 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon dioxide capture and utilization: using dinuclear catalysts to prepare polycarbonates†
N.
Yi
,
J.
Unruangsri
,
J.
Shaw
and
C. K.
Williams
*
Department of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: c.k.williams@imperial.ac.uk
First published on 6th October 2015
Abstract
The copolymerization of epoxides, including cyclohexene oxide and vinyl-cyclohexene oxide with carbon dioxide are presented. These processes are catalyzed using a homogeneous di-zinc complex that shows good activity and very high selectivities for polycarbonate polyol formation. The polymerizations are investigated in the presence of different amounts of exogenous reagents, including water, diols and diamines, as models for common contaminants in any carbon dioxide capture and utilization scenario.
Introduction
Using carbon dioxide to make useful products, including chemicals and fuels, is an attractive way to add value to a common waste gas and could be a useful method to reduce pollution.1–4 From a practical, implementation focused perspective, there are two primary considerations for any carbon capture and usage process: (1) the overall carbon dioxide emission must be reduced, which sounds obvious but requires careful consideration of the overall energies of both the process and embedded in the preparation of co-reagents; and (2) the product must be needed and viable, both in terms of the potential market volume and value.Amongst the myriad of possible carbon dioxide transformations, the production of polymers offers significant potential.5–10 Such polymers are prepared by the ring-opening copolymerization (ROCOP) of carbon dioxide and epoxides (Scheme 1), a process which requires a catalyst. The reaction has attracted considerable attention, with a number of different catalysts and processes being reported by both academic and industrial researchers worldwide.5–18
 | ||
| Scheme 1 Illustrates the ring opening copolymerizations using cyclohexene oxide (CHO) and vinyl-cyclohexene oxide (v-CHO). | ||
The products of the reaction are aliphatic polycarbonates; at low molecular weights these polymers are suitable replacements for petrochemical polyols used in polyurethane manufacturing.19,20 In the context of carbon dioxide reduction, the replacement of polyether polyols by poly(ether carbonate) polyols leads to significant reductions in greenhouse (GHG) emissions (∼20%).21 By selecting the correct catalyst, it is possible to substitute >40% of the epoxide with carbon dioxide. Such a substitution is attractive both economically and environmentally as it may further reduce GHG emissions. Thus, the first criterion can be met by the production of polymers from carbon dioxide.
The second criterion, that the products are valuable and needed/used on a large-scale, is also met by polymer production. Considering only polyols, there are currently >8 Mt of polyurethanes produced globally requiring 3–4 Mt of polyols in their manufacture.19,20 Therefore, the production of polymers from CO2 has the potential to offer a viable industrial route to add value to waste emissions. The process is critically dependent on the selection of the catalyst, with a range of very promising heterogeneous and homogeneous catalysts having been reported.5–18 However, so far there remain far fewer studies on the tolerance of such catalysts to a range of impurities, which are likely to be present in any carbon capture and usage scenario.22,23 We recently reported that our dinuclear metal catalysts are tolerant to a wide range of impurities found in captured carbon dioxide, including nitrogen, oxygen, oxides of carbon, NOx and SOx.23 Furthermore, using di-magnesium catalysts a wide-ranging study of common contaminants was undertaken which revealed remarkable catalyst stability, even in the presence of large amounts of contaminants.23 In that study, carbon dioxide captured from a UK power station was applied and showed near-equivalent performance compared to pure carbon dioxide.23
Our group have reported a series of homogeneous catalysts for CO2/epoxide copolymerization, based on dinuclear complexes of Zn(II), Mg(II), Co(II/III) and Fe(III).24–35 This discussion paper will focus on a di-zinc bis(trifluoroacetate) complex28 and the influence of the addition of known quantities of water, alcohols and amines to the polymerizations. The additives and monomers are investigated both to test tolerance of the catalyst and also to target particular polymer chain functionalities (end-groups, side-chain substituents) so as to widen the scope, and ultimately the scale, of future applications.
Results and discussion
The preparation and structure of catalyst 1 is shown in Scheme 2.28 | ||
| Scheme 2 Synthesis of the dizinc complex (LZn2(O2CCF3)2, 1). Reagents and conditions: (i) 2 equiv. Zn(O2CCF3)2·xH2O, MeOH, 25 °C, 16 h. | ||
The catalyst was synthesized from the ancillary ligand, H2L, which can itself be prepared in high yield (82%) from commercial materials in 3 steps. Its purity was assessed by 1H NMR spectroscopy (Fig. S1†) and by elemental analysis, which showed results in line with the literature.28
Catalyst 1 was applied in the copolymerization of cyclohexene oxide (CHO) or vinyl cyclohexene oxide (vCHO) and carbon dioxide (Table 1 and Scheme 2). It showed a good performance using both monomers, with the TOF for CHO being 25 h−1 and for v-CHO being 24 h−1, at 1 bar CO2 pressure, 0.1 mol% catalyst loading and 80 °C. The activity for CHO is in-line with the value reported in the literature.28 The activity using v-CHO has not previously been reported and is more difficult to quantify precisely due to significant overlap of resonances associated with monomer and polymer in the crude 1H NMR spectrum (Fig. S2 and S3†). For both monomers, the polycarbonates produced show a very high selectivity for carbonate linkages (≥99%) and there is ≤1% formation of a cyclic carbonate by-product.
| Entry | Monomer | Catalyst | Temp. (°C) | TONb | TOFc (h−1) | % carbonated | % cyclic carbonated | M n (g mol−1) | M w/Mne (Đ) |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: 0.1 mol% catalyst, in neat epoxide, for 6–24 h, 1 bar research grade CO2 unless stated otherwise. b TON = number of epoxide consumed per mole of catalyst. c TOF = TON per hour. d Determined by comparing the integrals of resonances at 4.65 ppm (PCHC) or 4.75–4.90 ppm (PVCHC), at 4.0 ppm (cyclic carbonate by-product) and at 3.45 ppm (ether linkages). e Determined by SEC, in THF using narrow Mw polystyrene standards. f Unpurified CHO. g Unpurified v-CHO. | |||||||||
| 1f | CHO | 1 | 80 | 608 | 25 | 99 | 3 | 1500 | 1.08 |
| 2 | CHO | 1 | 80 | 502 | 25 | 99 | 1 | 6600 | 1.18 |
| 3g | v-CHO | 1 | 80 | 429 | 20 | >99 | <1 | 5900 | 1.30 |
| 4 | v-CHO | 1 | 80 | 389 | 24 | >99 | <1 | 6700 | 1.18 |
The alternating polymers show moderate molecular weights, with values typically being lower than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, and narrow dispersities (Đ < 1.20). Copolymerizations using 1 show a linear evolution of molecular weight vs. CHO conversion and narrow dispersities: both are features of well-controlled/living polymerizations. Such behaviour is indeed typical of this class of dinuclear catalyst and indicates good polymerization control.30
000 g mol−1, and narrow dispersities (Đ < 1.20). Copolymerizations using 1 show a linear evolution of molecular weight vs. CHO conversion and narrow dispersities: both are features of well-controlled/living polymerizations. Such behaviour is indeed typical of this class of dinuclear catalyst and indicates good polymerization control.30
Controlled polymerizations are defined as exhibiting rapid and quantitative initiation leading to all chains propagating at the same rate and limited/no termination or chain transfer reaction. In such a polymerization, the molecular weight of the polymer should be predictable from the monomer conversion and catalyst/initiator concentration. However, the polymerizations using catalyst 1 result in the production of polymers that have molecular weights, determined by size exclusion chromatography, which are substantially lower than predicted; indeed the values obtained suggest that chain transfer reactions occur, and that approximately 8 chains grow per equivalent of catalyst. Thus, the polymerizations appear to be occurring under immortal conditions, whereby chains are rapidly being exchanged or transferred between the catalyst and a chain transfer agent (often an alcohol). Fig. 1 illustrates the key processes occurring during polymerization. It is proposed that 1,2-diols are present in the polymerizations and function as the chain transfer agents. Such chain transfer processes are commonly observed in this field using a range of different catalysts.14,27,30,36–38
 | ||
| Fig. 1 The proposed catalytic cycle for the copolymerization of CO2 and epoxides.8 | ||
A further consideration is that bimodal molecular weight distributions are commonly observed, but seldom explained, for a wide range of catalysts in the literature.27,29,30,37,39 Indeed, catalyst 1 also shows a distinctly bimodal Mw distribution for both PCHC and PvCHC (Fig. 2, blue trace, Fig. S4 and S5†). In order to study the observed bimodality, two fractions of PCHC containing the higher and lower Mn distributions were isolated by SEC, according to a preparative separation by elution time (see Experimental section). These fractions were analysed by 1H NMR spectroscopy, MALDI-ToF and SEC. For both fractions, the 1H NMR spectra are identical, indicating PCHC end-capped with hydroxyl groups (Fig. S6†). Furthermore 19F NMR spectroscopy does not show any signals consistent with a lack of trifluoroacetate end-groups on the polymer chains. The MALDI-ToF spectra show that both fractions contain a single series attributed to polycarbonates having di-hydroxyl end groups. SEC analyses showed a clear separation into two molecular weight distributions: the lower fraction has Mn of 3900 (Đ: 1.14), whilst the higher fraction has Mn of 9000 (Đ: 1.05). Based on these observations, it appears that catalyst 1 leads to the production of PCHC with narrow but bimodal molecular weight distributions, where all detectable chains are di-hydroxyl terminated (Fig. 3).
 | ||
| Fig. 2 Top: molecular weight distribution of the isolated (crude) PCHC polymer (Table 1 entry 2) and bottom: the separation of PCHC into two fractions of low and high Mw. | ||
 | ||
| Fig. 3 MALDI-ToF spectrum of the low Mn fraction of PC (Table 1 entry 2), a single series is observed corresponding to: [HO(C7H10O3)nC6H11O2]K+ = [(142.15)n + 116.16 + 39.1]. | ||
We have previously observed bimodal Mw distributions, due to chains end-capped by the initiating group (lower Mn series) and by the di-hydroxyl groups (high Mn series, Fig. S3†).30 Based on this previous data, it would be expected that catalyst 1 would show chains end-capped by both trifluoroacetate (from the catalyst) and di-hydroxyl groups (from 1,2-diols). As the polymerizations are well-controlled, it is expected that the chains propagate at the same rate, therefore the molecular weight of the polymer from the telechelic diol would be expected to be double that of polymers initiated from the trifluoroacetate group. Thus, the bimodality can be generally attributed to the two types of initiating group. In order to rationalize the lack of a trifluoroacetate end-group (by spectroscopy and MALDI-ToF), it is proposed that those chains are susceptible to end-group (trifluoroacetate) hydrolysis during work-up due to the strongly electron-withdrawing substituents on the carboxylate group. The hydrolysis would lead to a bimodal Mw distribution where both series of chains are end-capped by hydroxyl groups, in line with the experimental observations. Darensbourg and co-workers observed that salen-chromium(trifluoroacetate) catalysts also lead to di-hydroxyl terminated polycarbonates.40 They confirmed that the trifluoroacetate end-group was present during polymerization, using anhydrous electrospray mass spectrometry, but was hydrolysed during termination/work-up to selectively produce the polyol.40
The important outcome is that catalyst 1 leads to the highly selective formation of dihydroxyl end-capped polymers or polyols. This is potentially beneficial for further application in polyurethane manufacture or in the production of higher polymers. We have previously demonstrated that such polyols are viable initiators for lactide ring-opening polymerizations, upon addition of a ROP catalyst, so as to produce ABA type triblock copoly(ester–carbonate–esters).28 Given that selective production of polyols is feasible using catalyst 1, it is of interest to investigate its tolerance to the presence of various other chain transfer agents and also the potential to prepare mono-modal Mw distributions.
Polymerizations with added water
Chain transfer reactions are known to occur during these polymerizations if protic compounds are present. These reactions serve to ‘swap’ the growing polymer chain on/off the metal centre and result in more than one polymer chain growing per equivalent of catalyst (Fig. 1). Given that there is good control of the Mw and the dispersities are narrow, it is proposed that the chain transfer reactions occur more rapidly than propagation. Thus, it should be possible to control the molecular weights via the addition of chain transfer agents, the simplest and most cost-effective of these is probably water. Furthermore, water is of interest as it is a common contaminant of carbon dioxide and would be expected to be present in any gases captured from industrial processes. In order to investigate the tolerance of catalyst 1 to water, a series of copolymerizations were run with increasing quantities of water being added (Table 2).| Entry | Amount of water added (mol equiv.) | ppm H2Ob (by weight) | M n (g mol−1) | M w/Mnc (Đ) |
|---|---|---|---|---|
| a Polymerization conditions: 0.1 mol% of 1, 18 h reaction time, 1 bar research grade CO2, 80 °C. b The quantity of water (as ppm weight fraction) added, based on the overall quantities of catalyst 1, epoxide and additive. c Determined by SEC, in THF, using narrow molecular weight polystyrene standards to calibrate the instrument. | ||||
| 1 | 0.5 | 91 | 5700 | 1.17 |
| 2 | 1 | 182 | 5200 | 1.15 |
| 3 | 2 | 364 | 4600 | 1.12 |
| 4 | 4 | 728 | 3300 | 1.11 |
| 5 | 8 | 888 | 2500 | 1.10 |
| 6 | 16 | 2904 | 1800 | 1.10 |
| 7 | 32 | 5792 | 800 | 1.09 |
In all cases where water was added, the polymerizations were nearly as active and selective as polymerizations in the absence of any additive (Table 2). It is feasible to add large quantities of water to the polymerizations, including from ∼90–5700 ppm by weight, and still maintain effective polymerization. This is particularly notable as typical pipeline specifications for captured CO2 water content are approximately 100–1000 ppm.23
For the polymerizations using water as the chain transfer agent, the polymer MWs reduce as increasing quantities of water are added. Furthermore, monomodal Mw distributions are observed when excess water is added (Fig. 4). A representative MALDI-ToF spectrum (Fig. 4) confirms that only a single series of polycarbonate polyol chains is formed in all cases.
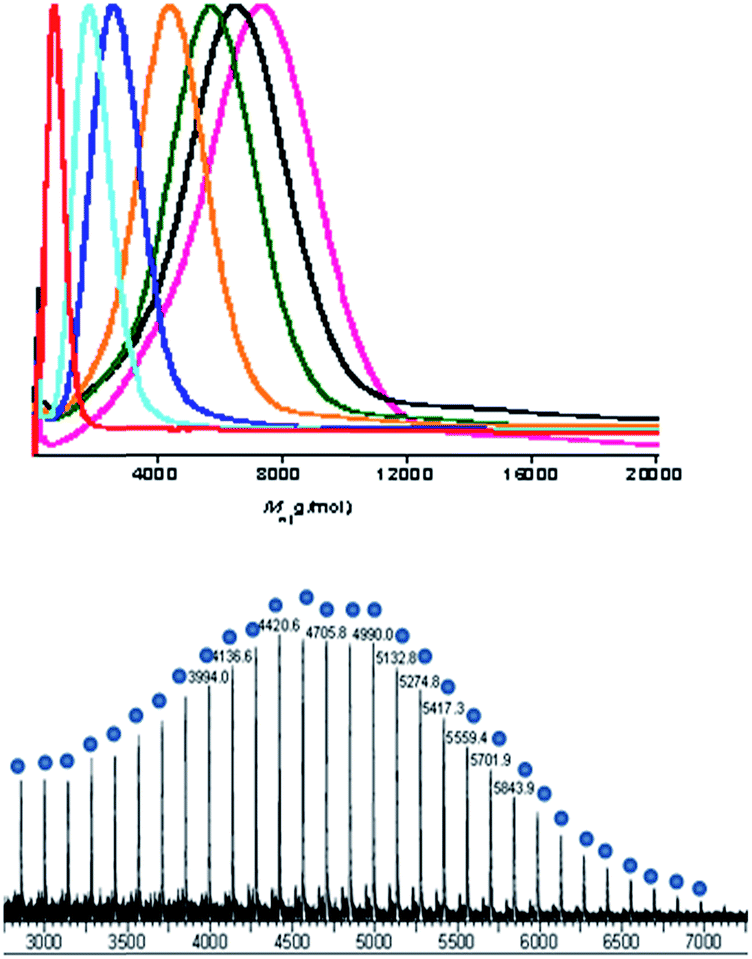 | ||
| Fig. 4 Top: SEC traces for the PCHC formed in Table 2. Colour code: entry 1: pink, 2: black, 3: green, 4: yellow, 5: dark blue, 6: pale blue, 7: red. Bottom: the MALDI-ToF spectrum obtained from Table 2, entry 2. | ||
Polymerizations with added alcohols
It has been proposed that water reacts with epoxides to produce cyclohexane-1,2-diol (CHD), such a process likely occurs immediately and may be metal catalyzed.27–30 Cyclohexane-1,2-diol would be expected to function as a chain transfer agent (CTA), leading to propagation from both of its secondary hydroxyl groups.41 Thus, it is of interest to investigate polymerizations in the presence of various equivalents of exogenous CHD (Table 3, entry 1–4). Using either CHO or vCHO resulted in successful polymerizations, and all the polymers contained high carbonate linkage contents (>99%) and low quantities of cyclic carbonates (∼5% for PCHC and <1% for PvCHC). Using a 5-fold excess of CHD, vs. catalyst, with CHO or v-CHO led to polymerizations occurring at near equivalent activity to those run in the absence of additives (Table 1, entries 2 and 4). Increasing the amount of CHD to a 10-fold excess also enabled successful polymerization with the selectivity remaining very high.| Entry | Monomer | Additiveb | Amount of additive addedc (mol equiv.) | ppm additivesd (by weight) | Conversione (%) | % selectivityf | M n (g mol−1) | M w/Mng (Đ) |
|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: 0.1 mol% of 1, in neat epoxide, for 16–72 h, 1 bar research grade CO2, 80 °C. b Weighed directly into reaction vessel. c Based on 1. d The quantity of additives (as ppm weight fraction) added, based on the overall quantities of catalyst 1, epoxide and additive. e Determined by 1H NMR spectroscopy, based on comparing integration values for the polymers (4.55–4.70 ppm, PCHC or 2.45 ppm, PvCHC) vs. epoxides (3.10 ppm, CHO or 5.62–5.80 ppm, vCHO). f Selectivity for polycarbonate vs. polyether, determined by comparing the integrals of resonances in the 1H NMR spectra at 4.65 ppm (PCHC) or 4.75–4.90 ppm (PVCHC) (corresponding to polymer carbonate linkages), and at 3.45 ppm (corresponding to polymer ether linkages). g Determined by SEC, in THF using narrow Mw polystyrene as standard to calibrate the instrument. | ||||||||
| 1 | CHO | CHD | 5 | 5831 | 53 | >99 | 4400 | 1.16 |
| 2 | CHO | CHD | 10 | 11598 |
53 | >99 | 2100 | 1.15 |
| 3 | v-CHO | CHD | 5 | 4623 | 40 | >99 | 7500 | 1.21 |
| 4 | v-CHO | CHD | 10 | 9231 | 59 | >99 | 5700 | 1.17 |
| 5 | CHO | TEA | 20 | 2905 | 49 | >99 | 1500 | 1.23 |
| 6 | CHO | H2NBn | 20 | 2116 | 22 | >99 | 950 | 1.10 |
| 7 | CHO | HNBn2 | 20 | 3834 | 33 | >99 | 3700 | 1.18 |
| 8 | CHO | MEA | 10 | 6068 | 65 | >99 | 2200 | 1.21 |
| 9 | CHO | EN | 10 | 6008 | 61 | >99 | 2900 | 1.21 |
As would be expected, increased quantities of CHD led to a decrease in polycarbonate Mn due to multiple polymer chains resulting per metal centre. As stated earlier, bimodal molecular weight distributions were observed for both PCHC and PvCHC. In contrast, the addition of an excess of CHD suppressed this bimodality and produced polycarbonates with monomodal molecular weight distributions (Fig. 5 and S8†).
 | ||
| Fig. 5 Top: the monomodal molecular weight distribution of PvCHC (Table 3 entry 4), as determined by SEC, using 10 equiv. of CHD in the polymerization. Bottom: MALDI-ToF spectrum of the PvCHC produced in Table 3 entry 4, using 10 equiv. of CHD. | ||
Based on the successful results using CHD (a bifunctional alcohol) as a chain transfer agent, it was of interest to investigate the addition of a tri-functional alcohol to produce branched polycarbonates (Table 3). The efficient copolymerization of CO2 and CHO was undertaken in the presence of 20 mol equiv. vs. catalyst 1, of triethanolamine (N(CH2CH2OH)3, TEA) (Table 3, entry 5). The polycarbonate was analysed by 1H and 31P{1H} NMR spectroscopy, MALDI-ToF and SEC; all of which confirmed the formation of a perfectly alternating copolymer. The synthesis of tri-functional, or branched, PCHC was confirmed from the 1H NMR spectrum (Fig. S9†), where the ethylene protons of the NCH2 and OCH2 groups of the TEA segment were observed at 2.86 and 4.17 ppm, respectively. Furthermore, only secondary alcohol groups (145.5 ppm) were identified using a 31P{1H} NMR spectroscopic analysis method (Fig. S10†). The method involves reacting a sample of the polymer with 2-chloro-4,4,5,5-tetramethyl dioxaphospholane, using bis(phenol)A as an internal standard (Scheme 3).42–44 It indicates that, within the detection limits of the technique, all polymerizations were successfully initiated from all the hydroxyl functionalities of TEA as all the primary alcohol groups were consumed and only secondary hydroxyl groups, due to cyclohexanol end groups were observed. The MALDI-ToF spectrum (Fig. S11†) showed a major series corresponding to the dihydroxyl end-capped PCHC. The absence of the expected series, corresponding to PCHC initiated from TEA, is proposed to be due to difficulties in branched chains. In this case, the rate of polymerization was relatively slower (TOF = 7 h−1), yielding a polymer with Mn of 1500 g mol−1 (Đ = 1.23).
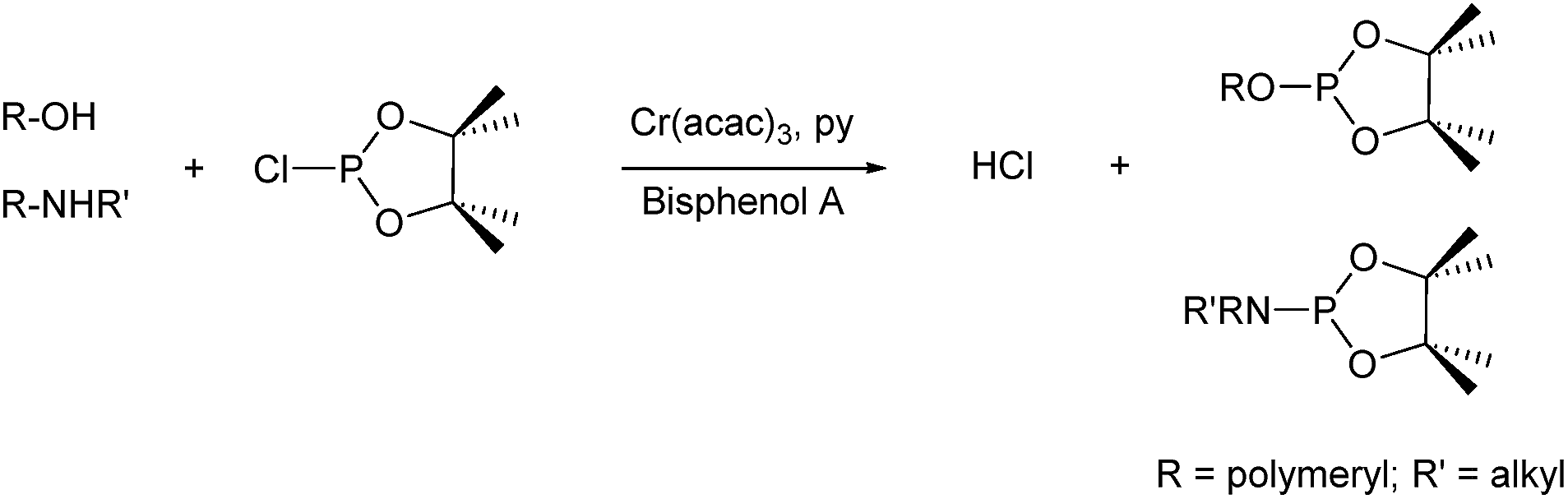 | ||
| Scheme 3 The reaction between alcohol or amine and 2-chloro-4,4,5,5-tetramethyl dioxaphospholane. | ||
Polymerizations with added amines
Amine based post combustion capture of CO2 has recently received much attention as a promising method for the reduction of atmospheric CO2 emissions.45 Approximately, 0.2 ppm of amine, namely methanolamine (MEA) and methylamine, in conjunction with 20 ppm ammonia are found as common contaminants. Denitrification is required to remove the amine-based contaminants in the production of reclaimed CO2.46,47It has already been established that the successful ROCOP of CHO and reclaimed CO2 occurs using analogous a di-magnesium catalyst (Table 1, entry 6).23 It was, therefore, of interest to establish whether LZn2(O2CCF3)2 (1) can successfully catalyze ROCOP in the presence of amine additives.
The polymerizations of CHO and CO2 with primary (H2NBn) and secondary (HNBn2) amine additives occurred successfully (Table 3, entries 6 and 7, respectively). Slightly lower polymerization rates (TOF < 20 h−1) were obtained in both cases compared to the polymerizations in the absence of any additives (TOF = 25 h−1, Table 1, entry 2), whereas in all cases the polymerization selectivities were high. The 1H NMR spectra of the isolated PCHC clearly indicate the incorporation of the amine reagents into the polymer chains. A representative 1H NMR spectrum of the PCHC from Table 3, entry 7 is shown in Fig. 6, illustrating the key resonances assigned to the benzyl protons at 7.32–7.22 and 4.37 ppm, respectively. In addition, SEC analysis using a UV detector also served to confirm the incorporation of the benzyl units as end-groups to the polymers (Fig. S12†).
 | ||
| Fig. 6 1H NMR spectrum (400.0 MHz, CDCl3-d1, 298 K) of the isolated PCHC produced in the polymerization with H2NBn additive (Table 3, entry 7). The asterisk denotes the residual protio-solvent. | ||
The MALDI-ToF mass spectra showed three series attributed to polycarbonates end-capped by: (1) dihydroxy groups, (2) amine/hydroxyl groups and (3) carbamate/hydroxyl groups (Fig. 7).
 | ||
| Fig. 7 MALDI-ToF spectrum of the PCHC obtained in Table 3 entry 7, using 20 equiv. of H2NBn. | ||
The presence of both amine and amide end groups must be due to the formation of both the carbamate alcohol (a, Scheme 4) and the amino alcohol (b, Scheme 4) from the reaction between amine, CHO and CO2, in the presence of catalyst 1. Other researchers have reported the reactions of amines, carbon dioxide and epoxides can lead to the formation of both products, although it is notable that in these cases more forcing conditions were generally required.48–50
 | ||
| Scheme 4 The synthesis of carbamate alcohol and amino alcohol. | ||
A control experiment was carried out to examine the behaviour of the amine reagents under the polymerization conditions which showed that polymerization only occurred in the presence of the catalyst, i.e. the amines themselves are not able to ‘catalyze’ polymerization. This observation reinforces the proposed metal-mediated ROCOP pathway, whereby monomers are activated through the coordination to the metal centres prior to reactions with the amines. The carbamate alcohol and amino alcohol intermediates are expected to act as chain transfer reagents in the polymerizations. These polymerizations occurred with slightly slower rates than in the absence of additives, this may be due to inhibition of monomer binding by the amines and/or slower initiation processes. The polymers were analyzed using MALDI-ToF MS and by 31P{1H} NMR spectroscopy, after reaction with 2-chloro-4,4,5,5-tetramethyl dioxaphospholane. The resonances were characteristic of polycarbonates with secondary hydroxyl end-groups, with peaks at 145.5 ppm in 31P{1H} NMR spectra (C and D, Fig. S10†). Whereas the reaction between dibenzylamine and the dioxaphospholane resulted in a peak at 142.5 ppm in the 31P{1H} NMR spectrum (B, Fig. S10†). Thus supporting the involvement of the NH groups in the polymerizations, as illustrated in Scheme 4.
The scope of polymerizations in the presence of amines was expanded to include bi-functional reagents, such as, monoethanolamine (MEA) and ethylenediamine (EN). The formation of a white suspension was observed during the initial stages of polymerizations using these additives (Table 3, entries 10 and 11, respectively). Such observations are consistent with the formation of ammonium salts, as part of equilibria between amines, carbon dioxide and carbamic acid.48–50 The white suspension dissolved as the polymerization proceeded, and the reactions resulted in the formation of low molecular weight PCHC, with a slightly broader dispersity (Mn = 2200 g mol−1, Đ = 1.21 for MEA and Mn = 2900 g mol−1, Đ = 1.21 for EN) compared to that obtained from the control experiment (Mn = 6600 g mol−1, Đ = 1.18). Once again, the polymerization activities were significantly lower (TOF = 9 h−1) than that of the control experiment. The polymer chain end group analysis, using the reaction with 2-chloro-4,4,5,5-tetramethyl dioxaphospholane, showed the formation of polycarbonates with secondary alcohol end-groups (E and F, Fig. S10†), however, the MALDI-ToF spectra were highly complex and as yet cannot be fully assigned. These findings indicate that although successful polymerizations did occur in the presence of primary amines, the precise polymer structures may be complicated by carbamate formation at the chain ends.
Experimental section
Materials and methods
All manipulations were carried out using standard Schlenk line or dry-box techniques under an inert atmosphere of nitrogen. All glassware was dried at 160 °C for 20 h and cooled under vacuum prior to use. Solvents were dried by passing through a column of appropriate drying agent, degassed and stored under nitrogen atmosphere. Chloroform-d3 was dried over CaH2, distilled under reduced pressure and stored over 4 Å molecular sieves, under nitrogen atmosphere.Ligand H2L and catalyst 1 were synthesised according to the previously published literature. Cyclohexene oxide and vinyl cyclohexene oxide were purchased from Acros and Sigma-Aldrich, respectively, and fractionally distilled from CaH2. Cyclohexene diol was recrystallised from ethyl acetate and stored under nitrogen atmosphere. Benzylamine (99%), dibenzylamine (97%), monoethanolamine (99.5%), ethylenediamine (99.5%) and triethanolamine (98%) were purchased from Sigma-Aldrich, degassed and stored under nitrogen atmosphere. High-purity CO2 was obtained by passing research grade CO2 (99.99995%) purchased from BOC (Linde Gas) through a high performance purifier (Drierite column).
1H, 13C{1H}, 31P{1H} NMR spectra were recorded using a Bruker AV400 MHz spectrometer at ambient temperature (unless stated otherwise). 1H, 13C{1H} NMR spectra were referenced internally to residue proteo-solvent (1H) or solvent (13C) resonances, and are reported relative to tetramethylsilane. SEC data were determined by a Shimadzu LC-20AD instrument using MALLS detector (Wyatt Dawn 8+), with THF as the eluent, at a flow rate of 1.0 mL min−1 at 30 °C. Two Mixed Bed PSS SDV linear S columns were used in series. The MALLS detector was calibrated using a polystyrene standard. MALDI-ToF MS experiments were carried out on Waters/Micromass MALDI micro MX spectrometer, using a dithranol matrix in THF at a 1:1 loading with potassium trifluoroacetate (KO2CCF3) as the cationizing agent. Elemental analyses were carried out using the Elemental Analysis Service at London Metropolitan University.
Synthetic procedures
Copolymerization of cyclohexene oxide or vinyl cyclohexene oxide and carbon dioxide. Cyclohexene oxide (1.67 mL, 17 mmol) or vinyl cyclohexene oxide (2.15 mL, 17 mmol) and LZn2(O2CCF3) (1, 15.0 mg, 0.017 mmol) were added into a Schlenk tube. The epoxide was degassed, before being left stirring under 1 atm CO2 at 80 °C for a pre-determined time period. The crude reaction mixture was dried in vacuo to remove unreacted epoxide. The polymer was purified by repeat precipitations from methylene chloride using methanol.
Separation of poly (cyclohexene carbonate) (PCHC) with a bimodal molecular weight distribution. 100 mg of PCHC in THF (1.5 mL) was analysed using the previously stated SEC equipment. The delay time of the instrument was calculated based on the flow rate and the column length. Different fractions were collected manually according to the pre-determined time (elution and delay time).
Polymerizaitons in the presence of chain transfer agents. The general procedure stated above was followed, whereby multi-equivalents of chain transfer agents were added to the reaction mixture prior to the degassing process.
Conclusions
In conclusion, a di-zinc trifluoroacetate catalyst was investigated for the copolymerization of cyclohexene oxide or vinyl-cyclohexene oxide and carbon dioxide. The catalyst shows moderate/good activity and productivity and very high selectivity for carbonate linkage formation. It also selectively produces polycarbonate polyols, with bimodal molecular weight distributions. The catalyst functions well in the presence of additives, including water, diols and amines. These added reagents lead to good catalytic performances and enable control over the molecular weight of the polymers and the production of polyols with monomodal molecular weight distributions. The catalyst shows a high tolerance to such contaminants, including the ability to selectively prepare polyols in the presence of >5000 ppm of water.Acknowledgements
The authors acknowledge the Engineering and Physical Sciences Research Council (EPSRC grants: EP/K035274/1, EP/L017393/1, EP/H046380) for funding.Notes and references
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281–7305 Search PubMed.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 Search PubMed.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 Search PubMed.
- D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS PubMed.
- D. J. Darensbourg and A. D. Yeung, Polym. Chem., 2014, 5, 3949–3962 RSC.
- M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- K. Nakano, K. Kobayashi, T. Ohkawara, H. Imoto and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 8456–8459 CrossRef CAS PubMed.
- M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A.-K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef CAS PubMed.
- J. K. Varghese, D. S. Park, J. Y. Jeon and B. Y. Lee, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4811–4818 CrossRef CAS.
- W. C. Ellis, Y. Jung, M. Mulzer, R. Di Girolamo, E. B. Lobkovsky and G. W. Coates, Chem. Sci., 2014, 5, 4004–4011 RSC.
- G.-P. Wu, D. J. Darensbourg and X.-B. Lu, J. Am. Chem. Soc., 2012, 134, 17739–17745 CrossRef CAS PubMed.
- Y. Liu, W. M. Ren, M. Wang, C. Liu and X. B. Lu, Angew. Chem., Int. Ed., 2015, 54, 2241–2244 CrossRef CAS PubMed.
- L. Pena Carrodeguas, J. Gonzalez-Fabra, F. Castro-Gomez, C. Bo and A. W. Kleij, Chem.–Eur. J., 2015, 21, 6115–6122 CrossRef CAS PubMed.
- P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 50, 4164–4167 RSC.
- S. H. Lee, A. Cyriac, J. Y. Jeon and B. Y. Lee, Polym. Chem., 2012, 3, 1215–1220 RSC.
- J. Langanke, A. Wolf, J. Hofmann, K. Bohm, M. A. Subhani, T. E. Muller, W. Leitner and C. Gurtler, Green Chem., 2014, 16, 1865–1870 RSC.
- N. Von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- D. J. Darensbourg, W.-C. Chung, K. Wang and H.-C. Zhou, ACS Catal., 2014, 4, 1511–1515 CrossRef CAS.
- A. C. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 1581–1588 CrossRef CAS.
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC.
- C. Romain, A. Thevenon, P. K. Saini and C. K. Williams, in Topics in Organometallic Chemistry: Carbon Dioxide and Organometallics, ed. X. B. Lu, Springer DE, 2015 Search PubMed.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- M. R. Kember, J. Copley, A. Buchard and C. K. Williams, Polym. Chem., 2012, 3, 1196–1201 RSC.
- A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredrickson and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS.
- M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535–9542 CrossRef CAS PubMed.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed.
- M. R. Kember, F. Jutz, A. Buchard, A. J. P. White and C. K. Williams, Chem. Sci., 2012, 3, 1245–1255 RSC.
- Y. Liu, W. M. Ren, K. K. He and X. B. Lu, Nat. Commun., 2014, 5, 1–7 RSC.
- R. K. Dean, L. N. Dawe and C. M. Kozak, Inorg. Chem., 2012, 51, 9095–9103 CrossRef CAS PubMed.
- M. W. Lehenmeier, C. Bruckmeier, S. Klaus, J. E. Dengler, P. Deglmann, A.-K. Ott and B. Rieger, Chem. – Eur. J., 2011, 17, 8858–8869 CrossRef CAS PubMed.
- K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972–6980 CrossRef CAS.
- D. J. Darensbourg and G.-P. Wu, Angew. Chem., 2013, 10796–10800 CrossRef.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- Y. Zhu, C. Romain, V. Poirier and C. K. Williams, Macromolecules, 2015, 48, 2407–2416 CrossRef CAS.
- E. Hatzakis, E. Archavlis and P. Dais, J. Am. Oil Chem. Soc., 2007, 84, 615–619 CrossRef CAS.
- A. Spyros, D. S. Argyropoulos and R. H. Marchessault, Macromolecules, 1997, 30, 327–329 CrossRef CAS.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 Search PubMed.
- IEAGHG, Gaseous emissions from amine based PCC processes and their deep removal, May, 2012.
- I. Hauser, A. Einbu, H. F. Stevendsen and K. Ostgaard, J. Exp. Clin. Med., 2014, 1, 1–5 Search PubMed.
- Y. Yoshida and S. Inoue, J. Chem. Soc., Perkin Trans. 1, 1979, 3146–3150 RSC.
- Y. Yoshida and S. Inoue, Chem. Lett., 1978, 7, 139–140 CrossRef.
- D. Chaturvedi, Tetrahedron, 2012, 68, 15–45 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5fd00073d |
| This journal is © The Royal Society of Chemistry 2015 |