauser and B. Hecht, Nat. Commun., 2010, 1, 150 CrossRef PubMed.
- T. Deckert-Gaudig, E. Rauls and V. Deckert, J. Phys. Chem. C, 2010, 114, 7412–7420 CAS.
- T. Deckert-Gaudig, F. Erver and V. Deckert, Langmuir, 2009, 25, 6032–6034 CrossRef CAS PubMed.
- P. Singh, T. Deckert-Gaudig, H. Schneidewind, K. Kirsch, E. M. van Schrojenstein Lantman, B. M. Weckhuysen and V. Deckert, Phys. Chem. Chem. Phys., 2015, 17, 2991–2995 RSC.
- E. M. van Schrojenstein Lantman, T. Deckert-Gaudig, A. J. G. Mank, V. Deckert and B. M. Weckhuysen, Nat. Nanotechnol., 2012, 7, 583–586 CrossRef CAS PubMed.
- A. V. Krasnoslobodtsev, A. M. Portillo, T. Deckert-Gaudig, V. Deckert and Y. L. Lyubchenko, Prion, 2010, 4, 265–274 CrossRef CAS PubMed.
- D. Kurouski, T. Deckert-Gaudig, V. Deckert and I. K. Lednev, J. Am. Chem. Soc., 2012, 134, 13323–13329 CrossRef CAS PubMed.
- M. Schleeger, C. C. vandenAkker, T. Deckert-Gaudig, V. Deckert, K. P. Velikov, G. Koenderink and M. Bonn, Polymer, 2013, 54, 2473–2488 CrossRef CAS PubMed.
- D. Kurouski, T. Deckert-Gaudig and V. Deckert, et al. , Biophys. J., 2014, 106, 263–271 CrossRef CAS PubMed.
- T. Deckert-Gaudig, E. Kämmer and V. Deckert, J. Biophotonics, 2012, 5, 215–219 CrossRef CAS PubMed.
- M. Paulite, C. Blum, T. Schmid, L. Opilik, K. Eyer, G. C. Walker and R. Zenobi, ACS Nano, 2013, 7, 911–920 CrossRef CAS PubMed.
- R. Treffer, X. Lin, E. Bailo, T. Deckert-Gaudig and V. Deckert, Beilstein J. Nanotechnol., 2011, 2, 628–637 CrossRef CAS PubMed.
- R. Treffer, R. Böhme, T. Deckert-Gaudig, K. Lau, S. Tiede, X. Lin and V. Deckert, Biochem. Soc. Trans., 2012, 40, 609–614 CrossRef CAS PubMed.
- V. Deckert and M. Zeisberger, register. dpma. de., 2013, DE102012024203A1.
- E. Bailo and V. Deckert, Angew. Chem., Int. Ed. Engl., 2008, 47, 1658–1661 CrossRef CAS PubMed.
- J. M. Klingsporn, M. D. Sonntag, T. Seideman and R. P. van Duyne, J. Phys. Chem. Lett., 2014, 5, 106–110 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |