 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly luminescent gold nanoparticles: effect of ruthenium distance for nanoprobes with enhanced lifetimes†
Shani A. M.
Osborne
and
Zoe
Pikramenou
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: z.pikramenou@bham.ac.uk
First published on 26th June 2015
Abstract
The photophysical properties of gold nanoparticles, AuNPs, with sizes of 13, 50 and 100 nm in diameter, coated with surface-active ruthenium complexes have been studied to investigate the effect of the distance of the ruthenium luminescent centre from the gold surface. Luminescence lifetimes of the three ruthenium probes, RuS1, RuS6 and RuS12, with different length spacer units between the surface active groups and the ruthenium centre were taken. The metal complexes were attached to AuNP13, AuNP50 and AuNP100via thiol groups using a method of precoating the nanoparticles with a fluorinated surfactant. The luminescence lifetime of the longer spacer unit complex, RuS12, was enhanced by 70% upon attachment to the AuNP when compared to the increase of the short and medium linker unit complexes, RuS1 (20%) and RuS6 (40%) respectively. The effect of the surfactant in the lifetime increase of the ruthenium coated AuNPs was shown to be larger for the medium spacer probe, RuS6. There was no effect of the change of the size of the AuNPs from 13 to 50 or 100 nm.
Introduction
Gold nanoparticles, AuNPs, are ideal probes for cellular imaging based on their high electron density, which allows multimodal imaging microscopies to be used and improved spatial resolution in detection as opposed to molecular probes. Although the labelling of AuNPs with luminescent probes has been reported for some time, a limitation of their use has been the quenching of the molecular fluorescence by different mechanisms involving the surface plasmon of the gold.1–3The distance of the fluorophore to the gold surface and the method of attachment are important factors to the luminescent properties of the particles and their study can provide an understanding of the mechanism involved in quenching of the fluorescence, as well as directing future molecular designs. The quenching of the fluorescence signal by plasmonic nanoparticles at short distances has been attributed to a “near-field” effect involving energy or electron transfer non-radiative pathways.4,5 In most cases it is shown that if the fluorophore is within 5 nm of the AuNP surface, it is close enough to electronically interact with the AuNP and the fluorophore's excited electron is donated to the gold.1,6 More recently, elegant approaches to examine the effect have involved methods for distancing the fluorophore from the gold surface either through an electrolyte film,7 using layer by layer assemblies,8 or through formation of silica shells around the gold.9–11 In many cases it has been shown that organic dyes' fluorescence can be enhanced with increasing the distance from the AuNP, but they only achieve lifetimes12 between the regions of a few ps to 50 ns or 8-fold overall fluorescence enhancement.8
We have been interested in the attachment of metal complexes on AuNPs to produce nanoprobes which bear the distinct optical signature of the metal complex, including large Stokes shift and high photostability. Luminescent europium coated AuNPs have been prepared and employed as cellular probes.13–15 To stabilise positively charged ruthenium polypyridyl coated nanoparticles we have used a fluorinated surfactant which has provided a method to stabilise luminescent ruthenium nanoparticles of sizes up to 100 nm.16 Enhancement of a NIR organic dye has been shown in a system of gold nanorods with silica shells of 17 nm.11
It has been shown that the luminescence of ruthenium complexes is quenched when attached to an AuNP.17–19 The adsorption of Ru(bpy)3Cl2 on the surface of a 10 nm AuNP has shown a luminescence lifetime decrease from 623 to 0.8 ns.6 It was found that even at a distance of 2 nm from the gold surface, a tris(bipyridine)ruthenium complex has a highly quenched luminescence lifetime, and an enhancement of 4-fold was seen at a distance of 50 nm via a silica shell.20
In our approach the fluorosurfactant coating of the particles has been shown to have the effect of protecting the ruthenium probe excited state from quenching by oxygen, increasing the lifetime of the complex on the nanoparticles. The use of surfactant has become increasingly popular for the increased stability of nanoprobes.21 In this study we examine the effect on the luminescence of the ruthenium probe by varying the distance of the attachment of the probe to the surface of the AuNP. We used three ruthenium probes, RuS1, RuS6 and RuS12 (Fig. 1), with different length spacer units between the surface active groups, previously developed in our group.16,22,23 We have also varied the size of the AuNP to examine if there is an influence on the luminescence lifetime of the probes. We describe herein an improved method for coating AuNPs, using fluorosurfactant stabilised AuNPs before ruthenium complex addition. In this study we establish the effect of the spacer length together with the fluorosurfactant interactions for the development of the most efficient design for the luminescent ruthenium nanoparticles.
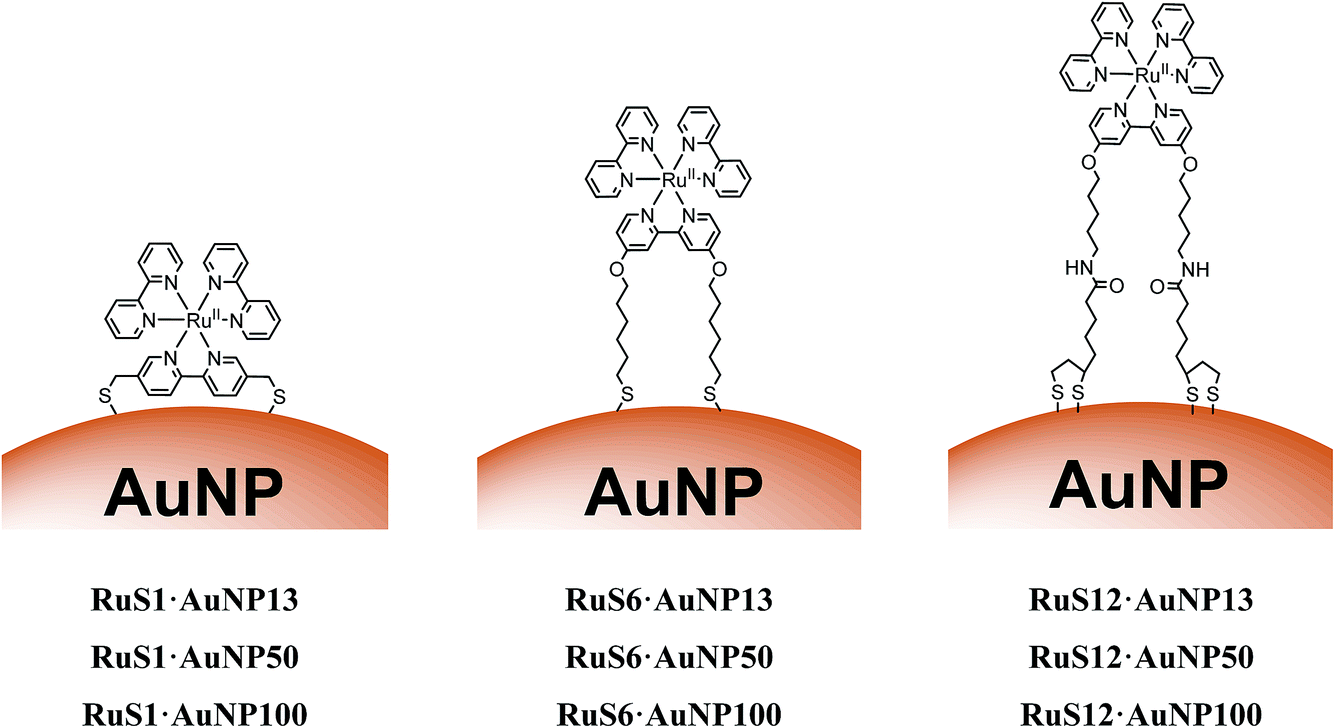 | ||
| Fig. 1 Schematic to show the structures of RuS1·AuNP13, RuS1·AuNP50, RuS1·AuNP100, RuS6·AuNP13, RuS6·AuNP50, RuS6·AuNP100, RuS12·AuNP13, RuS12·AuNP50 and RuS12·AuNP100. | ||
Results and discussion
Gold nanoparticle coating with surfactant and metal complex
Three ruthenium complexes with different sized linker units, RuS1, RuS6 and RuS12 (Fig. 1) for attachment to gold were synthesised and fully characterised following previously published methods.16,22,23 For each complex, ion exchange was used to convert the counter ion to chloride, for improved solubility in aqueous solutions, employed in the nanoparticle preparation.Monodispersed 13, 50 and 100 nm AuNPs (AuNP13, AuNP50 and AuNP100) were synthesised using slight modifications of previously published methods.24–26 The protocol involves synthesising AuNP13 seeds and stabilising them with citrate anions. The AuNPs were characterised by the specific surface plasmon resonance (SPR) band in the visible of the absorption spectrum, transmission electron microscopy (TEM), dynamic light scattering (DLS) sizing and zeta potential measurements (ESI†).
Solutions of AuNP13, AuNP50 and AuNP100 displayed a band with a maximum, λmax, at 517, 532 and 566 nm, respectively, characteristic of their SPR bands and in agreement with previously published data.27,28 DLS sizing confirmed the AuNP13, AuNP50 and AuNP100 to be 14 ± 4 nm (PDI = 0.09), 50 ± 12 nm (PDI = 0.04) and 100 ± 24 nm (PDI = 0.01) respectively. TEM images suggested the sizes of AuNP13, AuNP50 and AuNP100 to be 17, 60 and 120 nm respectively (ESI†) in good agreement with the DLS data.
Upon addition of the Zonyl surfactant to the AuNPs, a shift of 1 nm of the SPR band was observed. The surfactant coated particles Z·AuNP13 were isolated by centrifugation and they were then used for the titration of the ruthenium probe, monitoring the SPR band (Fig. 2).16
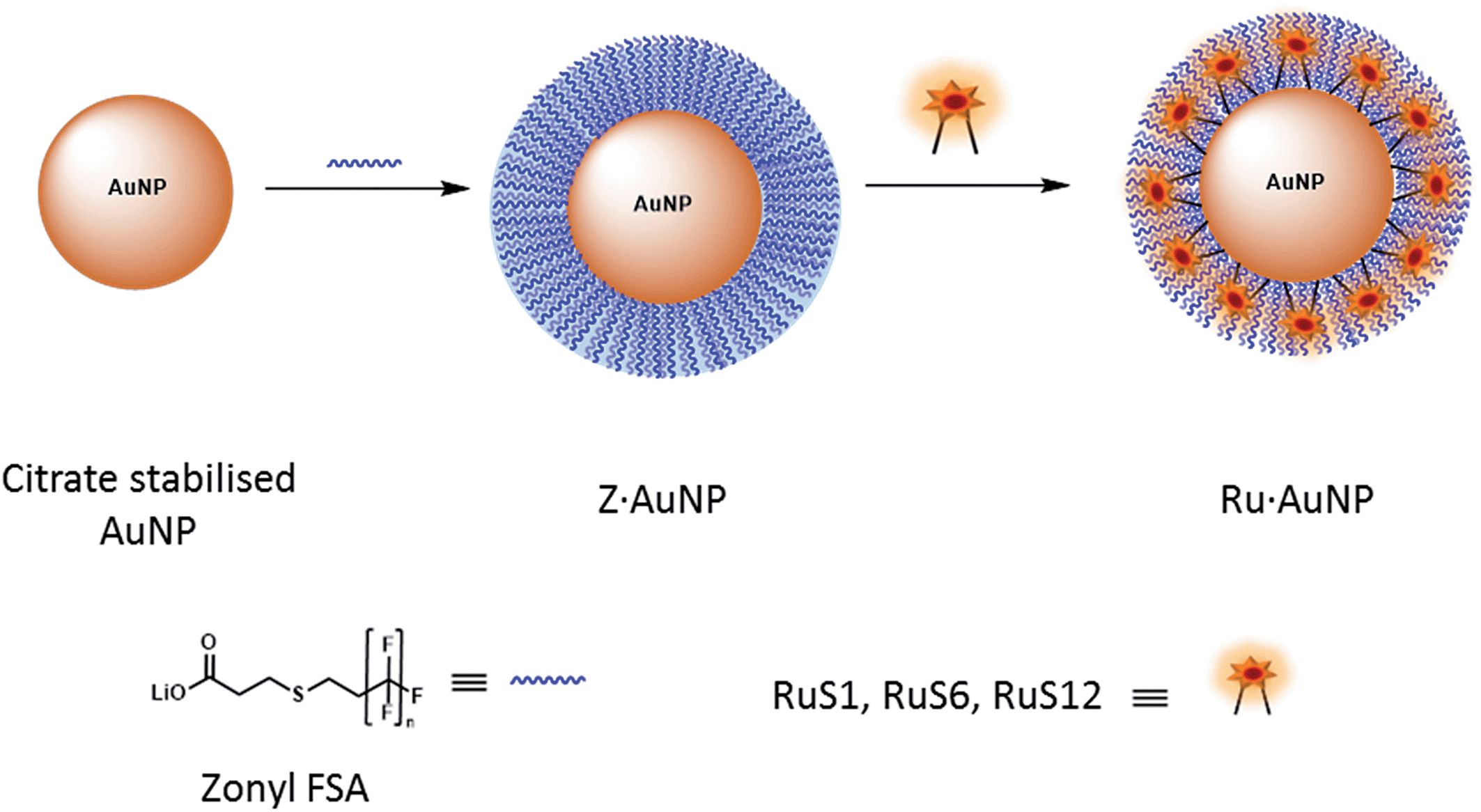 | ||
| Fig. 2 Schematic to show the attachment of the fluorinated surfactant, Zonyl FSA, and the ruthenium complex to an AuNP. | ||
For the coating of Z·AuNP13, aliquots (2 μL) of 1.19 mM RuS1, 0.95 mM RuS6 and 0.87 mM RuS12 were titrated into a 4.5 nM solution of Z·AuNP13 and the SPR shift was monitored using the change in λmax to determine the saturation of the AuNP surface (Fig. 3). As more probe was added, the SPR shifted to the red until the optimum coating was achieved and a shift was no longer observed. The addition of 12 μL 1.19 mM RuS1, 16 μL 0.95 mM RuS6 and 20 μL 0.87 mM RuS12 to 4.5 nM Z·AuNP13 resulted in a 4 (521 nm), 5 (522 nm) and 3 nm (520 nm) shift in λmax respectively (Table 1). All three probes caused a similar shift in the SPR band upon addition to the AuNPs. Analyses of the elemental composition of the nanoparticles by inductively coupled plasma mass spectrometry (ICP-MS) revealed a Ru![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratio of 1:160, suggesting a coating of 600 ruthenium complexes per AuNP13.
:Au ratio of 1:160, suggesting a coating of 600 ruthenium complexes per AuNP13.
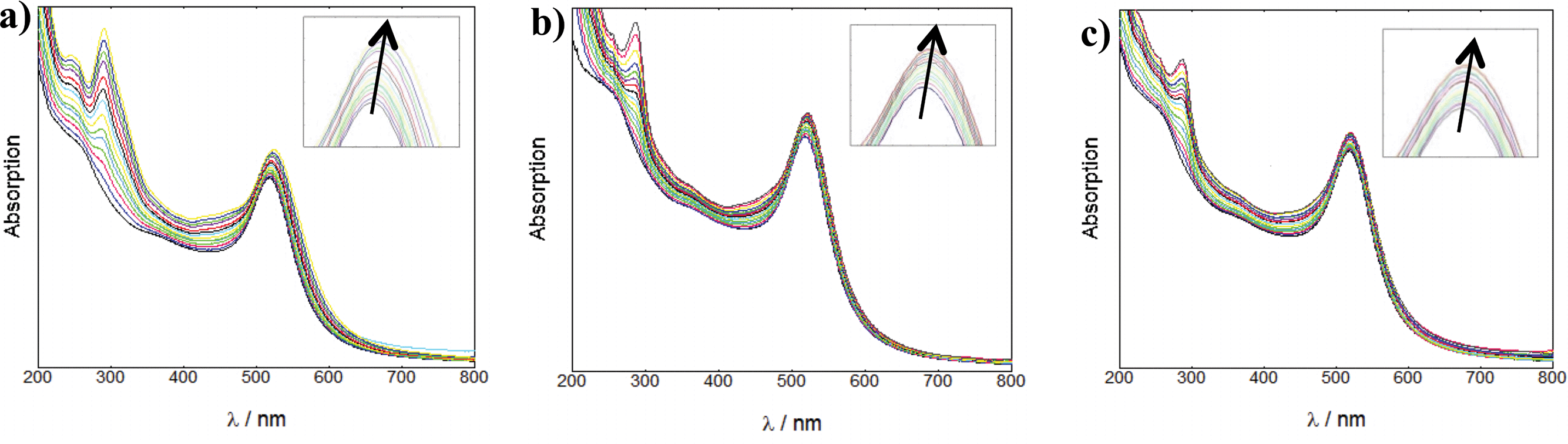 | ||
| Fig. 3 UV-Vis titrations of 1.19 mM RuS1 (a), 0.95 mM RuS6 (b) and 0.87 mM RuS12 (c) into 4.5 nM Z·AuNP13 in water. | ||
| λ max (nm) | Shift (nm) | λ max (nm) | Shift (nm) | λ max (nm) | Shift (nm) | |||
|---|---|---|---|---|---|---|---|---|
| AuNP13 | 517 | 0 | AuNP50 | 532 | 0 | AuNP100 | 566 | 0 |
| Z·AuNP13 | 518 | 1 | Z·AuNP50 | 533 | 1 | Z·AuNP100 | 567 | 1 |
| RuS1·AuNP13 | 521 | 4 | RuS1·AuNP50 | 537 | 5 | RuS1·AuNP100 | 569 | 3 |
| RuS6·AuNP13 | 522 | 5 | RuS6·AuNP50 | 536 | 4 | RuS6·AuNP100 | 569 | 3 |
| RuS12·AuNP13 | 520 | 3 | RuS12·AuNP50 | 537 | 5 | RuS12·AuNP100 | 569 | 3 |
The particles isolated following size exclusion chromatography showed the same λmax (Fig. 4) as the particles saturated with the ruthenium complex, formed during titration. This confirmed that the surface coating of the particles had not changed and only the excess molecular complex was removed during chromatography.
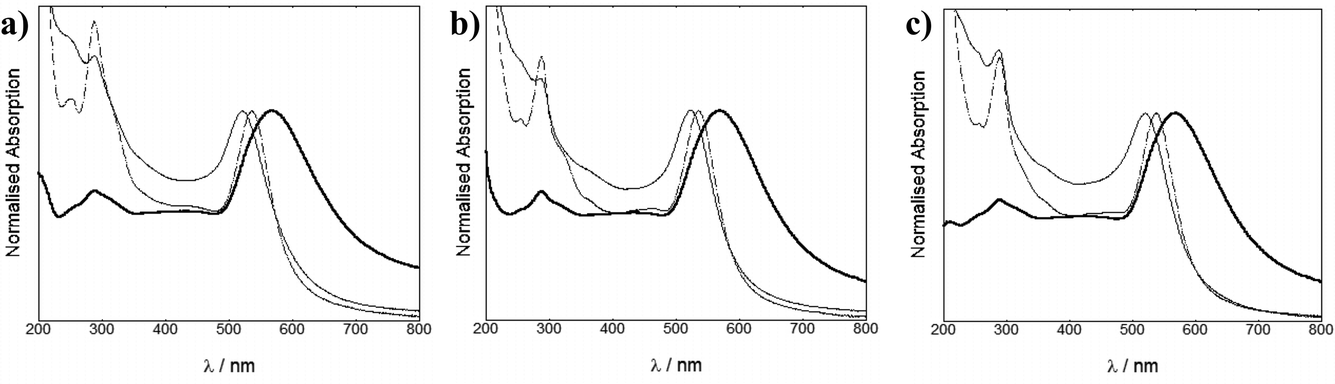 | ||
| Fig. 4 UV-Vis spectra of 4.5 nM RuS1·AuNP13 (thin solid line), 40 pM RuS1·AuNP50 (dotted line) and 20 pM RuS1·AuNP100 (thick solid line) (a), 4.5 nM RuS6·AuNP13, 40 pM RuS6·AuNP50 and 20 pM RuS6·AuNP100 (b) and 4.5 nM RuS12·AuNP13, 40 pM RuS12·AuNP50 and 20 pM RuS12·AuNP100 (c) in water. | ||
TEM and DLS studies show that the sizes of the AuNPs did not significantly change upon coating with the surfactant and the ruthenium complex. Images of the nanoparticles from TEM show monodispersed, uniform NPs with estimated sizes from the image of 17 nm for RuS1·AuNP13, RuS6·AuNP13 and RuS12·AuNP13, 60 nm for RuS1·AuNP50, RuS6·AuNP50 and RuS12·AuNP50 and 120 nm for RuS1·AuNP100, RuS6·AuNP100 and RuS12·AuNP100 (Fig. 5). The RuS12·AuNP100 were imaged as single nanoparticles using NanoSight tracking both through scatter and ruthenium emission detection in the red upon 488 nm excitation (ESI†).
 | ||
| Fig. 5 TEM images of RuS1·AuNP13 (a), RuS6·AuNP13 (b), RuS12·AuNP13 (c), RuS1·AuNP50 (d), RuS6·AuNP50 (e), RuS12·AuNP50 (f), RuS1·AuNP100 (g), RuS6·AuNP100 (h) and RuS12·AuNP100 (i). Images were taken on the Jeol 1200 EX TEM. | ||
Luminescent studies of ruthenium probe functionalised AuNPs
To characterise the luminescence properties of the probes attached to AuNPs, we used steady state and time-resolved emission spectroscopy. The luminescence spectra and lifetimes of the nanoprobes were recorded and compared with the molecular complexes in solution, in the presence and absence of the Zonyl surfactant (Fig. 6). There is no significant shift in the λmax of the emission peak upon addition of Zonyl to the complex or upon attachment of the complex to the Z·AuNP. We have previously found that attachment of the RuS12 complex to a gold surface causes a 15 nm blue shift in the λmax.22 This shift may not be present on the AuNP due to the presence of the surfactant or the different probe environment on the gold surface as compared with the nanoparticle.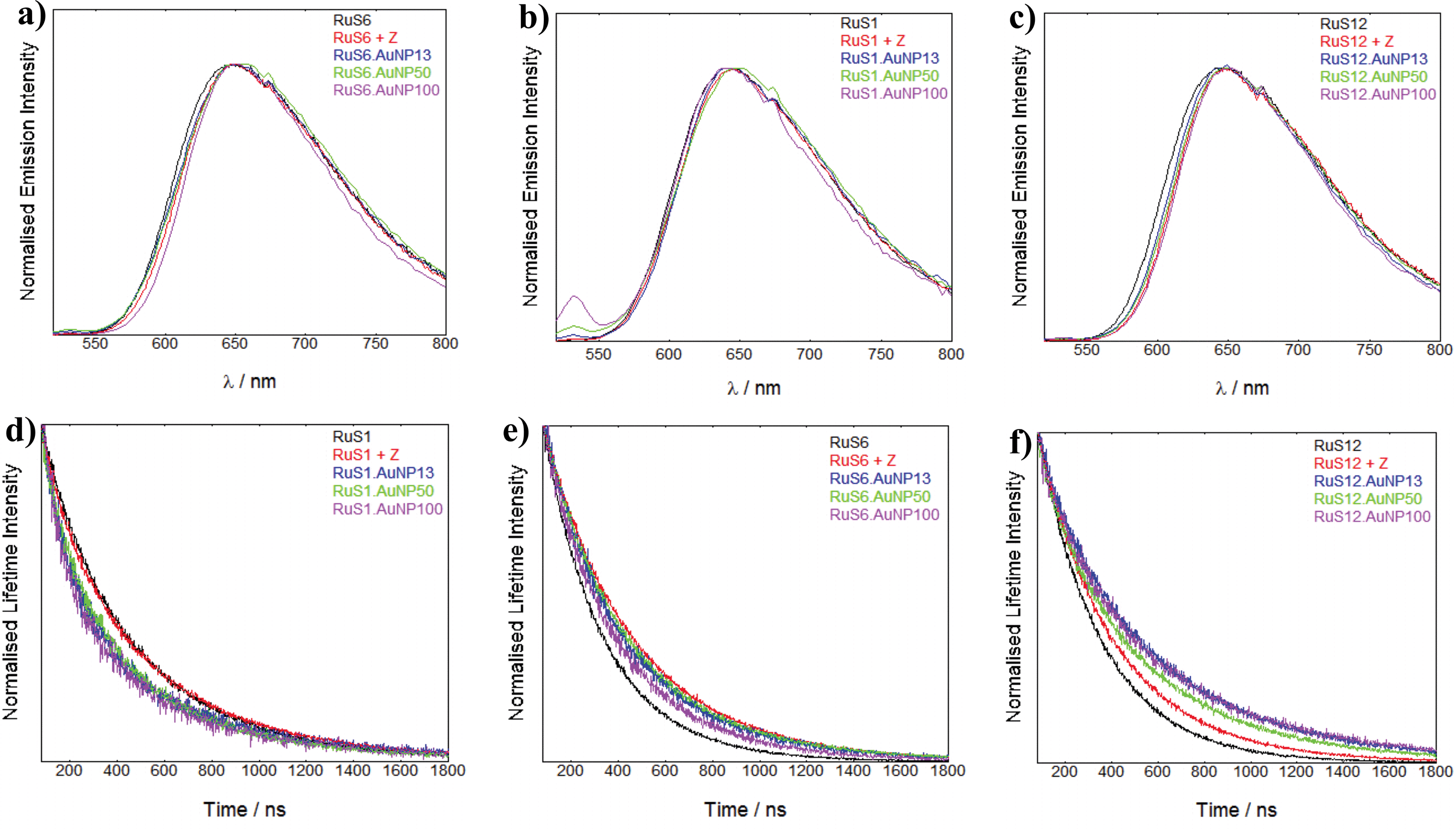 | ||
| Fig. 6 Luminescence emission data of RuS1, RuS1 + Z, RuS1·AuNP13, RuS1·AuNP50 and RuS1·AuNP100 (a), RuS6, RuS6 + Z, RuS6·AuNP13, RuS6·AuNP50 and RuS6·AuNP100 (b) RuS12, RuS12 + Z, RuS12·AuNP13, RuS12·AuNP50 and RuS12·AuNP100 (c). λexc = 450 nm and λdet = 650 nm. The spectra are taken from 520–800 nm. Luminescence lifetime data of RuS1, RuS1 + Z, RuS1·AuNP13, RuS1·AuNP50 and RuS1·AuNP100 (d), RuS6, RuS6 + Z, RuS6·AuNP13, RuS6·AuNP50 and RuS6·AuNP100 (e) RuS12, RuS12 + Z, RuS12·AuNP13, RuS12·AuNP50 and RuS12·AuNP100 (f). λexc = 445 nm and λdet = 650 nm. | ||
The luminescence lifetimes of the coated AuNPs are summarised in Table 2 and comparisons of the lifetime decays are presented to illustrate the changes in the lifetimes (Fig. 6).
| τ/ns | % | τ/ns | % | τ/ns | % | |||
|---|---|---|---|---|---|---|---|---|
| RuS1 | 420 | 0 | RuS6 | 240 | 0 | RuS12 | 280 | 0 |
| RuS1 + Z | 420 | 0 | RuS6 + Z | 400 | 70 | RuS12 + Z | 350 | 25 |
| RuS1·AuNP13 | 470 | 20 | RuS6·AuNP13 | 340 | 40 | RuS12·AuNP13 | 480 | 70 |
| RuS1·AuNP50 | 470 | 20 | RuS6·AuNP50 | 340 | 40 | RuS12·AuNP50 | 480 | 70 |
| RuS1·AuNP100 | 470 | 20 | RuS6·AuNP100 | 340 | 40 | RuS12·AuNP100 | 480 | 70 |
To examine the effect of the Zonyl surfactant on the luminescence properties, we compared the luminescence decays of each ruthenium probe (RuS1, RuS6 and RuS12) upon addition of Zonyl (10 μL of 10% in water). The luminescence lifetimes of both RuS6 and RuS12 increased upon addition of surfactant by 70% and 25% respectively, compared to the complex in solution (Fig. 6f and g). In contrast, the lifetime of RuS1 did not change upon addition of the Zonyl surfactant (Fig. 6d). We attribute the increase in lifetime of RuS6 and RuS12 to interaction of the surfactant with the molecular complex and consequent protection from 3O2 quenching. Increasing the hydrophobicity of a probe increases interaction with the surfactant and oxygen shielding allows for the largest change in luminescence lifetime upon addition of surfactant. Previous studies have shown that increasing the hydrophobicity of the ligands increased ruthenium complex binding to ionic and non-ionic surfactants.29–31 It was found that Ru(phen)2(CN)2 had a 10-fold increase in binding to the anionic sodium dodecyl sulfate surfactant when compared with the less hydrophobic Ru(bpy)2(CN)2 complex.30 The lifetime increased significantly more for RuS6 (240 to 400 ns, 70%) than with RuS12 (280 to 350 ns, 25%) in the presence of Zonyl. This is attributed to a less tight interaction of RuS12 with Zonyl, possibly due to the presence of the amide bonds on the aliphatic legs. The lack of increase in lifetime for RuS1 may be attributed to the absence of aliphatic legs for the surfactant to interact with, indicating the higher polarity of the complex compared to RuS6 and RuS12.
To compare the effect of the different sized AuNPs on the properties of the ruthenium probe we studied the luminescence lifetime decays for the isolated nanoparticles (AuNP13, AuNP50 and AuNP100) coated with ruthenium. The lifetime decays of RuS1·AuNP13, RuS1·AuNP50 and RuS1·AuNP100 overlap (Fig. 6d), showing that there is no difference in the effect of size of the AuNP on the luminescence lifetime of the probe (470 ns). Similar observations were made for the luminescence lifetime decays of RuS6·AuNP13, RuS6·AuNP50 and RuS6·AuNP100 (Fig. 6e) as well as those of RuS12·AuNP13, RuS12·AuNP50 and RuS12·AuNP100 (Fig. 6f). These results show that the size of the NP does not affect the luminescence lifetime of the three probes, RuS1, RuS6 and RuS12. It is worth noting here that for all the lifetime fittings of the coated AuNPs, we also observed a short component (50–100 ns) with a small percentage contribution (5–20%). From our measurements this was attributed to being a scattering artifact and only the long component is reported.
The luminescence lifetimes of RuS1, RuS6 and RuS12 upon attachment to the AuNPs showed increases of 20%, 40% and 70%, respectively, from the free complex (Table 2). These results show that there is an enhancement of the lifetime from the Zonyl-coated AuNP surface, which can be attributed to the interaction with the Zonyl surfactant or to enhancement by the AuNP surface. These results mirror the increase in quantum yields from 0.02 for the three complexes in solution to 0.02, 0.05 and 0.09 for RuS1·AuNP13, RuS6·AuNP13 and RuS12·AuNP13, respectively, showing this enhancement is due to an increase in radiative decay and decrease in non-radiative decay (ESI).† The enhancement of the RuS12 complex on the AuNPs is significantly larger than that of RuS1 and RuS6, even though the effect of the Zonyl surfactant is less pronounced than in RuS6. This larger enhancement can be attributed to an interaction of the AuNP electromagnetic field with the luminescent probe dipole, observed only for RuS12, located at a longer distance from the particle surface than the other complexes. It is expected that the closer the luminescent probe is to the surface, the larger the quenching effect. This agrees with previous research which states that a lumophore close to the gold surface is quenched due to electronically interacting with the surface's strong magnetic field.1 The effect is attributed to the excited electron being donated to the gold surface, quenching fluorescence by non-radiative pathways. In a study of a ruthenium complex with a similar chain to RuS6, a 60% quenching of luminescence was observed when attached to a gold surface.19 In our case it is clear that the effect of Zonyl is important at this distance from the surface. It is also possible that the induced rigidity upon attachment of the ruthenium complexes to the surfactant functionalised AuNPs contributes to the increase in luminescence lifetimes. Although there are examples of luminescence enhancement of lumophores on AuNPs, most are at distances greater than 5 nm. No reports to our knowledge have been made of an enhancement in luminescence at such short distances from the surface. The estimated distances of the ruthenium centres in RuS1, RuS6 and RuS12 are 0.7, 1.6 and 2.5 nm from the surface respectively. Rubinstein et al. observed a 4-fold increase in luminescence at a distance at 50 nm, but at 2 nm from the surface, which is equivalent to the RuS12 distance, they saw a large quenching in luminescence.20 Our previous studies have shown that the luminescence lifetime of RuS12 is not quenched when the complex is attached to a gold surface, supporting the results that this distance is ideal for gold surfaces.22 It is not surprising than for the AuNPs, the enhancement can be observed at this distance based on the nanoparticle induced characteristics.
Conclusions
We have demonstrated the effect of the distance of thiol-functionalised ruthenium complexes from the AuNP surface on the luminescence properties of the nanoparticles. The RuS12 complex is shown to display a greater enhancement of luminescence upon attachment to the AuNP, which is significantly higher than those of RuS1 and RuS6 due to its improved distance from the gold surface. Even at these rather close distances to the gold surface, all three probes show an enhancement of luminescence lifetime when attached to the AuNP. We have shown that the coating with the Zonyl surfactant is important in the enhancement of the luminescence lifetime, especially for the medium chain ruthenium complex. The increase of the size of the AuNP from 13 to 50 and 100 nm led to probes with the same lifetimes as the 13 nm particles. Our studies provide an insight into the design of functionalised nanoparticles with luminescent probes which can be adopted for other fluorophores.Experimental
Materials
Starting materials were purchased from Sigma Aldrich or Fisher Scientific.Synthesis of AuNPs
000g for 30 minutes. The supernatant was decanted and the pellet was redispersed in deionised water to form a 9 nM solution of AuNP13.
000g for 90 s. The supernatant was decanted and the pellet was redispersed in deionised water.
Ruthenium molecular complexes
The RuS1, RuS6 and RuS12 probes were prepared using previously published methods and all characterisation agreed with previous results.16,22,23 The counter ion was exchanged using Dowex 1 X 8 ion exchange chromatography and the final solutions to be used for coating were prepared in methanol as 1.19, 0.95 and 0.87 mM solutions of RuS1, RuS6 and RuS12, respectively. RuS6 was sonicated with NH4OH to produce a 0.63 mM solution.Attachment of probe to the NP
000g for 30 min, the supernatant was decanted and the pellet was resuspended in deionised water (1 mL) to form Z·AuNP13. λmax (H2O) = 518 nm (SPR). Diameter = 12 ± 4 nm (DLS number distribution). ζ-potential = −50 ± 8 mV.
:Au is 1:180, suggesting 550 complexes per AuNP13.
:Au is 1:180, suggesting 550 complexes per AuNP13.
:Au is 1:150, suggesting 690 complexes per AuNP13.
000g for 90 s, the supernatant was decanted and the pellet was resuspended in deionised water (1 mL) to form Z·AuNP100. λmax (H2O) = 567 nm (SPR). Diameter = 107 ± 27 nm (DLS number distribution). ζ-potential = −53 ± 11 mV.
Instrumentation
UV-Vis spectroscopy was carried out on a Varian Cary 50 spectrophotometer. UV-Vis spectra were collected using 1 cm path length quartz cuvettes. Luminescence spectroscopy was carried out on an Edinburgh Instruments FLS920 steady state and time-resolved spectrometer described elsewhere.22 Luminescence lifetime experiments were carried out using an Edinburgh Instruments EPL-445 laser as the excitation source. Lifetimes were fitted using Edinburgh Instruments FAST software, with errors of ±10%. Luminescence experiments were carried out using 1 cm path length quartz cuvettes. TEM images were carried out on a Jeol 1200 EX transmission electron microscope. DLS sizing and zeta potential measurements were carried out on a Malvern Zetasizer nano ZSP and flow imaging was carried out on a Malvern Nanosight NS300.Acknowledgements
We would like to thank John Ddungu for preliminary results. We wish to acknowledge EPSRC, The Leverhulme Trust (ZP) and the School of Chemistry, University of Birmingham for financial support. Some of the spectrometers used in this research were obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and partial funding from the European Regional Development Fund (ERDF). We would like to thank Philip Aston at the University of Warwick for assistance with the ICP-MS.References
- S. Eustis and M. A. El Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- E. Dulkeith, A. C. Morteani, T. Niedereichholz, T. A. Klar, J. Feldmann, S. A. Levi, F. C. J. M. van Veggel, D. N. Reinhoudt, M. Möller and D. I. Gittins, Phys. Rev. Lett., 2002, 89, 203002 CrossRef CAS.
- K. G. Thomas and P. V. Kamat, Acc. Chem. Res., 2003, 36, 888–898 CrossRef CAS PubMed.
- F. Liu and J. M. Nunzi, Appl. Phys. Lett., 2011, 99, 123302 CrossRef PubMed.
- J. Kümmerlen, A. Leitner, H. Brunner, F. R. Aussenegg and A. Wokaun, Mol. Phys., 1993, 80, 1031–1046 CrossRef PubMed.
- W. R. Glomm, S. J. Moses, M. K. Brennaman, J. M. Papanikolas and S. Franzen, J. Phys. Chem. B, 2004, 109, 804–810 CrossRef PubMed.
- H. Zhang, M. Cao, W. Wu, H. Xu, S. Cheng and L. J. Fan, Nanoscale, 2015, 7, 1374–1382 RSC.
- A. V. Sorokin, A. A. Zabolotskii, N. V. Pereverzev, I. I. Bespalova, S. L. Yefimova, Y. V. Malyukin and A. I. Plekhanov, J. Phys. Chem. C, 2015, 119, 2743–2751 CAS.
- I. O. Osorio-Román, A. R. Guerrero, P. Albella and R. F. Aroca, Anal. Chem., 2014, 86, 10246–10251 CrossRef PubMed.
- P. Reineck, D. Gómez, S. H. Ng, M. Karg, T. Bell, P. Mulvaney and U. Bach, ACS Nano, 2013, 7, 6636–6648 CrossRef CAS PubMed.
- N. S. Abadeer, M. R. Brennan, W. L. Wilson and C. J. Murphy, ACS Nano, 2014, 8, 8392–8406 CrossRef CAS PubMed.
- D. Lee and D. J. Jang, Polymer, 2014, 55, 5469–5476 CrossRef CAS PubMed.
- A. C. Savage and Z. Pikramenou, Chem. Commun., 2011, 47, 6431–6433 RSC.
- A. Davies, D. J. Lewis, S. P. Watson, S. G. Thomas and Z. Pikramenou, Proc. Nat. Acad. Sci., 2012, 109, 1862–1867 CrossRef CAS PubMed.
- S. Comby, E. M. Surender, O. Kotova, L. K. Truman, J. K. Molloy and T. Gunnlaugsson, Inorg. Chem., 2014, 53, 1867–1879 CrossRef CAS PubMed.
- N. J. Rogers, S. Claire, R. M. Harris, S. Farabi, G. Zikeli, I. B. Styles, N. J. Hodges and Z. Pikramenou, Chem. Commun., 2014, 50, 617–619 RSC.
- P. Zhang, J. Wang, H. Huang, H. Chen, R. Guan, Y. Chen, L. Ji and H. Chao, Biomaterials, 2014, 35, 9003–9011 CrossRef CAS PubMed.
- F. C. Leung, A. Y. Tam, V. K. Au, M. J. Li and V. W. Yam, ACS Appl. Mater. Interfaces, 2014, 6, 6644–6653 CAS.
- M. Jebb, P. K. Sudeep, P. Pramod, K. G. Thomas and P. V. Kamat, J. Phys. Chem. B, 2007, 111, 6839–6844 CrossRef CAS PubMed.
- O. Kedem, W. Wohlleben and I. Rubinstein, Nanoscale, 2014, 6, 15134–15143 RSC.
- K. Wee, M. K. Brennaman, L. Alibabaei, B. H. Farnum, B. Sherman, A. M. Lapides and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 13514–13517 CrossRef CAS PubMed.
- S. J. Adams, D. J. Lewis, J. A. Preece and Z. Pikramenou, ACS Appl. Mater. Interfaces, 2014, 6, 11598–11608 CAS.
- P. Bertoncello, E. T. Kefalas, Z. Pikramenou, P. R. Unwin and R. J. Forster, J. Phys. Chem. B, 2006, 110, 10063–10069 CrossRef CAS PubMed.
- C. Ziegler and A. Eychmüller, J. Phys. Chem. C, 2011, 115, 4502–4506 CAS.
- K. C. Grabar, R. G. Freeman, M. B. Hommer and M. J. Natan, Anal. Chem., 1995, 67, 735–743 CrossRef CAS.
- F. Schulz, T. Homolka, N. G. Bastús, V. Puntes, H. Weller and T. Vossmeyer, Langmuir, 2014, 30, 10779–10784 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- B. Factor, B. Muegge, S. Workman, E. Bolton, J. Bos and M. M. Richter, Anal. Chem., 2001, 73, 4621–4624 CrossRef CAS.
- S. W. Snyder, S. L. Buell, J. N. Demas and B. A. DeGraff, J. Phys. Chem., 1989, 93, 5265–5271 CrossRef CAS.
- W. J. Dressick, B. L. Hauenstein, T. B. Gilbert, J. N. Demas and B. A. DeGraff, J. Phys. Chem., 1984, 88, 3337–3340 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5fd00108k |
| This journal is © The Royal Society of Chemistry 2015 |