 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen deficient layered double perovskite as an active cathode for CO2 electrolysis using a solid oxide conductor
Tae Ho
Shin
*ac,
Jae-Ha
Myung
a,
Maarten
Verbraeken
a,
Guntae
Kim
b and
John T. S.
Irvine
*a
aSchool of Chemistry, University of St Andrews, St Andrews, Fife, KY16 9ST, Scotland, UK. E-mail: ths@st-andrews.ac.uk; ceramist95@gmail.com; Tel: +44-(0)759-676-2424
bDept. of Chemical and Energy Engineering, Ulsan National Institute of Science and Technology (UNIST), Korea
cElectronic Materials Convergence Division, Korea Institute of Ceramic Engineering & Technology (KICET), Korea
First published on 26th March 2015
Abstract
A-site ordered PrBaMn2O5+δ was investigated as a potential cathode for CO2 electrolysis using a La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM) electrolyte. The A-site ordered layered double perovskite, PrBaMn2O5+δ, was found to enhance electrocatalytic activity for CO2 reduction on the cathode side since it supports mixed valent transition metal cations such as Mn, which could provide high electrical conductivity and maintain a large oxygen vacancy content, contributing to fast oxygen ion diffusion. It was found that during the oxidation of the reduced PrBaMn2O5+δ (O5 phase) to PrBaMn2O6−δ (O6 phase), a reversible oxygen switchover in the lattice takes place. In addition, here the successful CO2 electrolysis was measured in LSGM electrolyte with this novel oxide electrode. It was found that this PrBaMn2O5+δ, layered perovskite cathode exhibits a performance with a current density of 0.85 A cm−2 at 1.5 V and 850 °C and the electrochemical properties were also evaluated by impedance spectroscopy.
1. Introduction
With growth of renewable energy technologies, it is necessary to store vast amounts of energy by, e.g., converting electricity overcapacity to chemical energy in the form of compounds such as hydrogen, methane, or methanol. In particular, the variety of products from high temperature electrolysis can be used as feedstocks for chemical synthesis or converted into highly dense electric power; it would provide a means of storing renewable electricity in a convenient, high energy-density form. Recently, hydrogen has attracted attention as an alternative mediator for energy conversion and storage because of its high gravimetric energy density and zero carbon emission.1–5 However, the utilization of hydrogen has been limited due to the practical engineering and economic limitations with respect to its generation and distribution.6 On the other hand hydrocarbon fuels and carbon monoxide cost-effectively match the existing energy infrastructure well because of their similarity to current fossil fuels; they would be effective energy carriers in a period of transition toward zero carbon emission.7–9 Recycling or reuse of CO2 could be an effective approach for accessing a new form of energy carrier and enabling a carbon-neutral cycle. In particular, electrochemical reduction of CO2 into CO fuel with renewable electricity has been proposed as an alternative method to store this energy, overcoming the inherent problems with intermittency and geographical distribution of renewable energy sources.10–13Solid oxide electrolysis cells (SOECs) have recently attracted a great deal of interest as a highly efficient electrolyser and a green energy technology. Current developments are mainly based upon steam electrolysis for hydrogen production. However, reduction of CO2 using a highly efficient solid oxide electrolyser is also feasible. This would be an alternative to carbon capture and advanced utilization of CO2.14,15 Solid oxide electrolysers with oxygen ion conducting electrolytes are able to separately produce CO and O2 from CO2 (CO2 → 1/2O2 + CO) at high and intermediate temperature (500–900 °C) with high efficiencies (as they are not limited by the Carnot cycle). Both products, CO and O2 may be useful in industrial processes. The electrolysis of CO2 in a solid oxide device is analogous to high temperature steam electrolysis by SOECs, i.e. CO2 molecules are electrochemically reduced into CO at the cathode (fuel electrode) and oxygen ions are transported through the oxygen-ion conducting electrolyte to the anode (oxygen electrode) where gaseous O2 is produced and released. High temperature solid oxide electrolysis reduces electricity demand, and offers fast electrode kinetics, reducing the overall cell internal resistance as a consequence and increasing electrolysis efficiency. Due to the operating conditions in SOECs using CO2, cathode materials require stability in a wide pO2 and temperature range, and additionally need to be tolerant towards carbon coking. To date, SOECs predominantly comprise materials and techniques from solid oxide fuel cells (SOFCs), but operated in reverse modes. This means that electrode materials have not been tailored specifically for use in electrolysis conditions, which can differ substantially from fuel cell conditions. Long term stability of solid oxide electrode materials is still one of the important challenging issues for both SOECs and SOFCs for commercialisation into the energy market.
Currently, high temperature electrolysis of CO2 has been mainly performed using Ni–YSZ cermet cathodes in oxygen ion conducting SOECs. However, Ni is easily deactivated by reoxidation or carbon formation when it is directly exposed to CO/CO2 or a hydrocarbon atmosphere.16–18 Therefore, a significant reduction atmosphere is required to flow over the Ni surface base electrode to avoid oxidation of Ni to NiO, which would cause a loss of electronic conductivity and the failure of the electrode. In the most serious circumstances, the electrode fails mechanically as a result of deposited coke or re-oxidation.19 Consequently, redox stability and reversibility have recently been emphasised as important requirements for the cathode (fuel electrode) material in SOECs. So far, several conductive perovskite oxides have been reported as potential anode materials in SOFCs, which may also be useful as cathode materials in SOECs. For more sturdy and superior materials in durable SOFC/SOEC systems, various mixed ionic and electronic conducting (MIEC) oxides have been explored. Among these, researchers have mainly demonstrated that La(Sr)MO3 (M = Cr, Mn, Fe, Ti) perovskite oxides20–22 can, in principle, be good fuel electrode materials with sufficient electronic conductivity. For instance, the efficient electrolysis of CO2 based on a ceramic cathode, La(Sr)Mn(Cr)O3 (LSCM) perovskite oxide have been recently demonstrated.20,23–25 However, electrochemical performance of the ceramic electrodes is still smaller than that using a Ni based electrode, due to insufficient electrical conductivity and low electrocatalytic activity. LSCM suffers from a drop in conductivity under the reducing SOEC cathode conditions.
Recently we have succeeded in synthesising the oxygen deficient layered phase, PrBaMn2O5+δ under reducing atmosphere. Whereas polycrystalline samples sintered in air adopt the cubic ABO3 perovskite structure, the A-site ordered layered perovskite PrBaMn2O5+δ, can be grown under reducing conditions. Furthermore, it was found that the double perovskite, PrBaMn2O5+δ exhibits high electrical conductivity and excellent redox and coking tolerance when used as an anode for an SOFC.26 In this work, PrBaMn2O5+δ, employed as a potential ceramic cathode for CO2 electrolysis SOEC with a configuration of PrBaMn2O5+δ|La doped ceria (LDC)|La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM)|La(Sr)Fe(Co)O3. The performance of the PrBaMn2O5+δ cathode and the electrochemical process of high temperature electrolysis of CO2 in a cell using LSGM as the electrolyte were investigated and evaluated.
2. Experimental
The Pr0.5Ba0.5MnO3−δ was prepared using the conventional solid-state reaction method and then A-site ordered layered PrBaMn2O5+δ is obtained by in situ annealing of Pr0.5Ba0.5MnO3−δ in reducing conditions using 5% H2 at temperatures higher than 800 °C. The crystalline phase characterization of the oxide powder was performed by X-ray diffraction (PANalytical Empyrean, Mo Kα radiation and Cu Kα1 radiation). Thermal gravimetric analysis (TGA) was carried out on a NETZSCH TG 209 instrument (NETZSCH Geraetebau GmbH, Selb, Germany) with a heating & cooling rate of 2–5 °C min−1 to evaluate the oxygen loss and incorporation on redox cycling in air and 5% H2 on the compositions synthesized and dilatometry was also performed at 100 °C to 800 °C with a heating/cooling rate of 2 °C min−1 in air and 5% H2 to characterize the thermo-physical properties. Electrical conductivity was measured on the sintered pellets by the standard four terminal DC method or van der Pauw method up to 900 °C in ambient air. A current of 50 mA (model: Keithley 220, Keithley Instruments Inc., U.S.A.) was applied in both directions, and resistance was calculated as a gradient of potential vs. current. These conductivity details were previously reported.26For solid oxide electrolyser experiments, a configuration of PrBaMn2O5+δ|LDC|La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM)|La(Sr)Fe(Co)O3 was fabricated using 200 μm thick LSGM electrolyte supports. Commercially available powder, La0.9Sr0.1Ga0.8Mg0.2O3−δ (LSGM, 99.9% Kceracell co., Ltd, South Korea) was used for the preparation of electrolyte. The LSGM electrolyte supports were prepared by dry-pressing powder into a circular green body followed by high-temperature sintering in air at 1450 °C for 6 h and mechanical polishing until 2 cm diameter and 200 μm thick electrolyte supports were obtained. Electrodes were deposited onto the faces of the LSGM electrolyte disk by screen-printing inks of the respective powders, followed by sintering at 1100 °C for 30 min (thickness of screen printed electrode, <25 μm). The active area of the cells, equal to the area to the cathode area (PrBaMn2O5+δ), was 0.5 cm2 and the area of the anode was same.
To evaluate the electrochemical properties, I–V curves and impedance were measured using a four terminal configuration with silver mesh/wire as the electrode current collectors. In order to perform electrochemical testing in fuel cell mode, a dried CO2/CO (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 vol) mixture was supplied into the cathode with a 100 ml min−1 flow rate while the anode side was exposed to constant oxygen flow as an oxidant gas (100 ml min−1). The cell was maintained in each condition for at least one hour to reach steady state, before carrying out any electrochemical tests. AC impedance spectroscopy was recorded using an IM6 Electrochemical Workstation (Zahner, Germany) with frequency ranged from 0.1 Hz to 100 kHz with an amplitude of 10 mV.
:30 vol) mixture was supplied into the cathode with a 100 ml min−1 flow rate while the anode side was exposed to constant oxygen flow as an oxidant gas (100 ml min−1). The cell was maintained in each condition for at least one hour to reach steady state, before carrying out any electrochemical tests. AC impedance spectroscopy was recorded using an IM6 Electrochemical Workstation (Zahner, Germany) with frequency ranged from 0.1 Hz to 100 kHz with an amplitude of 10 mV.
3. Results and discussion
The disordered cubic perovskite Pr0.5Ba0.5MnO3 was prepared by a typical solid state reaction. To avoid the complete oxidization of Mn3+ to Mn4+, which would favour the formation of BaMnO3, the powder was calcined in an atmosphere with lower oxygen partial pressure (using a mixture of Ar/air) below the synthesis temperature (<1300 °C). A-site ordering in the layered manganite perovskite would be partially hindered by the formation of the hexagonal related perovskite BaMnO3. Fig. 1 shows the XRD pattern for the cubic disordered perovskite (Fig. 1a), prepared by solid state reaction at 1200 °C in air; reduction in 5% H2 leads to A-site ordering and hence doubling of the unit cell, giving a layered phase with tetragonal symmetry (Fig. 1b). The XRD pattern of disordered cubic perovskite shown in Fig. 1a, suggests the presence of a small amount secondary hexagonal phase, as similarly observed in a previous study in which the powder was prepared by Pechini synthesis at lower temperature.26 However, they are not serious impurities but a single phase of Pr0.5Ba0.5MnO3 was mainly indicated. Furthermore, the A-site ordered layered perovskite PrBaMn2O5+δ could also be grown by annealing the disordered phase in reducing atmosphere in accordance with previous results.26 To fully understand the phase changes during redox cycling, we checked the phase transition from disordered cubic Pr0.5Ba0.5MnO3 phase (O3) to oxygen deficient layered PrBaMn2O5+δ (O5) by in situ variable temperature/atmosphere X-ray diffraction (Mo Kα1/Kα2), whilst performing reduction–oxidation cycles. Fig. 2a–c shows Rietveld refinement profiles of Pr0.5Ba0.5MnO3 phase (O3), PrBaMn2O5+δ (O5) and PrBaMn2O6−δ (O6) from in situ XRD data at 800 °C in various atmospheres. The refined unit cell parameters and refinement statistics are summarized in Tables 1 and 2 and show selected bond distances and angles.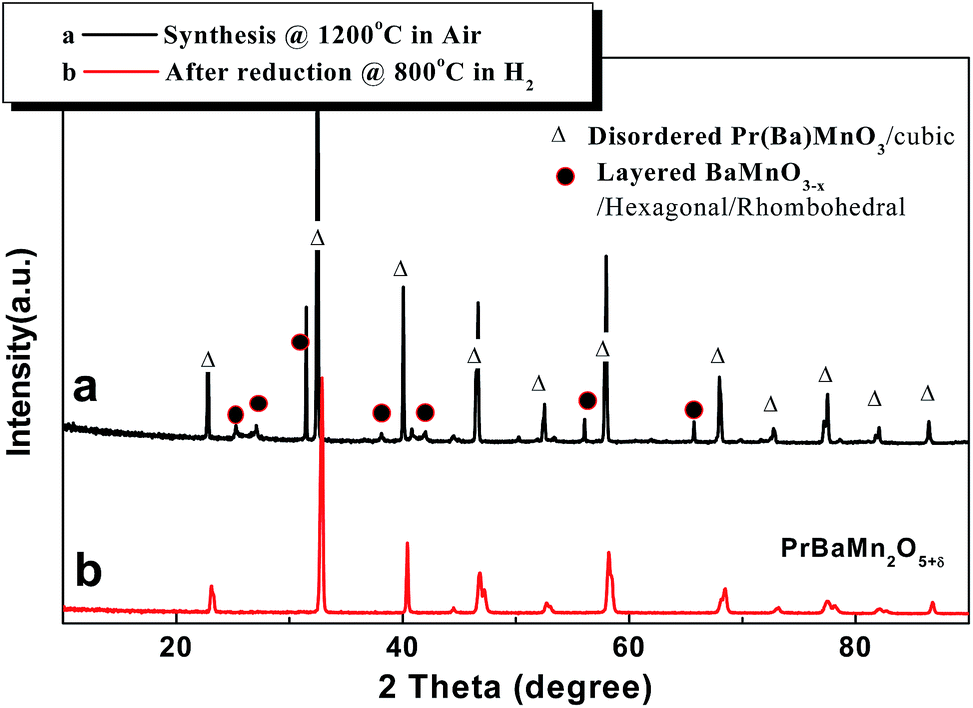 | ||
| Fig. 1 X-ray diffraction patterns of (a) Pr0.5Ba0.5MnO3 (O3) prepared by solid state reaction at 1200 °C in air, and (b) PrBaMn2O5+δ (O5) was obtained by reducing the as-prepared powder in 5% H2 at 800 °C for 4 h (Cu Kα radiation). | ||
 | ||
| Fig. 2 Rietveld-refined XRD profiles of (a) Pr0.5Ba0.5MnO3 (O3), (b) PrBaMn2O5+δ (O5) and (c) PrBaMn2O6−δ (O6) obtained by simultaneous fitting of in situ high temperature XRD data at 800 °C in air, 5% H2 and air, respectively. (Mo Kα1, Kα2 radiation). | ||
| Pr0.5Ba0.5MnO3 (O3) in air | PrBaMn2O5+δ (O5) in H2 | PrBaMn2O6−δ (O6) in air | |
|---|---|---|---|
| Space group |
Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (cubic) m (cubic) |
P4/nmm (tetra) | P4/mmm (tetra) |
| a = b (Å) | 3.93593(5) | 4.02405(9) | 3.93985(9) |
| c (Å) | — | 7.90618(24) | 7.84136(24) |
| 2a/c | 1.018 | 1.005 | |
| V (Å3) | 60.9735(13) | 128.024(6) | 121.717(5) |
| λ (Å) Mo Kα1 | 0.0793 | 0.7093 | 0.7093 |
| λ (Å) Mo Kα2 | 0.7136 | 0.7136 | 0.7136 |
| R p% | 7.93 | 6.43 | 6.06 |
| R wp% | 11.19 | 8.30 | 8.01 |
| Pr0.5Ba0.5MnO3 (O3) in air | PrBaMn2O5+δ (O5) in H2 | PrBaMn2O6−δ (O6) in air | |
|---|---|---|---|
| Distance (Å) | |||
| Mn–O1 | 1.96796(2) 6 | 2.067(5) × 1 | 2.03072(6) × 1 |
| Mn–O2 | — | 2.0405(18) × 4 | 1.97022(4) × 4 |
| Mn–O3 | — | — | 1.88996(6) × 1 |
| Mn–O (average) | 1.96796 | 2.0458 | 1.96693 |
| Angles (°) | |||
| Mn–O1–Mn | 180 | 180 | 180 |
| Mn–O2–Mn | — | 160.83(57) | 178.01680(10) |
| Mn–O3–Mn | — | — | 180 |
| O1–Mn–O3 | 180 | — | 180 |
| O2–Mn–O2 | 180 | 160.83(57) | 178.01680(10) |
The in situ high temperature X-ray diffraction study confirms cubic symmetry for the disordered O3 phase at 800 °C. Upon reduction at 800 °C, the A-site ordered O5 phase emerges with tetragonal symmetry, causing a relative shrinkage of the c-axis as compared to the a-axis. Refinement is successful using the P4/mmm space group. Subsequent re-oxidation of the A-site ordered O5 phase at 800 °C results in the emergence of the double perovskite, PrBaMn2O6−δ (O6) phase. This O6 phase also has tetragonal symmetry with space group P4/mmm. Re-oxidation of the O5 phase causes a decrease in tetragonality as evidenced by the drop in 2a/c ratio from 1.018 to 1.005. Despite this, direct evidence of A-site ordering and thus doubling of unit cell in both O5 and O6 phased from the XRD data is the (001) reflection found at 2θ ≈ 5°as shown in Fig. 2b and c. Despite the weak intensity of the (001) reflection due to small contrast between Pr3+ and Ba2+, its emergence on redox cycling is significant.
The fully oxidised ordered O6 phase shows A-site ordering along the c-axis, with a stacking pattern of alternating BaO–MnO2–PrO–MnO2–BaO layers. As expected from perovskite materials, Mn is six coordinated, whereas both Ba and Pr are twelve coordinated. The MnO6 octahedron is slightly distorted, causing the Mn and O2 sites to be closer to Pr than to Ba. The reduced O5 variant has an identical stacking sequence but additionally exhibits oxygen vacancy ordering, with the O3 site vacant, resulting in Pr layers without oxygen ions, shown in Fig. 3. In this structure Mn becomes square pyramidally coordinated, whereas Pr is now 8 coordinated to O2. Ba becomes less coordinated to O2, as the latter moves closer to Pr. The lattice volume is increased due to Mn reduction as shown in Tables 1 and 2.
 | ||
| Fig. 3 A schematic drawing of crystal structure model determined for A-site ordering layered PrBaMn2O5+δ (O5) and oxygen vacancy ordering in oxygen deficient O5 phase by Rietveld-refined XRD profile. | ||
Due to the reduction of Mn3+/Mn4+ in the O3/O6 phases to Mn2+/Mn3+ in O5 the Mn–O bond length at 800 °C increases from 1.89–2.03 to 2.04–2.07 Å. It is expected that the oxygen deficient PrOx layer provides a channel for fast oxygen transport and therefore A-site ordering is in fact enhancing the oxygen mobility, albeit only in two dimensions.27 The coexistence of Mn oxidation states is further expected to give rise to electronic conductivity, which is a requirement for any SOEC/SOFC electrode material. The mixed valence of transition metals such as Mn could give rise to different Mn–O bond lengths at various oxygen partial pressure conditions; the swollen Mn–O bonding length (ca. 2.046 Å) of the oxygen deficient phase could be expected from the reduced oxidation state of Mn (Mn3+ or Mn2+). The vacancy would be mainly located between reduced Mn3+/Mn2+ state cations, between pairs of square pyramidal MnO5. Thus oxygen vacancy ordering provides a mechanism to reduce the coordination number of the smaller Pr3+ ion without reducing the coordination number of the larger Ba2+ ion. Under reducing conditions, oxygen atoms in the PrOx plane can be partially or entirely removed, creating many oxygen vacant sites in the crystal sites. This might illustrate the close coupling between A-site ordering and oxygen vacancy ordering.27
What makes the layered perovskite-manganese oxide PrBaMn2O5+δ a promising candidate for high oxygen mobility is its remarkable variability in oxygen content under redox conditions. It was generally found that for ReBaMn2O5+δ (Re = Pr, Nd, La, Gd) the oxidation of the reduced (O5) phase PrBaMn2O6−δ (O6), in the case of ReBaMn2O5+δ, a reversible oxygen switchover in the lattice takes place. The reversibility of the phase change during redox cycle was also established by thermal analysis. The fully oxygen charged PrBaMn2O6−δ (O6) had been reduced in 5% H2/95% Ar at elevated temperature up to 800 °C and cooled in 5% H2/95% Ar to room temperature (2.5 °C min−1) and re-oxidized up to 700 °C in air. Fig. 4 shows the thermogravimetric analysis (TGA) in which a weight gain of ca. 3.58 wt% was observed on both reduction and re-oxidation corresponding to a loss/uptake of 1 formula unit of oxygen (δ = 1). This symmetry between reduction and oxidation cycles suggests excellent reversibility, which is in agreement with our previously reported dilatometry data.26
 | ||
| Fig. 4 TGA curves of PrBaMn2O5+δ in 5% H2/Ar from room temperature to 700 °C to room temperature and in re-oxidation cycle from room temperature to 700 °C. | ||
Since PrBaMn2O5+δ shows superior redox reversibility as well as potential electrochemical performance as oxide anode for SOFCs, application of PrBaMn2O5+δ for a potential cathode in CO2 electrolysis using solid oxide ion conductor, LSGM, was further studied. Fig. 5a shows the temperature dependent I–V curves for CO2 electrolysis between 750 °C and 850 °C under the following gas feed conditions: air on the anode side and a mixture of 70% CO2/30% CO on the cathode side with 100 ml min−1. It was found that PrBaMn2O5+δ is very effective for CO2 electrolysis with a remarkable cathodic current density of 0.84 A cm−2 at 1.55 V and 850 °C. At lower temperature (750 °C), it seems that there was no significant current flow at lower voltages suggesting a slow electrolysis reaction, which might be explained by the large overpotential on the cathode side; the cell voltage sharply increased with increasing current density at initial low current range. Fig. 5b shows the I–V curve at 850 °C, during current cycling, which was measured with increasing current up to the maximum current range and subsequent decreasing current, leaving long stabilisation times at every current step to attempt to attain equilibrium. However, it was observed that voltage recovery was slow and strongly time dependent. As shown in Fig. 5b, initial OCV (0.825 V) was slightly increased to 1.141 V after current cycling; the initial OCV was however recovered over time. The increased OCV after cycling could be related with momentary carbon deposition or carbon monoxide adsorption on the catalyst surface, as 1.14 V is very close to the theoretical OCV value for carbon fuel; slow surface kinetics of PrBaMn2O5+δ might explain such desorption/adsorption CO/CO2 or carbon species. Alternatively, poor catalytic activity for carbon oxidation or a Ba carbonation reaction might play a role too. The slow chemical kinetics are not fully understood at this stage, but a more detailed analysis using in situ exhausts gas analysis, catalyst additives and in situ surface analysis would be helpful to shed further light on the issue in future work. In particular, stable catalytic activity of PrBaMn2O5+δ for CO2 could be improved with other catalyst additives, whereas B-site doping may help stabilize the Ba from carbonisation.
 | ||
| Fig. 5 (a) Temperature dependent I–V curves for CO2 electrolysis under gas feed conditions with elevating temperature (750–850 °C) with the cell configuration of PrBaMn2O5+δ|LDC|La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM)|La(Sr)Fe(Co)O3 and (b) the I–V curves, voltages during steady current cycling at 850 °C. | ||
For the detailed reaction mechanisms, ac impedance spectra were measured. Fig. 6 shows complex impedance plots under open circuit conditions for internal resistance of the cell PrBaMn2O5+δ|LDC|La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM)|La(Sr)Fe(Co)O3. In general, it is necessary to have a reference electrode to separate anode and cathode losses. However, it should be recognized that improper placement of the reference electrode and working electrodes will give erroneous value; thus, exactly separating each electrode can be difficult even in a symmetric electrode geometry in a thin electrolyte support cell, when the kinetics of the two electrodes are significantly different.28–30 As shown in Fig. 6a, it is significant that the ohmic resistance in both impedance spectra are approximately the same, and almost exactly one-half of the total ohmic resistance of the cell, showing that the reference electrode is sampling the potential at the centre of the electrolyte. Thus, it can be roughly inferred that total polarization resistance was clearly dominated by cathodic electrode (PrBaMn2O5+δ) because cathodic impedance spectra was similar to the total cell impedance. It is obvious that at least two semicircles are present in the impedance plot. Considering the response frequencies of the two parallel RQ circuits, the resistance at higher frequency (Rp1) could be assigned to a charge transfer process, whereas the lower frequency process (Rp2) seems related to a diffusion process of the cathodic reaction. The contribution of Rp2 is much larger than that of the surface reaction step (Rp1), and so the observed impedance may indicate a slow CO2 diffusion, which may be due to slow chemical surface adsorption/desorption and/or slow surface-diffusion processes for CO2 reduction on the surface of PrBaMn2O5+δ at lower temperature. Although fast oxygen ion mobility would be expected due to a large concentration of ordered oxide ion vacancies in PrBaMn2O5+δ, it shows lower surface activity for CO2 reduction at lower temperature. However, fairly good performances were still achieved at temperatures higher than 800 °C and its surface activity for CO2 may be improved if other catalytic additives are effectively employed. Due to improved surface activity, various metal catalyst additives are now under study, and the results will be reported in the future. Fig. 7 shows SEM micrographs of the cathode of the PrBaMn2O5+δ|LDC|La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM)|La(Sr)Fe(Co)O3 cell before and after CO2 electrolysis. There appears to be some limited coking on the top surface that might be explained by Boudouard reaction at high temperature with CO2/CO mixture or slow response of PrBaMn2O5+δ for the chemical surface adsorption/desorption of CO2 molecules; however, there were no serious surface morphology changes. It was therefore concluded that A-site ordered layered perovskite, PrBaMn2O5+δ, could be used as a potential cathode in solid oxide electrolysis with excellent redox tolerance.
 | ||
| Fig. 6 Nyquist plot of the PrBaMn2O5+δ|LDC|La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM)|La(Sr)Fe(Co)O3 cell under open circuit: (a) impedance plot of cells, cathodic and anodic under 70% vol CO2 gas fuel condition at 850 °C, (b) comparison of cathodic impedance plot under H2 with 70% vol CO2, (c) temperature dependant impedance plot of the total cell under 70% vol CO2 gas fuel, (d) temperature dependant impedance plot of the total cell under H2 gas fuel. | ||
 | ||
| Fig. 7 SEM images of a PrBaMn2O5+δ|LDC|La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM)|La(Sr)Fe(Co)O3 cell after electrochemical tests, (a) cross section of the cell and (b) cathode parts include PrBaMn2O5+δ|LDC, (c) the top of the surface, and (d) high magnification of the cathode. | ||
4. Conclusion
In summary, PrBaMn2O5+δ was successfully prepared as a single phase cathode material from a disordered O3 perovskite phase after reduction treatment. The reduction to obtain layered PrBaMn2O5+δ can be performed in situ during SOEC/SOFC testing to limit processing steps. An in situ high temperature X-ray diffraction study confirmed the phase changes under reducing and oxidising conditions, showing that the A-site ordered layered perovskite can be formed from disordered cubic phase under reducing conditions at 800 °C with reversible phase change between PrBaMn2O5+δ (O5) and PrBaMn2O6−δ (O6) during redox cycles. The superior redox ability of PrBaMn2O5+δ (O5) was also observed by thermo-gravimetric methods, showing a reversible weight change during redox cycling corresponding to a change in oxygen stoichiometry of δ = 1.Moreover, the reduction of CO2via elevated-temperature (750–850 °C) electrolysis using an SOEC with LSGM as the electrolyte was studied using a PrBaMn2O5+δ (O5) cathode. High current density (0.845 A cm−2) was obtained at 850 °C and electrolysis potential of 1.55 V for a cell using the PrBaMn2O5+δ|LDC|La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM)|La(Sr)Fe(Co)O3. As a result, this study revealed that PrBaMn2O5+δ (O5) is a potential cathode material for SOECs.
Acknowledgements
We thank EPSRC (EP/I022570/1, EP/K015540/1, EP/I038950/1) and the Royal Society (Wolfson Research Merit award) for support.References
- J. L. Bernal-Agustín and R. Dufo-López, Int. J. Hydrogen Energy, 2008, 33, 6401 CrossRef PubMed.
- J. O. M. Bockris and T. N. Veziroglu, Int. J. Hydrogen Energy, 2007, 32, 1605 CrossRef CAS PubMed.
- S. H. Jensen, P. H. Larsen and M. Mogensen, Int. J. Hydrogen Energy, 2007, 32, 3253 CrossRef CAS PubMed.
- W. Doenitz, R. Schmidberger, E. Steinheil and R. Streicher, Int. J. Hydrogen Energy, 1980, 5, 55 CrossRef CAS.
- A. O. Isenberg, Solid State Ionics, 1981, 3–4, 431 CrossRef CAS.
- E. H. Seymour, L. Murray and R. Fernandes, Int. J. Hydrogen Energy, 2008, 33, 3015 CrossRef CAS PubMed.
- L. Schlapbach, Nature, 2009, 460, 809 CrossRef CAS PubMed.
- L. Wang and R. T. Yang, Energy Environ. Sci., 2008, 1, 268 CAS.
- B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121 CrossRef CAS PubMed.
- S. D. Ebbesen and M. Mogensen, J. Power Sources, 2009, 193, 349 CrossRef CAS PubMed.
- N. Q. Minh and M. B. Mogensen, Electrochem. Soc. Interface, 2013, 22, 55 CAS.
- C. Graves, S. D. Ebbesen and M. Mogensen, Solid State Ionics, 2011, 192, 398 CrossRef CAS PubMed.
- K. Xie, Y. Zhang, G. Meng and J. T. S. Irvine, J. Mater. Chem., 2011, 21, 195 RSC.
- C. Graves, S. D. Ebbesen, S. H. Jensen, S. B. Simonsen and M. B. Mogensen, Nat. Mater., 2015, 14, 239 CrossRef CAS PubMed.
- D. M. Bierschenk, J. R. Wilson and S. A. Barnett, Energy Environ. Sci., 2011, 4, 944 CAS.
- E. P. Murray, T. Tsai and S. A. Barnett, Nature, 1999, 400, 649 CrossRef CAS PubMed.
- P. Huang, A. Horky and A. Petric, J. Am. Ceram. Soc., 1999, 82, 2402 CrossRef CAS PubMed.
- B. C. H. Steele, I. Kelly, H. Middleton and R. Rudkin, Solid State Ionics, 1988, 28–30, 1547 CrossRef.
- S. McIntosh and R. J. Gorte, Chem. Rev., 2004, 104, 4845 CrossRef CAS.
- X. L. Yue and J. T. S. Irvine, J. Electrochem. Soc., 2012, 159, F442 CrossRef CAS PubMed.
- S. Wang, H. Tsuruta, M. Asanuma and T. Ishihara, Adv. Energy Mater., 2015, 5, 1401003 Search PubMed.
- Y. Li, J. Zhou, D. Dong, Y. Wang, J. Z. Jiang, H. Xiang and K. Xie, Phys. Chem. Chem. Phys., 2012, 14, 15547 RSC.
- X. Yue and J. T. S. Irvine, Electrochem. Solid-State Lett., 2012, 15, B31 CrossRef CAS PubMed.
- X. L. Yue and J. T. S. Irvine, Solid State Ionics, 2012, 225, 131 CrossRef CAS PubMed.
- F. Bidrawn, G. Kim, G. Corre, J. T. S. Irvine, J. M. Vohs and R. J. Gorte, Electrochem. Solid-State Lett., 2008, 11, B167 CrossRef CAS PubMed.
- S. Sengodan, S. Choi, A. Jun, T. H. Shin, Y.-W. Ju, H. Y. Jeong, J. Shin, J. T. S. Irvine and G. Kim, Nat. Mater., 2015, 14, 205 CrossRef CAS PubMed.
- M. T. Anderson, J. T. Vaughey and K. R. Poeppelmeier, Chem. Mater., 1993, 5, 151 CrossRef CAS.
- S. McIntosh and R. J. Gorte, Chem. Rev., 2004, 104, 4845 CrossRef CAS.
- S. B. Adler, J. Electrochem. Soc., 2002, 149, E166 CrossRef CAS PubMed.
- S. McIntosh, J. Vohs and R. Gorte, J. Electrochem. Soc., 2003, 150, A1305 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |