 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a diffuse reflectance infrared fourier transform spectroscopy (DRIFTS) cell for the in situ analysis of co-electrolysis in a solid oxide cell
Denis J.
Cumming
*a,
Christopher
Tumilson
ab,
S. F. Rebecca
Taylor
b,
Sarayute
Chansai
b,
Ann V.
Call
a,
Johan
Jacquemin
b,
Christopher
Hardacre
*b and
Rachael H.
Elder
*a
aDepartment of Chemical and Biological Engineering, University of Sheffield, Mappin St, Sheffield, S1 3JD, UK. E-mail: d.cumming @sheffield.ac.uk; r.elder@sheffield.ac.uk
bSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, N. Ireland. E-mail: c.hardacre@qub.ac.uk
First published on 12th March 2015
Abstract
Co-electrolysis of carbon dioxide and steam has been shown to be an efficient way to produce syngas, however further optimisation requires detailed understanding of the complex reactions, transport processes and degradation mechanisms occurring in the solid oxide cell (SOC) during operation. Whilst electrochemical measurements are currently conducted in situ, many analytical techniques can only be used ex situ and may even be destructive to the cell (e.g. SEM imaging of the microstructure). In order to fully understand and characterise co-electrolysis, in situ monitoring of the reactants, products and SOC is necessary. Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) is ideal for in situ monitoring of co-electrolysis as both gaseous and adsorbed CO and CO2 species can be detected, however it has previously not been used for this purpose. The challenges of designing an experimental rig which allows optical access alongside electrochemical measurements at high temperature and operates in a dual atmosphere are discussed. The rig developed has thus far been used for symmetric cell testing at temperatures from 450 °C to 600 °C. Under a CO atmosphere, significant changes in spectra were observed even over a simple Au|10Sc1CeSZ|Au SOC. The changes relate to a combination of CO oxidation, the water gas shift reaction, carbonate formation and decomposition processes, with the dominant process being both potential and temperature dependent.
1 Introduction
Electrochemical devices based on solid-state ceramic electrolytes have a wide range of applications from sensors to membrane reactors; batteries to fuel cells. One of the attractive features of these devices is their flexibility and efficiency when operated at high temperature. Solid oxide fuel cells have received much attention over the last four decades, resulting in many advances in the understanding of the technology. It was recognised very early on that solid oxide cells (SOC) were equally capable of reverse operation in electrolysis mode, for example the pioneering work of Doenitz et al.1,2 on steam electrolysis.Recent research interest has focused on the co-electrolysis of steam and carbon dioxide for the single-step production of syngas. This process is attractive because it represents an efficient route to producing synthetic liquid hydrocarbon fuels with all of the associated benefits of high energy density, transport infrastructure and ease of handling. In cases where the carbon dioxide used during electrolysis is recycled from the air or another point source, a closed carbon cycle is possible. Furthermore, the efficient conversion of electricity into an energy-dense chemical offers the possibility for commercially viable large-scale energy storage.
There are compelling reasons to pursue high temperature co-electrolysis, but as it stands the technology remains by-and-large in the laboratory. One of the key challenges facing co-electrolysis is the lack of understanding of fundamental reaction mechanisms. Knowledge of the rate-limiting reactions within a cell would aid rational design of materials and microstructures, leading to increased device efficiency and longevity. Comprehensive understanding of the relationship between the electrolysis, surface catalytic and water gas shift reactions would enable advancement of electrolysis technology. To achieve the level of detailed understanding required, the development of in situ analysis techniques is essential.
Typical in situ or operando testing of fuel cells and electrolysers has been limited to electrochemical methods such as impedance spectroscopy and direct current techniques. In situ spectroscopy could provide invaluable information about the surfaces and species that play a crucial role in the overall macroscopic reaction, along with information on the complex interplay of reactions occurring during co-electrolysis. Progress in the development and application of in situ spectroscopy methods to high temperature electrochemical devices has accelerated recently and techniques such as Raman3–11 and X-ray photoelectron spectroscopy7,12–15 now deliver valuable information from operational devices. Typically these techniques give potential-dependent structural or chemical information, but do not always yield direct information about the chemistry of the reactions, intermediates or surface interactions.
Infrared absorption spectroscopy can bridge the gap between the surface electronic/crystal structure and link to the chemistry of the gas-phase and absorbed species. It is particularly suited to this application because most molecular vibrational modes fall into the energy range of mid-infrared light (2.5–50 μm, 4000–200 cm−1). Fourier transform infrared (FTIR) spectroscopy has become routine in most analytical laboratories throughout the world and commercially available instruments tend to be highly adaptable, allowing numerous combinations of gas–solid interactions to be probed. This has made in situ IR techniques highly popular for heterogeneous catalysis research.16–18 The majority of early catalysis studies were carried out in transmission mode (TIR) whereby the incident beam passes through the sample/gases of interest. This requires that samples are self-supporting and not completely opaque to the incident beam. Whilst this method has been applied to the study of fuel cell materials,19,20operando studies are difficult and not representative of working conditions due to the requirement of transparent electrodes for cell polarization. An alternative is to use diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), another technique commonly employed in the catalysis community.16 The experimental configuration has the advantage of allowing the typical powder-basket arrangement to be replaced by a small solid oxide cell operated in a scenario very similar to most lab-scale electrochemical tests.
There are few reported studies using FTIR to probe SOCs and those that exist mainly focus on fuel cell operation. Lu et al. used IR emission spectroscopy to study the oxygen electrode material in SOCs, finding that two to three different oxygen ion species were present on the electrode surface. Murai et al.21 simplified the experiment by using symmetric Pt|YSZ|Pt cells to study the oxygen reduction reaction, finding electrode microstructure and silica contamination caused significant variation in both electrochemical performance and spectroscopic response. There have been very few IR studies to examine the fuel electrode in detail, with the exception of Kirtley et al.22 It is rather surprising that there are so few examples of in situ or operando research on the fuel electrode given the large number of examples of application of IR spectroscopy to low and high temperature water–gas shift reactions23–32 using metal catalyst particles on active catalyst supports (such as zirconia and ceria), which bear close resemblance to the reactions and materials systems in a SOC.
In this paper we report the development of a modified commercial DRIFTS unit for the study of SOCs under operating conditions. Herein, we describe the technical design of this apparatus, and demonstrate its viability and potential to provide surface specific information on the electrochemistry of polarized electrodes and ionic surfaces. This is illustrated by preliminary experiments using symmetric gold electrodes on scandia-stabilized zirconia substrates under a range of technologically relevant conditions.
2 Technical design of the DRIFTS apparatus
A schematic of the DRIFTS setup is shown in Fig. 1. The Spectra-Tech system fits an environmental chamber (from here on referred to as the base unit) into a demountable mirror assembly (Fig. 1D), which decouples the base unit from the mirror assembly allowing modifications to be made to the base unit design. Previous catalysis studies in our laboratory33 required modifications to the original factory base unit design (Fig. 1A) to improve sealing around the crucible and reduce the pressure drop through the catalyst bed (Fig. 1B). Further improvements to this design were incorporated to include electrical feed-throughs and gas ports (Fig 1C and detail in Fig. 2). There are several design requirements for the modified apparatus to operate SOCs effectively, which can be broken down into four main categories: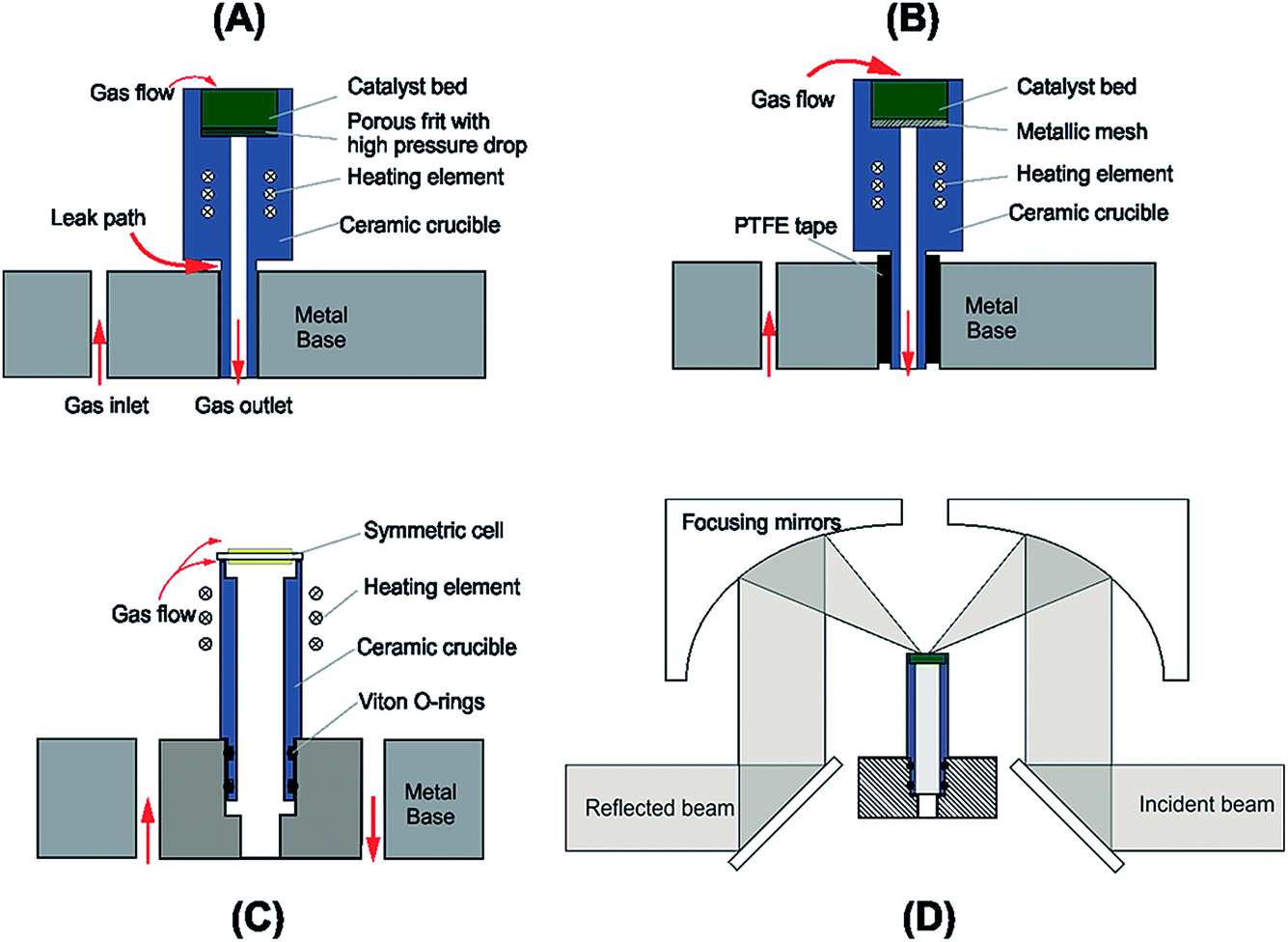 | ||
| Fig. 1 Schematic drawing the evolution of the base unit design and the optical path through the DRIFTS apparatus. (A) As purchased Spectra-Tech configuration. (B) Modification to reduce gas leak in chamber. (C) SOC operation configuration. (D) Full schematic view of DRIFTS rig showing mirror assembly and beam path. | ||
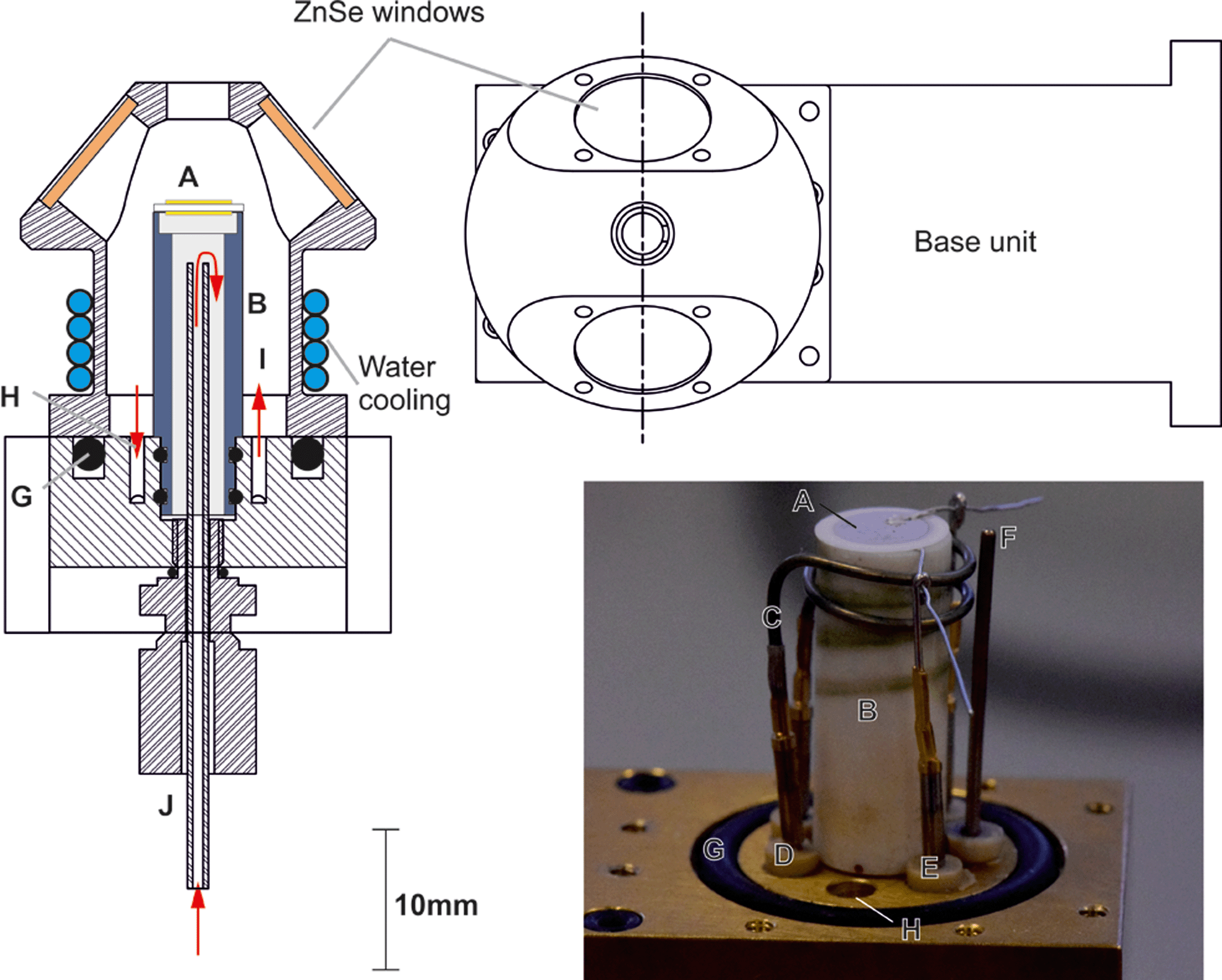 | ||
| Fig. 2 Detailed cross section of base and dome and a photograph of apparatus showing electrochemical cell (A), crucible (B) heating element (C), gold crimp connections for the heating wire (D) and the electrical cell connections (E), K-type thermocouple (F), dome Viton O-ring seal (G) cell, gas feed-throughs (H and I) and underside inlet port (J) (see text for more detail). | ||
• Temperature;
• Geometrical considerations;
• Electrical and spectroscopic measurement;
• Materials.
2.1 Overview of design criteria
To gather data relevant to solid oxide electrolysis cells, operation in the range of 500–800 °C is required. The temperature needs to be distributed evenly across the cell to ensure reproducible electrochemical performance and to correlate the spectra with the correct operational temperature. Heating is achieved by placing an electrical resistance wire as near to the sample as possible. This is commonly achieved by utilizing a wire-wrapped crucible33 or by installing a cartridge heater close the sample.34
The cell configuration design is only limited by the cell diameter, which has to be a minimum of 8.5 mm to fit the top of the cell and be supported by the crucible used in this rig (Fig. 2A). As with any electrochemical cell it must be self-supporting, but can be either electrode or electrolyte supported.
The previous version of this DRIFTS apparatus33 used a crucible with a narrow neck which was susceptible to cracking during sample loading or high temperature operation. More recent iterations of the design widened the crucible neck to accommodate an internal gas feed and which also reduced the likelihood of cell failure due to cracking at the crucible neck.
2.2 Detailed design of the cell used in these studies
Fig. 2 shows the base unit in detail: the electrochemical cell (Fig. 2A) is mounted on the crucible (Fig. 2B) which is machined from a commercial glass ceramic (Macor, RS Components). Previous crucible designs (Fig. 1B) had a very narrow neck which was susceptible to mechanical failure, causing loss of the gas-tight seal. In the design iteration reported herein, the wider base adds space for a gas tube to feed gas to the internal electrode of the cell. Introduction of a double Viton O-ring seal (Fig. 2G) prevents gas leakage between the crucible and the base. In another departure from previous reported designs,33 an exposed heater wire (Fig. 2C) was used which can be detached independently from the crucible. The heater consisted of a spiral-wound length of nichrome furnace wire, 0.8 mm in diameter and 16 cm in length with a total resistance of ∼0.7 Ω. It was attached to the feed-through pins (Fig. 2D) using gold plated crimp connectors (RS components). Temperature control was achieved within an accuracy of ±1 °C by means of a custom built 24 V system with a furnace controller (2216e, Eurotherm) with the input from a type K thermocouple (Fig. 2F) mounted as close to the cell as practically possible. Hot zone temperatures were limited to 600 °C as the low heater wire resistance means a very large current is drawn to deliver the power needed for operation above 600 °C. This heater configuration was retained as it allows significant flexibility in the chamber configuration. Future iterations are planned to increase the heater resistance to allow higher temperature operation.For the testing reported in this paper, a symmetric cell was operated in a single atmosphere. To achieve this, the outlet below the crucible (Fig. 2J) was closed and the cell placed on top of the crucible suspended by a few dots of silver paste to form a small gap below the cell, allowing gas flow across both electrodes. Electrode wires were connected to the same type of pins and crimp connectors (Fig. 2E) used for the heater wire. Gas feed-throughs consist of gold plated pins embedded in a barbed PEEK collar (Fig. 2H). Within the collar, the pin is soldered to a length of wire. To achieve a hermetic seal, the PEEK collar, along with the pin, is press fit into the base unit. Both the feed-though and the O-rings are all capable of withstanding temperatures up to 200 °C.
Water cooling of the base unit is required when the cell operating temperature is above 200 °C and is supplied via internal channels in the base unit, fed (via a peristaltic pump) into a tube coil wrapped around the neck of the dome (shown in Fig. 2). The base unit remains below 50 °C when the water-cooling is in operation up to hot zone temperatures of 600 °C. Addition of the gas feed-throughs, extra gas port and compression fitting on the underside of the base unit provide the capability to perform full cell testing while still fitting into the existing mirror assembly.
3 Experimental
3.1 Sample preparation
SOCs were prepared by brushing 6 mm diameter symmetric Au electrodes (C5756, Heraeus) onto commercial 10Sc1CeSZ electrolytes (Kerafol) before being dried at 120 °C for 1 h. Cells were then sintered in air at 600 °C for 1 h. Typical electrolyte thicknesses were 130 μm with electrode thicknesses of 90 μm. The cells were then placed on top of the crucible and Ag flying lead current collectors were attached to the electrodes using Ag paste (Gwent Electronic Materials), which were subsequently fired in situ at 600 °C for 1 h.3.2 Sample characterisation
The reactant gases, Ar (99.998% Ar), Kr (5% Kr in Ar), and CO (10% CO in Ar) supplied by BOC, were individually regulated by Aera™ FC-7700C mass flow controllers at a total flow rate of 40 cm3 min−1. The cell was heated to temperature in Ar at a rate of 3 °C min−1 and pre-cleaned by two potentiodynamic scans in Ar between ±2.0 V prior to testing. The experimental atmosphere was then applied for a minimum of 30 min to achieve steady state conditions. Infrared spectra were obtained using a Bruker Vertex 70 FTIR spectrometer equipped with a liquid N2 cooled detector. The in situ DRIFTS spectra were recorded twice at each voltage with a resolution of 4 cm−1. A background spectrum of the cell (56 scans averaged over 30 s) was taken in the reactant atmosphere at temperature, at open circuit, and has been subtracted from the results presented. All DRIFT spectra are displayed as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1/R where R is the relative reflectance (R = reflectance of the sample recorded in the gas feed at temperature and potential/reflectance of the sample recorded under gas feed at temperature and open circuit potential). During measurements potential was applied, starting at 0 V, in steps of 200 mV. The cell was held at each step voltage for a duration of 1 min to enable the data for two spectra to be recorded. Spectra were then analyzed using proprietary OPUS software. Electrochemical measurements were performed using a potentiostat (Ecochemie B.V., Autolab PGSTAT302) with the error in the applied potentials of ±10 mV. The reacted gas phase species were analyzed using an online Hiden Analytical HPR20 Mass Spectrometer (MS). Hermetic sealing of the system was verified by monitoring the levels of H2O (m/z = 18), O2 (m/z = 32) and CO2 (m/z = 44) using the MS.
1/R where R is the relative reflectance (R = reflectance of the sample recorded in the gas feed at temperature and potential/reflectance of the sample recorded under gas feed at temperature and open circuit potential). During measurements potential was applied, starting at 0 V, in steps of 200 mV. The cell was held at each step voltage for a duration of 1 min to enable the data for two spectra to be recorded. Spectra were then analyzed using proprietary OPUS software. Electrochemical measurements were performed using a potentiostat (Ecochemie B.V., Autolab PGSTAT302) with the error in the applied potentials of ±10 mV. The reacted gas phase species were analyzed using an online Hiden Analytical HPR20 Mass Spectrometer (MS). Hermetic sealing of the system was verified by monitoring the levels of H2O (m/z = 18), O2 (m/z = 32) and CO2 (m/z = 44) using the MS.
4 Results
The effect of potential on surface species during polarization (−2 V to +2 V) of the symmetric Au|10Sc1CeSZ|Au SOC as detected by in situ infrared spectroscopy under a gas feed of 5% CO in Ar is depicted in Fig. 3. For clarity, only steps at 0 V, ±0.8 V and ±1.6 V are shown. | ||
| Fig. 3 Comparison of DRIFTS spectra obtained for symmetric SOC utilizing a doped zirconia electrolyte and Au electrodes, operated in a 5% CO in Ar atmosphere under D.C. polarization at (A) 450 °C, (B) 550 °C and (C) 600 °C. | ||
DRIFTS data recorded at 450 °C (selected potentials shown in Fig. 3A) show distinct peaks at 3245, 2360 and 1320–1896 cm−1. An increase in the intensity of peaks at 3245 and 1320–1896 cm−1, attributed to surface hydroxyl species,19,20 is observed when a negative potential is applied. Interestingly, these hydroxyl species appear to accumulate with applied potential over time and the surface response does not return to base levels even at zero potential, indicating a non-reversible accumulation. In contrast, the strong signal associated with gas-phase CO2 (2360 cm−1) is clearly potential dependent. This band shows a negative feature when positive potential is applied, which is associated with a loss of CO2 from the gas phase, i.e. a reduction in the rate of CO2 formation compared with that formed at open circuit potential at 450 °C. This loss, still present on returning to 0 V, is then reversed under negative applied potential, resulting in an increase in the production rate of gas phase CO2 compared with the initial state of the catalyst.
When increasing the temperature from 450 °C to 550 °C (Fig. 3A and B) or to 600 °C (Fig. 3C), the peaks attributed to surface hydroxyls20 (3245 cm−1) and CO2 (2360 cm−1) persist. The intensity of the hydroxyl signal shows a distinct decrease in intensity with increasing temperature across the three experiments. A noticeable change in the potential dependence of the band associated with CO2 is observed by comparing spectra at 450 °C and 550 °C. At 550 °C (Fig. 3B) the intensity of the CO2 signal increases and then decreases under positive and negative potentials, respectively. Inspection of the 1200–1800 cm−1 region reveals several possible species with signals observed at 1694, 1540, 1400 and 1297 cm−1. These peaks are tentatively assigned to carbonate and bicarbonate19,20 species on the electrode surface with the positive peaks indicating formation and negative peaks removal from the surface. The positive signal detected at 1540 cm−1, assigned to bidentate carbonate species formation, shows the largest change in intensity under positive potentials, while bicarbonate species (1694, 1400 and 1297 cm−1) exhibited significant response under applied negative potential. In the latter case, both formation and removal are observed.
In contrast to observations at 450 °C (Fig. 3A) and 550 °C (Fig. 3B), the CO2 peak observed at 600 °C (Fig. 3C) remains positive throughout the experiment, showing an increased rate of gas phase CO2 production with respect to the initial open circuit potential state of the catalyst over time when a potential is applied. When comparing data collected at 550 °C and at 600 °C (Fig. 3Bvs.3C), the intensity of the signals associated with carbonate and bicarbonate species (1200–1800 cm−1) also increase with operating temperature. This indicates that the formation of carbonate and bicarbonate species is both potential and temperature dependent. Increased operating temperature induces the formation of carbonate and bicarbonate species while more complex effects such as formation/adsorption and removal are observed when a potential is applied.
5 Discussion
A rig for operando DRIFTS studies of SOCs has been designed and constructed and used successfully to analyse the surface and gas species under both fuel cell and electrolysis operating potentials.DRIFTS spectra are collected from the top electrode of the cell with the option to switch polarization between positive and negative potential, resulting in electrode dynamics which reflects either electrolysis (negative) or fuel cell (positive) operation. Since the atmosphere is swept continuously by the feed gas, it is unlikely that products from electrode reactions occurring at the bottom electrode are detected by the spectroscopy and that changes observed in the DRIFTS are only associated with the top electrode.
The results show three clear processes occurring during cell operation: (1) the potential dependent production of CO2, (2) the formation of complex carbonate species,19,20 and (3) the role of water in the form of an associated surface hydroxide species35 which are surface species that interact via hydrogen bonding giving rise to the broad hydroxyl peak in the 3500–3000 cm−1 range. There are frequent spectroscopic observations in the literature of carbonate formation in both the ceramic-only zircona system19,20 and the phenomena is more widely observed in the supported catalysis literature, particularly with reference to the water–gas shift reaction (WGS).36 Additionally, there is some debate in the literature as to the precise assignment of the carbonate species due to the possibility of the development of formate species.32 However, it should be noted that as well as the features between 1200–1800 cm−1, additional bands, just below 3000 cm−1, are also typically observed24 when formates are present. In the present study, these features were not present and thus it is thought that carbonate rather than formate is the predominant surface species.
For carbonates to form from CO, either water or a hydroxylated species must be present.35 Gas phase CO2 can then subsequently be formed by decomposition of the carbonate. The surface and gas phase species observed are not thought to originate from outgassing of organics from the electrode paste due to the combination of high temperature preprocessing of the electrodes in air (600 °C), long equilibration time in each atmosphere, and evidence in the literature suggesting that preparation methods using pastes were not considered an origin of carbonate and hydration products.37
It is possible that the hydroxyl species observed, which are prevalent throughout the experiments and continuously increase, may indicate some water ingress in the experimental apparatus. This is, however, unlikely due to the high purity of the feed gases, the very low water concentration detected by the mass spectrometer during the measurements and the gas-tight seal of the chamber. These species are, therefore, thought to arise from the fact that the surface and sub-surface of the zirconia electrolyte was partially hydroxylated35,38 before testing and the electrochemical cleaning procedure used was not sufficient to remove surface species. This is in agreement with previous studies which showed that pre-treatment of yttria-doped zirconia at 600 °C in 20% O2/Ar for 1 h did not completely remove surface hydroxyl species.20
Fig. 3A shows a large band at 450 °C associated with OH species (3245 cm−1). As the temperature increases this band became smaller in keeping with higher protonic (or hydroxylates species) surface diffusion observed in zirconia.39 At 450 °C, as the cell is polarized in the positive direction, there is a marked decrease in CO2(g). This is consistent with the formation of surface carbonate according to Fig. 4.
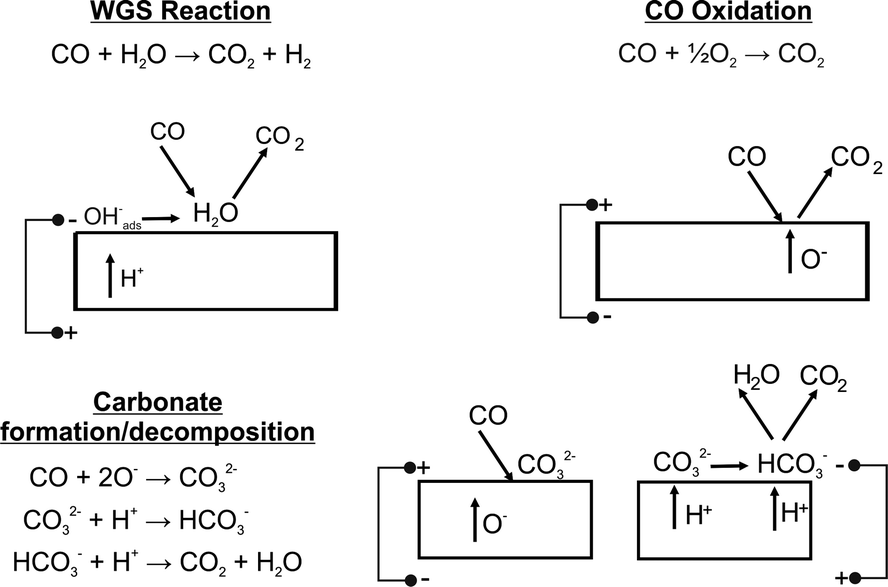 | ||
| Fig. 4 Proposed reaction mechanisms at the electrode surface. | ||
Electrochemically, a negative bias corresponds to pumping oxygen away from the spectroscopically probed surface (electrolysis mode). The changes observed can be attributed to both the thermal hydroxide/CO reaction, i.e. the water gas shift reaction, and electrochemical processes; however, it is not clear at this point which dominates. When the cell polarity is reversed, we see an increase in CO2 production. Once again this could be due to carbonate decomposition or the electrochemical oxidation of CO with oxygen ion transport. However, given the low temperature the oxygen ion conductivity is expected to be small, therefore, it is more likely that the heterogeneous catalytic reaction is controlled by the diffusion of protonic species and which is modified by the applied bias. At 550 °C (Fig. 3B), the OH bands are noticeably weaker. Carbonate species are removed throughout the period of the experiment, but the potential dependent change in the CO2 concentration has switched. The fuel cell mode produces CO2, presumably through the decomposition of remaining surface carbonates. There is no longer a sufficient amount of protonic species to allow carbonate formation during this process. During polarisation in electrolysis mode, very little CO2 production is observed, but carbonate removal continues. At 600 °C (Fig. 3C), the trend continues with the OH band decreasing, carbonate removal increasing, resulting in an increase in CO2 production, both from carbonate decomposition and CO oxidation.
There is some difficulty accounting for the presence of some of these species on the gold surface. Hydroxides and carbonates are often observed on the surfaces of metal oxides, but the mechanism for their existence on a pure metal surface is less well understood. Some studies have suggested that the penetration depth of an IR beam in a reflectance geometry is a few hundred microns33 suggesting subsurface detection would be possible. However, at 90 μm thick it is unlikely that the beam is passing through the electrode but further work is required to confirm this. It should also be noted that the observations from IR studies and the electrochemical behaviour of the cell do not necessarily match. This is because the majority of the electrochemistry occurring at the triple-phase boundaries is located at the Au|SSZ interface. Species and chemical responses that are observed in the IR spectra are, therefore, not necessarily dominated by intermediates or products of the electrochemical reactions. This is also why the only potential dependent species observed in this study is CO2, a by-product of the catalysis and electrochemical reactions.
One would also expect that, particularly in fuel cell mode when oxygen in pumped towards the spectroscopically active electrode, that a marked increase in CO oxidation would be observed. If the presence of hydroxides and carbonates preferentially react with electrochemically supplied oxygen or simply adsorb to sites active to CO oxidation at the phase boundary between gas|Au|SSZ, the absence of CO peaks at (2200–1950 cm−1) would be justified.
In summary, under negative bias the water gas shift reaction occurs at 450 °C but its importance decreases with temperature due to the fact that it is an exothermic equilibrium reaction. The changes with temperature under this bias are, therefore, likely to be due to the thermal/proton mediated decomposition of carbonates which increases in importance with temperature. Under positive bias, little CO oxidation occurs at 450 °C due to slow oxygen ion conduction; however, this process becomes more important as the temperature increases due to the increase in oxygen ion conduction. Hence at 450 °C, CO2 production only occurs at negative bias whereas at 600 °C it occurs under positive and negative bias.
6 Outlook
One of the main challenges facing any in situ measurement is to characterise the sample as close to realistic operational conditions as possible. Many of the limitations associated with this challenge have been overcome with this design of experimental apparatus, however, there remain some key aspects that need further development. Achieving the desired cell operating temperature and distribution remains a challenge as operating closer to 800 °C will induce even higher thermal gradients across the base unit components. The location of the heating element within the chamber atmosphere presents challenges to the integrity of the furnace wire but also introduces the risk of contamination of SOCs from volatile elements (such as chromium) in the heater wire. Future work will focus on a more improved thermal design to minimize these issues. Another limitation of the reflectance technique is difficulty in analysing the surface of electrodes that absorb strongly – which includes most state-of-the-art materials. Efforts are being focused on improved collection efficiency for the optical path and IR spectrometer.Despite careful control of gas inlet composition and design of a hermetically sealed chamber, the spectra showed that surface hydroxyl and complex carbonate species play a subtle but crucial role in the surface chemistry in a SOC. Although regularly discussed in the catalysis literature, the origin and nature of these species is not documented in the solid-state electrochemistry literature. From what has been observed in this study and those in literature, these species influence the water–gas shift reaction, an important reaction during co-electrolysis, and may also influence rate limiting intermediates during electrochemical reaction. These observations also highlight that IR spectroscopy will see all species present on the electrode surface meaning it is not only important to determine the region of the cell or electrode the IR signal originates from, but it is crucial to interpret which species are participating in the electrochemical reaction, which are influenced by those reactions and which are spectating. Once this is understood the key rate-limiting reactions may be identified and steps taken to improve cell chemistry and microstructure. This will be developed using analysis of the temporal behaviour of the electrochemistry and spectroscopic species by inducing a step change in potential or gas composition, for example.40,41
The above observations also highlight the importance of cell cleanliness. What is the origin of the observed surface species? Have they formed recently or is their occurrence based of sample history? Factors such as sample processing (e.g. electrode pastes) and the previous testing history are very relevant to cell cleanliness. Based on the results presented here, and from other IR studies in the literature, it is essential to monitor and tightly control the moisture in the system. A suitable cleaning routine, which could include electrochemical pre-treatment, is necessary to form a benchmark during future testing.
In an effort to better understand the role of some of the species observed in this work there is significant scope for use of alternative materials, such as doped ceria instead of zirconia for some electrode components. There is a large body of catalysis literature that shows the mechanism of carbonate formation and decomposition is highly dependent on the how easily the cations can be reduced in the ionic conductor support material. Comparison with the zirconia system and the influence of electrochemical bias will give further insights into the carbonate cycle.
The inability to determine the exact depth profile of the DRIFTS beam has meant there is some uncertainty surrounding the precise location of the observed species. By switching from pure metal electrodes to composite electrodes containing an ionic conductor the electrochemically active region will be extended away from the electrode–electrolyte interface to make it more optically accessible. Other in situ experiments have overcome this problem by using well defined patterned electrodes, however this reduces the cell performance to a level that is difficult to reconcile with a real cell. Another issue with this approach is the low concentration of surface species which will decrease the available spectroscopic information.
Finally, the main thrust of future work should be focused on the operation of full SOCs in situ. The apparatus we present here has the capability to do this and future publications will cover this area. There are important reactions on both the fuel and oxygen electrode which will benefit from full cell testing. This paper demonstrates the sensitivity of DRIFTS and it is anticipated that full cell testing will enable detailed operando study of degradation and poisoning mechanisms.
7 Conclusions
A new diffuse reflectance spectroscopic setup has been developed to enable the surface reactions within a SOC under bias and at temperature to be probed. Under a CO atmosphere, even over a simple Au|10Sc1CeSZ|Au significant changes were observed related to a combination of CO oxidation, the water–gas shift reaction and carbonate decomposition processes. The dominant process was found to be both potential and temperature dependent. This is a powerful technique with which to understand SOC in electrolysis and fuel cell mode and is currently being developed to allow the use of asymmetric cells in the presence of different atmospheres.Acknowledgements
This work was carried out as part of the “4CU” programme grant, aimed at sustainable conversion of carbon dioxide into fuels, led by The University of Sheffield and carried out in collaboration with The University of Manchester, Queens University Belfast and University College London. The authors acknowledge gratefully the Engineering and Physical Sciences Research Council (EPSRC) for supporting this work financially (Grant no. EP/K001329/1).References
- W. Doenitz and R. Schmidberger, Int. J. Hydrogen Energy, 1982, 7, 321–330 CrossRef CAS.
- W. Doenitz, R. Schmidberger, E. Steinheil and R. Streicher, Int. J. Hydrogen Energy, 1980, 5, 55–63 CrossRef CAS.
- M. B. Pomfret, J. C. Owrutsky and R. A. Walker, J. Phys. Chem. B, 2006, 110, 17305–17308 CrossRef CAS PubMed.
- R. C. Maher, L. F. Cohen, P. Lohsoontorn, D. J. Brett and N. P. Brandon, J. Phys. Chem. A, 2008, 112, 1497–1501 CrossRef CAS PubMed.
- J. Malzbender, R. W. Steinbrech and L. Singheiser, Fuel Cells, 2009, 9, 785–793 CrossRef CAS PubMed.
- D. J. L. Brett, A. R. Kucernak, P. Aguiar, S. C. Atkins, N. P. Brandon, R. Clague, L. F. Cohen, G. Hinds, C. Kalyvas, G. J. Offer, B. Ladewig, R. Maher, A. Marquis, P. Shearing, N. Vasileiadis and V. Vesovic, ChemPhysChem, 2010, 11, 2714–2731 CrossRef CAS PubMed.
- M. B. Pomfret, J. C. Owrutsky and R. A. Walker, in Annual Review of Analytical Chemistry, 2010, vol. 3, pp. 151–174 Search PubMed.
- R. C. Maher, G. Offer, N. P. Brandon and L. F. Cohen, 2011 MRS Fall Meeting, Boston, MA, 2012 Search PubMed.
- J. E. A. Saunders and M. H. Davy, Int. J. Hydrogen Energy, 2012, 37, 3403–3414 CrossRef CAS PubMed.
- B. C. Eigenbrodt and R. A. Walker, Spectroscopy, 2014, 29, 24–30 Search PubMed.
- X. Li, M. Liu, S. Y. Lai, D. Ding, M. Gong, J. P. Lee, K. S. Blinn, Y. Bu, Z. Wang, L. A. Bottomley, F. M. Alamgir and M. Liu, Chem. Mater., 2015, 27, 822–828 CrossRef CAS.
- S. C. DeCaluwe, G. S. Jackson, R. L. Farrow, A. H. McDaniel, F. El Gabaly, K. F. McCarty, S. Nie, M. A. Linne, H. Bluhm, J. T. Newberg, Z. Liu and Z. Hussain, Solid State Ionic Devices 6-Nanoionics - 214th ECS Meeting, Honolulu, HI, 2008 Search PubMed.
- M. Yang, E. Bucher and W. Sitte, J. Power Sources, 2011, 196, 7313–7317 CrossRef CAS PubMed.
- B. Bozzini, M. Amati, L. Gregoratti, C. Mele, M. K. Abyaneh, M. Prasciolu and M. Kiskinova, Electrochem. Commun., 2012, 24, 104–107 CrossRef CAS PubMed.
- Y. Yu, B. Mao, A. Geller, R. Chang, K. Gaskell, Z. Liu and B. W. Eichhorn, Phys. Chem. Chem. Phys., 2014, 16, 11633–11639 RSC.
- F. Zaera, Chem. Soc. Rev., 2014, 43, 7624–7663 RSC.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
- J. Ryczkowski, Catal. Today, 2001, 68, 263–381 CrossRef CAS.
- E.-M. Köck, M. Kogler, R. Pramsoler, B. Klötzer and S. Penner, Rev. Sci. Instrum., 2014, 85, 084102 CrossRef PubMed.
- E. M. Kock, M. Kogler, T. Bielz, B. Klotzer and S. Penner, J. Phys. Chem. C, 2013, 117, 17666–17673 Search PubMed.
- T. Murai, K. Yashiro, A. Kaimai, T. Otake, H. Matsumoto, T. Kawada and J. Mizusaki, 30th Symposium on Solid State Ionics in Japan, 2005, 176, 2399–2403 CrossRef CAS PubMed.
- J. D. Kirtley, D. A. Steinhurst, J. C. Owrutsky, M. B. Pomfret and R. A. Walker, Phys. Chem. Chem. Phys., 2014, 16, 227–236 RSC.
- F. C. Meunier, A. Goguet, C. Hardacre, R. Burch and D. Thompsett, J. Catal., 2007, 252, 18–22 CrossRef CAS PubMed.
- F. C. Meunier, D. Reid, A. Goguet, S. Shekhtman, C. Hardacre, R. Burch, W. Deng and M. Flytzani-Stephanopoulos, J. Catal., 2007, 247, 277–287 CrossRef CAS PubMed.
- D. Tibiletti, F. C. Meunier, A. Goguet, D. Reid, R. Burch, M. Boaro, M. Vicario and A. Trovarelli, J. Catal., 2006, 244, 183–191 CrossRef CAS PubMed.
- F. C. Meunier, D. Tibiletti, A. Goguet and R. Burch, Oil Gas Sci. Tech., 2006, 61, 497–502 CrossRef CAS.
- G. Jacobs, P. M. Patterson, L. Williams, E. Chenu, D. Sparks, G. Thomas and B. H. Davis, Appl. Catal., A, 2004, 262, 177–187 CrossRef CAS PubMed.
- E. Chenu, G. Jacobs, A. C. Crawford, R. A. Keogh, P. M. Patterson, D. E. Sparks and B. H. Davis, Appl. Catal., B, 2005, 59, 45–56 CrossRef CAS PubMed.
- G. Jacobs, U. M. Graham, E. Chenu, P. M. Patterson, A. Dozier and B. H. Davis, J. Catal., 2005, 229, 499–512 CrossRef CAS PubMed.
- T. Tabakova, F. Boccuzzi, M. Manzoli and D. Andreeva, Appl. Catal., A, 2003, 252, 385–397 CrossRef CAS.
- T. Shido and Y. Iwasawa, J. Catal., 1993, 141, 71–81 CrossRef CAS.
- G. Jacobs, E. Chenu, P. M. Patterson, L. Williams, D. Sparks, G. Thomas and B. H. Davis, Appl. Catal., A, 2004, 258, 203–214 CrossRef CAS PubMed.
- F. C. Meunier, A. Goguet, S. Shekhtman, D. Rooney and H. Daly, Appl. Catal., A, 2008, 340, 196–202 CrossRef CAS PubMed.
- X. Lu, P. W. Faguy and M. Liu, J. Electrochem. Soc., 2002, 149, A1293–A1298 CrossRef CAS PubMed.
- M. Kogler, E. M. Kock, T. Bielz, K. Pfaller, B. Klotzer, D. Schmidmair, L. Perfler and S. Penner, J. Phys. Chem. C, 2014, 118, 8435–8444 CAS.
- S. Hilaire, X. Wang, T. Luo, R. J. Gorte and J. Wagner, Appl. Catal., A, 2001, 215, 271–278 CrossRef CAS.
- P. Vernoux, L. Lizarraga, M. N. Tsampas, F. M. Sapountzi, A. De Lucas-Consuegra, J. L. Valverde, S. Souentie, C. G. Vayenas, D. Tsiplakides, S. Balomenou and E. A. Baranova, Chem. Rev., 2013, 113, 8192–8260 CrossRef CAS PubMed.
- D. L. Hoang and H. Lieske, Catal. Lett., 1994, 27, 33–42 CrossRef CAS.
- B. Scherrer, M. V. F. Schlupp, D. Stender, J. Martynczuk, J. G. Grolig, H. Ma, P. Kocher, T. Lippert, M. Prestat and L. J. Gauckler, Adv. Funct. Mater., 2013, 23, 1957–1964 CrossRef CAS PubMed.
- S. Chansai, R. Burch, C. Hardacre, J. Breen and F. Meunier, J. Catal., 2011, 281, 98–105 CrossRef CAS PubMed.
- S. Chansai, R. Burch and C. Hardacre, J. Catal., 2012, 295, 223–231 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |