 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of ammonia directly from wet air using Sm0.6Ba0.4Fe0.8Cu0.2O3−δ as the catalyst
Rong
Lan
a,
Khaled A.
Alkhazmi
a,
Ibrahim A.
Amar
a and
Shanwen
Tao
*ab
aDepartment of Chemical & Process Engineering, University of Strathclyde, Glasgow G1 1XJ, UK
bSchool of Engineering, University of Warwick, Coventry, CV4 7AL, UK. E-mail: S.Tao.1@warwick.ac.uk; Fax: +44 (0) 24 764 18922; Tel: +44 (0) 24 761 51680
First published on 8th April 2015
Abstract
Ammonia was directly synthesised from wet air at 400 °C at atmospheric pressure. A new perovskite Sm0.6Ba0.4Fe0.8Cu0.2O3−δ was used as the electrocatalyst for electrochemical synthesis of ammonia. Ammonia formation rates of 9.19 × 10−7 mol s−1 m−2 and 1.53 × 10−6 mol s−1 m−2 were obtained at 400 °C when wet air and wet N2 were introduced into a simple single chamber reactor, respectively. The perovskite catalyst is low cost compared to the previously reported Ru/MgO and Pt/C catalysts. This experiment indicates that ammonia can be directly synthesised from wet air, a very promising simple technology for sustainable synthesis of ammonia in the future.
1 Introduction
Nitrogen fertiliser has supported approximately 27% of the world's population over the last century, equivalent to around 4 billion people born (or 42% of the estimated total births) since 1908.1 Although over 78% of the atmosphere is composed of nitrogen, it is difficult to use N2 directly from plants as plants can only use fixed nitrogen. The breakthrough in nitrogen fixation took place a century ago, the well-known Haber–Bosch process, operating at high temperatures (∼500 °C) and high pressures (150–300 bar).2 Fritz Haber and Carl Bosch won the Nobel Prize in Chemistry in 1918 and 1931, respectively, for their contributions in industrial production of ammonia. This innovation became a key driver in the development of the industrialized society and is still securing the nutrition of billions of people today. In 2007, the Nobel Prize in Chemistry was awarded to Gerhard Ertl for his studies of chemical processes on solid surfaces. A major contribution to this prize was Gerhard Ertl’s understanding of the reaction mechanism of H2 and N2 on solid ammonia synthesis catalysts.3In the Haber–Bosch process, the required hydrogen is produced through the steam reforming of natural gases or coal of which consumed more than 1% of world power generation.4 In addition, CO2 emitted from the ammonia industry is equivalent to 0.77% of the world total CO2 emission. In order to reduce the current dependency on fossil fuels and to reduce carbon emissions associated with their use, it is essential to introduce new ammonia synthesis processes and break the link between fossil fuels and the ammonia industry.5
To reduce global CO2 emission, renewable electricity generated from solar, wind, marine and other sources are becoming more and more important. Due to the intermittent nature of those renewable resources, energy storage, particularly renewable electricity storage has become a big challenge. One of the possible solutions for renewable electricity storage is to produce basic chemical feedstocks such as ammonia. The electrochemical synthesis processes are among the promising alternatives in particular if the hydrogen required for ammonia synthesis is produced from non-fossil fuel resources.6–8 Recently, it has been reported that ammonia can be directly synthesised from air and water at ambient conditions.2 It has been widely reported that ammonia can be synthesised from N2 and H2 (or steam) at a temperature around 500 °C.9–15 The possible reason is that, the decomposition of ammonia at high temperature is kinetically slow thus ammonia can still be collected if it is removed from the reactor in time. Ammonia will be able to be synthesised from air and water at elevated temperatures if the oxidation of ammonia is also kinetically slow.
In our previous papers, we reported the electrochemical synthesis of ammonia based on electrochemical cells using a Ce0.8Sm0.2O2−δ–(Li,Na,K)2CO3 composite electrolyte.12 The eutectic point of (Li,Na,K)2CO3 ternary molten salts is 396 °C.16,17 The low melting point of mixed (Li,Na,K)2CO3 salts causes the composite to exhibit high ionic conductivity at relatively low temperatures which can minimise the operating temperature of the cell, reducing the thermal decomposition and oxidation of ammonia. It has been reported that the ionic conductivity of the Ce0.8Sm0.2O2−δ–(Li,Na,K)2CO3 composite reached 0.1 S cm−1 at a temperature around 400 °C.18 The total ionic conductivity includes those from Li+, Na+, K+, H+, HCO3−, CO32− and O2− ions. Good fuel cell performance based on a Ce0.8Sm0.2O2−δ–(Li,Na,K)2CO3 composite electrolyte indicates higher H+/O2− ionic conductivity in this type of composite material.18–20 Either H+ or O2− ionic conduction can be used for electrochemical synthesis of ammonia.6 Therefore a similar composite electrolyte Ce0.8Gd0.2O2−δ–(Li,Na,K)2CO3 was used as the electrolyte in this study for ammonia synthesis. Recently, it has been reported that ammonia was successfully synthesised directly from wet air using La0.8Cs0.2Fe0.8Ni0.2O3−δ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 or Pr0.6Ba0.4Fe0.8Cu0.2O3−δ22 as the catalyst. It has been reported that Cu and Ni co-doped SmFeO3, SmFe0.7Cu0.1Ni0.2O3−δ is a very good ammonia synthesis catalyst and an ammonia formation rate of 1.13 × 10−8 mol cm−2 s−1 was observed at 80 °C when N2 and H2 were used as the reactants.23 This inspired us to use doped SmFeO3 catalysts for the electrochemical synthesis of ammonia. On the other hand, BaO is a typical promoter of ammonia synthesis catalysts.24,25 However, BaO itself is unstable in wet air because of the reaction between BaO and H2O to form Ba(OH)2. Therefore, in this paper, we introduce some Ba at the A-site of perovskite oxide SmFeO3. A new perovskite, Sm0.6Ba0.4Fe0.8Cu0.2O3−δ (SBFCu), was synthesised as an electro-catalyst for the synthesis of ammonia from air/N2 and water. Sm0.6Ba0.4Fe0.8Cu0.2O3−δ was used as both the cathode and anode and a single chamber reactor was used for the synthesis.
21 or Pr0.6Ba0.4Fe0.8Cu0.2O3−δ22 as the catalyst. It has been reported that Cu and Ni co-doped SmFeO3, SmFe0.7Cu0.1Ni0.2O3−δ is a very good ammonia synthesis catalyst and an ammonia formation rate of 1.13 × 10−8 mol cm−2 s−1 was observed at 80 °C when N2 and H2 were used as the reactants.23 This inspired us to use doped SmFeO3 catalysts for the electrochemical synthesis of ammonia. On the other hand, BaO is a typical promoter of ammonia synthesis catalysts.24,25 However, BaO itself is unstable in wet air because of the reaction between BaO and H2O to form Ba(OH)2. Therefore, in this paper, we introduce some Ba at the A-site of perovskite oxide SmFeO3. A new perovskite, Sm0.6Ba0.4Fe0.8Cu0.2O3−δ (SBFCu), was synthesised as an electro-catalyst for the synthesis of ammonia from air/N2 and water. Sm0.6Ba0.4Fe0.8Cu0.2O3−δ was used as both the cathode and anode and a single chamber reactor was used for the synthesis.
2 Experimental
Materials synthesis
Calculated amounts of Gd2O3 were dissolved in hot dilute nitric acid to form a gadolinium nitrate aqueous solution. Then an appropriate amount of Ce(NO3)3·6H2O was added to the solution. A 1 M (NH4)2CO3 solution was added dropwise with vigorous stirring until the pH value reached 7–8, after that vigorous stirring was continued for another 30 min. The formed precipitate was then collected by vacuum filtration and washed several times with deionised water then dried at 100 °C overnight. The obtained powder was calcined in air at 650 °C for 2 hours with a heating/cooling rate of 2 °C min−1. Finally, ultrafine powder of GDC was obtained.Ce0.8Gd0.2O2−δ and ternary carbonate mixture (Li,Na,K)2CO3 (32.1 wt% Li2CO3; 34.5 wt% K2CO3; 33.4 wt% Na2CO3) powders were mixed together with a weight ratio of oxide to carbonate of 7:3. The mixture was mixed and ground with the use of 25 ml isopropanol. Then the mixture was put in a ball miller (Planetary Mono Mill Pulverisette 6) for mixing with a speed of 400 rpm for 4 hours. The weight ratio of balls to powders was roughly 1:1. Then the mixture was heated by a hot plate magnetically stirred at 150 °C to release and remove the isopropanol. The powder was heated at 600 °C for one hour then quenched in air to room temperature. The as-prepared oxide–carbonate composite electrolyte will be used for ammonia synthesis.
Samarium(III) oxide was dissolved in diluted nitric acid to form samarium nitrate under heating and stirring. Calculated amounts of nitrates Ba(NO3)2, Fe(NO3)3·9H2O and Cu (NO3)3·2.5H2O were dissolved in deionised water and were added to the above prepared solution. Appropriate amounts of citric acid and EDTA (ethylenediaminetetraacetic acid) were then added as complexing agents with a molar ratio of citric acid:EDTA:metal cations of 1.5:1:1. Dilute aqueous ammonia solution was then added to the mixed solution to adjust the pH value to around 6 and a dark green clear solution was obtained. The mixed solution was evaporated on a hot-plate at 200–250 °C under stirring which gradually changed into a dark sticky gel before complete drying. By further heating the gel converted to a black colour. The brown ash was grounded and subsequently calcined in air at 900 °C for 2 hours with heating/cooling rates of 5 °C min−1 to obtain a single phase of Sm0.6Ba0.4Fe0.8Cu0.2O3−δ.
Materials characterisation
X-ray data were collected on a PANanalytical X' Pert Pro in the Bragg–Brentano reflection geometry with a Ni-filtered Cu Kα source (1.5405 Å), fitted with a X'Celerator detector and an Empyrean CuLFF XRD tube. Absolute scans in the 2θ range of 5–100° with step sizes of 0.0167° were used during data collection. The surface area of Sm0.6Ba0.4Fe0.8Cu0.2O3−δ powder was measured by a N2 adsorption method using Micromeritics ASAP 2420.TG/DSC analyses were performed using a Stanton Redcroft STA/TGH series STA 1500 operating through a Rheometric Scientific system interface controlled by the software RSI Orchestrator in flowing air or N2 at a flow rate of 50 ml min−1.
Total conductivity measurements were carried out by a pseudo-four-terminal method using a computer-controlled Solartron Analytical® SI 1470A electrochemical interface by applying a constant current. The Sm0.6Ba0.4Fe0.8Cu0.2O3−δ powder fired at 900 °C was pressed into pellets with diameters of 13 mm and thicknesses of around 2 mm which were then fired at 1100 °C for 5 hours. A silver coated pellet was fitted into the measuring apparatus and the conductivity measurement was carried out in air between room temperature and 750 °C.
Fabrication of the single cell and ammonia synthesis measurements
The cell was placed in a self-designed single chamber reactor as described elsewhere.22 The electrolytic cell for ammonia synthesis was a tri-layer single cell which was fabricated by a cost-effective one-step dry-pressing and co-firing process. The anode and cathode material was 0.8 g Sm0.6Ba0.4Fe0.8Cu0.2O3−δ plus 0.2 g composite electrolyte to form composite electrode materials. The composite anode, composite electrolyte and composite cathode (SBFCu/CGO–(Li,Na,K)2CO3/SBFCu) were fed into the die, layer by layer, with the aid of a sieve to ensure uniform powder distribution, and then unixally pressed at a pressure of 300 MPa by cold pressing into 13 mm pellets. The pellets were sintered at 600 °C for 2 h. The thickness of the anode, electrolyte and cathode was approximately 0.6, 1.0 and 0.6 mm, respectively. The catalyst surface area of the cathode and anode was 1.281 cm2. Silver paste was painted on each electrode surface of the cell as a current collector. Ag wires were used as output terminals for both electrodes. It has been reported that silver itself has negligible catalytic effects on ammonia synthesis.9Compressed air or N2 was passed through room temperature water before inputting into the single chamber reactor as described in previous reports.21,22 The flow rate of air (or N2) was 50 ml min−1. The steam concentration in air (or N2) was 3 mol%. A DC voltage was applied to the cell using a Solartron 1287A electrochemical interface. The synthesised ammonia was absorbed by 25 ml of diluted sulphuric acid (0.001 M) for 30 min as described before.2,12 The concentration of NH4+ in the absorbed solution was analysed using the Orion Application solution for low adjusting ISA. The produced ammonia was detected using an ammonia meter (ISE Thermo Scientific Orion Star A214) and the rate of ammonia formation was calculated using the following equation.2
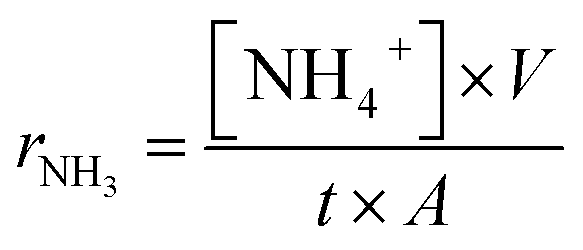 | (1) |
3 Results and discussion
Thermal analysis of the brown ash was carried out in air (Fig. 1A). It was found that a weight loss of ∼52% occurred at a temperature between 300 and 430 °C. This is due to the decomposition of the ash and loss of organic components. The sample weight became stable at a temperature above 800 °C indicating the minimum formation temperature for Sm0.6Ba0.4Fe0.8Cu0.2O3−δ is 800 °C. To guarantee the formation of a single phase, we choose 900 °C as the firing temperature for Sm0.6Ba0.4Fe0.8Cu0.2O3−δ. The ash was fired at 900 °C for 2 hours to form a perovskite phase. XRD analysis of the powder after firing at 900 °C indicates it is a single phase (Fig. 2). It exhibits an cubic structure with a space group of Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m(221); a = 3.9148(1) Å, V = 60.00(1) Å3 (Table 1). In our previous report, perovskite oxide La0.6Sr0.4Fe0.8Cu0.2O3−δ exhibits a cubic structure with a space group of Pm–3m(221).12 SEM pictures indicate that the prepared Sm0.6Ba0.4Fe0.8Cu0.2O3−δ powders agglomerated together with a secondary particle size of ∼0.1 μm (Fig. 3). The surface area of the La0.6Sr0.4Fe0.8Cu0.2O3−δ prepared at 900 °C was 7.94(1) m2 g−1.
m(221); a = 3.9148(1) Å, V = 60.00(1) Å3 (Table 1). In our previous report, perovskite oxide La0.6Sr0.4Fe0.8Cu0.2O3−δ exhibits a cubic structure with a space group of Pm–3m(221).12 SEM pictures indicate that the prepared Sm0.6Ba0.4Fe0.8Cu0.2O3−δ powders agglomerated together with a secondary particle size of ∼0.1 μm (Fig. 3). The surface area of the La0.6Sr0.4Fe0.8Cu0.2O3−δ prepared at 900 °C was 7.94(1) m2 g−1.
 | ||
| Fig. 1 The STA analysis of the Sm0.6Ba0.4Fe0.8Cu0.2O3−δ ash (A) and the Sm0.6Ba0.4Fe0.8Cu0.2O3−δ powder in N2 (B). | ||
 | ||
| Fig. 2 XRD pattern of Sm0.6Ba0.4Fe0.8Cu0.2O3−δ at room temperature. | ||
| Atom | Site | Occupancy | x | y | z | U iso/Å2 |
|---|---|---|---|---|---|---|
|
a Space group Pmm(221); a = 3.9148(1) Å, V = 60.00(1) Å3. Rwp = 6.11%, Rp = 4.48%, χred2 = 2.52.
|
||||||
| Sm | 1a | 0.6 | 0 | 0 | 0 | 0.0181(5) |
| Ba | 1a | 0.4 | 0 | 0 | 0 | 0.0181(5) |
| Fe | 1b | 0.8 | 0.5 | 0.5 | 0.5 | 0.0171(9) |
| Cu | 1b | 0.2 | 0.5 | 0.5 | 0.5 | 0.0171(9) |
| O | 3c | 1 | 0 | 0.5 | 0.5 | 0.796(27) |
 | ||
| Fig. 3 SEM images of Sm0.6Ba0.4Fe0.8Cu0.2O3−δ powder after calcining at 900 °C for 2 hours. (A) at ×2000 magnification, (B) at 10000 magnification. | ||
The electronic conductivity of Sm0.6Ba0.4Fe0.8Cu0.2O3−δ is very important in order to be used as both the cathode and anode for the electrochemical cells for ammonia synthesis. The DC conductivity measurement indicated that the conductivity increased before 550 °C, reaching a value of 2.29 S cm−1 and then started to decrease (Fig. 4). This is probably due to the semi-conductor to metal transition which is a common phenomenon in perovskite oxides.26 The conductivity of Sm0.6Ba0.4Fe0.8Cu0.2O3−δ was 1.88 S cm−1 at 400 °C. Regarding the operating temperature of the cell, the lowest working temperature of the CGO–(Li,Na,K)2CO3 composite electrolyte (400 °C) was used in order to minimise possible ammonia decomposition and oxidation.
 | ||
| Fig. 4 The DC conductivity of Sm0.6Ba0.4Fe0.8Cu0.2O3−δ in air. | ||
During the ammonia synthesis process, Sm0.6Ba0.4Fe0.8Cu0.2O3−δ was exposed to wet N2 or wet air. Therefore the stability of Sm0.6Ba0.4Fe0.8Cu0.2O3−δ in N2 is very important. The STA analysis of Sm0.6Ba0.4Fe0.8Cu0.2O3−δ in N2 is shown in Fig. 1B. It was found that the sample kept losing weight until the measured 600 °C. The initial weight loss below is due to the desorption of adsorbed water and gases. Further weight loss at higher temperatures is probably due to the lattice oxygen, forming oxygen vacancies. No obvious thermal effect was observed on the DSC curve indicating that there is no obvious chemical reaction between Sm0.6Ba0.4Fe0.8Cu0.2O3−δ and N2 at a temperature below 600 °C.
For ammonia synthesis, wet air was fed into the single chamber reactor as described elsewhere.22 The gas composition was approximately 3 mol% H2O, 97 mol% air. The effect of the applied potential on ammonia formation rate was investigated at 400 °C and the order for applied voltage was from low to high, between 1.2 and 1.7 V with and interval of 0.1 V. Fig. 5A shows the recorded current change against time for the electrolytic cell during the ammonia synthesis process at different potentials at 400 °C over a period of 3600 seconds (was 3300 seconds due to an interruption when applying 1.6 V). As can be seen, the current density decreased at the beginning but tended to become stable. The ammonia produced in the single chamber reactor was absorbed by 20 ml of dilute H2SO4 (0.001 M) for 60 minutes. It is surprising that ammonia was successfully synthesised at atmospheric pressure from wet air (∼3% H2O). As can be seen in Fig. 6A the ammonia formation rate increased significantly with increasing the applied potential and reached a maximum value at 1.4 V (9.19 × 10−7 mol s−1 m−2 at 400 °C), showing that 1.4 V was the optimum potential for ammonia synthesis in this study. In terms of catalyst weight, the ammonia formation rate was 1.48 × 10−10 mol s−1 gcat−1. This is two orders of magnitude higher than the reported ammonia formation rates (around 1.0 × 10−12 mol s−1 gcat−1 at ∼600 °C) when either a H+ (SrCe0.95Yb0.05O3−δ) or O2− (8 mol% yttria-stabilized zirconia) conductor was used as the electrolyte with an industrial Ru/MgO catalyst used at the cathode.9 It was 400 °C in our experiment which is roughly 200 °C lower than that reported by Skodra.9 Higher working temperatures may cause the thermal decomposition or oxidation of the produced ammonia, particularly at a temperature above 500 °C.2 The Sm0.6Ba0.4Fe0.8Cu0.2O3−δ catalyst used in this study is much cheaper than the Ru/MgO catalyst reported by Skodra and Stoukides9 and the Pt/C catalyst used for low temperature synthesis of ammonia from air and water.2,4
 | ||
| Fig. 5 The current density across the cell in wet air (A) and wet N2 (B) when applied with different voltages. | ||
 | ||
| Fig. 6 The ammonia formation rate (A) and Faraday efficiency (B) at 400 °C in wet air and wet N2 when applied with different voltages. | ||
For comparison, ammonia was synthesised from wet nitrogen using the same cell. Fig. 5B shows the recorded current change against time for the electrolytic cell during the ammonia synthesis process at different potentials at 400 °C over a period of 60 minutes. As can be seen, the electrolytic cell was stable when the applied voltage was over 1.4 V. As expected, ammonia was also successfully synthesised at 400 °C under atmospheric pressure from ∼3% H2O and N2 (Fig. 6A). Again the ammonia formation rate increased significantly by increasing the applied potential and reached a maximum value at 1.4 V (1.53 × 10−6 mol s−1 m−2 at 400 °C), showing that 1.4 V was the optimum potential for ammonia synthesis in this study. The same tendency was observed for both wet air and wet N2. This could be attributed to nitrogen chemisorption hindered by the high rate of electrochemically supplied H+, which in turn poisoned the catalyst (cathode surface).6,27 In terms of catalyst weight, the ammonia formation rate was 2.45 × 10−10 mol s−1 gcat−1 which is more than two orders of magnitude higher than the values reported by Skodra.9 When applying 1.7 V, the ammonia formation rates for wet air and wet N2 are comparable (Fig. 6A) while slightly more ammonia was produced at lower voltages when wet N2 was introduced into the single chamber reactor. The observed ammonia formation rates in this study were lower than the reported value when SmFe0.7Cu0.1Ni0.2O3−δ was used as the cathode catalyst but the experiments were operated under different conditions.23 H2 and N2 were used as the reactant while we used wet N2/air as the precursor. The cell operating temperature was 400 °C while it was near room temperature in a previous report.23
The Faraday efficiencies of the electrochemical synthesis process under different conditions are also calculated. As shown in Fig. 6B, when wet air was used, the highest Faraday efficiency was 0.74% when a voltage of 1.7 V was applied. In the case of wet N2, the highest Faraday efficiency was 1.19% at a voltage of 1.7 V. At lower applied voltages, the Faraday efficiencies were comparable for wet air and wet N2, indicating the possibility to use wet air instead of wet N2 for direct synthesis of ammonia at temperature or below 400 °C. Although the Faraday efficiencies in our experiments are lower than the high efficiency of 78% when H2 and N2 was used as the precursor,13 steam instead of H2, and air instead of N2 were used as the precursors in this study. It is believed that both the formation rate and Faraday efficiency can be improved when better electro-catalysts are identified.
4 Conclusion
In conclusion, after our previous reports on ammonia synthesis directly from air and water at ambient temperature up to 80 °C,2,4 ammonia was also synthesised directly from wet air at a higher temperature (400 °C). Perovskite oxide, Sm0.6Ba0.4Fe0.8Cu0.2O3−δ, was successfully synthesised and showed a conductivity of 1.88 S cm−1 at 400 °C. Ammonia was successfully synthesised at 400 °C under atmospheric pressure from both wet air and wet nitrogen using Sm0.6Ba0.4Fe0.8Cu0.2O3−δ as the anode and cathode catalyst. Ammonia formation rates of 9.19 × 10−7 mol s−1 m−2 and 1.53 × 10−6 mol s−1 m−2 were obtained at 400 °C when wet air and wet N2 were introduced into a simple single chamber reactor, respectively. These values are more than two orders of magnitude higher than the reported ammonia formation rates when synthesised from N2 and H2O at 600 °C.9 Our perovskite catalyst is also low cost compared to the Ru/MgO catalyst in a previous report9 and Pt/C catalysts in our previous study on ammonia synthesis from air and water.2,4 Although the ammonia formation rates were not high enough for mass production, this is a very simple process using low-cost materials. After further investigation and optimisation, we believe ammonia can be synthesised directly from wet air or wet N2 on a large scale to feed the growing world population in the future.Acknowledgements
The authors thank the EPSRC SuperGen XIV ‘Delivery of Sustainable Hydrogen’ project (Grant no. EP/G01244X/1) for funding.References
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS PubMed.
- R. Lan, J. T. S. Irvine and S. W. Tao, Sci. Rep., 2013, 3, 1145 CrossRef PubMed.
- http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2007/ .
- R. Lan and S. W. Tao, RSC Adv., 2013, 3, 18016–18021 RSC.
- R. Lan, J. T. S. Irvine and S. W. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS PubMed.
- I. A. Amar, R. Lan, C. T. Petit and S. W. Tao, J. Solid State Electrochem., 2011, 15, 1845–1860 CrossRef CAS.
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS PubMed.
- I. Garagounis, V. Kyriakou, A. Skodra, E. Vasileiou and M. Stoukides, Front. Energy Res., 2014, 2, 1–10 Search PubMed.
- A. Skodra and M. Stoukides, Solid State Ionics, 2009, 180, 1332–1336 CrossRef CAS PubMed.
- C. Chen and G. L. Ma, J. Mater. Sci., 2008, 43, 5109–5114 CrossRef CAS.
- I. A. Amar, R. Lan, C. T. Petit, V. Arrighi and S. W. Tao, Solid State Ionics, 2011, 182, 133–138 CrossRef CAS PubMed.
- I. A. Amar, C. T. Petit, L. Zhang, R. Lan, P. J. Skabara and S. W. Tao, Solid State Ionics, 2011, 201, 94–100 CrossRef CAS PubMed.
- G. Marnellos and M. Stoukides, Science, 1998, 282, 98–100 CrossRef CAS.
- G. Marnellos, S. Zisekas and M. Stoukides, J. Catal., 2000, 193, 80–87 CrossRef CAS.
- I. A. Amar, C. T. G. Petit, G. Mann, R. Lan, P. J. Skabara and S. W. Tao, Int. J. Hydrogen Energy, 2014, 39, 4322–4330 CrossRef CAS PubMed.
- G. J. Janz and M. R. Lorenz, J. Chem. Eng. Data, 1961, 6, 321–323 CrossRef CAS.
- R. I. Olivares, C. Chen and S. Wright, J. Sol. Energy Eng., 2012, 134 Search PubMed.
- C. Xia, L. Li, Y. Tian, Q. Liu, Y. Zhao, L. Jia and Y. Li, J. Power Sources, 2009, 188, 156–162 CrossRef CAS PubMed.
- L. Fan, C. Wang, M. Chen and B. Zhu, J. Power Sources, 2013, 234, 154–174 CrossRef CAS PubMed.
- L. Zhang, R. Lan, A. Kraft and S. W. Tao, Electrochem. Commun., 2011, 13, 582–585 CrossRef CAS PubMed.
- R. Lan, K. A. Alkhazmi, I. A. Amar and S. W. Tao, Electrochim. Acta, 2014, 123, 582–587 CrossRef CAS PubMed.
- R. Lan, K. A. Alkhazmi, I. A. Amar and S. W. Tao, Appl. Catal., B, 2014, 152–153, 212–217 CrossRef CAS PubMed.
- G. Xu, R. Liu and J. Wang, Sci. China, Ser. B: Chem., 2009, 52, 1171–1175 CrossRef CAS.
- C. H. Liang, Z. B. Wei, Q. Xin and C. Li, Appl. Catal., A, 2001, 208, 193–201 CrossRef CAS.
- W. Rarog-Pilecka, E. Miskiewicz, L. Kepinski, Z. Kaszkur, K. Kielar and Z. Kowalczyk, J. Catal., 2007, 249, 24–33 CrossRef CAS PubMed.
- T. Sugahara and M. Ohtaki, Appl. Phys. Lett., 2011, 99, 062107 CrossRef PubMed.
- W. Wang, X. Cao, W. Gao, F. Zhang, H. Wang and G. Ma, J. Membr. Sci., 2010, 360, 397–403 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |