 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photosynthetic water oxidation: binding and activation of substrate waters for O–O bond formation
David J.
Vinyard
,
Sahr
Khan
and
Gary W.
Brudvig
*
Department of Chemistry, Yale University, New Haven, CT, United States. E-mail: gary.brudvig@yale.edu
First published on 1st June 2015
Abstract
Photosynthetic water oxidation occurs at the oxygen-evolving complex (OEC) of Photosystem II (PSII). The OEC, which contains a Mn4CaO5 inorganic cluster ligated by oxides, waters and amino-acid residues, cycles through five redox intermediates known as Si states (i = 0–4). The electronic and structural properties of the transient S4 intermediate that forms the O–O bond are not well understood. In order to gain insight into how water is activated for O–O bond formation in the S4 intermediate, we have performed a detailed analysis of S-state dependent substrate water binding kinetics taking into consideration data from Mn coordination complexes. This analysis supports a model in which the substrate waters are both bound as terminal ligands and react via a water-nucleophile attack mechanism.
1. Introduction
The climate, biology and geology of Earth were transformed by the evolution of oxygenic photosynthesis approximately three billion years ago.1 All photosynthetic organisms, from the earliest cyanobacteria to modern vascular plants, use solar energy to oxidize water to molecular oxygen (O2), protons and electrons at the oxygen-evolving complex (OEC) of Photosystem II (PSII).2,3 PSII is a membrane-bound pigment–protein complex that generates a solar light-induced charge separation in order to oxidize water and reduce plastoquinone. The charge separation is initiated by the primary chlorophyll-a electron donor, P680, forming a powerful oxidant, P680+, that advances the oxidation state of the Mn4CaO5 cluster in the OEC (Fig. 1) via a redox-active tyrosine, YZ, that mediates electron transfer from the OEC to P680+. The overall four-electron process leads to the formation of O2 that is released as a byproduct and has accumulated in the biosphere and the release of protons that contribute to the trans-membrane proton motive force.4 The availability of O2 as an electron sink has since powered oxygenic respiration to give rise to Earth's current biological diversity.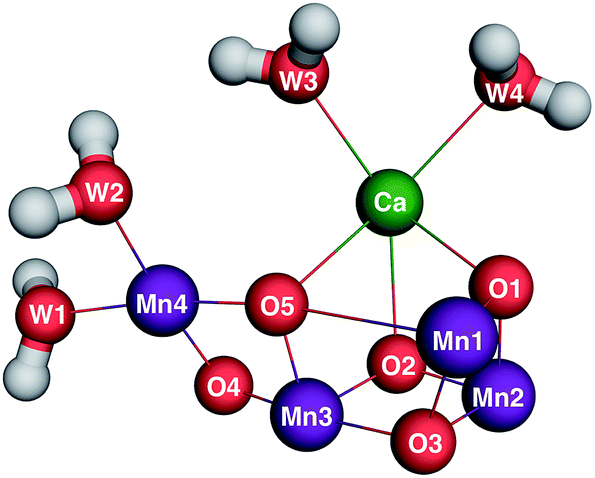 | ||
| Fig. 1 Quantum mechanics/molecular mechanics (QM/MM) optimized S1 structure of the OEC5 based on the 1.9 Å resolution crystal structure by Shen and coworkers.6 | ||
The catalytic cycle of the OEC involves five metastable redox intermediates known as Si states (i = 0–4), as first described by Kok and coworkers.7,8 The dark-stable intermediate, S1, contains the Mn oxidation state pattern (III,IV,IV,III)9 (order of oxidation states is based on the numbering of the Mn ions in Fig. 1) and is converted to S2 upon the loss of one electron. S2 is present as two spin isomers with oxidation states of (III,IV,IV,IV) (S = 1/2) or (IV,IV,IV,III) (S = 5/2).10 The next oxidation event results in the loss of one proton and one electron from the OEC to form S3, in which all four Mn ions are in the +4 oxidation state (S = 3).11,12 Oxidation of YZ following the next charge separation causes release of a proton from S3 to form a modified YZ˙ S3′ state,13 which then forms the S4 state. The structures of S3′ and S4 are poorly understood. S4 spontaneously produces O2, binds substrate water(s), and releases a proton to form S0, thus resetting the catalytic cycle.
The general structure of the OEC has been revealed through a series of X-ray diffraction (XRD) studies.6,14–17 However, the atomic structure of any single Kok cycle intermediate has not been determined through these experiments. Early XRD structures of the OEC suffered from X-ray induced reduction of the high-valent Mn centers.18 Nevertheless, the availability of metal–metal distances from extended X-ray absorption fine structure (EXAFS) spectroscopy19,20 allowed computational studies (QM/MM and density functional theory (DFT)) to refine XRD models to structures corrected for radiation damage.5,21 At present, the PSII research community is in general (but not universal3) agreement on the structures of the S0,5 S1,5,10 and S210 intermediates and the Mn oxidation states of all S states.9
The problem of X-ray induced Mn reduction can be completely avoided by using femtosecond pulses from X-ray free electron laser (XFEL) radiation sources.22,23 XFEL studies using PSII nanocrystals have produced structures of dark-adapted PSII at 6.5–4.9 Å resolution.24–27 Structures of S2,25,26 S3,26,27 and S026 have also been reported at low resolution, which limits their interpretation. However, QM/MM methods combined with electron density map analysis have shown that the S1 → S2 transition involves significant displacement (and oxidation) of Mn4 (also referred to as the “dangler” Mn).28 A 1.95 Å resolution XFEL structure of dark-adapted PSII was recently reported by Shen and coworkers.29 However, the resulting XRD model of the OEC does not agree well with EXAFS data of S1,19 which is likely the result of inaccuracy in positioning of the bridging oxo ligands and significant accumulation of S0.30
The S4 state decays faster than it is formed and, therefore, cannot be observed as a kinetic intermediate. Consequently, no direct experimental evidence for the nature of O–O bond formation in S4 has been produced. However, insights from inorganic and computational chemistry have produced two competing mechanisms: water-nucleophile attack and oxo–oxyl radical coupling.
In the oxo–oxyl radical coupling mechanism,31–33 a Mn(IV)–oxyl radical reacts with a Mn-bridging oxo to generate O2 (Fig. 2A). Extensive calculations by Siegbahn suggest that one of the substrate waters first binds during the S2 → S3 transition and then is oxidized to an oxyl radical in S4 to carry out the reaction.32
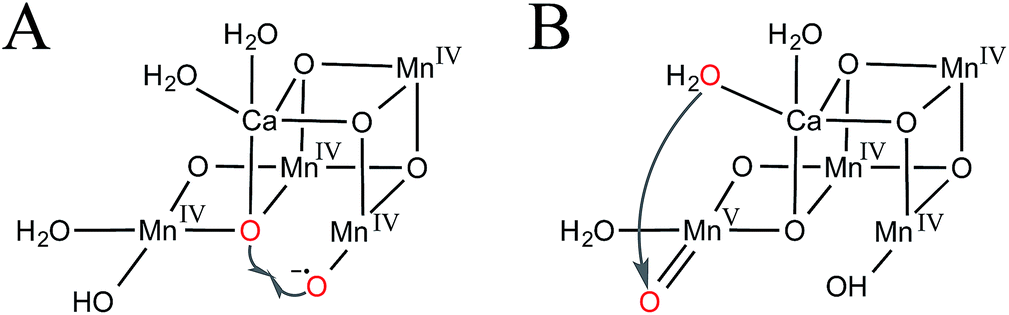 | ||
| Fig. 2 Proposed mechanisms of O–O bond formation in the S4 intermediate. (A) Oxo–oxyl radical coupling and (B) water-nucleophile attack. Substrate waters are shown in red. | ||
A water-nucleophile attack mechanism for the OEC34–37 is most consistent with synthetic water oxidation catalysts.38 As shown in Fig. 2B, such a mechanism would involve a terminal water-nucleophile (preferably bound to calcium, see discussion below) and a formally Mn(V)–oxo electrophile.
These mechanisms can be distinguished by the positions of the substrate waters involved. Kinetics of substrate water exchange throughout the Kok cycle have been determined by membrane inlet mass spectrometry (MIMS) measurements following rapid mixing of PSII with H218O. This technique, which was pioneered in the Wydrzynski laboratory, allows 34O2 and 36O2 release by PSII in specific S states (advanced by single-turnover flashes) to be monitored as a function of incubation time with H218O.39,40 The resulting rates of substrate water exchange are summarized in Table 1. In S0, only one slowly exchanging water (Ws) is resolved with a rate constant of approximately 10 s−1. The exchange rate of Ws is then dramatically slowed to 0.02 s−1 in S1. In S2 and S3, two phases of substrate water exchange kinetics are resolved; the slower and faster exchanging waters are referred to as Ws and Wf, respectively. Ws exchanges at a nearly identical rate in S2 and S3, but the rate is much faster than in S1. Wf exchanges at decreasing rates in S1, S2, and S3.
| k s, s−1 | k f, s−1 | |
|---|---|---|
| S0 | ∼10 | >120 |
| S1 | ∼0.02 | >120 |
| S2 | ∼2 | ∼120 |
| S3 | ∼2 | ∼40 |
Determining the molecular mechanism of photosynthetic water oxidation is crucial for applying the principles of Nature's design to synthetic systems for solar fuel production. In the following analysis, we detail models for substrate binding and O–O bond formation in the OEC in light of experimental evidence and comparisons to well-studied inorganic systems.
2. Where are the substrate binding sites?
Kinetics and mechanism of terminal water vs. μ-oxo ligand exchange from model chemistry
Given our current understanding of the structure of the OEC in the S0, S1, and S2 states, we can predict that the substrate waters that give rise to O2 are bound either as terminal ligands to Mn4 and Ca2+, or as μ-oxo/hydroxo bridges. In order to understand the substrate water exchange kinetics of the OEC, we first look to inorganic model systems.The exchange rate of a terminal water ligand on Mn depends greatly on the oxidation state, protonation, ancillary ligands, and geometry. As shown in Table 2, both the water exchange rate and pKa of a terminal aqua ligand decrease dramatically with increasing oxidation state from Mn2+ to Mn3+ to Mn4+. The hexaaqua Mn(IV) complex has an especially slow water ligand exchange rate (<10−4 s−1), as expected for a high-spin d3 ion with octahedral geometry for which the ligand field stabilization energy disfavors ligand dissociation leading to very slow ligand exchange. However, exchange of the terminal water ligands of the [Mn4IV,IV,IV,IV(μ-O)5(terpy)4(H2O)2]6+ (terpy = 2,2′:6′,2′′-terpyridine) complex occurs faster than the mixing time for the mass spectrometry measurement (∼10 s, kex > 10−1 s−1), despite the high-spin d3 configuration of the Mn(IV) ions. This can be explained by the reduced symmetry of the Mn(IV) ions in the tetrameric oxomanganese–terpy complex. Deviations from octahedral geometry of the MnIV centers resulting from the <90° bite angle of the tridentate terpy ligand and the asymmetry associated with oxo and pyridyl ligands result in loss of degeneracy of the t2g and eg orbitals, and can lower the barrier for exchange of the terminal water ligands. Indeed, computational studies have found low reaction barriers for the dissociative exchange of terminal waters in μ-O bridged Mn(IV) dimers.42
| pKa | Reference | k ex, s−1 | Reference | |
|---|---|---|---|---|
| a mes-terpy = 4′-mesityl-2,2′:6′,2′′-terpyridine. b terpy = 2,2′:6′,2′′-terpyridine. c L = 2-hydroxy-1,3-bis(3,5-X2-salicylideneamino)propane. d nd = not determined. | ||||
| [Ca(H2O)6]2+ | 12.8 | 43 | ∼108 | 44 |
| [Sr(H2O)6]2+ | 13.2 | 43 | ∼109 | 44 |
| [Mn(H2O)6]2+ | 10.6 | 45 | 2 × 107 | 46 |
| 1 × 107 | 47 | |||
| [Mn(H2O)6]3+ | 0.7 | 45 | 2 × 103 | 47 |
| 0.01–1 | 39 and 48 | |||
| [Mn(H2O)6]4+ | <0 | 10−6 to 10−4 | 39 | |
| 10−7 to 10−8 | 48 | |||
| [Mn2III,IV(μ-O)2(mes-terpy)2(H2O)2]3+a | ndd | >10−1 | 49 | |
| [Mn4IV,IV,IV,IV(μ-O)5(terpy)4(H2O)2]6+b | nd | >10−1 | 49 | |
| [Mn2IV,IV(μ-O)2(terpy)2(H2O)2]4+ | 1.8 | 50 | nd | |
| [Mn2III,III(L)2(H2O)]+c | 19–20 | 51 | nd | |
| [Mn2III,IV(L)2(H2O)]2+c | 10–11 | 51 | nd | |
Generally, ligand exchange for high-valent first-row transition ions involves a dissociative mechanism, especially for Mn(IV) which is normally six-coordinate and has a small ionic radius that makes an associative mechanism unfavorable. For terminal water ligands, dissociation involves removing a polar but neutral ligand from the cationic metal center, which has a modest barrier.52 However, dissociative exchange of a μ-oxo ligand involves removing an anionic ligand from the cationic metal center, which has a much higher barrier. Therefore, the exchange of μ-oxo ligands with bulk water is thermodynamically more challenging than the exchange of terminal water ligands.
The mechanism of μ-oxo exchange has been investigated for di-μ-oxo di-Mn(III,IV) complexes.53 These studies have shown that μ-oxo exchange requires protonation of the oxo before dissociation and exchange; therefore, μ-oxo pKas are good predictors of their rates of exchange. The μ-oxo pKa decreases dramatically with increasing Mn oxidation state in Mn model complexes (Table 3). The rate of μ-oxo exchange is very fast for Mn(III,III) dimers, owing to the high pKa of the μ-oxo ligands. Indeed, di-μ-oxo di-Mn(III,III) complexes are generally not stable to hydrolysis. For di-μ-oxo di-Mn(III,IV) complexes, the rate of μ-oxo exchange is on a timescale of minutes (t1/2 = 21 min for the [Mn2III,IV(μ-O)2(bpy)4]3+ complex, bpy = 2,2′-bipyridine).53 However, no μ-oxo exchange could be detected on a one-day timescale for all-Mn(IV) complexes, such as [Mn4IV,IV,IV,IV(μ-O)5(terpy)4(H2O)2]6+, reflecting the very low pKa of the μ-oxo ligands in all-Mn(IV) complexes (Table 3).
The [Mn2III,IV(μ-O)2(terpy)2(H2O)2]3+ complex contains terminal water ligands adjacent to the μ-oxo ligands. It has been found that the terminal water ligands accelerate the rate of μ-oxo exchange.53 Because the pKa of the water ligand bound to a high-valent Mn center is lower than solvent water (Table 2), a μ-oxo can be more readily protonated by ligated water, which opens up the μ-oxo bridge and lowers the energetic barrier for dissociation.53 In this mechanism, the terminal water and μ-oxo are in dynamic equilibrium with bulk water. For Mn dimers without terminal water ligands, the exchange is much slower, such as in [Mn2III,IV(μ-O)2(bpy)4]3+, because intramolecular proton transfer between terminal-bound water and μ-oxo bridges is eliminated.53
There have also been measurements of terminal water and μ-oxo ligand exchange of the di-Mn center in Mn-catalase.54 The active site of Mn-catalase contains a redox-active di-Mn center for which ligand exchange has been studied in the di-μ-oxo di-Mn(III,IV) state. It was found that the terminal water ligand exchanges within the mixing time of one minute, but the μ-oxo ligands require over one hour for exchange. These results parallel those obtained for Mn model complexes in solution, although the exchange rate of the μ-oxo ligands is much lower in the protein active site.
In summary, electrostatics play a dominant role in the dissociative ligand exchange reactions of high-valent oxo–manganese complexes. There is a large barrier for μ-oxo exchange due to the high negative charge of the oxo ligand. Thus, protonation of the μ-oxo ligand is required to promote ligand exchange. Because of the large pKa shift of the μ-oxo ligands between di-μ-oxo di-Mn(III,IV) and di-μ-oxo di-Mn(IV,IV) complexes, μ-oxo ligands exchange at a modest rate for di-μ-oxo di-Mn(III,IV) complexes but do not exchange for di-μ-oxo di-Mn(IV,IV) centers. On the other hand, terminal water ligands exhibit fast rates of ligand exchange for both Mn(III) and Mn(IV) centers. Moreover, the rate of terminal water ligand exchange is similar for Mn(III) and Mn(IV) because both have similar electrostatic potential (ESP) charges.52
Ammonia does not compete with substrate water
The water analog ammonia inhibits the OEC by binding to two sites in PSII. The first is in the outer coordination sphere of the OEC and upon binding of ammonia, alters the hydrogen-bonding networks in such a way that the S2 state S = 5/2 isomer is favored.56 The binding of ammonia to this site is competitive with chloride57,58 and can be accessed by larger amines,56,58 but its specific location has not been resolved.Ammonia also binds directly to Mn in the OEC in the S2 state resulting in the formation of an altered multiline g = 2 EPR signal.59 However, the substrate water exchange kinetics of S2 do not change when ammonia is bound.60 Therefore, the binding site of ammonia cannot be the binding site of a substrate water.
Lubitz and coworkers have proposed that ammonia binds to Mn4 trans to O5 based on 17O-ELDOR-detected NMR (EDNMR) measurements.60 In these experiments, dark-adapted PSII (poised in S1) is incubated with H217O for tens of minutes and perturbations in the 17O-EDNMR signal are detected when ammonia (as NH4Cl at pH 7.6) is present following advancement to the S2 state by 200 K illumination and subsequent annealing at 260 K. In accompanying 1H-ENDOR measurements, no change in the proton environment around the OEC was observed.60
Other lines of evidence have suggested that ammonia binds as a bridging ligand between two Mn ions. This motif was first proposed by Britt and coworkers based on ESEEM measurements of the ammonia-bound S2 state in higher plant PSII.61 The quadrupole coupling between the S = 1/2 S2 state of the OEC and bound 14NH3 was resolved as 1.61 MHz with η = 0.59. A very similar coupling was observed in cyanobacterial PSII (1.52 MHz, η = 0.47).60 For comparison, a nitrogen nucleus with purely axial symmetry is characterized by η = 0, while ammonia (amino) ligands display η ≤ 0.3. Therefore, the measured quadrupole coupling of ammonia to S2 is highly deviant from axial symmetry and could represent a deprotonated bridging ligand such as an imido (NH2−) or nitrido (N3−). However, Britt and coworkers have very recently revealed the origin of this anisotropy. In wild type cyanobacterial PSII, they measured the 14NH3 coupling as 1.62 MHz with η = 0.40, but in a D1-D61A mutant, the interaction was nearly completely axial (1.54 MHz, η = 0.04).62 This work provides convincing evidence that bound ammonia has a strong hydrogen bond to Asp61 and is, therefore, a terminal ligand to Mn4.
In a recent study using QM/MM methods and EXAFS simulations, we have proposed that instead of replacing a terminal water or μ-oxo group, ammonia binds as a sixth ligand to Mn4 resulting in a complete octahedral coordination sphere.63 In this model (Fig. 3), ammonia binds to the S = 5/2 S2 state but then induces a redox switch stabilizing the S = 1/2 S2 state by protonating O5 and deprotonating W2. This additional ligand would lower the reduction potential of ammonia-bound S2 as has been observed by both thermoluminescence64 and flash O2 measurements.65
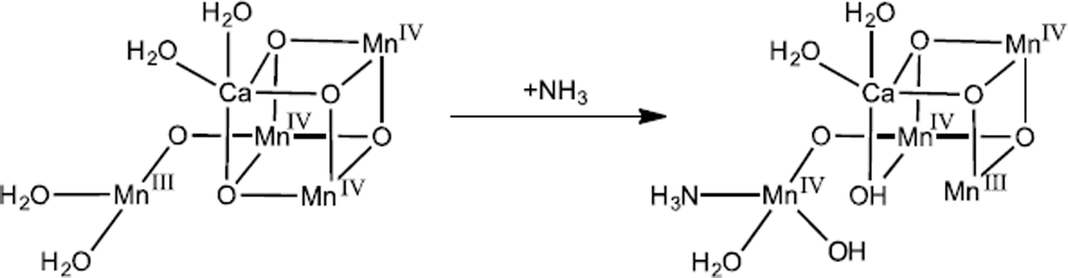 | ||
| Fig. 3 Proposed mechanism of ammonia binding in the S2 intermediate. Ammonia binds as an additional ligand to the dangler Mn in the S = 5/2 S2 state, which induces a redox switch to the S = 1/2 state. Adapted from ref. 63. | ||
While both substrate waters are already bound in S2,41 an additional water may bind during the S2→S3 transition as computationally predicted.32 We have proposed that ammonia, a “harder” Lewis base than water, binds in S2 to the dangler Mn4 in an analogous site as water binds in S3 to the dangler Mn4.63 The addition of a ligand to Mn4 in either S2 or S3trans to O5 would cause the other terminal water ligands (W1 and W2) to shift their positions towards the cuboidal core of the OEC. In this model of ammonia binding, both water–nucleophile attack and oxo–oxyl radical coupling O–O bond formation mechanisms are feasible.
A model for substrate water exchange in the OEC
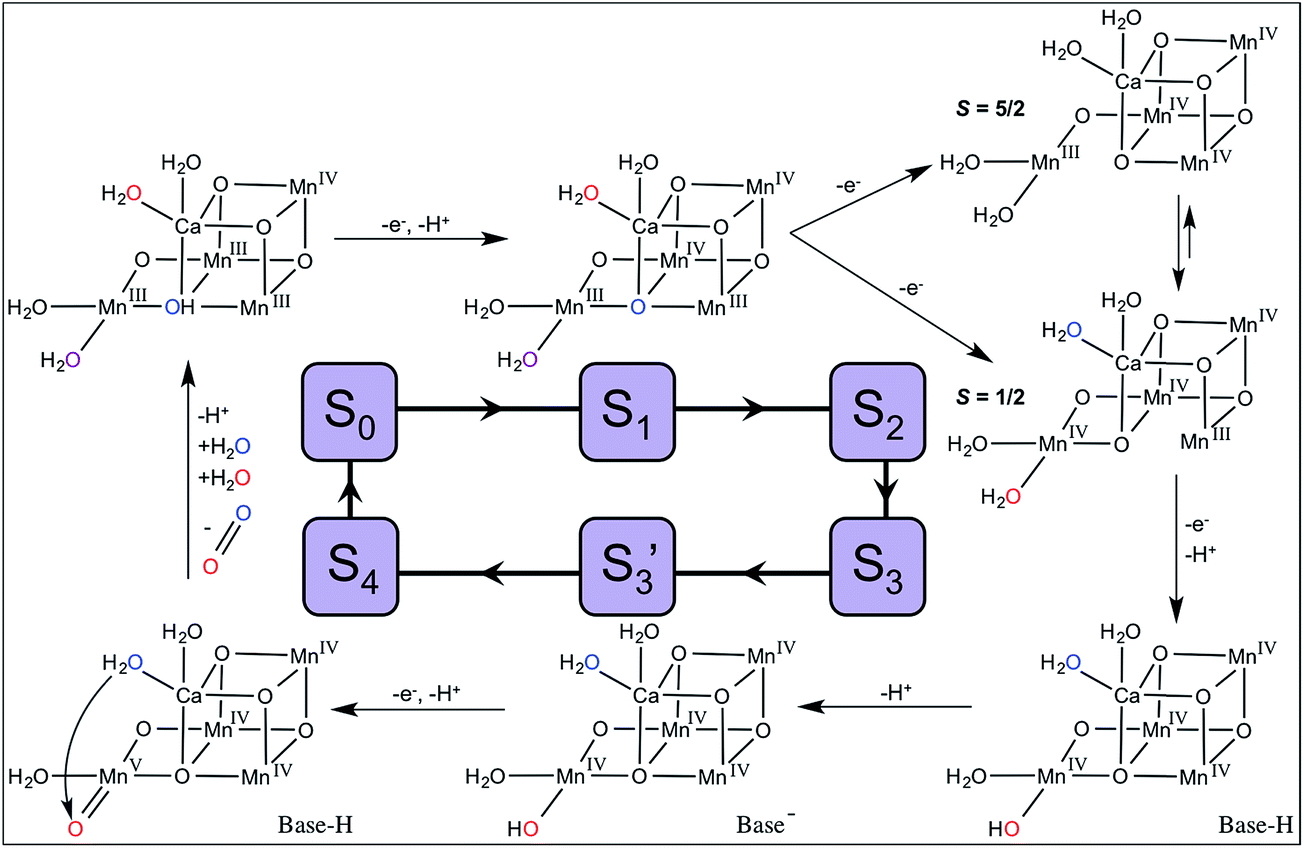 | ||
| Fig. 4 Proposed mechanism of OEC turnover and substrate water exchange. Ws is shown in blue and Wf in red (in S0 and S1, W2 is shown in purple as it contributes to both the Ws and Wf kinetics). | ||
For the water-nucleophile attack mechanism, the substrate waters are W2 and W3. W3, which is bound to Ca2+, is also expected to exchange rapidly (specifically in S0 and S1, see discussion below). Therefore, the Wf kinetic (measured by monitoring 34O2) may reflect contributions from both W2 and W3.
The exchange rate of W2 is first resolved after the S1 → S2 transition with a value in the S2 state of kf ∼120 s−1. As discussed above, S2 is present as two spin isomers. Given the changes seen in XFEL experiments at the Mn4 position measured at physiological temperature, we have asserted that the S = 1/2 isomer is dominant.28 Therefore, Wf would be bound as a terminal Mn4+-aqua species in the S2 state and exchanges at a rate comparable to those measured in model complexes (Table 2).
As modeled in Fig. 4, the formation of S3 involves the release of a proton from W2 and a 3-fold slowing of its exchange rate (Table 1). This change is qualitatively consistent with the differences expected between Mn4+-aqua and Mn4+-hydroxo species.
It has been determined that only one of the μ-oxo bridges denoted O5 slowly exchanges with H217O in the dark-stable S1 state over tens of seconds.54,69 We assume that O5 also exchanges with bulk water in S0 (oxidation states III,IV,III,III). QM/MM models of the S0 state5 predict that O5 is protonated, as expected if its pKa is compared to Mn(III,III) dimers (Table 3). Consequently, O5 exchange would be expected to occur relatively rapidly in S0 (10 s−1) because it is already protonated at physiological pH. During exchange, protonated O5 may dissociate from Mn3 becoming a terminal water ligand to Mn4. Thus, in the S0 state, W2 (purple, Fig. 4) and O5 (blue, Fig. 4) are expected to be in rapid equilibrium with each other.
In S1 (oxidation states III,IV,IV,III), O5 is present in a chemical environment analogous to Mn(III,IV) dimer complexes and the pKa of O5 in S1 is predicted to be much lower than in S0 (Table 3). Based on QM/MM models, O5 is not protonated in the S1 state5,21 and, thus, would have a higher energetic barrier for exchange. This change is proposed to be the reason for the 500-fold decrease in the exchange rate of Ws observed in S1 as compared to S0.
Wf is not resolved in S0 and S1 and reflects the rapid exchange of W2 (and W3, discussed below). Because μ-oxo exchange involves subsequent dissociation, binding, and association steps,53 isotopically labeled substrate will appear in both the O5 and W2 positions. Therefore, W2 is labeled with two rates. The first is rapid (>120 s−1) and reflects direct exchange with bulk water. The second kinetic is slow (0.02–10 s−1) and reflects the interconversion of labeled O5 with W2. Crucially, this mechanism implies that the rate of Wsexchange is controlled by the rate of O5 exchange in S0and S1, but that O5 is not necessarily a substrate water.
For the dominant S2 state S = 1/2 spin isomer (oxidation states IV,IV,IV,III), the O5 chemical environment is analogous to Mn(IV,IV) dimer complexes in which the μ-oxo pKa is very low (Table 3). Given the large energetic barrier for μ-oxo exchange, the measured rate of Ws in S2 (2 s−1) is too fast to be assigned to O5.
When Ca2+ is substituted with Sr2+, only Ws is significantly affected: ks is three- to four-fold faster in S1, S2, and S3 when Sr2+ is present.71 Both O5 and W3 are adjacent to Ca2+/Sr2+ and are, therefore, reasonable candidates for Ws. Because Ca2+ and Sr2+ have very similar Lewis acidities (similar pKas of the aqua ion, Table 2), W3 is expected to be fully protonated throughout the Kok cycle providing an adequate nucleophile in the water-nucleophile attack mechanism.36 However, Sr2+ is considerably larger than Ca2+ (ionic radii are 1.12 Å and 0.99 Å, respectively72) resulting in a weaker electrostatic interaction between Sr2+ and its ligands. This difference is reflected in the faster exchange rate of the water ligands bound to [Sr(H2O)6]2+ compared to [Ca(H2O)6]2+ (Table 2), and is also consistent with the faster exchange of Ws when Sr2+ is substituted for Ca2+ in the OEC.
In our model for OEC turnover and substrate water exchange shown in Fig. 4, the observed Ws kinetics reflect O5 exchanging with W2 in S0 and S1, and W3 exchange in S2 and S3. The Wf kinetic reflects both W2 and W3 exchange in S0 and S1, and only W2 in S2 and S3. This new substrate exchange model provides a complete description of all of the kinetic phases of substrate exchange throughout the S-state cycle and is consistent with data from model high-valent oxomanganese complexes. It, thus, provides a framework from which to design additional experimental measurements to test and validate this hypothesis.
3. Mechanism of O–O bond formation
The structure of the S4 intermediate will determine if the mechanism of photosynthetic water oxidation involves water-nucleophile attack or oxo–oxyl radical coupling. Unfortunately, few experimental clues about S4 are available. Given that the Mn oxidation states in S3 are (IV,IV,IV,IV), the S4 state is predicted to contain either a MnIV–oxyl radical or MnV–oxo species.A MnIV–oxyl radical species has been suggested through computational studies by Siegbahn and supports an oxo–oxyl radical coupling mechanism. On the other hand, a high-spin MnV–oxo is predicted to serve as the electrophile in a water-nucleophile attack mechanism. Borovik and coworkers have recently reported experimental support for the latter option through studies of [MnVH3buea(O)] (where buea = tris[(N′-tert-butylureaylato)-N-ethylene]aminato).73,74 The buea ligand imposes C3 symmetry around Mn and stabilizes the Mn–oxo group through intramolecular hydrogen bonding. [MnVH3buea(O)] is a high-spin S = 1 system as confirmed by EPR spectroscopy and supported by DFT calculations.73 Therefore, the dxz and dyz orbitals in [MnVH3buea(O)] are degenerate and the Mn–oxo bond is likely to be weaker (and more reactive) than corresponding MnV–oxo species with tetragonal symmetry (S = 0). By quantifying the hyperfine contributions from 17O-labeled [MnVH3buea(O)] using EPR spectroscopy, Borovik and coworkers determined that 0.45 spins reside on the oxo group. Therefore, the MnV–oxo group is strongly covalent, but does not contain an oxyl radical. For [MnIIIH3buea(O)]2−, only 0.30 spins reside on the oxo group, suggesting that the Mn–O bond becomes more strongly covalent (and the unit more electrophilic) with increasing Mn oxidation state.74
To date, no experimental evidence of a MnIV–oxyl radical species has been found, calling into question the accuracy of the oxo–oxyl radical coupling mechanism. However, if a reactive high-spin MnV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species is present in the S4 state, analogous to that found in [MnVH3buea(O)], a water-nucleophile attack mechanism would lead to O–O bond formation.
O species is present in the S4 state, analogous to that found in [MnVH3buea(O)], a water-nucleophile attack mechanism would lead to O–O bond formation.
4. Conclusions
Determining the molecular mechanism of O–O bond formation in photosynthetic water oxidation remains one of the great challenges in bioinorganic chemistry. In the absence of direct experimental evidence of the S4 intermediate, we can predict its structure and, therefore, the mechanism of O–O bond formation by comparing the OEC to model Mn complexes and by determining where the substrate waters bind throughout the catalytic cycle.In our proposed model of OEC turnover shown in Fig. 4, the substrate waters are W2 (terminal water on Mn4) and W3 (terminal water on Ca2+). The substrate water exchange kinetics in S0 and S1 are complicated by the dynamic equilibrium between W2 and the slowly exchanging μ-oxo/hydroxo bridge, O5. This assignment of substrates dictates that O–O bond formation occurs via a water-nucleophilic attack of W3 (as water) on W2 (as a high-spin MnVO species).
Acknowledgements
Funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (Grant no. DE-FG02-05ER15646). We thank Mikhail Askerka and Dr. Leslie Vogt for helpful discussions.References
- S. A. Crowe, L. N. Dossing, N. J. Beukes, M. Bau, S. J. Kruger, R. Frei and D. E. Canfield, Nature, 2013, 501, 535–538 CrossRef CAS PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- D. J. Vinyard, G. M. Ananyev and G. C. Dismukes, Annu. Rev. Biochem., 2013, 82, 577–606 CrossRef CAS PubMed.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Wiley-Blackwell, 2nd edn, 2014 Search PubMed.
- R. Pal, C. F. A. Negre, L. Vogt, R. Pokhrel, M. Z. Ertem, G. W. Brudvig and V. S. Batista, Biochemistry, 2013, 52, 7703–7706 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
- B. Forbush, B. Kok and M. P. McGloin, Photochem. Photobiol., 1971, 14, 307–321 CrossRef CAS PubMed.
- V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed.
- A. Boussac, M. Sugiura, A. W. Rutherford and P. Dorlet, J. Am. Chem. Soc., 2009, 131, 5050–5051 CrossRef CAS PubMed.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804–808 CrossRef CAS PubMed.
- A. Klauss, M. Haumann and H. Dau, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16035–16040 CrossRef CAS PubMed.
- N. Kamiya and J. R. Shen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 98–103 CrossRef CAS PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef CAS PubMed.
- A. Guskov, J. Kern, A. Gabdulkhakov, M. Broser, A. Zouni and W. Saenger, Nat. Struct. Mol. Biol., 2009, 16, 334–342 CAS.
- J. Yano, J. Kern, K. D. Irrgang, M. J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12047–12052 CrossRef CAS PubMed.
- A. Grundmeier and H. Dau, Biochim. Biophys. Acta, 2012, 1817, 88–105 CrossRef CAS PubMed.
- K. Sauer, J. Yano and V. K. Yachandra, Coord. Chem. Rev., 2008, 252, 318–335 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Chem.–Eur. J., 2008, 14, 8290–8302 CrossRef CAS PubMed.
- R. Neutze, R. Wouts, D. van der Spoel, E. Weckert and J. Hajdu, Nature, 2000, 406, 752–757 CrossRef CAS PubMed.
- H. N. Chapman, P. Fromme, A. Barty, T. A. White, R. A. Kirian, A. Aquila, M. S. Hunter, J. Schulz, D. P. DePonte, U. Weierstall, R. B. Doak, F. R. N. C. Maia, A. V. Martin, I. Schlichting, L. Lomb, N. Coppola, R. L. Shoeman, S. W. Epp, R. Hartmann, D. Rolles, A. Rudenko, L. Foucar, N. Kimmel, G. Weidenspointner, P. Holl, M. Liang, M. Barthelmess, C. Caleman, S. Boutet, M. J. Bogan, J. Krzywinski, C. Bostedt, S. Bajt, L. Gumprecht, B. Rudek, B. Erk, C. Schmidt, A. Homke, C. Reich, D. Pietschner, L. Struder, G. Hauser, H. Gorke, J. Ullrich, S. Herrmann, G. Schaller, F. Schopper, H. Soltau, K.-U. Kuhnel, M. Messerschmidt, J. D. Bozek, S. P. Hau-Riege, M. Frank, C. Y. Hampton, R. G. Sierra, D. Starodub, G. J. Williams, J. Hajdu, N. Timneanu, M. M. Seibert, J. Andreasson, A. Rocker, O. Jonsson, M. Svenda, S. Stern, K. Nass, R. Andritschke, C.-D. Schroter, F. Krasniqi, M. Bott, K. E. Schmidt, X. Wang, I. Grotjohann, J. M. Holton, T. R. M. Barends, R. Neutze, S. Marchesini, R. Fromme, S. Schorb, D. Rupp, M. Adolph, T. Gorkhover, I. Andersson, H. Hirsemann, G. Potdevin, H. Graafsma, B. Nilsson and J. C. H. Spence, Nature, 2011, 470, 73–77 CrossRef CAS PubMed.
- J. Kern, R. Alonso-Mori, J. Hellmich, R. Tran, J. Hattne, H. Laksmono, C. Glöckner, N. Echols, R. G. Sierra, J. Sellberg, B. Lassalle-Kaiser, R. J. Gildea, P. Glatzel, R. W. Grosse-Kunstleve, M. J. Latimer, T. A. McQueen, D. DiFiore, A. R. Fry, M. Messerschmidt, A. Miahnahri, D. W. Schafer, M. M. Seibert, D. Sokaras, T.-C. Weng, P. H. Zwart, W. E. White, P. D. Adams, M. J. Bogan, S. Boutet, G. J. Williams, J. Messinger, N. K. Sauter, A. Zouni, U. Bergmann, J. Yano and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9721–9726 CrossRef CAS PubMed.
- J. Kern, R. Alonso-Mori, R. Tran, J. Hattne, R. J. Gildea, N. Echols, C. Glöckner, J. Hellmich, H. Laksmono, R. G. Sierra, B. Lassalle-Kaiser, S. Koroidov, A. Lampe, G. Han, S. Gul, D. DiFiore, D. Milathianaki, A. R. Fry, A. Miahnahri, D. W. Schafer, M. Messerschmidt, M. M. Seibert, J. E. Koglin, D. Sokaras, T.-C. Weng, J. Sellberg, M. J. Latimer, R. W. Grosse-Kunstleve, P. H. Zwart, W. E. White, P. Glatzel, P. D. Adams, M. J. Bogan, G. J. Williams, S. Boutet, J. Messinger, A. Zouni, N. K. Sauter, V. K. Yachandra, U. Bergmann and J. Yano, Science, 2013, 340, 491–495 CrossRef CAS PubMed.
- J. Kern, R. Tran, R. Alonso-Mori, S. Koroidov, N. Echols, J. Hattne, M. Ibrahim, S. Gul, H. Laksmono, R. G. Sierra, R. J. Gildea, G. Han, J. Hellmich, B. Lassalle-Kaiser, R. Chatterjee, A. S. Brewster, C. A. Stan, C. Glöckner, A. Lampe, D. DiFiore, D. Milathianaki, A. R. Fry, M. M. Seibert, J. E. Koglin, E. Gallo, J. Uhlig, D. Sokaras, T.-C. Weng, P. H. Zwart, D. E. Skinner, M. J. Bogan, M. Messerschmidt, P. Glatzel, G. J. Williams, S. Boutet, P. D. Adams, A. Zouni, J. Messinger, N. K. Sauter, U. Bergmann, J. Yano and V. K. Yachandra, Nat. Commun., 2014, 5, 4371 CAS.
- C. Kupitz, S. Basu, I. Grotjohann, R. Fromme, N. A. Zatsepin, K. N. Rendek, M. S. Hunter, R. L. Shoeman, T. A. White, D. Wang, D. James, J.-H. Yang, D. E. Cobb, B. Reeder, R. G. Sierra, H. Liu, A. Barty, A. L. Aquila, D. Deponte, R. A. Kirian, S. Bari, J. J. Bergkamp, K. R. Beyerlein, M. J. Bogan, C. Caleman, T.-C. Chao, C. E. Conrad, K. M. Davis, H. Fleckenstein, L. Galli, S. P. Hau-Riege, S. Kassemeyer, H. Laksmono, M. Liang, L. Lomb, S. Marchesini, A. V. Martin, M. Messerschmidt, D. Milathianaki, K. Nass, A. Ros, S. Roy-Chowdhury, K. Schmidt, M. Seibert, J. Steinbrener, F. Stellato, L. Yan, C. Yoon, T. A. Moore, A. L. Moore, Y. Pushkar, G. J. Williams, S. Boutet, R. B. Doak, U. Weierstall, M. Frank, H. N. Chapman, J. C. H. Spence and P. Fromme, Nature, 2014, 513, 261–265 CrossRef CAS PubMed.
- M. Askerka, J. Wang, G. W. Brudvig and V. S. Batista, Biochemistry, 2014, 53, 6860–6862 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Nature, 2014, 517, 99–103 CrossRef PubMed.
- M. Askerka, D. J. Vinyard, J. Wang, G. W. Brudvig and V. S. Batista, Biochemistry, 2015, 54, 1713–1716 CrossRef CAS PubMed.
- V. K. Yachandra, K. Sauer and M. P. Klein, Chem. Rev., 1996, 96, 2927–2950 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Biochim. Biophys. Acta, 2013, 1827, 1003–1019 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Chem.–Eur. J., 2006, 12, 9217–9227 CrossRef CAS PubMed.
- J. Messinger, M. Badger and T. Wydrzynski, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3209–3213 CrossRef CAS.
- V. L. Pecoraro, M. J. Baldwin, M. T. Caudle, W.-Y. Hsieh and N. A. Law, Pure Appl. Chem., 1998, 70, 925–929 CrossRef CAS.
- G. W. Brudvig, Philos. Trans. R. Soc., B, 2008, 363, 1211–1219 CrossRef CAS PubMed.
- V. A. Szalai, D. A. Stone and G. W. Brudvig, in Photosynthesis: Mechanisms and Effects, vol. 2, ed. G. Garab, Kluwer Academic, Dordrecht, The Netherlands, 1998, pp. 1403–1406 Search PubMed.
- S. Romain, L. Vigara and A. Llobet, Acc. Chem. Res., 2009, 42, 1944–1953 CrossRef CAS PubMed.
- W. Hillier and T. Wydrzynski, Biochim. Biophys. Acta, 2001, 1503, 197–209 CrossRef CAS.
- W. Hillier and T. Wydrzynski, Coord. Chem. Rev., 2008, 252, 306–317 CrossRef CAS PubMed.
- W. Hillier and T. Wydrzynski, Phys. Chem. Chem. Phys., 2004, 6, 4882–4889 RSC.
- M. Lundberg, M. R. A. Blomberg and P. E. M. Siegbahn, Theor. Chem. Acc., 2003, 110, 130–143 CrossRef CAS.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw Hill Book Co., New York, 1985 Search PubMed.
- J. Burgess, Ions in Solution: Basic Principles of Chemical Interactions, Halsted Press, New York, 1988 Search PubMed.
- D. T. Richens, The Chemistry of Aqua Ions, Wiley, Chichester, U.K., 1997 Search PubMed.
- Y. Ducommun, K. E. Newman and A. E. Merbach, Inorg. Chem., 1980, 19, 3696–3703 CrossRef CAS.
- F. P. Rotzinger, J. Am. Chem. Soc., 1997, 119, 5230–5238 CrossRef CAS.
- D. Kuzek and R. J. Pace, Biochim. Biophys. Acta, 2001, 1503, 123–137 CrossRef CAS.
- R. Tagore, H. Y. Chen, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2006, 128, 9457–9465 CrossRef CAS PubMed.
- C. W. Cady, K. E. Shinopoulos, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2010, 39, 3985–3989 RSC.
- M. T. Caudle and V. L. Pecoraro, J. Am. Chem. Soc., 1997, 119, 3415–3416 CrossRef CAS.
- E. M. Sproviero, K. Shinopoulos, J. A. Gascón, J. P. McEvoy, G. W. Brudvig and V. S. Batista, Philos. Trans. R. Soc., B, 2008, 363, 1149–1156 CrossRef CAS PubMed.
- R. Tagore, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2007, 46, 2193–2203 CrossRef CAS PubMed.
- I. L. McConnell, V. M. Grigoryants, C. P. Scholes, W. K. Myers, P.-Y. Chen, J. W. Whittaker and G. W. Brudvig, J. Am. Chem. Soc., 2012, 134, 1504–1512 CrossRef CAS PubMed.
- M. Amin, L. Vogt, S. Vassiliev, I. Rivalta, M. M. Sultan, D. Bruce, G. W. Brudvig, V. S. Batista and M. R. Gunner, J. Phys. Chem. B, 2013, 117, 6217–6226 CrossRef CAS PubMed.
- W. F. Beck and G. W. Brudvig, Biochemistry, 1986, 25, 6479–6486 CrossRef CAS.
- P. O. Sandusky and C. F. Yocum, Biochim. Biophys. Acta, 1984, 766, 603–611 CrossRef CAS.
- P. O. Sandusky and C. F. Yocum, Biochim. Biophys. Acta, 1986, 849, 85–93 CrossRef CAS.
- W. F. Beck, J. C. de Paula and G. W. Brudvig, J. Am. Chem. Soc., 1986, 108, 4018–4022 CrossRef CAS.
- M. Pérez Navarro, W. M. Ames, H. Nilsson, T. Lohmiller, D. A. Pantazis, L. Rapatskiy, M. M. Nowaczyk, F. Neese, A. Boussac, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15561–15566 CrossRef PubMed.
- R. D. Britt, J. L. Zimmermann, K. Sauer and M. P. Klein, J. Am. Chem. Soc., 1989, 111, 3522–3532 CrossRef CAS.
- P. H. Oyala, T. A. Stich, R. J. Debus and R. D. Britt, J. Am. Chem. Soc., 2015, 137, 8829–8837 CrossRef CAS PubMed.
- M. Askerka, D. J. Vinyard, G. W. Brudvig and V. S. Batista, Biochemistry, 2015, 54, 5783–5786 CrossRef CAS PubMed.
- T.-A. Ono and Y. Inoue, Arch. Biochem. Biophys., 1988, 264, 82–92 CrossRef CAS.
- D. J. Vinyard and G. W. Brudvig, Biochemistry, 2015, 54, 622–628 CrossRef CAS PubMed.
- H. Nilsson, F. Rappaport, A. Boussac and J. Messinger, Nat. Commun., 2014, 5, 4305 CAS.
- N. Cox and J. Messinger, Biochim. Biophys. Acta, 2013, 1827, 1020–1030 CrossRef CAS PubMed.
- H. Nilsson, T. Krupnik, J. Kargul and J. Messinger, Biochim. Biophys. Acta, 2014, 1837, 1257–1262 CrossRef CAS PubMed.
- L. Rapatskiy, N. Cox, A. Savitsky, W. M. Ames, J. Sander, M. M. Nowaczyk, M. Rögner, A. Boussac, F. Neese, J. Messinger and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 16619–16634 CrossRef CAS PubMed.
- A. Klauss, M. Haumann and H. Dau, J. Phys. Chem. B, 2015, 119, 2677–2689 CrossRef CAS PubMed.
- G. Hendry and T. Wydrzynski, Biochemistry, 2003, 42, 6209–6217 CrossRef CAS PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press/Taylor and Francis, Boca Raton, FL, 88th (Internet Version 2008) edn, 2008 Search PubMed.
- T. Taguchi, R. Gupta, B. Lassalle-Kaiser, D. W. Boyce, V. K. Yachandra, W. B. Tolman, J. Yano, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2012, 134, 1996–1999 CrossRef CAS PubMed.
- R. Gupta, T. Taguchi, B. Lassalle-Kaiser, E. L. Bominaar, J. Yano, M. P. Hendrich and A. S. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5319–5324 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |