 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular photostabilization via triplet-state quenching: design principles to make organic fluorophores “self-healing”†
Jasper H. M.
van der Velde
a,
Jaakko J.
Uusitalo
b,
Lourens-Jan
Ugen
a,
Eliza M.
Warszawik
c,
Andreas
Herrmann
c,
Siewert J.
Marrink
b and
Thorben
Cordes
*a
aMolecular Microscopy Research Group & Single-molecule Biophysics, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: t.m.cordes@rug.nl
bGroningen Biomolecular Sciences and Biotechnology Institute, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands
cDepartment of Polymer Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
First published on 9th October 2015
Abstract
Covalent linkage of fluorophores and photostabilizers was recently revived as a strategy to make organic fluorophores “self-healing” via triplet-state quenching. Although Lüttke and co-workers pioneered this strategy already in the 1980s, the general design principles still remain elusive. In this contribution, we combine experiments and theory to understand what determines the photostabilization efficiency in dye–photostabilizer conjugates. Our results from single-molecule microscopy and molecular dynamics simulations of different Cy5-derivatives suggest that the distance and relative geometry between the fluorophore and photostabilizer are more important than the chemical nature of the photostabilizer, e.g. its redox potential, which is known to influence electron-transfer rates. We hypothesize that the efficiency of photostabilization scales directly with the contact rate of the fluorophore and photostabilizer. This study represents an important step in the understanding of the molecular mechanism of intramolecular photostabilization and can pave the way for further development of stable emitters for various applications.
Introduction
Fluorescence has become an essential tool for spectroscopic studies, biomedical sensing, and for imaging of (dynamic) processes and structures across various scientific disciplines. The photophysical performance of the employed fluorescent reporters – signal stability, intensity and duration – is still one of the limiting factors for many applications.1 Recently, intramolecular photostabilization2–4 was revived5–9 as a tool to impart organic fluorophores with “self-healing” properties.7 The covalent linkage of photostabilizers, e.g. triplet-state quenchers or redox-active molecules, to organic fluorophores allows suppression of irreversible destruction and transient excursions to dark-states (triplet- or radical-states). While bleaching pathways are not always triplet-based,10,11 they in fact represent one of the most common reasons for photobleaching.The underlying mechanism of intramolecular photostabilization via triplet-state quenching, along with highly desirable design principles to make any organic fluorophore “self-healing”, are still under active debate.5,7–9,12 Furthermore, all self-healing fluorophores described so far show less efficient photostabilization5,8,9 compared to the use of the same photostabilizers as a solution-additive13–17 – an approach that is experimentally much simpler compared to the synthesis of photostabilizer–dye conjugates needed for self-healing.18,19 It should be emphasized, however, that intramolecular photostabilization overcomes various limitations of solution-based healing approaches since it uses only high local concentrations of the photostabilizer. This reduces the general toxicity, reduces alterations of the biological structure, and makes photostabilization even possible in vivo or in situations where diffusion-based collisions between photostabilizer and fluorophore are prohibited. Considering these advantages, it can be suggested that intramolecular photostabilization is a superior method – once as efficient as solution-based approaches. The rational design of photostabilizer–dye conjugates, combinations with solution-based approaches, and mechanistic understanding should hence be primary goals of research in this area.
Collisions between the fluorophore and photostabilizer are essential for triplet-state quenching, independent of the mechanism (inter- or intramolecular). In dye–photostabilizer conjugates, the actual distance dependence seems, however, ambiguous. The performance of triplet-state quenchers, such as cyclooctatetraene (COT), scales directly with proximity,5,6 however, redox-active compounds, such as nitrophenyl-groups, show a more complicated behaviour.9,19 Here, cyanine-based photostabilizer–dye conjugates comprising nitrophenyl-groups via constrained linkers show a relatively moderate performance with a ∼10-fold increase in photostability when using an unnatural amino acid with an aromatic nitro group.19 A different conjugation between the same photostabilizer and fluorophore, having a larger spacing and increased structural flexibility, shows a significantly higher photostability with a ∼100-fold improvement compared to the non-stabilized fluorophore9 – a value that is close to the results of pristine photostabilizers in solution (∼200-fold improvement in the photostability).9
To mechanistically understand the differences between triplet-state quenchers and oxidants in intramolecular photostabilization and ultimately to establish design principles for making organic fluorophores “self-healing”, a detailed photophysical characterization of cyanine-based photostabilizer–dye conjugates is presented in this paper. Single molecule fluorescence microscopy in combination with molecular dynamics simulations allows us to identify critical parameters that determine the efficiency of intramolecular photostabilization. Therefore, we explored the consequences of structural modifications in oxidizing photostabilizers, i.e. different aromatic nitro moieties used for intramolecular photostabilization of cyanine fluorophores, such as Cy5. We further used molecular dynamics simulations to probe the distance and orientation dependence between the fluorophore and photostabilizer. The obtained results suggest that the distance and possible orientations between the fluorophore and photostabilizer are the crucial parameters for efficient intramolecular healing via triplet-state quenching rather than the chemical structure of the photostabilizer. We also show that the orientation of the dye and photostabilizer is largely governed by their local chemical environment (in our case DNA), which allows for end-stacking or binding to grooves. This study represents an important step in the understanding of the mechanism and working principles of intramolecular photostabilization and hence paves the way to obtain improved fluorophores for spectroscopic studies and imaging when triplet-based damage is the major route for photobleaching.
Results and discussion
After the pioneering work of Lüttke and co-workers in the 1980s,2–4 the Blanchard lab started to use both triplet-state quenchers, e.g. COT, as well as (anti)oxidants, such as trolox (TX) and nitrobenzoic alcohol (NBA), for intramolecular photostabilization via triplet-state quenching.5,6,12,18 In our studies, we focused on the mechanistic understanding of intramolecular photostabilization via (anti)oxidants7–9 and more recently on its practical implementation. For that purpose, a simple and versatile scaffolding strategy for photostabilizer–dye conjugates based on unnatural amino acids was developed.19 The resulting conjugates can be readily attached to various biomolecular targets (Fig. 1a, left panel, amino acid scaffolding). More details on the chemical synthesis and applicability of intramolecular photostabilization for various fluorophore classes (rhodamines, carbopyronines and cyanines) via amino acid scaffolding are given in ref. 19.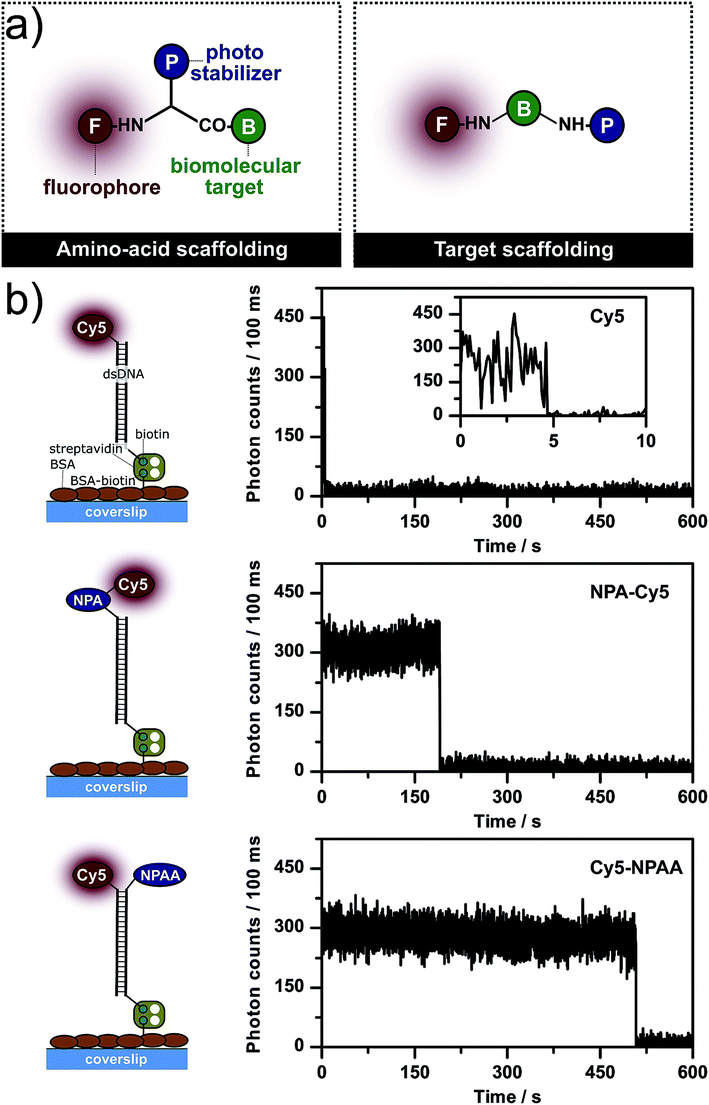 | ||
| Fig. 1 (a) Chemical strategies to scaffold photostabilizer–dye conjugates using the different residues of (unnatural) amino acids or strategic labelling of the biomolecular target to create permanent proximity. (b) Experimental strategy to immobilize dsDNA on microscope coverslips to study the photophysics of different Cy5 derivatives: parent fluorophore Cy5, photostabilizer and fluorophore linked via amino acid scaffolding (NPA–Cy5), and proximal attachment of the photostabilizer and fluorophore (Cy5–NPAA). The figure also shows representative fluorescence time traces of individual molecules from TIRF microscopy of the respective compounds. Additional data for each compound in panel (b) are shown in Fig. S1–S3.† | ||
In demanding fluorescence applications, such as single-molecule microscopy, intramolecular photostabilization via triplet-state quenching turned out to be less efficient than the use of solution-based stabilizers, i.e. the addition of similar compounds to the imaging buffer at high (∼mM) concentrations.7–9 Furthermore, the effect of intramolecular photostabilization for both triplet-state quenchers (COT) as well as (anti)oxidants (TX, NBA) seemed to correlate with their spatial separation. This was shown by using target scaffolding (Fig. 1a, right panel) where the photostabilizer and fluorophore are bound to different complimentary DNA-strands with varying attachment points for the emitter and stabilizer. After hybridization, it was found that a decreasing distance resulted in an increased photostability.6,12,18 This motivated us to develop more efficient photostabilizers for intramolecular triplet-state quenching to better compete with solution-based approaches.9 Notably, these experiments showed that a molecule containing an aromatic nitro group can show surprisingly different photostabilization efficiencies for the same fluorophore depending on the attachment geometry: a proximal attachment of the photostabilizer resulted in extremely stable fluorescence (Fig. 1b, bottom, Cy5–NPAA) while direct conjugation of the photostabilizer and fluorophore showed less photostabilization (Fig. 1b, middle, NPA–Cy5); the qualitative increases in the photostability can be seen by a comparison of the trace length of individual fluorophores (Fig. 1b).
To understand these effects, the photostability, fluorescence count rate, total number of detected photons and the signal-to-noise ratio (SNR) were determined for different surface-immobilized Cy5-derivatives that were linked to double-stranded DNA (dsDNA, Fig. 1 and 2) using camera-based imaging. Surface-immobilization was done according to published procedures on a BSA–biotin–streptavidin-coated microscope coverslip binding to a 3′-terminal biotin unit of the same dsDNA (Fig. 1b). Experiments were performed in the absence of oxygen and without photostabilizers in solution to minimize the convolution of inter- and intramolecular photostabilization. We performed single-molecule TIRF microscopy with excitation at 640 nm (intensities of ∼50–100 W cm−2) with a 100 ms integration time. Typical camera frames of single Cy5 fluorophores are shown in the ESI, e.g. Fig. S1–S3.† To quantify the photostability, the number of fluorescent molecules per video frame was determined and the decay in absolute numbers over subsequent image frames was fitted with an exponential decay to obtain the photobleaching lifetime, τbleach (Fig. 2a). From the videos, we extracted background-corrected single-molecule time traces to determine the fluorescence count rate in kHz, the signal-to-noise ratio, and the total number of detected photons before photobleaching (Fig. 2b, Ntotal = brightness × τbleach). The mean and standard deviation of all values was derived from multiple (n ≥ 3) independent experiments.
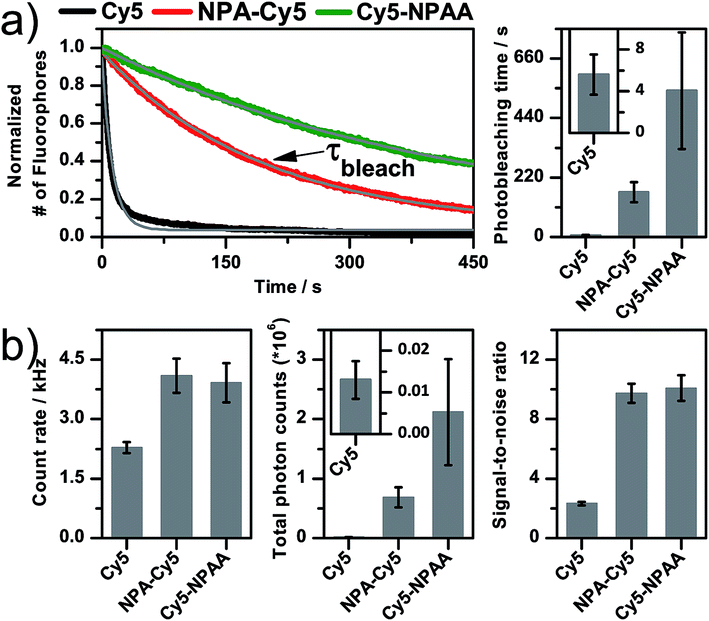 | ||
| Fig. 2 Quantitative photophysical characterization of photostabilizer–dye conjugates of Cy5 derived from single-molecule TIRF microscopy. Data was recorded in aqueous PBS buffer at pH 7.4 in the absence of oxygen under continuous 640 nm excitation with ∼50 W cm−2. (a) Decay curve of the number of fluorophores as a function of time. Fitting an exponential function (grey) to this decay resulted in the photobleaching lifetime. The curves shown were obtained by averaging over >5 TIRF movies. (b) Background-corrected intensity traces were used to derive the total number of photons (Ntotal), the average signal in kHz (brightness) and the signal-to-noise ratio (SNR). For further details of the experimental techniques, data acquisition and analysis see the Experimental section. | ||
The analysis of Cy5 and two of its derivatives, comprising either a photostabilizer covalently attached or in close proximity, is summarized in Fig. 2. It is apparent that Cy5 shows a low photostability (τbleach of 5.6 ± 1.9 s), count rate (2.3 ± 0.1 kHz), and SNR (2.3 ± 0.1), and hence has little potential for applicability without a photostabilizer (see also traces in Fig. 1b and S1†).
Adding the nitrophenyl-moiety via amino acid or target scaffolding improves all values drastically up to a τbleach value of ∼550 s, count rates of ∼4 kHz and SNR of ∼10, allowing us to collect more than 2 × 106 photons from individual molecules (Fig. 2) with little signal fluctuations (see fluorescence time traces in Fig. S2 and S3†). The experiments revealed, however, a substantial difference in photostability, i.e. τbleach and Nphot for NPA–Cy5 and Cy5–NPAA (Fig. 1 and 2).
This comparison alone does not, however, allow a satisfying conclusion about the (molecular) reasons for the observed difference. Unfortunately, in NPA–Cy5 and Cy5–NPAA, multiple aspects of the dye–photostabilizer conjugate are altered at the same time: (i) the chemical structure of the aromatic nitro-group is different for both compounds, potentially changing its redox-properties20via the electron-withdrawing or -donating characteristic of the different substituents21 and hence altering the rates for photo-induced electron transfer;22 (ii) the fluorophore and the photostabilizer are linked differently, which changes both their spatial separation and biochemical environment. It is known from other studies of cyanine dyes on DNA23–25 that the local environment has a drastic influence on the photophysical properties, such as the lifetime, spectrum, quantum yield and others, mainly via end-stacking or groove-binding with the DNA.26,27 Our approach in this paper was to de-convolute both effects via a combination of experiments on different dye–photostabilizer conjugates and to use molecular dynamics simulations to understand the possible modes of interactions in NPA–Cy5 and Cy5–NPAA.
First, we systematically varied the chemical structure of the aromatic nitro compound while keeping it at a fixed distance to the Cy5-fluorophore. Fig. 3a shows the chemical structures and photophysical properties of three different dye–photostabilizer conjugates that use nitrophenyl-acetic acid (NPAA), nitrophenyl-propanoic acid (NPP) or nitrophenylalanine (NPA) as the photostabilizer. As seen in Fig. 3a, the chemical structure of the conjugates shows variations in the linking element between the nitrophenyl and amide group. In NPAA and in NPP, a methylene group and an ethyl spacer are present, respectively, whereas NPA exhibits an additional amino-group that could have effects on the photophysical properties of fluorophores.4
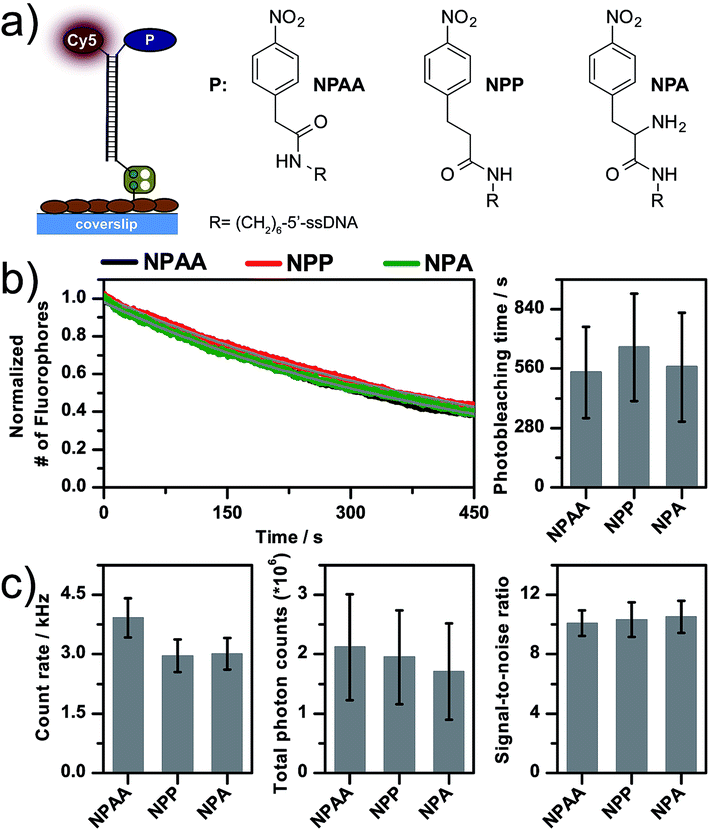 | ||
| Fig. 3 (a) Scheme of the linking and immobilization strategy of different photostabilizer–dye conjugates of Cy5 including the chemical structure of the aromatic nitro moieties. (b and c) Quantitative photophysical characterization of the photostabilizer–dye conjugates. Data was recorded in aqueous PBS buffer at pH 7.4 in the absence of oxygen under continuous 640 nm excitation with ∼50 W cm−2. (b) Decay curve of the number of fluorophores as a function of time. Fitting an exponential function (grey) to this decay resulted in the photobleaching lifetime. The curves shown were obtained by averaging over >5 TIRF movies. (c) Background-corrected intensity traces were used to derive the total number of photons (Ntotal), the average signal in kHz (brightness) and the signal-to-noise ratio (SNR). For further details of the experimental techniques, data acquisition and analysis see the Experimental section. | ||
The photophysical properties of all three compounds appeared to be strikingly similar (Fig. 3). Besides variations in the fluorescence count rate from 3–5 kHz (Fig. 3c), all other parameters were found in a very similar range given the experimental errors: the photobleaching lifetimes were in the range of ∼550 s, the total detected photon numbers were around ∼2 × 106 and the SNR was ∼10. The difference in the count rates indicates that indeed the redox-potential of the compounds might be different when comparing NPAA with NPP/NPA. The lower count-rate of the latter might indicate different degrees of singlet-quenching and hence a reduced quantum yield.28 Interestingly, the photostabilizing effects are identical for all compounds. Since the photostability is mainly improved via depletion of the reactive triplet-state, we propose that the rate of triplet-quenching is governed by interactions of the photostabilizer and fluorophore with the local chemical environment, which dictates the collision rate between the photostabilizer and fluorophore. Our results indicate that the overall performance of intramolecular photostabilization with aromatic nitro groups does not strongly depend on the actual chemical structure but instead on the interactions between the photostabilizer and fluorophore.
To understand the molecular details of these processes, we performed molecular dynamics simulations to (i) map the specific local environment of the fluorophore and photostabilizer, (ii) determine the possible interactions/binding modes of both including (iii) their collision rate. These three factors could account for the differences in the fluorophore brightness, lifetime (i/ii) and photostability (iii).
In Fig. 4, we show the simulation results for proximally attached NPAA (Cy5–NPAA, Fig. 4a), a directly conjugated fluorophore (NPA–Cy5, Fig. 4b), and a derivative of NPA–Cy5 that features a longer PEG-linker of the form (O–CH2–CH2)4 between the fluorophore and photostabilizer (Fig. 4c). The latter should mimic the superior interaction possibilities of Cy5–NPAA but at the same time use the practically more relevant attachment geometry of NPA–Cy5.19 For each system, the panels from left to right show the type of attachment as a cartoon (left), the most frequently occupied volumes of Cy5 and the photostabilizer around dsDNA, the distances between the different moieties measured during the simulation, and the volume-maps of the photostabilizer around Cy5 (right).
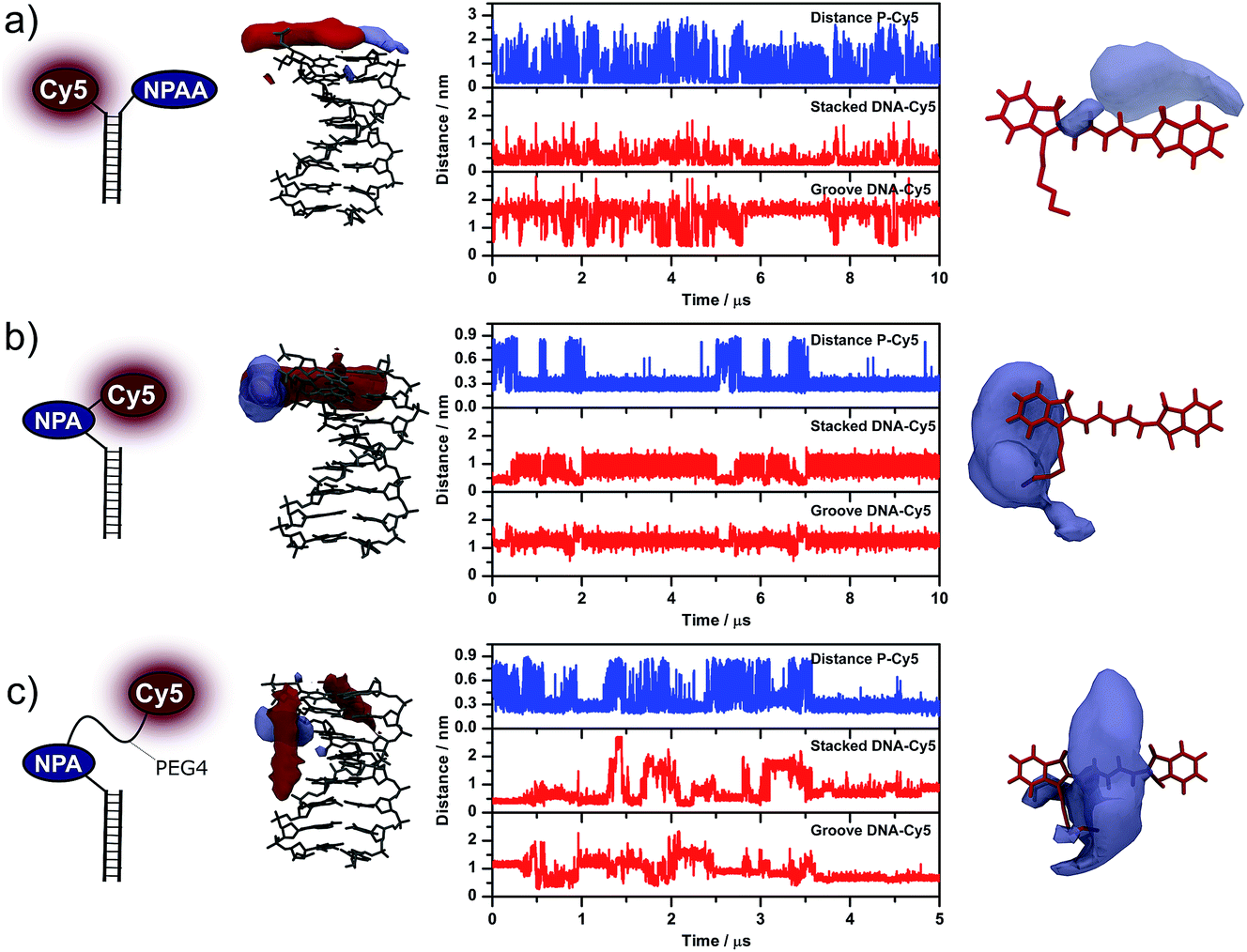 | ||
| Fig. 4 Relative orientation of Cy5, the photostabilizer and DNA during the simulations of the photostabilizer–dye conjugates: (a) Cy5–NPAA, (b) NPA–Cy5 and (c) NPA–PEG4–Cy5. The first column on the left shows the attachment of the molecules to DNA. The most common locations of Cy5 (shown in red) and the photostabilizer (shown in blue) relative to DNA are shown in the second column. The distances between Cy5 and the photostabilizer as well as Cy5 and the top and groove of DNA are presented in the third column. The fourth column shows the most common location of the photostabilizer relative to the fluorophore Cy5. | ||
The second column shows the most frequently occupied volumes of both Cy5 (shown in red) and the photostabilizer (shown in blue). In both cases, the volumes enclose all points relative to the DNA that are occupied in at least 2% of the simulation frames. In the case of Cy5–NPAA, the most likely location for both Cy5 and NPAA is on top of the DNA. The same is true for Cy5 in the absence of the photostabilizer, where pronounced end-stacking can be observed (Fig. S6†). For NPA–Cy5, both NPA and Cy5 are neither in a conformation that is directly on top of DNA nor in a DNA groove. The conformation shown in Fig. 4b is, however, fairly stable. When extending the linker between NPA and Cy5, both Cy5 and NPA are most likely located in a DNA groove. Similar conclusions can be drawn when looking at the distances between Cy5 and the photostabilizer (P–Cy5), as well as the distance of Cy5 from the top of the DNA (stacked DNA–Cy5) and the DNA groove (groove DNA–Cy5). A detailed description of how each distance was measured from the simulations is given in the Experimental section.
In the fourth column, the blue volume map encloses all the points relative to Cy5 that are occupied by the photostabilizer in at least 0.6% of the simulation frames. It is evident from the volume maps that in Cy5–NPAA and NPA–Cy5 the photostabilizer can interact with different parts of Cy5. Whereas in Cy5–NPAA interactions with both the aromatic part and the polymethine chain of Cy5 are observed, in NPA–Cy5 the interactions are limited to a small aromatic region at the linker attachment point of Cy5. Increasing the linker length (NPA–PEG4–Cy5) allows the NPA to interact with a larger surface region of Cy5.
While this data reveals clear differences for the interaction of the photostabilizer and fluorophore as well as their location within the chemical environment, the meaning of these results for photostabilization efficiency remains unclear. Our current model for intramolecular photostabilization via photo-induced electron transfer assumes a ping-pong mechanism7,8 with two subsequent electron transfer steps (Fig. 5). In the case of NPA-based conjugates, fluorophore oxidation results in the formation of two radical species, i.e. a fluorophore radical cation and a photostabilizer radical anion. Since both species cannot be separated by diffusive processes, both ultimately recombine in a second redox-reaction. Such a double-electron transfer (“geminate recombination”) is also postulated as a mechanism for diffusion-based photostabilization of organic fluorophores via thiol-compounds.29
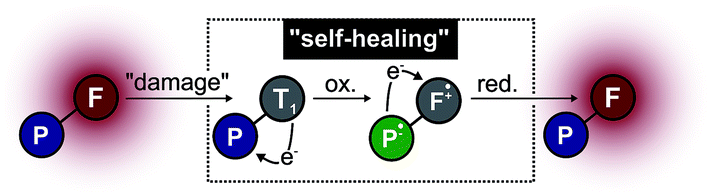 | ||
| Fig. 5 Proposed model for the depletion of the triplet-state via intramolecular photostabilization with aromatic nitro groups: (photo)oxidation of the fluorophore and subsequent recombination of two radicals forms both the fluorophore and photostabilizer in their neutral chemical forms. The redox-cascade is referred to as “self-healing”. | ||
Since it is not straightforward to predict whether the exact contact-mode of the fluorophore and photostabilizer (Fig. 4) influences the rate for electron transfer, we determined the “kinetics” of the collisions between Cy5 and the photostabilizer (Fig. 6, collision frequency). Cy5 and the photostabilizer were classified as having collided and being in contact when the distance between their centres of mass was smaller than 0.5 nm. Besides the actual collision frequency, and the mean and median times for the duration of being associated (Fig. 6, mean collision time), we also calculated the ratio of time they spent in contact (bound) versus unbound during the simulations (Fig. 6, bound/unbound ratio).
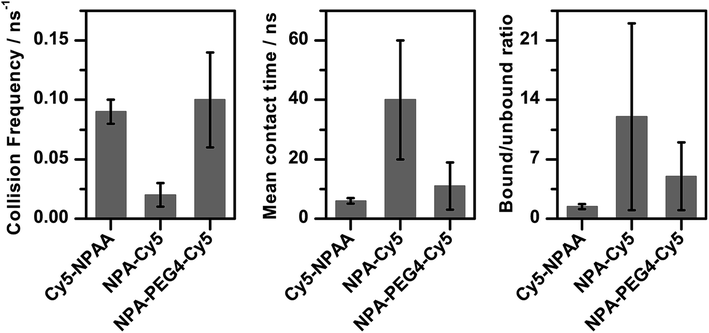 | ||
| Fig. 6 Interactions of Cy5 and the photostabilizer during the simulations for Cy5–NPAA, NPA–Cy5 and NPA–PEG4–Cy5: collision frequencies (left), the mean contact times for each system (middle) and the bound/unbound ratio (right). All three metrics show that NPA–Cy5 interacts in a different manner compared to the other two derivatives. | ||
For Cy5–NPAA, our data shows a high contact frequency with short association times (Fig. 6 and Table S1†). Although Cy5 and NPAA were frequently in contact, NPAA also explored other regions frequently while Cy5 was mostly located on top of the DNA and was in the groove only in 11% of the simulation frames. The collision frequency is 0.09 ± 0.01 ns−1, the mean and median contact times are 6 ± 1 ns and 2.3 ± 0.2 ns, respectively, and the bound/unbound ratio is 1.4 ± 0.3.
Inverse behaviour was found for NPA–Cy5, which shows a significantly lower collision frequency and a long association time (Fig. 6). Both the photostabilizer and fluorophore remain in contact in 87% of the simulation frames and the duration of the contacts is often long, while the position of Cy5 is somewhere between being on top of the DNA and in the groove of the DNA. In this case, the collision frequency is 0.02 ± 0.01 ns−1 and the bound/unbound ratio is 12 ± 11. The mean and median contact times are 40 ± 20 ns and 2 ± 1 ns, respectively. Due to the long contact times, the simulation results shown here are not long enough to accurately determine the exact bound/unbound ratio or the mean or median contact times, although these values are clearly larger than in the case of proximally attached NPAA. This is also reflected in the error estimates.
In the case of NPA–PEG4–Cy5, the increased mobility of NPA with respect to DNA is exhibited in values similar to those for Cy5–NPAA (Fig. 6). Furthermore, the longer linker increases the range of movement of Cy5 and it is frequently located in the grooves of the DNA. The collision frequency between NPA and Cy5 is 0.10 ± 0.04 ns−1 and the bound/unbound ratio is 5 ± 4. The mean and median contact times are 11 ± 8 ns and 0.8 ± 0.1 ns, respectively. Again, the simulation time is not long enough to give accurate estimates of the mean and median contact times.
These results indicate that a high collision frequency in combination with short association times of the photostabilizer and fluorophore results in the highest efficiency for photostabilization. This strongly suggests that the binding modes for the different compounds shown in Fig. 4 are not necessarily relevant for triplet-state quenching via photo-induced electron transfer since their duration does not scale with photostability. The contact rate, however, seems to follow this trend and only future studies with compounds like NPA–PEG4–Cy5 can show whether our hypothesis holds.
Conclusions
This article establishes a mechanistic basis for intramolecular triplet-state quenching and the resulting photostabilization using an oxidizing aromatic nitro group. We investigated the relationship between the chemical structure of the photostabilizer as well as possible interactions with its chemical surrounding and the fluorophore to be protected. Using single-molecule microscopy, we found that the chemical structure and its associated changes in functional properties (redox-potential) play only a minor role in the ability to stabilize organic fluorophores via intramolecular triplet-state quenching. Molecular dynamics simulations suggest, however, that interactions of the fluorophore and the photostabilizer with each other and the chemical surroundings govern efficient photostabilization. We suggest that a high collision rate is needed in combination with short interaction times of the photostabilizer and fluorophore. The data indicates that a longer interaction period does not contribute towards an increased photostabilization. Our study represents an important step in understanding the molecular mechanism of intramolecular photostabilization, but also calls for further studies with e.g. systematic variation of the linker length and the photostabilizer.Finally, it has to be stressed that intramolecular photostabilization is less efficient compared to the use of solution additives as photostabilizers. It remains, however, the only possible method for photostabilization when photostabilizers cannot be added to the system (e.g. under live-cell conditions), or when the addition of a diffusion-based photostabilizer is ineffective due to missing collisions or not tolerated by the system of interest.
Experimental
Sample preparation and surface immobilization
Immobilization and investigation of single fluorophores was achieved using a dsDNA scaffold comprising two 40-mer oligonucleotides, i.e. ssDNA–fluorophore and ssDNA–biotin. Sequences of both oligomers were adapted from ref. 16, 30 and 31. As a non-stabilized control fluorophore, we used ssDNA–fluorophore (Cy5-C6-5′-TAA TAT TCG ATT CCT TAC ACT TAT ATT GCA TAG CTA TAC G-3′, used as received from Eurofins and IBA, Germany). Single immobilized fluorophore molecules were studied in LabTek 8-well chambered cover slides (Nunc/VWR, The Netherlands) with a volume of 750 μL, as described in ref. 22. After cleaning with 0.1% HF and washing with PBS buffer (one PBS tablet was dissolved in deionized water containing 10 mM phosphate buffer, 2.7 mM potassium chloride and 137 mM sodium chloride at pH 7.4; Sigma-Aldrich, The Netherlands), each chamber was incubated with a mixture of 5 mg/800 mL BSA and 1 mg/800 mL BSA/biotin (Sigma Aldrich, The Netherlands) at 4 °C in PBS buffer overnight. After rinsing with PBS buffer, a 0.2 mg mL−1 solution of streptavidin was incubated for 10 minutes and subsequently rinsed with PBS buffer.The immobilization of dsDNA was achieved via a biotin–streptavidin interaction using pre-annealed dsDNA with the aim of observing single emitters for prolonged periods of time and allowing free rotation of the fluorophores. For this, 5–50 μL of a 1 μM solution of ssDNA–fluorophore or ssDNA–NPA–fluorophore was mixed with the complementary ssDNA–biotin at the same concentration (biotin-5′-CGT ATA GCT ATG CAA TAT AAG TGT AAG GAA TCG AAT ATT A-3′, used as received from IBA, Germany). The respective mixtures of the two oligomers were heated to 98 °C for 4 minutes and cooled down to 4 °C with a rate of 1 °C min−1 in annealing buffer (500 mM sodium chloride, 20 mM Tris–HCL, and 1 mM EDTA at pH = 8). The treated LabTek cover slides were incubated with a 50–100 pM solution of pre-annealed dsDNA for 1–2 minutes, leading to a typical surface coverage of fluorophore-labelled dsDNA as shown in the ESI, Fig. S1–S5.†
All single-molecule experiments were carried out at room temperature (22 ± 1 °C). Oxygen was removed from the buffer system utilizing an oxygen-scavenging system (PBS, pH = 7.4, containing 10% (wt/vol) glucose and 10% (vol/vol) glycerine, 50 μg mL−1 glucose-oxidase, 100–200 μg mL−1 catalase, and 0.1 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP)). As shown before, such low concentrations of the reducer TCEP have no noticeable effect on the photophysics of organic fluorophores31,32 and hence do not convolute with the effects from intramolecular stabilization. Glucose-oxidase catalase (GOC)22,33 was used instead of a combination of protocatechuic acid and protocatechuate-3,4-dioxygenase (PCA/PCD28) to avoid convolution of inter- and intramolecular photostabilization with PCA.34
TIRF-microscopy including data analysis
Wide-field fluorescence microscopy imaging was performed on an inverted microscope (Olympus IX-71 with UPlanSApo 100x, NA 1.49, Olympus, Germany) with an objective type total-internal-reflection fluorescence (TIRF) configuration, as described in ref. 8. Images were collected with a back-illuminated emCCD camera (512 × 512 pixel, C9100-13, Hamamatsu, Japan, filtered by ET700/75, AHF Analysentechnik, Germany). Excitation from a diode laser (Cube, Coherent, Germany, filtered by ZET640/10, Chroma, USA) was at 640 nm with ≈50 W cm−2 at the focal plane. To quantitatively characterize the photostability of the different photostabilized Cy5-derivatives, we imaged an area of 42 × 58 μm containing ≥100 molecules. Each area was illuminated for 600 s and recorded with an integration time of 100 ms. Fluorescence time traces of individual molecules were obtained by identifying those pixels that were at least 80–150 counts above the background noise in the standard deviation plot (standard deviation over all pixels over the complete movie) and averaging the intensity within a 3 × 3 pixel area in each recorded frame. Neighbouring peaks within a 5 pixels radius were excluded. The signal-to-noise ratio (SNR) was determined using the obtained fluorescence time traces after background correction by dividing the average fluorescence intensity of the signal before photobleaching by the standard deviation over the signal. The fluorescence intensity of the signal of >900 single emitters per Cy5-derivative was used to obtain quantitative information on the count rate. The number of fluorescent bursts in each frame image was determined using an absolute threshold criterion, i.e. the signal had to be higher than 800 counts (arbitrary units in the raw movie). The number of fluorescent emitters per frame was then plotted as a function of time and fitted to a mono-exponential decay: y(t) = C + Ae−c/t (with c = 1/τbleach and τbleach being the characteristic photobleaching time constant). Photobleaching times and the associated standard deviations were derived from multiple repeats of the experiment on different days with each ≥5 movies. For each Cy5-derivative, the total number of detected photons before photobleaching was calculated by a multiplication of the obtained count rate and photobleaching lifetime.Functionalization of oligonucleotides
The procedures for the functionalization of oligonucleotides were established and described in detail in ref. 9 and 19. NPA–Cy5 is fully characterized in ref. 19 and Cy5–NPAA in ref. 9.The synthesis of new ssDNA–photostabilizer conjugates was performed according to the following protocol: single-stranded DNA (ssDNA) was used as received from IBA, Germany (NH2-C6-5′-TAA TAT TCG ATT CCT CTG GAC GTAA TAT TCG ATT CCT TAC ACT TAT ATT GCA TAG CTA TAC G-3′). The lyophilized ssDNA was resuspended in MilliQ water and adjusted to 80 μM in 0.2 M NaHCO3 buffer (pH 8.35). To this solution, the same volume of a 20 mg mL−1 solution of a reactive NHS-ester (see below) dissolved in DMF was added and the mixture was vortexed thoroughly. If necessary, additional DMF was added in 10 μL portions to obtain a clear solution. After reaction at room temperature overnight, the functionalized oligonucleotides were purified on an illustra NAP 5 column with Sephadex G-25 DNA Grade material obtained from GE Healthcare (triethylammonium acetate buffer 50 mM, pH 7.0 was used as the eluent for purification on the illustra NAP 5 columns. MilliQ water was used for ssDNA–NPA–Fmoc to prevent partial deprotection of the Fmoc protecting group). Purification was performed by preparative reversed phase HPLC (rp-HPLC) (vide supra) to yield ssDNA–(NPP/NPA–Fmoc). The HPLC time was 18.7 minutes for ssDNA–NPP and 39.8 minutes for NPA–Fmoc.
Fmoc deprotection of ssDNA–NPA–Fmoc was performed as follows. The HPLC-purified and lyophilized oligonucleotide was resuspended in 50 μL of 50 mM triethylammonium acetate buffer (pH 7.0). Deprotection was achieved by the addition of 40 μL of DMF and 10 μL of piperidine. The mixture was vortexed and allowed to react for 2 hours at room temperature. The deprotected oligonucleotide was purified on an illustra NAP 5 column (vide supra) and lyophilised to yield ssDNA–NPA–NH2. The HPLC time was 34.5 minutes for this sample.
rp-HPLC analysis and the preparative purifications (isolations) were performed on a Shimadzu LC-10AD VP machine equipped with a Waters Xterra MS C18 column (3.0 × 150 mm, particle size 3.5 μm) and Waters Xterra MS C18 prep column (7.8 × 150 mm, particle size 10 μm) using a gradient of acetonitrile/triethylammonium acetate buffer 50 mM, pH 7.0. Gradient: 5/95 for 0 to 10 min, 65/35 at 60 min, 75/25 at 65 min, and 5/95 at 75 min for 15 min.
Synthesis of the reactive photostabilizer-derivatives
Molecular dynamics simulations
The molecular dynamics simulations were performed using the Gromacs 4.6 simulation package.35 All simulations used the GROMOS 53A6 force field.36 The topologies for Cy5 and NPA were created using the Automated Topology Builder37 then manually curated and attached to the DNA topologies created using the Gromacs tool pdb2gmx. An 8 bp long DNA with a sequence TAATATTC was used. During the simulation, the DNA structure was kept static using position restraints on all its atoms with a force constant of 1000 kJ (mol nm2)−1 in each direction. Each system was simulated in a dodecahedron box with around 5400 water molecules, 150 mM NaCl as well as counterions. The SPC water model was used while the ions were from the GROMOS 53A6 force field.36Simulations were run using a 2 fs time step. All bonds were constrained using the LINCS algorithm.38 A twin-range cut-off of 0.8 nm and 1.4 nm was used. For the electrostatics, a reaction field was used with a 1.4 nm cut-off and an epsilon_rf of 62. Lennard–Jones interactions used a simple cut-off of 1.4 nm with a dispersion correction. The temperature was kept at 298 K using a Berendsen thermostat39 with τT = 0.1 ps. The pressure was kept at a reference pressure of 1 bar with a Berendsen barostat,39τp = 0.5 ps and compressibility of 4.6 × 10−5 bar.
Each system was first energy minimized for 100 steps using the steepest descent algorithm. They were further equilibrated for 500 ps. The system with Cy5 and NPA attached to opposing sides was then simulated for 1 μs to obtain conformations with Cy5 both on top of DNA as well as in the groove. Two production simulations of 5 μs each were then started, one from each conformation. An identical procedure was followed for the NPA–Cy5 system. The system with a 4 monomer long PEG oligomer added between NPA and Cy5 was simulated first for 100 ns starting from an intermediate conformation and then a 5 μs production run was performed. The system with Cy5 only was simulated for 2 μs starting from each conformation obtained from a 1 μs equilibration simulation.
Before analysis, the simulation trajectories started from different conformations were concatenated and analysed together. The volume maps were produced using the program VMD40 by root mean square deviation (RMSD) fitting the trajectory on either DNA and Cy5 and then calculating the distribution of Cy5 and NPA or only NPA. When calculating the volume maps around DNA, the volume enclosed points where the molecule is in more than 2% of the simulation frames. For the Cy5 case, this value was 0.6%.
The movements of Cy5 and NPA were studied by measuring the distances between them and DNA. For Cy5 and NPA, the measurements were performed from the center of mass (COM) of the molecule, while the top of the DNA was represented by the COM of the N1 and N3 atoms of the top base pair of DNA. The groove of the DNA was defined by the COM of the N1 and N3 atoms of the fifth base pair from the top.
Acknowledgements
This work was financed by the Zernike Institute for Advanced Materials, the Centre for Synthetic Biology (University of Groningen, start-up grant to T. Cordes). J. H. M. van der Velde acknowledges Ubbo-Emmius funding (University of Groningen). J. J. Uusitalo is supported by a young scientist scholarship from the Emil Aaltonen foundation.References
- T. Ha and P. Tinnefeld, Annu. Rev. Phys. Chem., 2012, 63, 595–617 CrossRef CAS PubMed.
- B. Liphardt, B. Liphardt and W. Lüttke, Opt. Commun., 1981, 38, 207–210 CrossRef CAS.
- B. Liphardt and W. Lüttke, Liebigs Ann. Chem., 1981, 1118–1138 CrossRef CAS PubMed.
- B. Liphardt, B. Liphardt and W. Lüttke, Opt. Commun., 1983, 48, 129–133 CrossRef CAS.
- R. B. Altman, D. S. Terry, Z. Zhou, Q. Zheng, P. Geggier, R. A. Kolster, Y. Zhao, J. A. Javitch, J. D. Warren and S. C. Blanchard, Nat. Methods, 2012, 9, 68–71 CrossRef CAS PubMed.
- R. B. Altman, Q. Zheng, Z. Zhou, D. S. Terry, J. D. Warren and S. C. Blanchard, Nat. Methods, 2012, 9, 428–429 CrossRef CAS PubMed.
- P. Tinnefeld and T. Cordes, Nat. Methods, 2012, 9, 426–427 CrossRef CAS PubMed.
- J. H. M. van der Velde, E. Ploetz, M. Hiermaier, J. Oelerich, J. W. de Vries, G. Roelfes and T. Cordes, ChemPhysChem, 2013, 14, 4084–4093 CrossRef CAS PubMed.
- J. H. M. van der Velde, J. Oelerich, J. Huang, J. H. Smit, M. Hiermaier, E. Ploetz, A. Herrmann, G. Roelfes and T. Cordes, J. Phys. Chem. Lett., 2014, 5, 3792–3798 CrossRef CAS PubMed.
- R. Gronheid, J. Hofkens, F. Köhn, T. Weil, E. Reuther, K. Müllen and F. C. De Schryver, J. Am. Chem. Soc., 2002, 124, 2418–2419 CrossRef CAS PubMed.
- P. Tinnefeld, J. Hofkens, D.-P. Herten, S. Masuo, T. Vosch, M. Cotlet, S. Habuchi, K. Müllen, F. C. De Schryver and M. Sauer, ChemPhysChem, 2004, 5, 1786–1790 CrossRef CAS PubMed.
- Q. Zheng, S. Jockusch, Z. Zhou, R. B. Altman, J. D. Warren, N. J. Turro and S. C. Blanchard, J. Phys. Chem. Lett., 2012, 3, 2200–2203 CrossRef CAS PubMed.
- I. Rasnik, S. A. McKinney and T. Ha, Nat. Methods, 2006, 3, 891–893 CrossRef CAS PubMed.
- C. Eggeling, J. Widengren, L. Brand, J. Schaffer, S. Felekyan and C. A. Seidel, J. Phys. Chem. A, 2006, 110, 2979–2995 CrossRef CAS PubMed.
- J. Widengren, A. Chmyrov, C. Eggeling, P.-A. Löfdahl and C. A. M. Seidel, J. Phys. Chem. A, 2007, 111, 429–440 CrossRef CAS PubMed.
- T. Cordes, J. Vogelsang and P. Tinnefeld, J. Am. Chem. Soc., 2009, 131, 5018–5019 CrossRef CAS PubMed.
- L. A. Campos, J. Liu, X. Wang, R. Ramanathan, D. S. English and V. Muñoz, Nat. Methods, 2011, 8, 143–146 CrossRef CAS PubMed.
- Q. Zheng, M. F. Juette, S. Jockusch, M. R. Wasserman, Z. Zhou, R. B. Altman and S. C. Blanchard, Chem. Soc. Rev., 2014, 43, 1044–1056 RSC.
- J. H. M. van der Velde, J. Oelerich, J. Huang, A. Aminian, J. H. Smit, S. Galiani, K. Kolmakov, G. Gouridis, C. Eggeling, A. Herrmann, G. Roelfes and T. Cordes, 2015, paper in revision.
- W. H. Smith and A. J. Bard, J. Am. Chem. Soc., 1975, 97, 5203–5210 CrossRef CAS.
- C. A. Briehn, M.-S. Schiedel, E. M. Bonsen, W. Schuhmann and P. Bäuerle, Angew. Chem., Int. Ed., 2001, 40, 4680–4683 CrossRef CAS.
- J. Vogelsang, R. Kasper, C. Steinhauer, B. Person, M. Heilemann, M. Sauer and P. Tinnefeld, Angew. Chem., Int. Ed., 2008, 47, 5465–5469 CrossRef CAS PubMed.
- M. E. Sanborn, B. K. Connolly, K. Gurunathan and M. Levitus, J. Phys. Chem. B, 2007, 111, 11064–11074 CrossRef CAS PubMed.
- B. J. Harvey, C. Perez and M. Levitus, Photochem. Photobiol. Sci., 2009, 8, 1105–1110 CAS.
- J. Spiriti, J. K. Binder, M. Levitus and A. van der Vaart, Biophys. J., 2011, 100, 1049–1057 CrossRef CAS PubMed.
- M. Levitus and S. Ranjit, Q. Rev. Biophys., 2011, 44, 123–151 CrossRef CAS PubMed.
- E. M. S. Stennett, M. A. Ciuba and M. Levitus, Chem. Soc. Rev., 2014, 43, 1057–1075 RSC.
- J. Hu and C. Zhang, Anal. Chem., 2013, 85, 2000–2004 CrossRef CAS PubMed.
- P. Holzmeister, A. Gietl and P. Tinnefeld, Angew. Chem., Int. Ed., 2014, 126, 5792–5796 CrossRef PubMed.
- J. Vogelsang, T. Cordes, C. Forthmann, C. Steinhauer and P. Tinnefeld, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8107–8112 CrossRef CAS PubMed.
- J. Vogelsang, T. Cordes and P. Tinnefeld, Photochem. Photobiol. Sci., 2009, 8, 486–496 CAS.
- J. C. Vaughan, G. T. Dempsey, E. Sun and X. Zhuang, J. Am. Chem. Soc., 2013, 135, 1197–1200 CrossRef CAS PubMed.
- C. E. Aitken, R. A. Marshall and J. D. Puglisi, Biophys. J., 2008, 94, 1826–1835 CrossRef CAS PubMed.
- A. Le Gall, D. Dulin, G. Clavier, R. Méallet-Renault, P. Bouyer, K. Perronet and N. Westbrook, ChemPhysChem, 2011, 12, 1657–1660 CrossRef CAS PubMed.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. Van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- K. B. Koziara, M. Stroet, A. K. Malde and A. E. Mark, J. Comput.-Aided Mol. Des., 2014, 28, 221–233 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional single-molecule and simulation data. See DOI: 10.1039/c5fd00114e |
| This journal is © The Royal Society of Chemistry 2015 |