 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-association of organic solutes in solution: a NEXAFS study of aqueous imidazole†
M. J.
Thomason
a,
C. R.
Seabourne
b,
B. M.
Sattelle
a,
G. A.
Hembury
a,
J. S.
Stevens
a,
A. J.
Scott
b,
E. F.
Aziz
cd and
S. L. M.
Schroeder
*ab
aThe University of Manchester, School of Chemistry, School of Chemical Engineering and Analytical Science, Manchester, M13 9PL, UK. E-mail: slm.schroeder@outlook.com
bUniversity of Leeds, School of Chemical and Process Engineering, Leeds, LS2 9JT, UK
cJoint Ultrafast Dynamics Lab in Solutions and at Interfaces (JULiq), Helmholtz-Zentrum Berlin für Materialien und Energie, Albert Einstein Strasse 15, 12489 Berlin, Germany
dFreie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany
First published on 28th January 2015
Abstract
N K-edge near-edge X-ray absorption fine-structure (NEXAFS) spectra of imidazole in concentrated aqueous solutions have been acquired. The NEXAFS spectra of the solution species differ significantly from those of imidazole monomers in the gas phase and in the solid state of imidazole, demonstrating the strong sensitivity of NEXAFS to the local chemical and structural environment. In a concentration range from 0.5 to 8.2 mol L−1 the NEXAFS spectrum of aqueous imidazole does not change strongly, confirming previous suggestions that imidazole self-associates are already present at concentrations more dilute than the range investigated here. We show that various types of electronic structure calculations (Gaussian, StoBe, CASTEP) provide a consistent and complete interpretation of all features in the gas phase and solid state spectra based on ground state electronic structure. This suggests that such computational modelling of experimental NEXAFS will permit an incisive analysis of the molecular interactions of organic solutes in solutions. It is confirmed that microhydrated clusters with a single imidazole molecule are poor models of imidazole in aqueous solution. Our analysis indicates that models including both a hydrogen-bonded network of hydrate molecules, and imidazole–imidazole interactions, are necessary to explain the electronic structure evident in the NEXAFS spectra.
Introduction
Volmer's concept of the nucleation stage of crystallization1 resulted in the development of what is now known as classical nucleation theory (CNT). CNT assumes that the nucleation process occurs in two distinct steps. First, molecules in a supersaturated solution aggregate into nuclei, thereby developing an interface with the surrounding solution. The stability of these nuclei is size-dependent, reflecting the free energy balance between an interfacial tension penalty and cohesive energy stabilisation. Second, once the nuclei have grown beyond a critical size, above which the cohesive term outweighs the interfacial destabilisation, the total free energy decreases continuously as a function of size and crystal growth becomes the favourable process.2,3 Whilst a proven and useful concept for describing and predicting nucleation and crystal growth phenomena, often quantitatively, CNT remains a description of nucleation that does not explicitly consider intermolecular interactions or the precise structural nature of the pre-crystalline state; this limits its predictive power for many systems.4,5 Researchers are therefore using a variety of experimental techniques to obtain molecular level information about the solute species in solution, how they associate and assemble during nucleation, and whether the pre-crystalline structures relate to those in the crystalline products.4,6–10 Examples of techniques commonly employed include nuclear magnetic resonance (NMR),11–13 small- and wide-angle X-ray scattering (SAXS/WAXS),14 optical microscopy,15 and vibrational spectroscopies (infrared and Raman),16 as well as grazing incidence X-ray diffraction (GIXD) of interfacial species.17In this paper we introduce another technique for probing the molecular properties of organic solute species in concentrated solutions; namely, near-edge X-ray absorption fine-structure (NEXAFS) spectroscopy,18,19 which is also often referred to as X-ray absorption near-edge structure (XANES). Our application of NEXAFS to concentrated solutions builds on the recent realisation that chemical shifts in atomic core level binding energies, which can be measured by X-ray photoelectron spectroscopy (XPS),20–22 provide incisive information about the influence of hydrogen bonding and proton transfer on the structure of the organic solid state.23–28 High resolution X-ray spectroscopies that probe core levels are generally sensitive to the local electronic structure around, and bonding by, the element that is excited in the atomic core. The X-ray absorption process underlying NEXAFS is the excitation of atomic core electrons (for light elements such as C, N and O the 1s electrons) to unoccupied valence orbitals; this can be readily interpreted by the use of molecular orbital calculations. Due to its sensitivity to unoccupied valence orbitals, NEXAFS is chemically and structurally more incisive than core level binding energy measurements by XPS, and we have recently demonstrated the level of detail that can be obtained by combining XPS, NEXAFS and density functional theory, to examine local bonding both in the organic solid state29 and by solute species in solutions.30 Here we apply the same conceptual framework and extend it to the analysis of local bonding by imidazole in aqueous solution.
1H-Imidazole (for the remainder of this paper referred to as ‘imidazole’) crystallises in a monoclinic crystal structure with the space group P21/c.31–33 There are four molecules in the unit cell, and the main structural features are infinite chains of hydrogen-bonded imidazole molecules along the c-axis. The IUPAC numbering of the ring atoms in imidazole is indicated in Fig. 1. There is considerable interest in a deep understanding of the local interactions of imidazole in aqueous media, due to its biological importance as the side chain of the amino acid histidine. Experimental evidence for the self-association of imidazole in aqueous solutions at concentrations above 10−4 mol L−1 has been available for more than 70 years.34 π–π interactions between imidazole molecules have been invoked to explain self-association through the formation of molecule stacks.35,36 Such stack models contrast with the dominant motif of chain-formation through hydrogen bonding in solid imidazole.32,33 Recent molecular dynamics simulations of high concentrations (>0.5 mol L−1) of imidazole in aqueous solutions indicate that there is in fact a concentration-dependent balance, between π–π-assisted hydrogen-bonded stack structures and chains of hydrogen-bonded imidazole molecules37 that are similar to those found in liquid38 and solid32,33 imidazole.
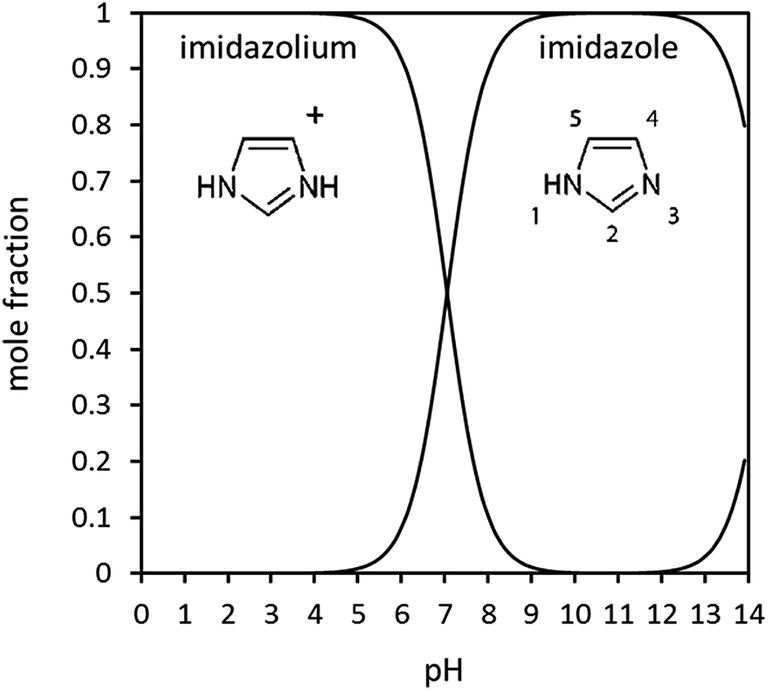 | ||
| Fig. 1 Imidazole speciation in aqueous solution as a function of pH, calculated from pKa values. Also indicated in the figure is the IUPAC numbering of the ring atoms. | ||
As a first step to experimentally probing these properties, we contrast here the N 1s NEXAFS spectra of (i) monomeric gas phase imidazole and (ii) crystalline imidazole with (iii) imidazole as a solute in concentrated aqueous solution. This approach provides an opportunity to begin building up a systematic picture of the electronic structure variations in the imidazole molecule, that arise from the three different environments. The aim of the investigations here is to establish whether available methods for electronic structure calculations correctly reproduce the experimental spectra, and whether the computational analysis of experimental core level spectra can realistically provide a quantitative insight into complex solution systems. For example, an important question arising in this context is to what extent the electronic transitions in the experimental excitation spectra are dominated by ground state (‘initial state’) properties of the imidazole molecules, and whether excited state properties (‘final state effects’) influence the spectra significantly.
Materials and methods
Imidazole was obtained from Sigma Aldrich as ACS grade ≥99% (titration) with total impurities of ≤0.2% water. The solubility of imidazole in water is very high, with saturation around 11 mol L−1 at 298.15 K.39 For this study, aqueous solutions with concentrations from 0.5 to 8.2 mol L−1 were prepared using laboratory grade deionised water. The pH of the solutions was monitored using a micro-tipped pH electrode. Before any measurements were taken, the pH electrode was calibrated using three various buffered solutions (3, 6 and 9). As expected for a weak base, it was found that all imidazole solutions were basic, with a pH around 10.5, indicating that the solutions contained >99% neutral imidazole species. The expected speciation of imidazole as a function of pH can be calculated from the known pKa values (7.05, 14.52) and is displayed in Fig. 1. We note that the measured pH agreed well with the expected pH (as calculated from the pKa value) at concentrations up to approximately 1 mol L−1. The experimental pH values were consistently somewhat lower (ΔpH ∼ 0.1) than expected at higher concentrations. This may be an indication of an increasing degree of self-association by imidazole–imidazole hydrogen bonding, which would be expected to reduce the OH− anion concentration. However, the effect is small, and the predicted pH is close to the estimated error of the pH electrode. Furthermore, it is not clear whether the response of the pH electrode may be affected by high concentrations of hydrogen-bonding solutes.NEXAFS measurements were undertaken at beamline U41-PGM of the BESSY-II synchrotron radiation storage ring of the Helmholtz Zentrum Berlin. The LIQUIDROM experimental chamber has been specifically designed for analysis of solution systems under vacuum or an inert helium atmosphere. Light from the storage ring passes through a differential stage and enters into the chamber through a thin Si3N4 membrane. To minimise radiation damage of the solution, a flow cell40 with a peristaltic pump was used, in which the X-rays impinge onto the solution through a Si3N4 membrane. The N K-edge spectra were recorded via fluorescence/luminescence yield detection with a GaAsP 5 × 5 mm2 photodiode.
The flow cell setup allows analysis of a series of solutions without opening the analysis chamber. During sample changes, the system was purged for 30 min with deionised water. A control measurement of the spectrum was then performed to ensure that the cell was clean. During experiments the flow rate was typically set to 40 mL min−1. The flowing system was allowed to equilibrate for at least 20 min before three NEXAFS spectra, each taking about 20 min to acquire, were measured.
StoBe-DeMon (‘StoBe’) calculations in this paper were carried out as previously described.40,41 The software uses a hybrid density functional theory (DFT)42–44 with a double zeta, local spin density exchange and correlation functional by Vosko and Wilk,45 and has been described in detail in several papers.42,43,46–48 For the calculation of the solid state N K-edge absorption spectrum, a 6-molecule cluster cut-out from the crystal structure of imidazole was used.
Gaussian03 (ref. 49) was used to carry out single point energy calculations, minimising the total energy of the gas phase molecule by geometry optimisation. The output provided molecular orbitals, their orbital energies, and populations. For the monomer structure, a geometry-optimised imidazole monomer from Gaussian03 (ref. 49) was used, which was obtained using the B3LYP/6-31G* basis set. A geometry-optimised stack structure, of three imidazole molecules coordinated with three water molecules, was generated using Gaussian03 at the BHANDH/6-311G** level of theory.
Density functional theory (DFT) was used to predict core-level spectroscopy results. Initially, the CASTEP code was used.50 CASTEP is a member of the linear-augmented plane-wave (LAPW) class of DFT codes. It is a pseudopotential code, whereby approximations are used to model the atomic-like core states, with the code only actively calculating valence states. Developments to the code have allowed core-level spectroscopy calculations to be carried out, as detailed in the literature.51–53 The generalized gradient approximation (GGA)54,55 was used throughout the calculations, applying a methodology previously developed in the context of analysing Al K-edges.56
The first parameter to converge was the basis set size (as defined by the kinetic energy cut-off). This was changed in intervals of 100 eV, with convergence being determined against the predicted core-level spectroscopy result (for the ‘pyridine’ N3 nitrogen atom), initially in the ground electronic state. Upon varying the parameter from value ‘A’ to value ‘B’, for each point on the energy axis (from −5.0 eV to 50 eV in steps of 0.05 eV) the modulus of the difference in intensity was found (with a nominal Gaussian broadening applied to the predicted results). The average percentage change relative to result A was then found across all the energy axis points. When this was less than 15% (meaning that no meaningful change could be observed by eye upon the application of a physically realistic broadening scheme) the results were considered to be converged. A similar process of convergence was carried out for the other key DFT code parameter – the quality of sampling in reciprocal space – as defined by the Monkhorst–Pack k-point grid.55 Ultimately the number of k-points in each reciprocal space dimension was doubled until convergence was achieved. This led to parameters as detailed in Table 1.
For the CASTEP predicted spectra, supercells were constructed, and 0.5 electron core-holes used at the two nitrogen positions. It was determined that a 2 × 2 × 1 supercell was sufficiently large to avoid artificial core-hole interactions (i.e. a minimum core-hole separation distance of 9.779 Å).
To complement the CASTEP results, the WIEN2K DFT code57,58 was used. WIEN2K, like CASTEP is a member of the LAPW family of codes. WIEN2K works by splitting the space in the theoretical cell into ‘muffin-tin’ regions centred on atoms, and interstitial space, with different exact modelling methodologies being used in each of those regions. WIEN2K is an all-electron code, and therefore it is possible to predict the onset energy difference (ΔIPs) between the N1 and N3 nitrogen atoms in imidazole. In this instance, convergence was based on the determined energy onset difference between the N1 and N3 positions. The key DFT code parameters (basis set size, defined in WIEN2K by the RKMAX value and the density of k-points in reciprocal space) were converged such that the energy difference between the two nitrogen positions was accurate to the nearest 0.1 eV.
| Cell details33 | Kinetic energy cut-off/eV | Minimum k-point separation in reciprocal space |
|---|---|---|
| a = 7.732 Å | 725 | 0.073 Å−1 |
| b = 5.458 Å | ||
| c = 9.779 Å | ||
| α = γ = 90° | ||
| β = 117.26° |
Results and discussion
Monomeric gas phase imidazole
Before embarking on the presentation and discussion of crystalline and aqueous imidazole, it is instructive to establish the origin of the features in the N K-edge core level excitation spectra of an isolated imidazole molecule. The energies of the atomic 1s core levels, the occupied molecular orbitals, and the three lowest unoccupied (‘virtual’) molecular orbitals obtained by a Gaussian03 ground state calculation for the gas phase monomer are all summarised in Table 2. From these data we can construct a schematic molecular orbital diagram summarising the atomic and molecular orbital energies relevant for the N K-edge NEXAFS spectrum (Fig. 2). It can be seen that the N 1s core level binding energy difference between N1 and N3 in the ground state of imidazole is 2.3 eV. Due to the aromatic nature of imidazole, the excitation of N 1s electrons to unoccupied π* states is expected to be the dominant absorption feature in the N K-edge NEXAFS.18 As can be seen in Table 2 and Fig. 2, there are two low-lying unoccupied π* states available. First, transitions into the lowest unoccupied molecular orbital (LUMO) occur, through the transitions labelled N1 1s → 1π* and N3 1s → 1π*, and possibly an additional absorption feature due to a transition of a N 1s electrons to the 2π* virtual orbital. In Fig. 3 the 1π* LUMO (#19 in Table 2), is visualised together with the Gaussian-derived second- (1σ*, #20) and third-lowest (2π*, #21) unoccupied MOs. It can be seen that the 2π* orbital has no density of states (DOS) at the N1 atom, so a significant N1 1s → 2π* transition can be excluded. The 2π* orbital has local DOS at the N3 centre, so a N3 1s → 2π* transition may arise in the experimental spectrum. This transition is therefore also included in the scheme in Fig. 2. Electronic transitions from the N 1s core levels to the 1σ* orbital (#20) should be weak and are therefore unlikely to be evident in the experimental spectrum.| Orbital # | Assignment | Energy/eV | |
|---|---|---|---|
| 1 | N1 1s | −391.602 | Atomic core orbitals |
| 2 | N3 1s | −389.337 | |
| 3 | C2 1s | −278.531 | |
| 4 | C4 1s | −277.833 | |
| 5 | C5 1s | −277.349 | |
| 6 | σ | −27.449 | Occupied molecular orbitals |
| 7 | σ | −23.540 | |
| 8 | σ | −20.117 | |
| 9 | σ | −16.638 | |
| 10 | σ | −15.952 | |
| 11 | σ | −15.265 | |
| 12 | σ | −11.869 | |
| 13 | σ | −11.761 | |
| 14 | π | −11.549 | |
| 15 | σ | −11.059 | |
| 16 | π | −7.529 | |
| 17 | σ | −7.101 | |
| 18 | π(HOMO) | −6.129 | |
| 19 | 1π*(LUMO) | 0.921 | Virtual (unoccupied) molecular orbitals |
| 20 | 1σ* | 1.998 | |
| 21 | 2π* | 2.065 |
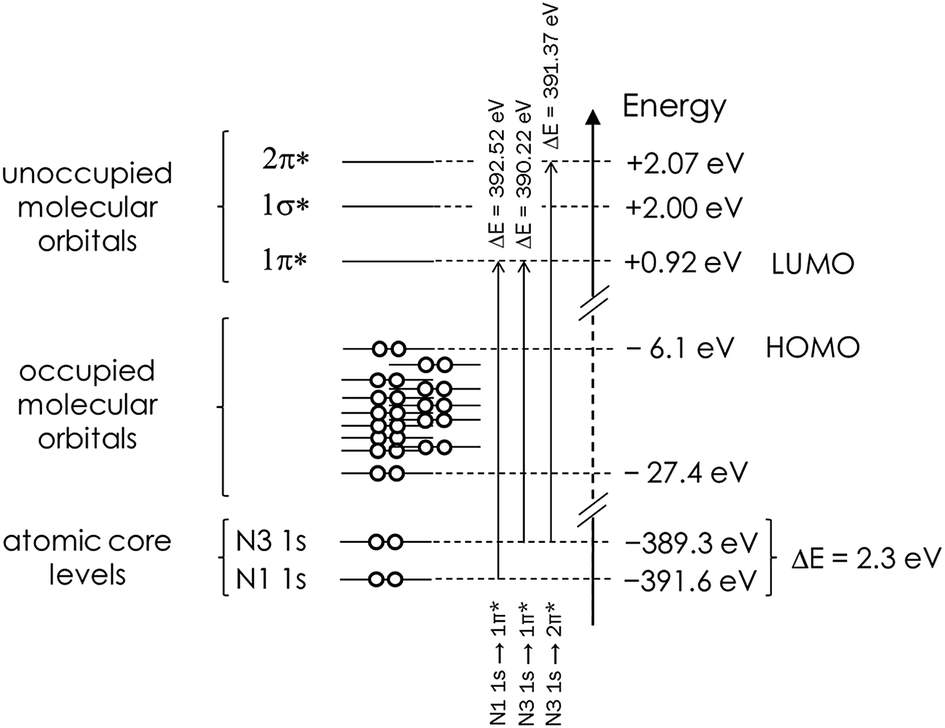 | ||
| Fig. 2 Schematic representation of the Gaussian03-derived energies of atomic core levels, as well as of the occupied and unoccupied molecular orbitals in a monomeric isolated (gas phase) imidazole molecule. The N 1s core level binding energy difference and predicted energies associated with the 1s → π* transitions are also indicated. Note that the energetic positions of the occupied MOs are a schematic illustration and not an accurate reflection of the actual energies in Table 2. | ||
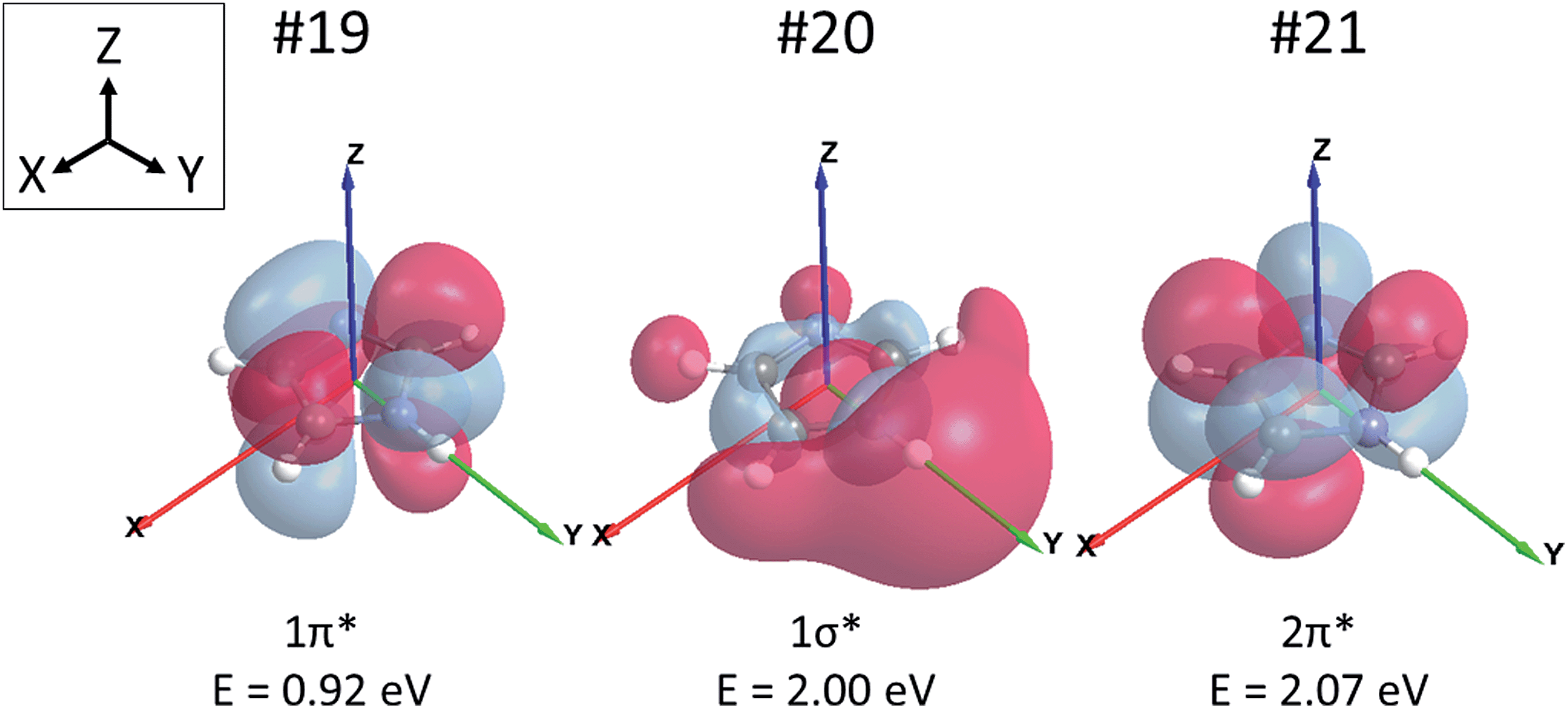 | ||
| Fig. 3 The lowest unoccupied molecular orbital (LUMO) #19 (left), the second-lowest unoccupied molecular orbital #20 (middle), and the third-lowest unoccupied molecular orbital #21 (right) of imidazole. The molecule is in the XY-plane with the N–H bond axis aligned in the direction of the Y vector. Orbital energies are noted below each orbital. | ||
Having established the expected transitions in the N K-edge NEXAFS of an isolated imidazole molecule, we can now turn to the experimental N K-edge gas phase spectrum (Fig. 4) as previously determined by Apen et al.59 using inner-shell electron energy loss spectroscopy (ISEELS). All features in the spectrum have been modelled through a least-squares curve fitting procedure, using Gaussian functions for transitions to bound states and arctan edge step functions for the ionisation potentials (IPs), i.e., the energies required for removal of the N 1s core electrons from the atom by photoemission into a continuum state. Beyond the IPs there are only multiple scattering resonances, such as the broad σ* shape resonance around 406.6 eV. The centroid energies of the fitted curve components in Fig. 4 are summarised in Table 3 and compared with values predicted by Gaussian03 (taken from Table 2 and Fig. 2) and a StoBe calculation of the N K-edge spectrum, taking the effect of the core hole state into account.
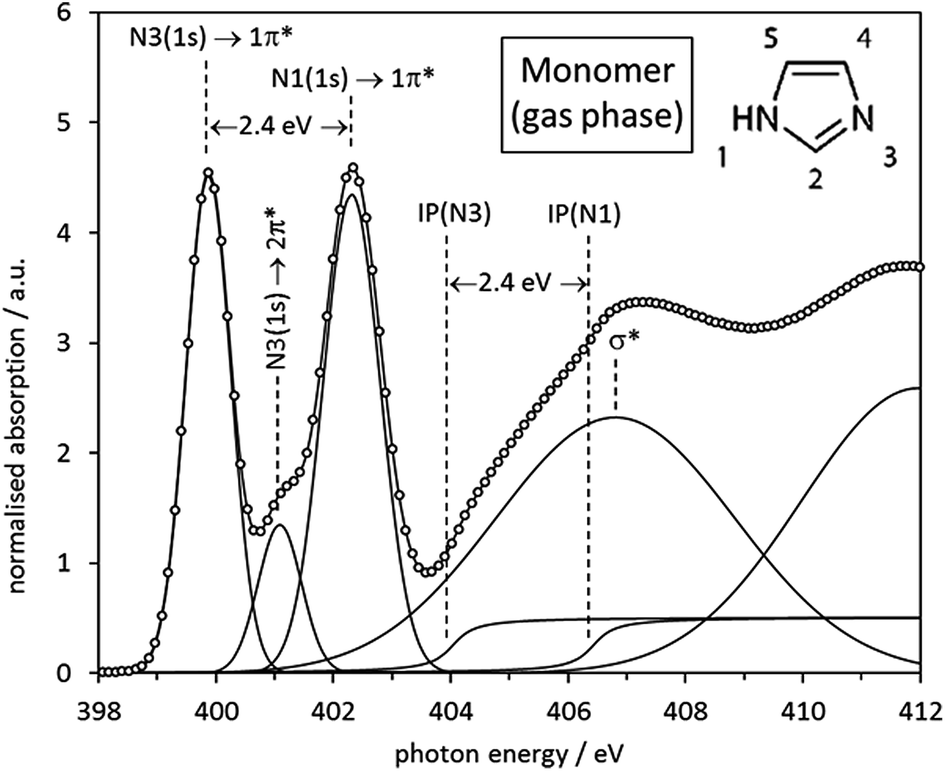 | ||
| Fig. 4 Experimental ISEELS N K-edge spectrum of imidazole monomer (gas phase) species with all spectral features fitted by Gaussian and arctan functions. The origin of each feature is indicated by the annotations in the figure. | ||
| Experim./eV | Calc. (StoBe)/eV | Calc. (Gaussian)/eV | |
|---|---|---|---|
| IP(N1) | 406.2 | 406.6 | 391.6 |
| IP(N3) | 403.9 | 404.4 | 389.3 |
| ΔIP | 2.4 | 2.2 | 2.3 |
| E(N1 1s → 1π*) | 402.3 | 402.3 | 392.5 |
| E(N3 1s → 1π*) | 399.9 | 400.1 | 390.2 |
| ΔE(1π*) | 2.4 | 2.2 | 2.3 |
| E(N3 1s → 2π*) | 401.3 | 401.2 | 391.4 |
| E(2π*) − E(1π*) | 1.4 | 1.1 | 1.2 |
It can be seen that the N1 and N3 IPs for the gas phase molecule agree well with those predicted by StoBe. The Gaussian03-derived values are approximately 15 eV lower, as a result of neglecting the core hole state in the Gaussian03 ground state calculation. However, for both ground state and excited state calculations the energy difference (ΔIP) between the two IPs is in almost quantitative agreement with the experimental value (2.4 eV), with a value of 2.3 eV for Gaussian03 and 2.2 eV for StoBe.
Also clearly visible are the two distinct near-edge peaks that stem from the predicted N 1s → 1π* transitions at the N1 and N3 centres, which are almost quantitatively reproduced by the StoBe calculation. The Gaussian03 ground state calculation predicts values that are approximately 10 eV lower, due to neglecting the core hole effect. It can also be seen that there is a weak peak at 401.3 eV in the experimental spectrum arising from the predicted N3 1s → 2π* transition, which is also well reproduced by the StoBe calculation. There is excellent agreement between the IP difference and the energy difference between the 1s → 1π* transitions at N1 and N3, for experimental as well as both computational values; i.e., with or without taking the core hole into account. This strongly indicates that there is no differential core hole relaxation effect in the isolated molecule, which had been previously invoked to explain an apparent discrepancy between experimentally determined IPs and the 1s → π* shifts.59
Overall, we can conclude from the analysis that the relative energetic positions of all features observed in the N K-edge spectrum are almost entirely determined by the ground state electronic properties of the molecule. The deviations between predicted and experimentally observed energetic shifts are at maximum on the order of 0.2 eV, even for the virtual 2π* state.
Finally, good agreement between experiment and theory was also achieved by the CASTEP analysis of the imidazole electronic structure. It was shown that a 15 Å theoretical ‘box’ was sufficiently large to simulate the molecule in the gas phase (as determined by comparing the predicted result for the N3 atom to that for smaller boxes). For the two nitrogen positions, predicted spectra were calculated, using a 0.5 electron core-hole, with spin allowed. Before comparing results with experiment,59 the results had a lifetime-broadening scheme individually applied, before a rigid shift of +2.4 eV was applied to the N1 result and averaging of the spectra was performed.60 The results for the two positions, normalised to the Fermi level, are shown in Fig. 5, along with comparisons to experiment. It can be seen that the calculated spectrum based on the CASTEP analysis is in excellent agreement with the experimental spectrum.59 The secondary peak visible at about 401 eV in the calculated spectrum is due to an overestimated transition to the 2π* MO.
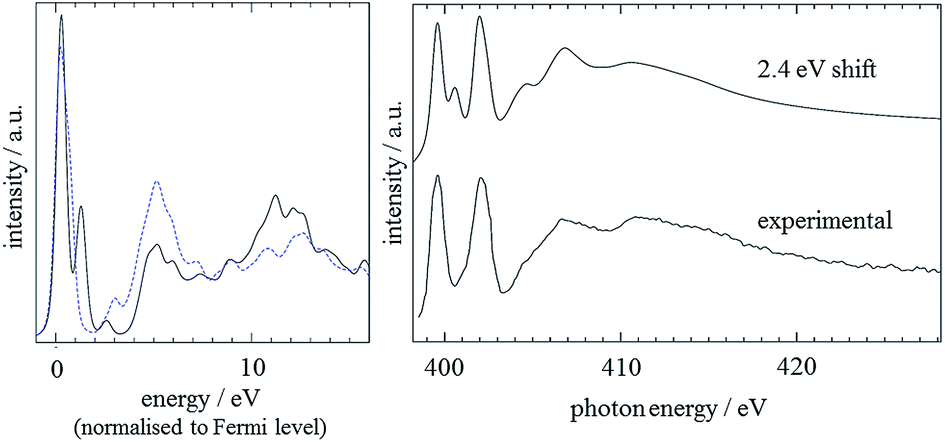 | ||
| Fig. 5 Left: CASTEP monomer predictions for the N3 (black line) and N1 (blue dashed line) nitrogens. A 0.2 eV Gaussian broadening was applied and the spectra were aligned to the Fermi level. Right: Comparison of predicted N K edge with experimental data, with the spectra of the two N atoms shifted relatively by the experimental value of 2.4 eV. An energy-dependent lifetime broadening, as described in the text, was applied and the energy scale of the spectrum adjusted to match with the experimental data. | ||
Imidazole crystal
The same curve fitting procedure as for the gas phase monomer was applied to the spectrum of crystalline imidazole, which was previously determined by Apen et al. via electron-yield detection.59 The most noticeable difference between the experimental spectra of gas phase and crystalline imidazole is a strong reduction of the energy split between the IPs and the 1s → 1π* transitions of the N1 and the N3 moieties, from 2.4 eV in the gas phase monomers (Fig. 4, Table 3) to 1.5 eV in the spectrum of the solid (Fig. 6). This difference arises from intermolecular N–H⋯N hydrogen bonding in the chains of imidazole molecules in the crystal structure. This causes intermolecular redistribution of electron density from N3 to N1 sites through weakening of the N1–H bond, and a partial levelling of the electronic structure difference around the two N centres in each molecule.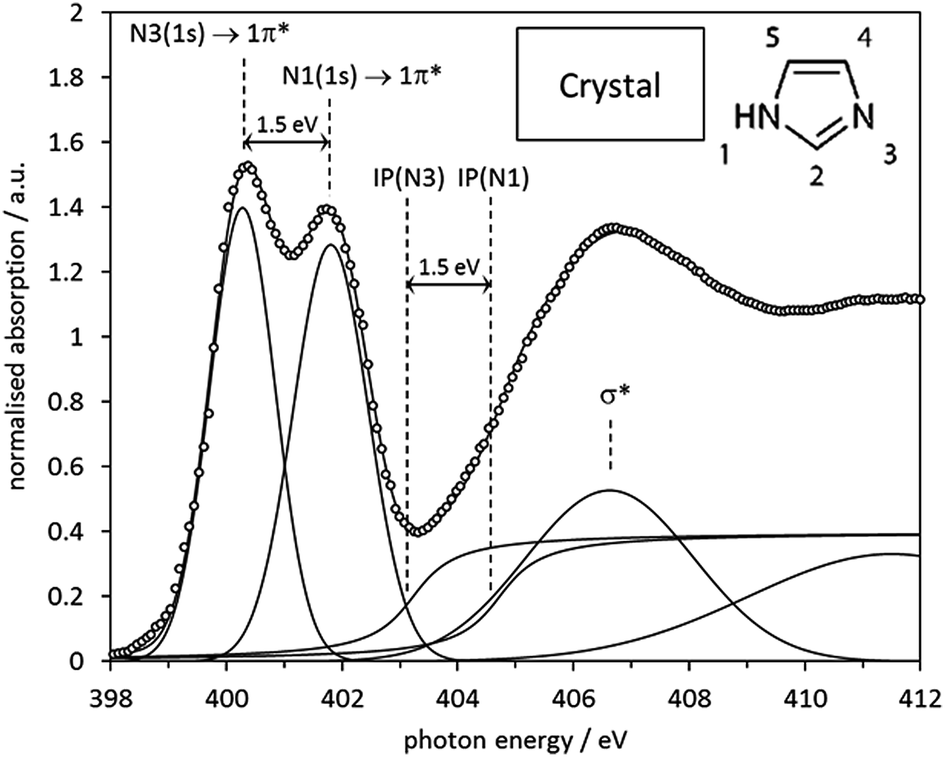 | ||
| Fig. 6 Experimental electron-yield N K-edge spectrum of crystalline imidazole, with all spectral features fitted by Gaussian and arctan step functions. The origin of each feature is indicated by the annotations in the figure. | ||
The spectrum predicted by StoBe for the six-molecule cluster from the imidazole crystal structure is shown in the middle of Fig. 7, displayed above the experimental spectrum. The calculated spectrum of the central imidazole molecule has an N3 1s → 1π* transition at an energy of 400.3 eV, and a small shoulder due to the 1s → 2π* transition at 401.2 eV. The N1 (NH) 1s → 1π* transition is evident as a single peak at 401.6 eV, with no noticeable shoulder contributions.
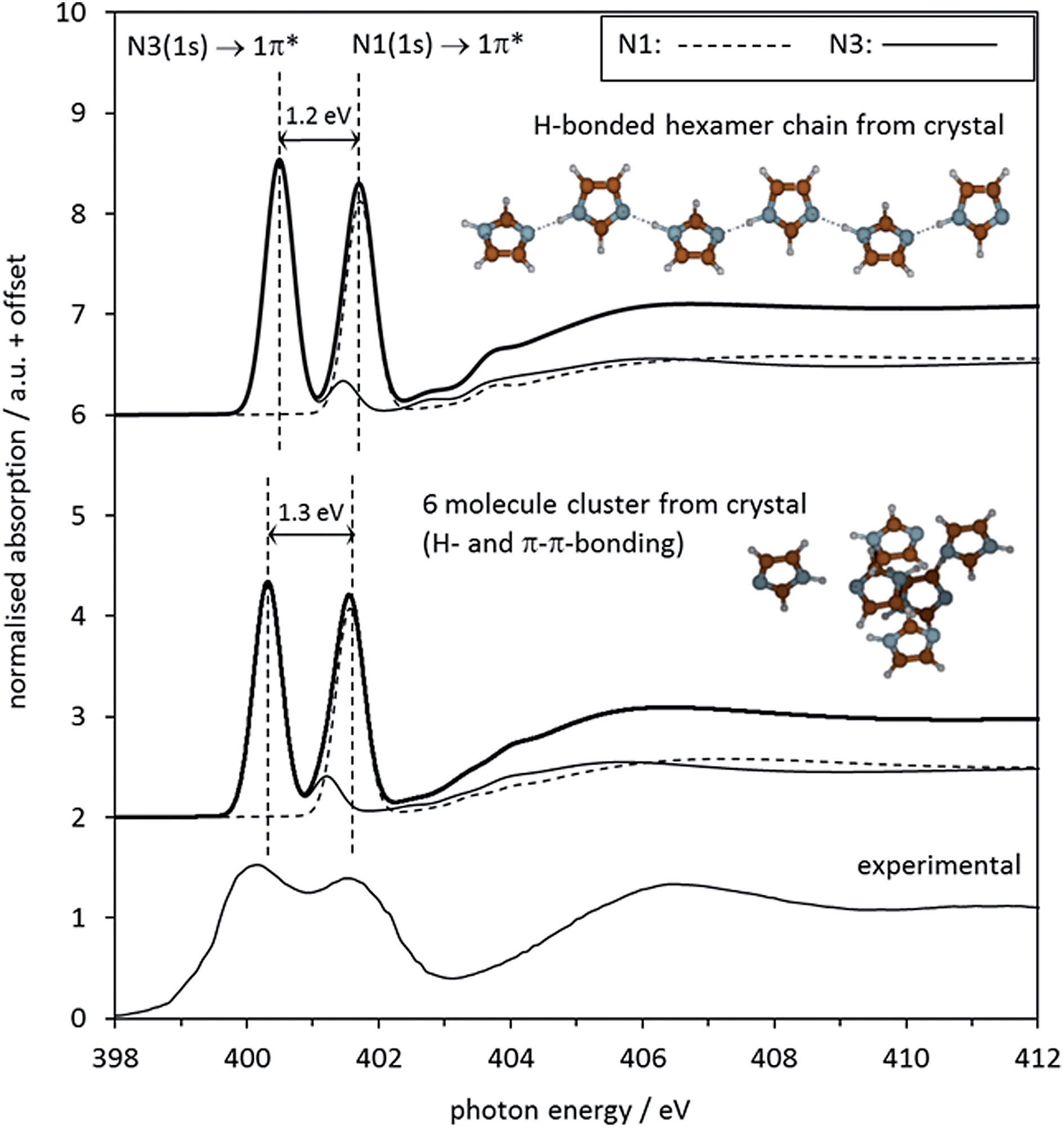 | ||
| Fig. 7 Top: Middle: Calculated N K-edge NEXAFS spectrum of solid imidazole, as predicted by the StoBe code for the central molecule in a cluster of 6 molecules cut out from the crystal structure of imidazole. For both calculated spectra, the two individual absorption spectra of the two nitrogen moieties N1 and N3 in the central N atoms of the displayed molecular structures are shown alongside the combined spectrum. Bottom: Experimental observed electron-yield N K-edge spectrum from Fig. 6. | ||
The parameters describing the features in the experimental and the calculated spectra are summarised in Table 4. The overall agreement between calculated and experimental data is good. The reduced energy split ΔE(1π*) of 1.5 eV between the two 1s → 1π* transitions in the experimental data is reproduced almost quantitatively by the StoBe cluster calculation. Comparing the StoBe results to those obtained for the gas phase monomer (Table 2) reveals that most of the reduction in the energy split between the gas and solid state is due to a decrease of 1.1 eV in the IP of N1 (its N 1s core level binding energy), from 406.6 eV to 405.5 eV. This indicates that the formation of intermolecular hydrogen bonds (N1–H⋯N3) increases electron density at the N1 centre considerably, while the IP of the N3 centre is much less affected. This reflects the fact that the donation of electron density at N3 stems from the aromatic π system of the molecule, resulting in a delocalisation of the electron density loss across the whole ring. In contrast, the electron density gain at the N1 centre takes place through the local N–H bond, which does not permit delocalisation of charge into the aromatic system.
| Experimental/eV | Calculated (StoBe)/eV | |
|---|---|---|
| IP(N1) | 404.7 | 405.5 |
| IP(N3) | 403.2 | 404.3 |
| ΔIP | 1.5 | 1.2 |
| E(N1 1s → 1π*) | 401.8 | 401.6 |
| E(N3 1s → 1π*) | 400.3 | 400.3 |
| ΔE(1π*) | 1.5 | 1.3 |
Besides hydrogen bonding, additional interactions are expected in the solid state structure of imidazole. For example, π–π interactions between the sheets of hydrogen-bonded imidazole chains are expected. To examine the influence of such interactions on the spectra, we examined a chain (hexamer) of hydrogen-bonded imidazole molecules, retaining its geometry in the crystal structure. The absorption spectrum of the N1 and N3 moieties calculated by StoBe (Fig. 7, top spectrum) is almost identical to the spectrum calculated for the 3-dimensional arrangement of six imidazole molecules taken as a model of the crystal structure. This indicates that the impact of N1–H⋯N3 hydrogen bonding dominates the electronic structure of imidazole in the solid, with additional interactions leading only to secondary changes in the electronic structure.
It is interesting to note that recent core level binding energy measurements found an N 1s core level shift of 1.6 eV for the two nitrogen ring moieties in the neutral imidazole side chain of solid histidine.61 This shift is comparable to the IP difference observed in the experimental and calculated NEXAFS of solid imidazole, although the hydrogen bonding experienced by the imidazole system is fundamentally different. In the crystal structure of histidine both N centres of the imidazole ring take part in hydrogen bonding, with the N1 (NH) centre donating its hydrogen to a more electronegative carboxylate acceptor (O), while the N3 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) centre is accepting from the protonated primary amine group of the zwitterion.62
N) centre is accepting from the protonated primary amine group of the zwitterion.62
Fig. 8 shows the CASTEP-predicted absorption spectra for the N3 and N1 centres in the crystal structure. Compared to the gas phase monomer (Fig. 5) we observe a relative similarity in the predicted absorption spectra, as a consequence of the hydrogen bonding in the crystal structure levelling the differences between the nitrogen environments.
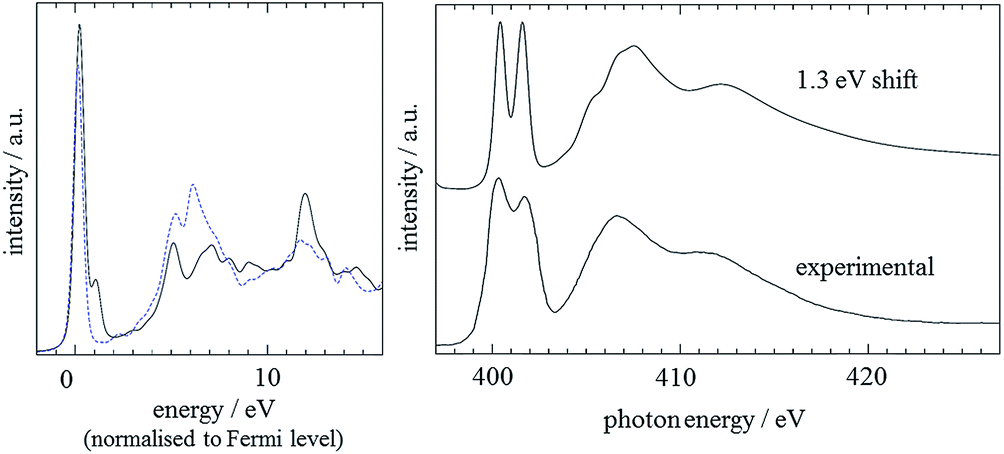 | ||
| Fig. 8 Left: CASTEP results for N3 (black solid line) and N1 (blue dashed line) in the imidazole crystal. A Gaussian broadening of 0.2 eV has been applied and the calculated spectra are aligned to the Fermi level. Right: Comparison of experimental and predicted spectra, with an energetic shift of 1.3 eV between the two N 1s core level binding energies applied. The calculated spectrum includes an energy-dependent lifetime broadening, as described in the main text; the energy scale of the spectrum was adjusted to match with the experimental data. | ||
To model the onset energy difference between the two nitrogen positions, the 1.3 eV energy difference derived by StoBe was utilised, and the individual N1 and N3 spectra were lifetime broadened as described above. As can be seen, this leads to excellent agreement with the experimental spectrum.59
Aqueous imidazole solutions
Shown in Fig. 9 are the obtained aqueous imidazole N K-edge NEXAFS spectra covering a concentration range from 0.50 mol L−1 (∼100H2O molecules per imidazole molecule) to the saturation concentration of 8.20 M (∼7H2O molecules per imidazole molecule). It can be seen that variations between the spectra as a function of concentration are minor, indicating that the average local coordination of imidazole molecules in this concentration range does not vary significantly. This observation suggests two possible scenarios. Either self-association involves only secondary interactions between individually hydrated imidazole molecules, which do not manifest themselves strongly in the N K-edge spectra; or self-association of imidazole is already dominant at the lower end of the concentration range investigated, so that any imidazole molecules added to the solution increase the volume fraction of such assemblies. Of course, the two scenarios are not mutually exclusive and may occur simultaneously.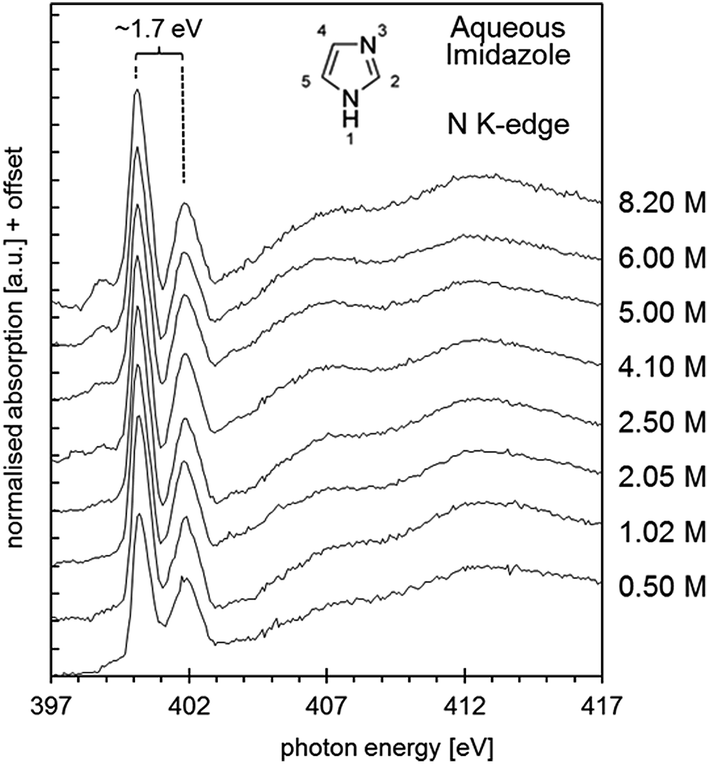 | ||
| Fig. 9 N K-edge near edge X-ray absorption fine structure (NEXAFS) spectra of aqueous imidazole solutions as a function of concentration, from 0.5 mol L−1 to 8.2 mol L−1. | ||
Previous photoelectron spectroscopy investigations41,63,64 of aqueous imidazole solutions focused on solvent interactions, i.e., omitting imidazole–imidazole interactions and other self-association effects from the analysis. Insight into the electronic structure of imidazole molecules in aqueous solution as a function of pH was gained, and the sensitivity of C 1s and N 1s core level spectroscopy to pH-induced protonation of imidazole was clearly demonstrated by detection of electronically equivalent N atoms in the imidazolium cation under acidic conditions.41,63 An attempt was made to explain the electronic structure of neutral imidazole species in solution, through calculations of the electronic properties of microhydrated gas-phase clusters comprising a single imidazole molecule and up to five water molecules.64 This provided some insight into the effect of the nearest-neighbour water coordination shells on the imidazole molecules. The study concluded that longer-range effects in the solvent probably need to be taken into account to obtain a good agreement between theory and experiment.
As already observed for the gas phase monomer and the crystalline state, the solution NEXAFS spectra are dominated by the 1s → 1π* transitions of the N1 and N3 moieties in the imidazole ring, which now appear at photon energies of approximately 400.2 eV and 401.9 eV (Fig. 10). The photon energy difference of 1.7 eV is lower than the value observed for the gas phase monomer (2.4 eV) and slightly higher than in crystalline imidazole (1.5 eV). That the solution value is closer to that of the hydrogen-bonded solid state structure suggests that significant hydrogen bonding with surrounding water or imidazole molecules takes place. Since the extent of proton transfer is insignificant in the pH range of the solutions (pH ∼ 10.5) it is reasonable to suggest that the observed energy difference of 1.7 eV is due to imidazole–water and/or imidazole–imidazole interactions.
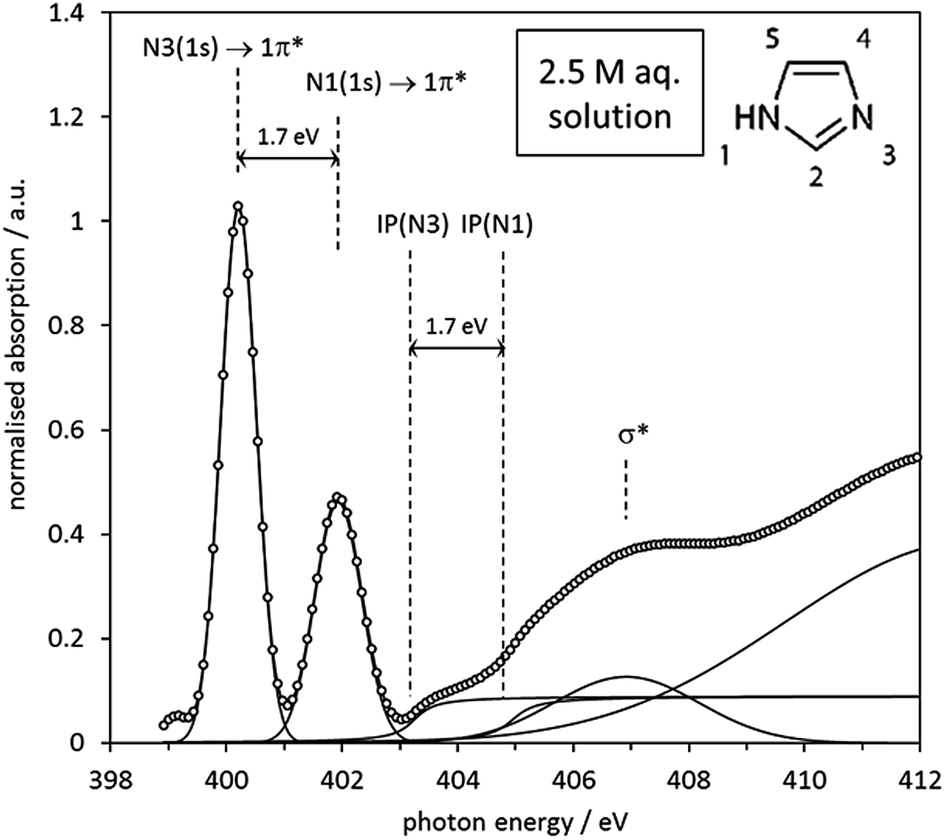 | ||
| Fig. 10 Experimental N K-edge spectrum of 2.5 mol L−1 aqueous imidazole solution, with all spectral features fitted by Gaussian and arctan step functions. The origin of each feature is indicated by the annotations in the figure. | ||
The observed 1.7 eV difference between the 1s → 1π* transitions at the N1 and N3 centres matches the previously reported difference between the N 1s photoemission peaks of aqueous imidazole solutions.41 Since the N 1s photoemission peak shifts are identical to the IP difference between the two N moieties (Fig. 10) we can conclude that the chemical shift between the two N 1s → 1π* transitions is primarily determined by the IP difference, just as observed in the above analysis of the imidazole monomer and crystal spectra, as well as in a similar analysis of the solid state of p-aminobenzoic acid.29 The relative energetic positions of the transitions appear to be determined by ground state core level binding energy differences, and additional differences due to final state effects are negligible.
The effect of imidazole–water interactions was explored more systematically by calculating the N K-edge NEXAFS of the microhydrated cluster structures used previously to interpret the photoelectron spectra of imidazole in aqueous solution.64 The structures of these clusters are displayed in the ESI (Figs. S1–S9†). Starting with a hydrogen-bonded HOH⋯N3 water–imidazole dimer #1, it can be seen that the effect of the hydrogen bond on the calculated NEXAFS spectrum is negligible (Table 5): the N1/N3 1s → 1π* peak energy difference is essentially the same as in the imidazole monomer (Table 3). The core level binding energies of the N3 (N) and N1 (NH) centres remain very similar to the monomer binding energies, with only a slight shift of 0.3 eV to 406.9 eV for N1 and 404.7 eV for N3. This reflects the donation of electron density from the aromatic system to the water molecule.
’) 1s → 1π* NEXAFS bands for the imidazole N atoms in the gas phase monomer (#0, see Table 3) and the geometry-optimised gas phase imidazole–water clusters of Jagoda-Cwiklik et al.64 The structures of the imidazole–water clusters are visualised in Fig. S1–S9 of the ESI
| Clustera | IP/eV | ΔIP/eV | ΔE(1s → 1π*)/eV | ||
|---|---|---|---|---|---|
| N1 | N3 | ||||
| a Clusters are visualised in the ESI. | |||||
| #0 | imi·0H2O | 406.6 | 404.4 | 2.2 | 2.2 |
| #1 | imi·1H2O | 406.9 | 404.7 | 2.2 | 2.1 |
| #2 | imi·2H2O | 406.7 | 404.5 | 2.2 | 2.0 |
| #3a | imi·3H2O | 406.5 | 404.7 | 1.8 | 1.8 |
| #3b | 406.0 | 404.3 | 1.7 | 1.6 | |
| #4a | imi·4H2O | 406.1 | 404.6 | 1.5 | 1.5 |
| #4b | 406.1 | 404.6 | 1.5 | 1.5 | |
| #4c | 406.0 | 404.4 | 1.6 | 1.5 | |
| #4d | 406.2 | 404.8 | 1.4 | 1.4 | |
| #5 | imi·5H2O | 406.0 | 404.6 | 1.4 | 1.3 |
The addition of another water molecule leads to cluster #2, which exhibits some subtle differences to the monomer. Although the spectral features are similar, there is a reduction of the N1/N3 1s → 1π* peak energy difference by 0.1 eV compared to the monomer. Interestingly, it appears that the N1 contribution has shifted by 0.1 eV with the addition of the extra bound water in the vicinity of the N3 centre.
Adding a third water molecule to the cluster led to equilibrium structures #3a and #3b.64 These clusters exhibit the first significant changes compared to the monomer. The calculated N1/N3 1s → 1π* peak energy differences are reduced significantly relative to the monomer value of 2.1 eV, to 1.8 eV and 1.6 eV, respectively. These values, along with the corresponding N 1s core level binding energy differences of 1.8 eV and 1.7 eV, are actually in good agreement with the NEXAFS data, and with the N 1s binding energies previously reported.41 However, this seemingly excellent agreement should not be overinterpreted, for the following reasons. First, the electronic structure of these clusters does not satisfactorily model the vertical ionisation potentials.64 Second, it seems unlikely that imidazole in aqueous solution is solvated by only three water molecules. Third, we observed that adding more water molecules to the microhydrated clusters continues the trend of reducing the IP difference between the N1 and the N3 centres. As can be seen in Table 5, adding additional water molecules to yield hydrate shells with 4 (clusters #4a–#4d) and 5 (cluster #5) water molecules has the effect of further reducing the N1/N3 1s → 1π* peak energy difference, with the 5 water hydration shell producing a peak split of 1.3 eV and an IP difference of 1.4 eV. These are both significantly below the experimentally observed value of 1.7 eV. In fact, the system approaches the values calculated for the extensively hydrogen-bonded crystal, suggesting that hydrogen bonding of monomeric imidazole to water molecules alone does not correctly represent the structure of the system. Inclusion of larger hydration shells leads to a stronger than experimentally observed effect on the levelling of the N 1s core level energy difference between the two nitrogen centres. It may be speculated that one contribution to this result may be the asymmetric and incomplete coordination of the imidazole molecules in models invoking so few water molecules. In real solutions, the electron density variations induced by hydrate coordination from one side of the molecule are counterbalanced by coordination from the other, leading perhaps to an overall weaker net effect on the electronic structure of the central imidazole molecule. It remains to be examined whether this and other possible effects (such as longer range polarisation of the hydrogen bonded water network) in more extended hydration clusters may weaken hydrogen bonding to the imidazole molecule.
There is, of course, strong previous evidence that self-association of imidazole takes place in the range of aqueous concentrations investigated here. It is likely that either secondary interactions between hydrated clusters take place, or that even direct imidazole–imidazole interactions exist in solution, perhaps in hydrogen-bonded chain-and-stack structures such as those recently predicted by molecular dynamics simulations.37 Indeed, a previous X-ray scattering analysis of aqueous imidazole solutions suggested that self-association involves the formation of molecule stacks held together by hydrogen-bonding through water molecules.35 To examine whether such a model would lead to a more correct reproduction of the NEXAFS data, we set up a more complex geometry-optimised structure model in Gaussian03. A geometry-optimised stack structure of three imidazole molecules was generated at the BHANDH/6-311G** level of theory, involving three water molecules coordinating the central imidazole molecule. The resulting structure and its predicted absorption spectrum are shown in Fig. 11. It can be seen that the geometry-optimised cluster exhibits hydrogen bonding interactions for one of the water molecules linking two of the imidazole species, just as in the model previously generated by X-ray scattering.35 The energy difference between the N 1s → 1π* resonances in the spectra is 1.5 eV, significantly more in agreement with experiment than the microsolvated cluster models involving only one imidazole molecule.
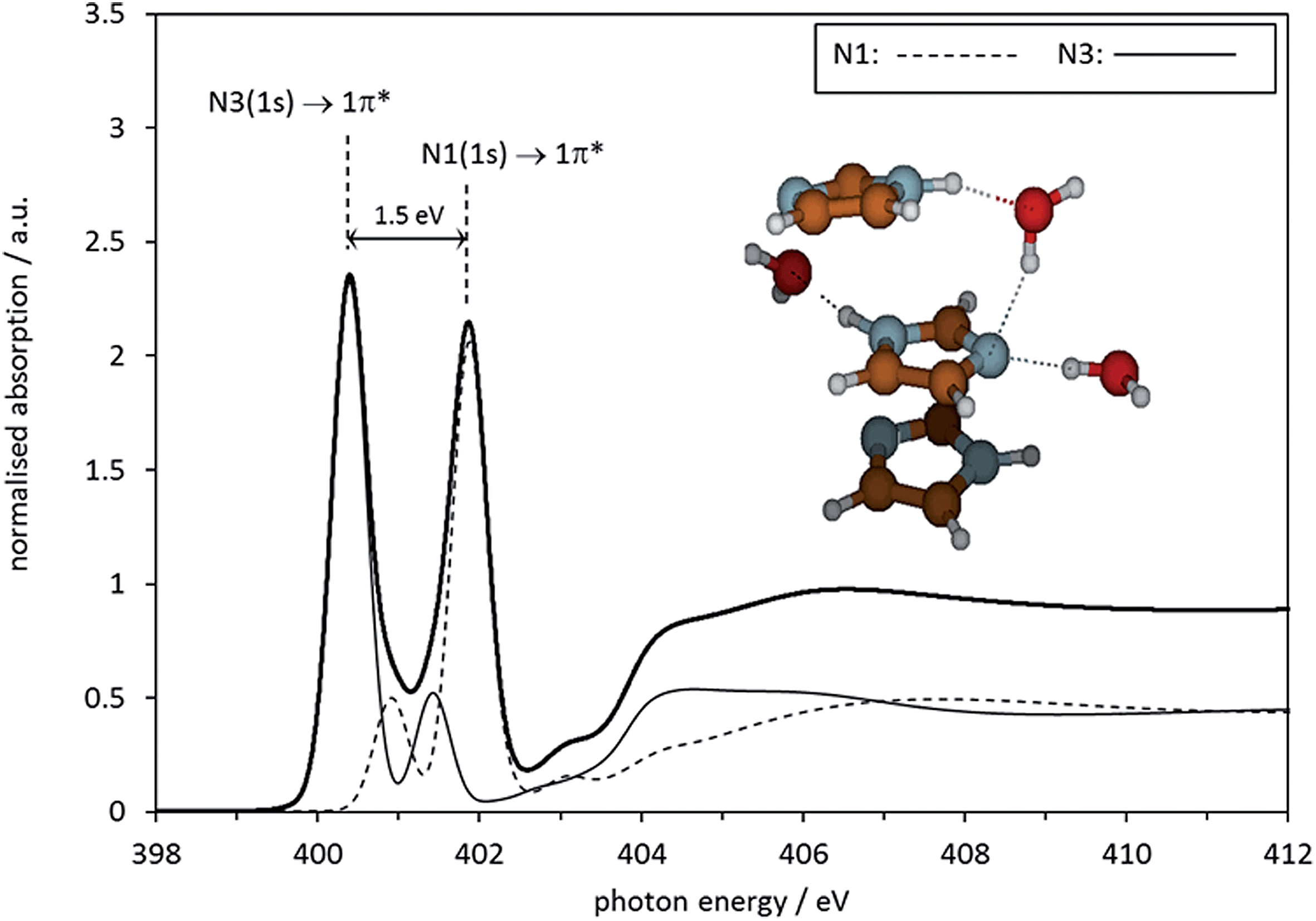 | ||
| Fig. 11 Calculated N K-edge absorption spectrum for the central molecule in a microhydrated Gaussian 03 optimised stack of 3 imidazole molecules. | ||
This result suggests that self-association involves complex synergistic hydrogen bonding interactions mediated by water molecules alongside imidazole–imidazole interactions. This model is compatible with the idea of π–π-assisted hydrogen-bonded stack structures, in competition with chains of hydrogen-bonded imidazole molecules37 that are similar to those found in liquid38 and solid32,33 imidazole. We should mention that the calculated absorption spectra of the central imidazole in these structures are quite sensitive to conformational detail in the evaluated cluster, resulting in varying degrees of agreement with experimental data. Clearly, more complex structure models, including more water and imidazole molecules, need to be more systematically evaluated before firm conclusions about the limitations of the micro-cluster system modelling approach can be drawn. Moreover, combination with other techniques sensitive to local structure in solution (X-ray and neutron scattering, NMR, vibrational spectroscopies, also C K-edge NEXAFS) should be explored to generate a more complete picture. However, the insight already obtained by the modelling of the NEXAFS N K-edge data, and the observed sensitivity of NEXAFS to local structure, provide some confidence that the technique could start playing a role as a tool for validating computationally derived structural predictions. In the context of nucleation studies, a particularly valuable objective for further development will be the setting up of an experimental infrastructure that permits studies of supersaturated solutions.
Conclusions
We have carried out a feasibility study examining the possibility of obtaining N K-edge near-edge X-ray absorption fine-structure (NEXAFS) spectra of a solute at high concentrations in aqueous solution. NEXAFS has been shown to be able to distinguish between different chemical environments of imidazole, including gas phase, solid state and the solution state. We have shown that the analysis of known structures (monomer, crystal) provides valuable insight for unravelling the more complex origins of electronic structure variations in the solution system. We have also shown that different types of electronic structure calculations (Gaussian, StoBe, CASTEP) can be used to provide a consistent interpretation of NEXAFS data, which, especially in combination with information from other experimental techniques, should enable more incisive analytical approaches to determining the structure and properties of organic solutes in solution. The results obtained so far have confirmed that microhydrated clusters with a single imidazole molecule are poor models for aqueous imidazole, and that more complex models, which include hydrogen-bonded hydrate molecules as well as imidazole–imidazole interactions lead to better agreement with experimental data.Acknowledgements
This research was supported by EPSRC grants GR/S85801/01, EP/D023807/1, GR/R59069/01, EP/I014446/1 and EP/I013563/1. MJT gratefully acknowledges support by EPSRC through a DTA PhD studentship. We thank the Helmholtz Zentrum Berlin (HZB) for a BESSY-II beamtime award (2007_61140). This work made use of the facilities of N8 HPC, provided and funded by the N8 consortium and EPSRC (Grant no. EP/K000225/1). The Centre is coordinated by the Universities of Leeds and Manchester. The authors would also like to acknowledge the usage of the ARC1 computing facility at the University of Leeds, and the usage of Hartree Centre resources for this work. The STFC Hartree Centre is a research collaboration funded by the UK's investment in e-Infrastructure. CRS would like to acknowledge the support of the High Performance Computing (HPC) team at the University of Leeds, for their design of the advanced research computing 1 (ARC1) facility, and assistance in code compilation and optimization. We thank Dr Luanga N. Nchari and Dr Angela M. Beesley for assistance during the NEXAFS measurements. We thank Professor Pavel Jungwirth for sharing the microhydrated cluster model structures.Notes and references
- M. Volmer, in Kinetik der Phasenbildung, Steinkopff, Leipzig, Germany, 1939 Search PubMed.
- P. G. Debenedetti, Nature, 2006, 441, 168–169 CrossRef CAS PubMed.
- A. C. Pan, T. J. Rappl, D. Chandler and N. P. Balsara, J. Phys. Chem. B, 2006, 110, 3692–3696 CrossRef CAS PubMed.
- R. J. Davey, S. L. M. Schroeder and J. H. ter Horst, Angew. Chem., Int. Ed., 2013, 52, 2166–2179 CrossRef CAS PubMed.
- D. W. Oxtoby, Acc. Chem. Res., 1998, 31, 91–97 CrossRef CAS.
- D. Kashchiev and G. M. van Rosmalen, Cryst. Res. Technol., 2003, 38, 555–574 CrossRef CAS PubMed.
- D. Kashchiev, in Nucleation: Basic Theory with Applications, Butterworth-Heinemann, Oxford, 2000 Search PubMed.
- P. G. Vekilov, Cryst. Growth Des., 2010, 10, 5007–5019 CAS.
- D. Gebauer and H. Cölfen, Nano Today, 2011, 6, 564–584 CrossRef CAS PubMed.
- J. Bernstein, in Polymorphism in Molecular Crystals, Clarendon, Oxford, 2002 Search PubMed.
- A. Spitaleri, C. A. Hunter, J. F. McCabe, M. J. Packer and S. L. Cockroft, CrystEngComm, 2004, 6, 489–493 RSC.
- C. A. Hunter, C. M. R. Low, M. J. Packer, S. E. Spey, J. G. Vinter, M. O. Vysotsky and C. Zonta, Angew. Chem., Int. Ed., 2001, 40, 2678–2682 CrossRef CAS.
- J. Bernstein, R. J. Davey and J. O. Henck, Angew. Chem., Int. Ed., 1999, 38, 3441–3461 CrossRef CAS.
- H. G. Alison, R. J. Davey, J. Garside, M. J. Quayle, G. J. T. Tiddy, D. T. Clarke and G. R. Jones, Phys. Chem. Chem. Phys., 2003, 5, 4998–5000 RSC.
- P. E. Bonnett, K. J. Carpenter, S. Dawson and R. J. Davey, J. Chem. Soc., Chem. Commun., 2003, 698–699 RSC.
- R. J. Davey, G. Dent, R. K. Mughal and S. Parveen, Cryst. Growth Des., 2006, 6, 1788–1796 CAS.
- I. Weissbuch, M. Lahav and L. Leiserowitz, Cryst. Growth Des., 2003, 3, 125–150 CAS.
- J. Stöhr, in NEXAFS Spectroscopy (Springer Series in Surface Sciences 25), Springer, Berlin, Heidelberg, 1992 Search PubMed.
- G. Hähner, Chem. Soc. Rev., 2006, 35, 1244–1255 RSC.
- Practical Surface Analysis, Auger and X-ray Photoelectron Spectroscopy, ed. D. Briggs and M. P. Seah, John Wiley & Sons, Chichester, New York, Brisbane, Toronto, Singapore, 2 edn, 1990, vol. 1 Search PubMed.
- J. W. Niemantsverdriet, in Spectroscopy in Catalysis, VCH, Weinheim, 1993 Search PubMed.
- S. Hüfner, in Photoelectron Spectroscopy. Principles and Applications, Springer Verlag, Berlin, Heidelberg, New York, 2 edn, 1996 Search PubMed.
- J. S. Stevens, S. J. Byard and S. L. M. Schroeder, Cryst. Growth Des., 2010, 10, 1435–1442 CAS.
- J. S. Stevens, S. J. Byard and S. L. M. Schroeder, J. Pharm. Sci., 2010, 99, 4453–4457 CrossRef CAS PubMed.
- J. S. Stevens, S. J. Byard, C. A. Muryn and S. L. M. Schroeder, J. Phys. Chem. B, 2010, 114, 13961–13969 CrossRef CAS PubMed.
- J. S. Stevens, S. J. Byard, C. C. Seaton, G. Sadiq, R. J. Davey and S. L. M. Schroeder, Angew. Chem., Int. Ed., 2011, 50, 9916–9918 CrossRef CAS PubMed.
- J. S. Stevens, S. J. Byard, E. Zlotnikov and S. L. M. Schroeder, J. Pharm. Sci., 2011, 100, 942–948 CrossRef CAS PubMed.
- J. S. Stevens, S. J. Byard, C. C. Seaton, G. Sadiq, R. J. Davey and S. L. M. Schroeder, Phys. Chem. Chem. Phys., 2014, 16, 1150–1160 RSC.
- J. S. Stevens, C. R. Seabourne, Ch. Jaye, D. A. Fischer, A. J. Scott and S. L. M. Schroeder, J. Phys. Chem. B, 2014, 118, 12121–12129 CrossRef CAS PubMed.
- J. S. Stevens, A. Gainar, E. Suljoti, J. Xiao, R. Golnak, E. F. Aziz and S. L. M. Schroeder, Chem.–Eur. J., 2015, 21 DOI:10.1002/chem.201405635.
- G. Will, Nature, 1963, 198, 575 CrossRef CAS PubMed.
- S. Martinez, Acta Crystallogr., 1966, 20, 783 CrossRef.
- B. M. Craven, R. K. Mcmullan, J. D. Bell and H. C. Freeman, Acta Crystallogr., Sect. B: Struct. Sci., 1977, 33, 2585–2589 CrossRef.
- W. Hückel, J. Datow and E. Simmersbach, Z. Phys. Chem., Abt. A, 1940, 186, 129–179 Search PubMed.
- L. Gontrani, R. Caminiti, L. Bencivenni and C. Sadun, Chem. Phys. Lett., 1999, 301, 131–137 CrossRef CAS.
- F. Peral and E. Gallego, J. Mol. Struct., 1997, 415, 187–196 CrossRef CAS.
- S. Y. Liem, M. S. Shaik and P. L. A. Popelier, J. Phys. Chem. B, 2011, 115, 11389–11398 CrossRef CAS PubMed.
- M. S. Shaik, S. Y. Liem, Y. N. Yuan and P. L. A. Popelier, Phys. Chem. Chem. Phys., 2010, 12, 15040–15055 RSC.
- U. Domanska, M. K. Kozlowska and M. Rogalski, J. Chem. Eng. Data, 2002, 47, 456–466 CrossRef CAS.
- E. F. Aziz, PhD Dissertation, Freie Universität Berlin, 2007.
- D. Nolting, N. Ottosson, M. Faubel, I. V. Hertel and B. Winter, J. Am. Chem. Soc., 2008, 130, 8150–8151 CrossRef CAS PubMed.
- L. Triguero and L. G. M. Pettersson, Surf. Sci., 1998, 398, 70–83 CrossRef CAS.
- L. Triguero, L. G. M. Pettersson, B. F. Minaev and H. Agren, J. Chem. Phys., 1998, 108, 1193–1205 CrossRef CAS PubMed.
- K. Herrmann, L. G. M. Pettersson, M. E. Casida, C. Daul, A. Goursot, A. Koester, E. Proynov, A. St-Amant and D. R. Salahub, StoBe-deMon version 2.2, Berlin, Stockholm, 2006 Search PubMed.
- S. H. Vosko and L. Wilk, Phys. Rev. B, 1980, 22, 3812–3815 CrossRef CAS.
- C. Kolczewski, R. Püttner, O. Plashkevych, H. Agren, V. Staemmler, M. Martins, G. Snell, A. S. Schlachter, M. Sant'Anna, G. Kaindl and L. G. M. Pettersson, J. Chem. Phys., 2001, 115, 6426–6437 CrossRef CAS PubMed.
- L. Triguero, L. G. M. Pettersson and H. Agren, J. Phys. Chem. A, 1998, 102, 10599–10607 CrossRef CAS.
- L. Triguero, L. G. M. Pettersson and H. Agren, Phys. Rev. B, 1998, 58, 8097–8110 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Wallingford, CT, Gaussian, Inc., 2004 Search PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, J. Crystallogr., 2005, 220, 567–570 CAS.
- S. P. Gao, C. J. Pickard, A. Perlov and V. Milman, J. Phys.: Condens. Matter, 2009, 21, 104203 CrossRef PubMed.
- S. P. Gao, C. J. Pickard, M. C. Payne, J. Zhu and J. Yuan, Phys. Rev. B, 2008, 77, 115122 CrossRef.
- C. J. Pickard, PhD thesis, University of Cambridge, 2007.
- J. D. Pack and H. J. Monkhorst, Phys. Rev. B, 1977, 16, 1748–1749 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- C. R. Seabourne, A. J. Scott, R. Brydson and R. J. Nicholls, Ultramicroscopy, 2009, 109, 1374–1388 CrossRef CAS PubMed.
- P. Blaha, K. Schwarz, P. Sorantin and S. B. Trickey, Comput. Phys. Commun., 1990, 59, 399–415 CrossRef CAS.
- K. Schwarz, P. Blaha and S. B. Trickey, Mol. Phys., 2010, 108, 3147–3166 CrossRef CAS PubMed.
- E. Apen, A. P. Hitchcock and J. L. Gland, J. Phys. Chem., 1993, 97, 6859–6866 CrossRef CAS.
- P. Moreau, F. Boucher, G. Goglio, D. Foy, V. Mauchamp and G. Ouvrard, Phys. Rev. B, 2006, 73, 195111 CrossRef.
- J. S. Stevens, A. C. de Luca, M. Pelendritis, G. Terenghi, S. Downes and S. L. M. Schroeder, Surf. Interface Anal., 2013, 45, 1238–1246 CrossRef CAS PubMed.
- P. Edington and M. M. Harding, Acta Crystallogr., Sect. B: Struct. Sci., 1974, 30, 204–206 CrossRef CAS.
- B. Jagoda-Cwiklik, P. Slavícek, D. Nolting, B. Winter and P. Jungwirth, J. Phys. Chem. B, 2008, 112, 7355–7358 CrossRef CAS PubMed.
- B. Jagoda-Cwiklik, P. Slavícek, L. Cwiklik, D. Nolting, B. Winter and P. Jungwirth, J. Phys. Chem. A, 2008, 112, 3499–3505 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Microhydrated cluster models – Fig. S1–S9. See DOI: 10.1039/c5fd00005j |
| This journal is © The Royal Society of Chemistry 2015 |