 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of solvent on crystal nucleation of risperidone†
Donal
Mealey
a,
Jacek
Zeglinski
*a,
Dikshitkumar
Khamar
a and
Åke C.
Rasmuson
*ab
aMaterials and Surface Science Institute, Chemical and Environmental Science Department, University of Limerick, Limerick, Ireland. E-mail: ake.rasmuson@ul.ie; jacek.zeglinski@ul.ie; Tel: +353 61 23 46 17 Tel: +353 61 23 41 57
bDepartment of Chemical Engineering and Technology, KTH Royal Institute of Technology, Stockholm, Sweden
First published on 3rd February 2015
Abstract
Over 2100 induction time experiments were carried out for the medium-sized, antipsychotic drug molecule, risperidone in seven different organic solvents. To reach the same induction time the required driving force increases in the order: cumene, toluene, acetone, ethyl acetate, methanol, propanol, and butanol, which reasonably well correlates to the interfacial energies as determined within classical nucleation theory. FTIR spectroscopy has been used to investigate any shifts in the spectra and to estimate the interaction of solute and solvent at the corresponding site. The solution condition has also been investigated by Density Functional Theory (DFT) calculations over (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent–solute binding interactions at 8 different sites on the risperidone molecule. The DFT computational results agree with the spectroscopic data suggesting that these methods do capture the binding strength of solvent molecules to the risperidone molecule. The difficulty of nucleation correlates reasonably to the DFT computations and the spectroscopic measurements. The results of the different measurements suggest that the stronger the solvent binds to the risperidone molecule in solution, the slower the nucleation becomes.
:1) solvent–solute binding interactions at 8 different sites on the risperidone molecule. The DFT computational results agree with the spectroscopic data suggesting that these methods do capture the binding strength of solvent molecules to the risperidone molecule. The difficulty of nucleation correlates reasonably to the DFT computations and the spectroscopic measurements. The results of the different measurements suggest that the stronger the solvent binds to the risperidone molecule in solution, the slower the nucleation becomes.
1. Introduction
Crystal nucleation from solution is a key step in crystallization processes and solution crystallization is widely used in the pharmaceutical, food, chemical and agrochemical industries. Crystal nucleation is influenced by the solvent and within the Classical Nucleation Theory (CNT) framework this is captured in the interfacial energy1,2 and in the pre-exponential factor.3 The influence on the kinetics of nucleation is assumed to be partly related to an influence on the type and the size of molecular assemblies (clusters) in the solution. Solution structuring of solute molecules has been investigated by various spectroscopic methods (IR,4 Raman,5 NMR,6 UV-visible7) to establish a link with the crystal structure of the nucleating solid phase. In some cases such a link could be established and in others cases not.8,9 In addition to the type of aggregates6,10 in the solutions, the type of solvent–solute interaction can also dictate the morphology11 as well as the kinetics of nucleation.12 Lohani et al.13 showed the type of solvent–solute interaction affecting the diffusion coefficient, resulting in the preferential crystallization of one form over another. The hydrogen bonding capabilities of a solvent and the nature of interaction prevailing between solute and solvent resulting in the nucleation of a particular form has also been a subject of a number of studies.5,14Application of the CNT framework on experimentally measured nucleation rate data helps to determine the nucleation work and the interfacial energy in different solvents. The influence of solvent on the ease or difficulty of nucleation can be used to probe the nucleation process on the molecular level. Our previous work12 on a small molecule, salicylic acid (138.12 g mol−1) reveals the role of the solvent in the nucleation of this compound. By employing solution calorimetry, vibrational spectroscopy and quantum-chemical calculations over one-to-one solvent–solute molecule interactions as well as over the complete first solvation shell, it was consistently observed that the stronger the solvent binds to salicylic acid, the greater is the difficulty of nucleation. On a similar note, the effect of solvent was also recently highlighted by Sullivan et al.15 in which the authors suggest the process of desolvation affects the kinetics of nucleation and thus governs the nucleation process. Zhao et al.16 studied the effect of solvent on the crystallization of nesquehonite (MgCO3·2H2O). They reported faster nucleation kinetics with the addition of dimethyl formamide (DMF) to the water which they attributed to the kinetic factors accelerating the cluster aggregation in a non-classical mechanism.
In spite of the critical importance of the choice of solvent in solution crystallization, there is only a limited amount of nucleation rate data available for medium- to large-sized organic molecules in a range of organic solvents. Much of the previously published work is carried out either in one solvent only or for small-sized molecules only. The size and complexity of the solute molecule can certainly add complications to an already complex nucleation process. In this work, we use a medium sized (410.49 g mol−1), fairly complex and conformationally flexible pharmaceutical molecule (API), risperidone, and use the influence of the solvent as a probe to investigate the mechanisms of nucleation. The results are analysed directly without the use of the CNT framework as well as within that framework. The strength of solvent–solute interaction is quantified using DFT calculations and is examined by solution spectroscopy for particular interaction sites. The molecular structure of risperidone is shown in Fig. 1. There are two reported polymorphs of risperidone and both structures, form I (WASTEP) and form II (WASTEP01) are reported in the Cambridge Structure Database.
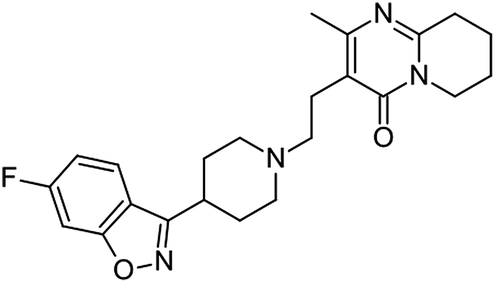 | ||
| Fig. 1 Molecular structure of risperidone. | ||
2. Materials and methods
2.1. Materials
Risperidone (>99.95%), form I, was kindly provided by Janssen Pharmaceuticals, Ireland, and used without further purification. All solvents were purchased from VWR International; methanol (99.8% purity by volume), 1-propanol (99.8%), 1-butanol (99.8%), ethyl acetate (99.8%), acetone (99.8%), toluene (99.8%), and cumene (99.8%).2.2. Induction time measurement
The induction time of nucleation of risperidone was measured in seven solvent systems with a saturation temperature of 50 °C. Over 2160 nucleation events were observed at 27 different conditions.Stock solutions were prepared in 500 ml sealed glass bottles by weighing appropriate amounts of risperidone and solvent according to previously published solubility data.9 An equilibration period of 12 h at 55 °C was allowed, with agitation provided with a PTFE (polytetrafluoroethylene) coated magnetic stirrer (38 × 10 mm, 3 × 11 mm pivot ring) at 500 rpm. Solutions were dispensed via preheated syringes and 0.2 μm solvent compatible filters (PTFE or Nylon), into twenty 20 ml glass vials (70 × 20 mm, VWR International). A magnetic stirring bar (13 × 3 mm) was added to each vial prior to sealing with a plastic screw cap with a PTFE seal. Solutions were then subjected to a second equilibration period at 55 °C overnight before the crystallization experiments were performed.
An experimental apparatus identical to that described by Mealey et al.17 was employed to simultaneously monitor 20 individual nucleation experiments. Solutions were transferred from bath A at 55 °C to bath B held at a temperature where the solutions were supersaturated (Tsupersat). Agitation was provided at 400 rpm via a 60 point submersible magnetic stirring plate (2Mag). Immediately after the transfer, the time to nucleate (temperature equilibration time + isothermal nucleation time) was measured with a high definition video camera observing the transition from a clear to cloudy solution. The approach to temperature equilibrium was measured separately with a control tube of pure solvent and an in situ calibrated temperature probe (Dostmann P600). Once all tubes had nucleated they were transferred back to water bath A where complete dissolution occurred and equilibration was allowed overnight. The cycle was repeated four times until approximately 80 nucleation events were recorded for a given driving force and solvent.
To avoid the possible effects of degradation of the risperidone molecule each batch of 20 tubes were subjected to just four temperature cycles. It was observed that upon extended time at elevated temperature an obvious colour change would occur in the solutions. A new batch of tubes was prepared for each separate driving force. Along with monitoring solutions visually, after four cycles solutions were spotted onto silica gel thin layer chromatography (TLC) plates using methanol as a mobile phase and viewed under UV light to ensure there were no detectable impurities.
Solid samples were isolated by filtration, moments after nucleation for both an early nucleation and a late nucleation event, at both high and low driving forces. The polymorphic form was determined by reflectance powder X-ray diffraction (PXRD, Philips PANalytical X'Pert MP PRO) in the range 5–35° (2θ) with a wavelength of 1.54 Å (40 kV, 35 mA). In all cases the solid phase analysed was pure form I. The theoretical PXRD patterns generated from the crystallographic information files for both form I and form II along with the samples taken from risperidone nucleation in toluene are shown in the ESI.†
2.3. Computational methods
Density Functional Theory (DFT) calculations have been applied to investigate (1:1) solvent–solute pair interactions. For this purpose, a risperidone molecule has been extracted from the crystal lattice of its form I (WASTEP) and optimized in isolation at the B97-D3/6-31G(d,p) level. The molecular geometry changed only slightly upon gas-phase optimization, preserving the original crystal-like conformation (cf. Fig. S4, ESI†). The seven solvents used in our experimental work can be divided into three groups: polar protic (alcohols), polar aprotic (acetone and ethyl acetate), and nonpolar (toluene and cumene). Three solvents (one from each group), representing different polarity and H-bond capability, were chosen for the modelling part, i.e. 1-propanol, acetone, and toluene. These solvents posses different bonding mechanisms. 1-Propanol is both a H-bond donor and acceptor, acetone is only capable of accepting H-bonds, while toluene is not capable of making strong H-bonds, although it can interact through π-electrons located over its benzene ring. The (1:1) solvent–solute interactions are probed at eight sites of the risperidone molecule chosen with aid of the electrostatic potential map (EPM) as shown in Fig. 2. The EPM indicates higher electron deficiency (dark blue colour) at the hydrogens labelled H1, H2, and H3, while the electron rich regions, having H-bond accepting capabilities, are highlighted in red/yellow at the heteroatoms O1, O2, and N1–N3 (Fig. S2, ESI,† shows more projections of the EPM). Based on the electron distribution, one can expect that in the crystal, the higher electron deficiency at the ring 1 hydrogens will be complemented by neighbouring electron rich polar atoms resulting in relatively strong intermolecular bonding. This can indeed be observed in the risperidone crystal structure (cf.Fig. 2b) and thus validates the electrostatic potential map presented.
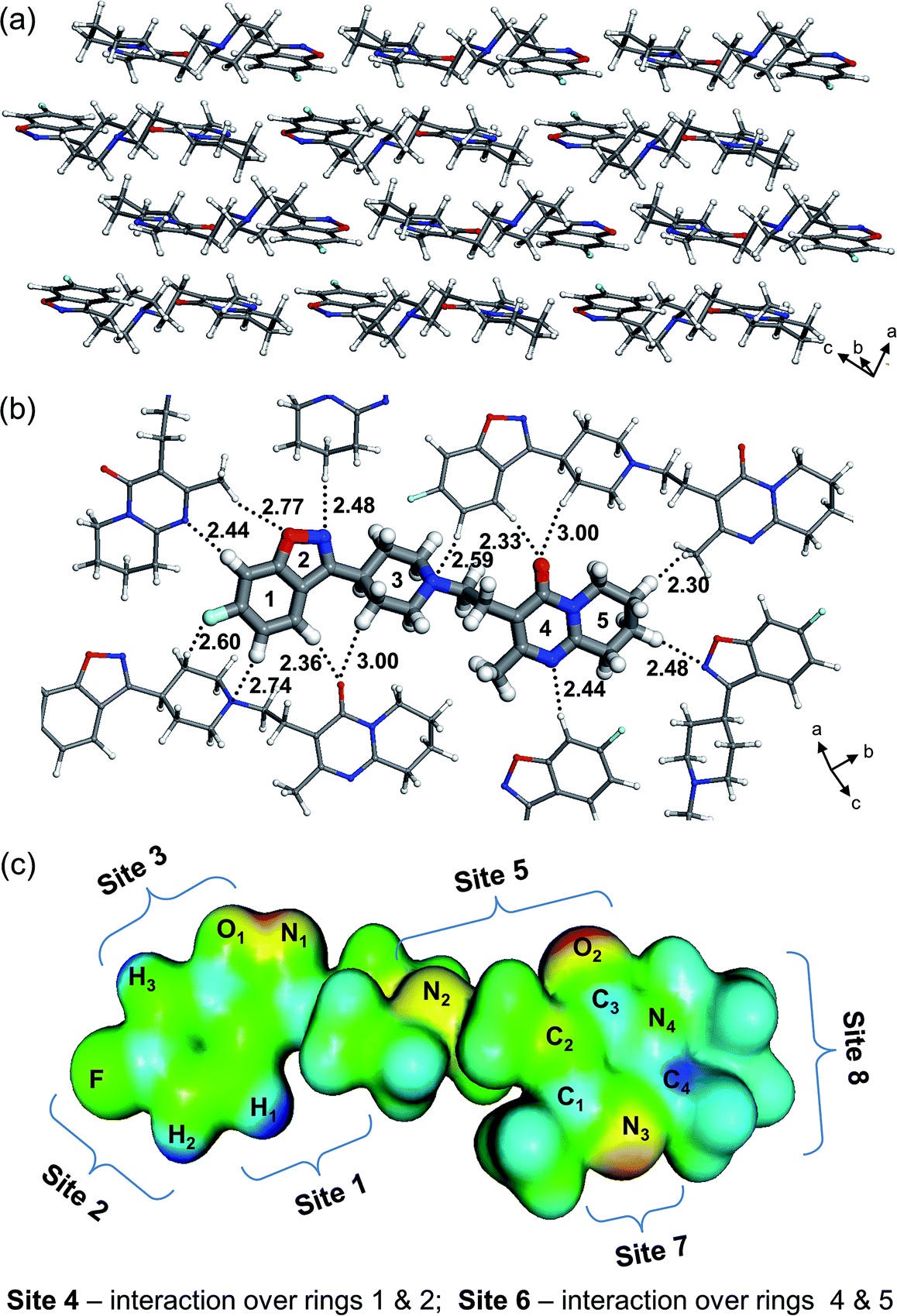 | ||
| Fig. 2 Molecular arrangement in the risperidone crystal (a); chief interactions and intermolecular distances (in Angstroms) between molecules neighbouring in a monolayer (b); electrostatic potential isosurface and definition of interaction sites in risperidone molecule for probing solvent–solute bonding (c) (blue – positive, red – negative, green – neutral potential). | ||
The considered eight sites represent all the important binding features of the risperidone molecule such as interaction at (i) polar sites containing O and N heteroatoms (H-bond acceptors; sites 3, 5, and 7), (ii) non-polar C–H-terminated sites 1 and 8, and (iii) the connected rings 1–2 (site 4) and 4–5 (site 6). In addition, for all the seven solvents we have modelled the (1:1) risperidone–solvent associates at site 5; this site combines two electron-rich atoms, i.e. carbonyl oxygen and tertiary amine nitrogen, and thus it is expected to yield strong interactions. Another reason for choosing site 5 was the ability of comparing the calculated solvent–solute binding energies of the risperidone carbonyl group across all the seven solvents with the respective experimentally determined C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O shifts in the FTIR spectrum.
O shifts in the FTIR spectrum.
The equilibrium geometries of the (1:1) dimers are calculated with a B97-D3 Grimme's functional,18 and a Gaussian-type 6-31G(d,p) basis set.19 Binding energies in the dimers are calculated for the lowest energy configurations, after probing a set of sensible molecular orientations, using a double hybrid B2PLYP-D3 functional,20 which combines exact Hartree–Fock exchange with an MP2-like correlation and long-range dispersion corrections; here we use a basis set of quadruple-ζ valence quality (def2-QZVP).21 The binding energy is calculated as follows:
| ΔEbind = EA–B − (EA + EB) | (1) |
Conformational analysis of the risperidone molecule has been carried out at the DFT B97-D3/6-31G(d,p)//B2PLYP-D3/def2-QZVP level (gas-phase, 0 K), with energy barriers to rotation calculated for four rotational centres (single bonds) of the risperidone molecule (Fig. S5, ESI†). We restricted our calculations to electronic energies only (corrections for zero point energy and other vibrational contributions are not included). A relaxed potential energy surface (PES) scan over 360°, with the step interval of 5°, has been performed for the respective dihedral angles. Four new low energy conformers were identified upon PES scanning and their stabilities have been compared relative to the relaxed crystal-like conformation.
Calculations were performed using the GAUSSIAN 09 package.22 Electrostatic potential maps were generated with the MOLEKEL 5.4 software.23
2.4. Spectroscopic methods
Solution spectroscopy was carried out for risperidone in all the seven solvents that were used for nucleation work. Raman spectra were collected using Kaiser Raman Rxn2 analyser with an Invictus 785 nm excitation laser. For liquid samples, a short focus immersion probe of 1/4 inch diameter was used and the solid samples were analysed using a non-contact optic probe. Each spectrum was collected for a minimum of 15 s exposure time and five accumulations in the spectral region of 3400–200 cm−1 using Mettler Toledo iC Raman software version 4.1.IR spectrum of the solid material was collected using Perkin Elmer spectrum 100 spectrometer with Universal ATR accessory in the spectral region of 4000–650 cm−1 with 4 cm−1 resolution. IR spectra of solutions were collected using Mettler Toledo ReactIR 10 fitted with a fibre optic probe and a diamond composite tip. Due to the nature of the fibre optic probe, the solution spectra were collected in the fingerprint region of 2000–650 cm−1 with 4 cm−1 resolution using iC IR software version 4.3. All the spectral data were collected at ambient temperature (20–22 °C).
3. Results
3.1. Rate of nucleation in different solvents
Induction time distributions from toluene (a) and ethyl acetate (b) are shown in Fig. 3.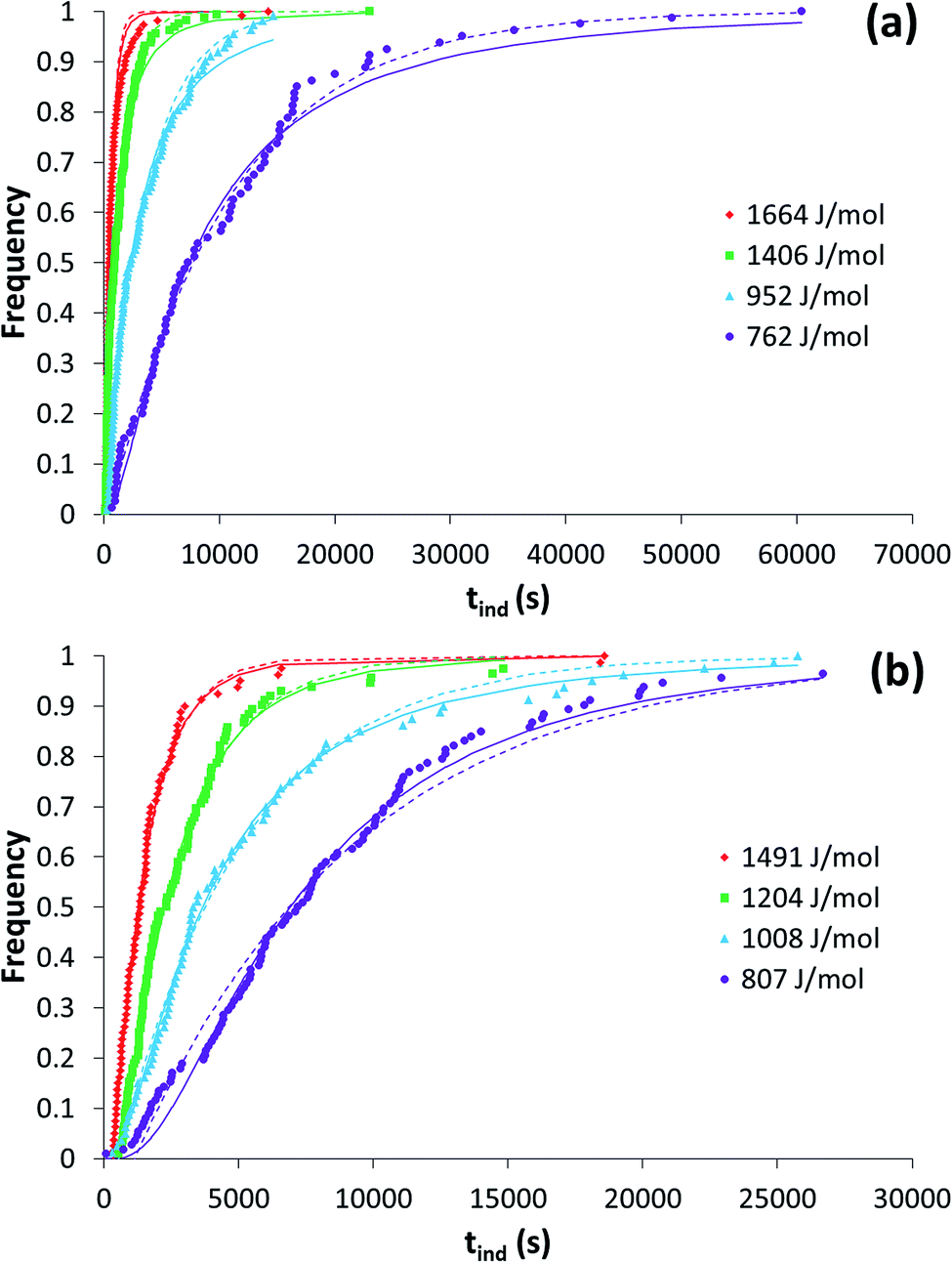 | ||
| Fig. 3 Induction time distributions from toluene (a) and ethyl acetate (b) at various driving forces. Also shown is the fit of the LCDF (solid lines) and the exponential distribution function24 (dashed lines). | ||
The use of various distribution functions to examine induction time data has previously been examined.17,24 Here we have used the lognormal cumulative distribution function (LCDF) and the exponential-based function of Jiang and ter Horst.24 Both distributions provide reasonably similar fits to the data, and results are collected in Table 1. The calculated nucleation rates using both methods display similar trends with driving force. Fitting the LCDF produced coefficients of determination greater than 0.97 in all instances while the fits of the exponential function were similar with one exception resulting in a coefficient of determination of 0.94.
| Solvent |
RTlnS [J mol−1] |
η* (s) ×∕ σ* | J (m−3 s−1) | N* | J a (m−3 s−1) |
|---|---|---|---|---|---|
| Cumene | 757 | 4682 ×∕ 3.75 | 10.68 | 24 | 6.67 |
| 940 | 1343 ×∕ 1.93 | 37.25 | 13 | 43.48 | |
| 1120 | 732 ×∕ 1.83 | 68.29 | 7 | 87.22 | |
| Toluene | 762 | 7397 ×∕ 2.84 | 6.76 | 23 | 4.78 |
| 952 | 2286 ×∕ 3.24 | 21.87 | 12 | 14.28 | |
| 1406 | 886 ×∕ 3.15 | 56.44 | 4 | 38.00 | |
| 1664 | 489 ×∕ 2.19 | 102.28 | 2 | 105.12 | |
| Acetone | 526 | 18374 ×∕ 3.22 | 2.72 | 78 | 1.99 |
| 858 | 5528 ×∕ 2.36 | 9.05 | 18 | 7.43 | |
| 1074 | 2472 ×∕ 2.02 | 20.23 | 9 | 18.25 | |
| 1287 | 1036 ×∕ 2.17 | 48.28 | 5 | 46.65 | |
| Ethyl acetate | 807 | 6914 ×∕ 2.21 | 7.23 | 15 | 6.05 |
| 1008 | 3666 ×∕ 2.55 | 13.64 | 8 | 10.78 | |
| 1204 | 2247 ×∕ 2.20 | 22.25 | 5 | 21.16 | |
| 1491 | 1265 ×∕ 2.17 | 39.51 | 2 | 37.46 | |
| Methanol | 1338 | 3166 ×∕ 2.07 | 15.79 | 9 | 14.66 |
| 1553 | 2289 ×∕ 2.68 | 21.85 | 6 | 16.42 | |
| 1897 | 1514 ×∕ 3.45 | 33.02 | 3 | 20.20 | |
| 2224 | 673 ×∕ 2.41 | 74.31 | 2 | 64.33 | |
| 1-Propanol | 1072 | 16549 ×∕ 3.52 | 3.02 | 19 | 1.81 |
| 1632 | 3216 ×∕ 3.43 | 15.55 | 5 | 9.54 | |
| 2013 | 1172 ×∕ 2.76 | 42.65 | 3 | 31.99 | |
| 2378 | 654 ×∕ 2.39 | 76.48 | 2 | 68.52 | |
| Butanol | 1377 | 4362 ×∕ 2.86 | 11.46 | 6 | 8.41 |
| 1654 | 2963 ×∕ 2.51 | 16.88 | 4 | 13.84 | |
| 2025 | 2262 ×∕ 2.52 | 22.10 | 2 | 18.97 | |
| 2370 | 1239 ×∕ 2.01 | 40.37 | 1 | 42.53 | |
The fitting to the LCDF includes no assumption of a particular mechanism of nucleation. By examining the relation of the induction time distributions to the driving force in Fig. 4 across the various solvents, it is obvious that nucleation is faster in non-polar (toluene and cumene) and polar aprotic (acetone and ethyl acetate) solvents than in the alcohols (1-propanol, methanol, and butanol). To reach equal median induction time, in the range where data is available for all solvents, the driving force required increases in the order: cumene, toluene, acetone, ethyl acetate, methanol, propanol and butanol (Table 2).
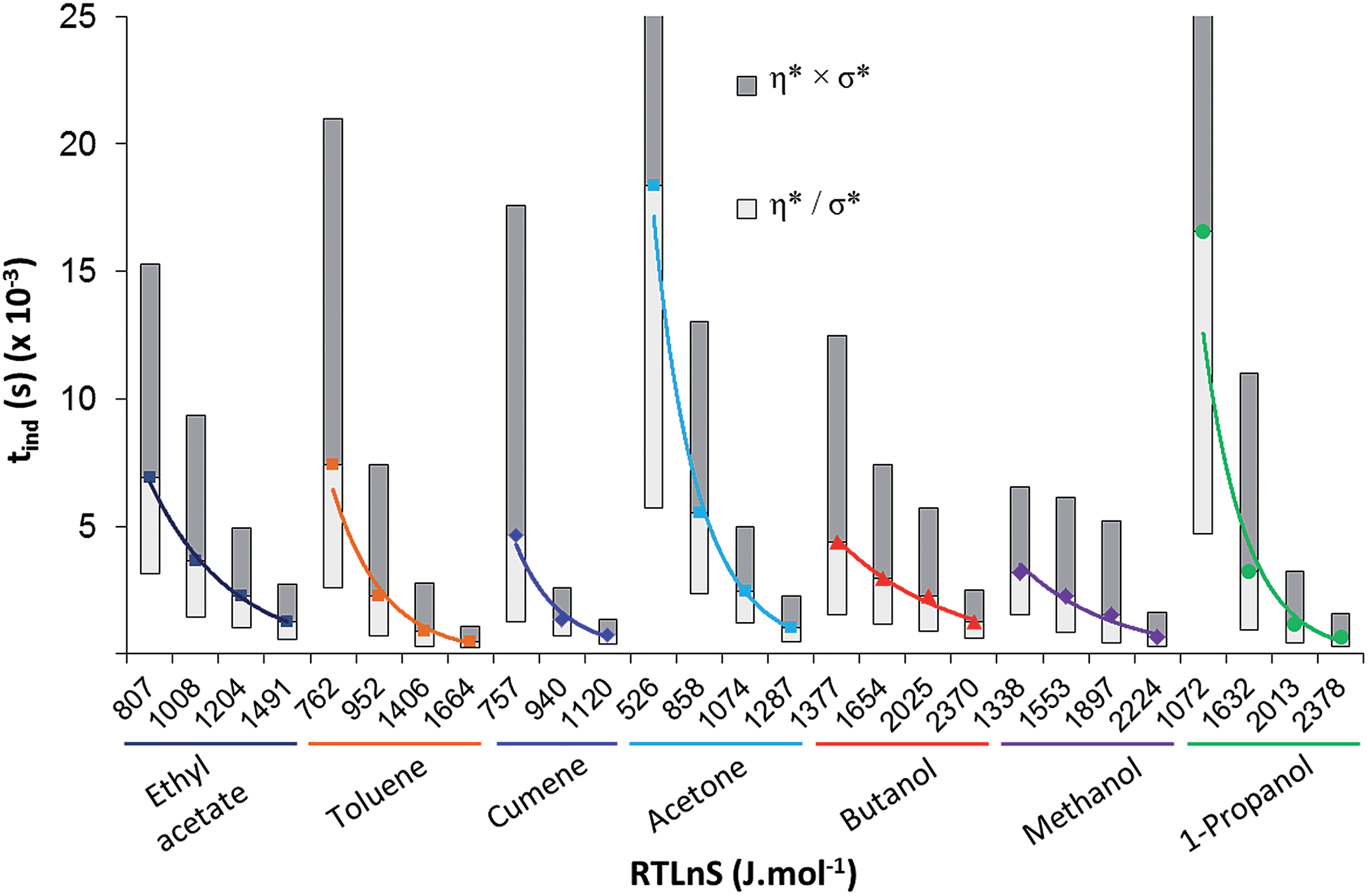 | ||
| Fig. 4 Induction time distributions. Data points represent the geometric mean (η*) of the LCDF while the shaded regions represent one geometric standard deviation (σ*) from this mean. | ||
| Solvent | Driving force (RTlnS, J mol−1) required to reach median induction time |
Driving force order | |
|---|---|---|---|
| 2000 s | 3000 s | ||
| Cumene | 854 | 784 | 1 |
| Toluene | 1015 | 841 | 2 |
| Acetone | 1055 | 954 | 3 |
| Ethyl acetate | 1288 | 1112 | 4 |
| Methanol | 1639 | 1370 | 5 |
| Propanol | 1829 | 1657 | 6 |
| Butanol | 1976 | 1671 | 7 |
Based on the geometric mean induction times reported in Table 1, Fig. 5 presents the standard plot for evaluation of the induction time data within the classical nucleation theory. By relating the induction time (tind) to the nucleation rate (J) it is possible to estimate the pre-exponential factor (A) and the interfacial energy (φ) using eqn (2) and (3):
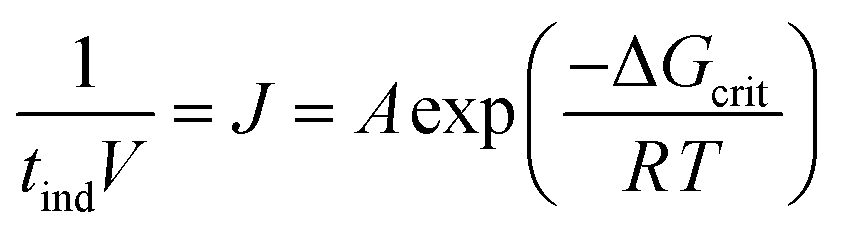 | (2) |
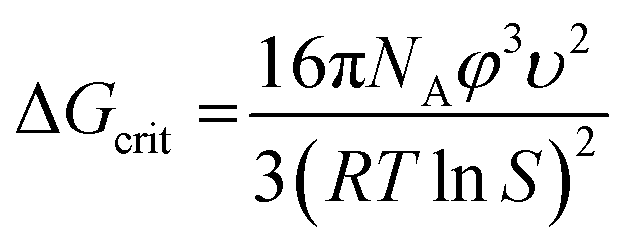 | (3) |
lnS is the supersaturation driving force where S is the supersaturation ratio in terms of the mole fraction. The solid liquid interfacial energies and the preexponential factors are reported in Table 3.
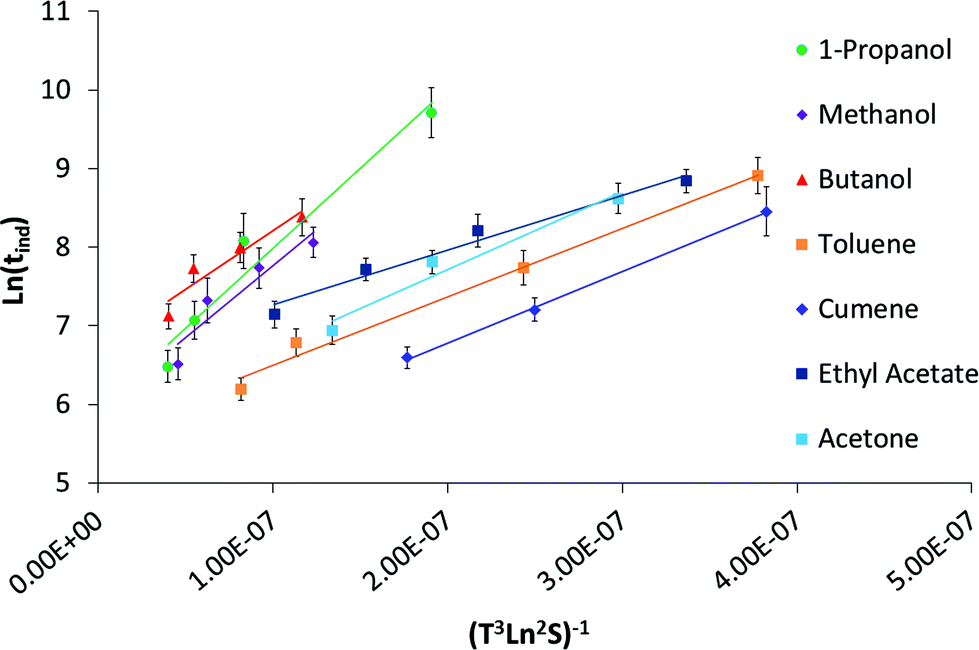 | ||
| Fig. 5 Log geometric mean induction times plotted against driving force to determine the interfacial energy and preexponential factor within classical nucleation theory. | ||
| Solvent | Driving force order from Table 2 | Interfacial energy (mJ m−2) | Pre-exponential factor, A (m−3 s−1) | Solubility at 25 °C | χ 12 | Solvent viscosity (mPa S) | |
|---|---|---|---|---|---|---|---|
| (MF × 103) | (mol l−1) | ||||||
| Cumene | 1 | 1.72 | 348 | 2.60 | 18.68 | 2.05 | 0.78 |
| Toluene | 2 | 1.70 | 181 | 6.67 | 63.17 | 1.43 | 0.59 |
| Acetone | 3 | 1.77 | 161 | 1.75 | 23.79 | 1.18 | 0.31 |
| Ethyl acetate | 4 | 1.58 | 71 | 2.16 | 22.01 | 1.32 | 0.426 |
| Methanol | 5 | 2.18 | 134 | 3.96 | 98.12 | 1.44 | 0.59 |
| 1-Propanol | 6 | 2.25 | 129 | 3.05 | 40.86 | 1.35 | 1.94 |
| Butanol | 7 | 2.04 | 61 | 3.47 | 38.01 | 1.46 | 2.54 |
No direct relation can be found between the interfacial energy in the different solvents and the solid–liquid solubility, regardless of units of the latter which agrees with our previous findings.3,12,25 There is neither a clear correlation with the Hansen solubility parameter nor with the viscosity of the solvent.
3.2. DFT calculations
By running the potential energy surface scan over the dihedral angles of the four single bonds that serve as rotational centres, we have found that the relevant energy barriers to interconversion between the different conformational isomers range from 10 kJ mol−1 to 26 kJ mol−1 (ESI†). There is one new isomer identified for each rotational centre being structurally different from those constituting known conformations in the crystal structure of risperidone form I (and form II). Three of the new conformers are linear and the fourth is U-shaped. Two of the linear conformers are slightly more stable (lower in energy by 2–3 kJ mol−1), while the U-shaped molecule is higher in energy by 6.3 kJ mol−1 as compared to the crystal-like starting conformer; this suggests the least favourable geometry. It can be assumed that only kinetically stable conformers, i.e. those separated by higher energy barriers, could significantly affect the nucleation process. The relatively low rotational barriers along with small energy differences of the three gas-phase linear conformations vs. the crystal structure conformations indicate absence of one kinetically stable conformation that could be unambiguously chosen as a model conformation to study interactions of risperidone with solvent molecules. For this reason, the linear conformation appearing in the crystal structure of risperidone is used consistently in our modelling work. The calculated low energy barriers to rotation indicate high conformational flexibility of the risperidone molecule and suggest a variety of different conformations are present in solution.The relatively large and complex risperidone molecule can possess a variety of binding configurations with solvent molecules. Here we consider eight binding sites, where the interactions at the risperidone molecule are probed with three representative solvents, i.e. 1-propanol, acetone and toluene. The DFT binding energy is calculated for each of the 24 solvent–solute (1:1) heterodimers (Fig. 6). One solvent molecule can interact with a number of atoms of the risperidone molecule. In case of site 1 and site 8, only C–H hydrogens are involved in bonding. The other sites feature a combined interaction of hydrogens and heteroatoms.
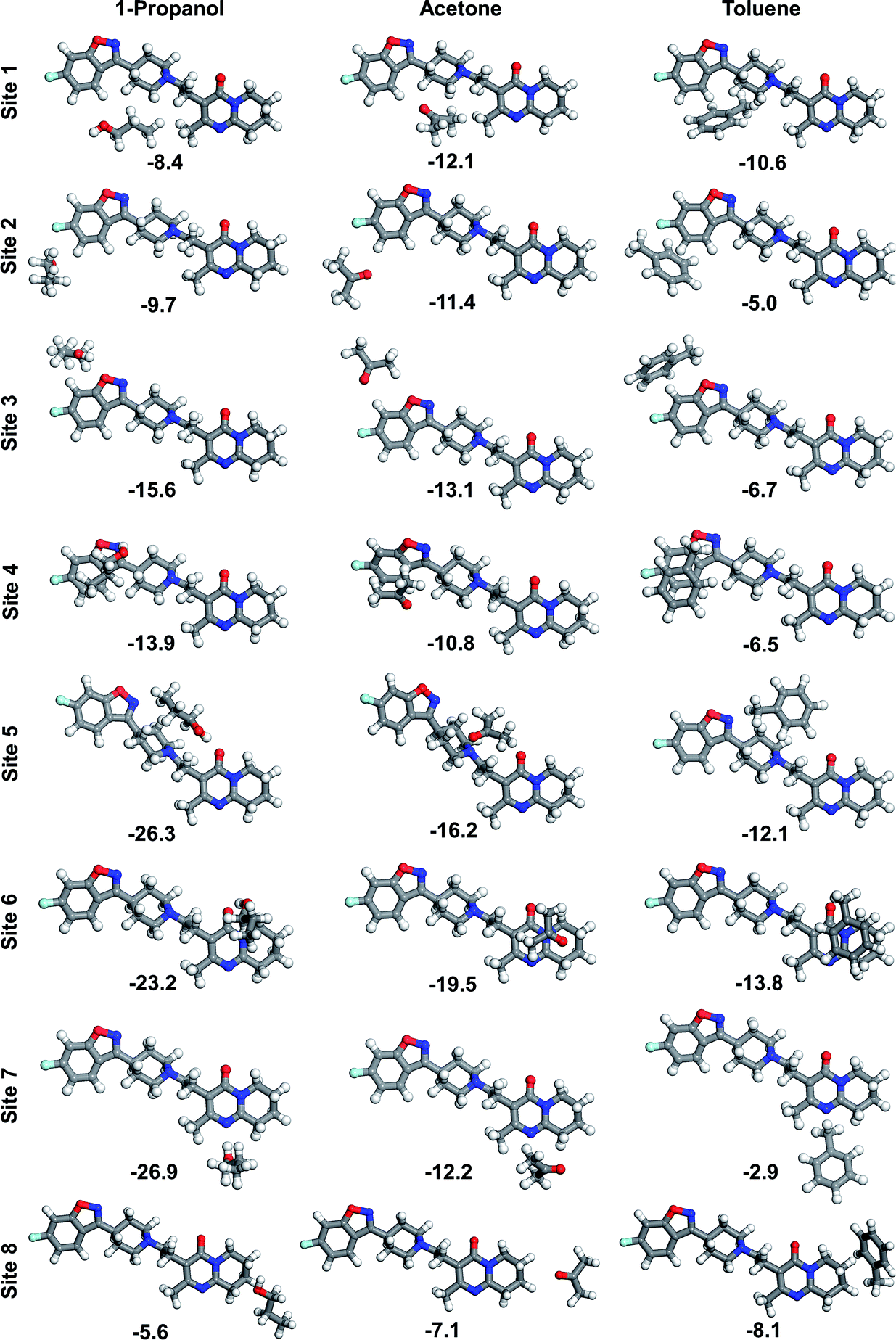 | ||
| Fig. 6 Optimized geometry of risperidone–1-propanol, risperidone–acetone, and risperidone–toluene (1:1) molecular associates (B97-D3/6-31G(d,p) level). Binding energy in kJ mol−1, calculated at B2PLYP-D3/def2-QZVP level. Hydrogen – white, carbon – grey, oxygen – red, nitrogen – blue, fluorine – pale blue. | ||
Our calculations indicate that 1-propanol does not establish strong bonding at the nonpolar sites 1 and 8 of risperidone, being up to 30% weaker than the respective bonding to acetone and toluene. However, the propanol molecule binds stronger at sites 3–7, where it forms H-bonds with polar heteroatoms of the solute molecule. The strongest binding of −26.9 kJ mol−1 and −26.3 kJ mol−1 is observed for site 7 and 5, respectively. The interaction at site 2, which combines binding to hydrogen and fluorine ring atoms, is relatively weak and decreases in order: acetone > 1-propanol > toluene. Acetone and toluene molecules yield the strongest binding of −19.5 kJ mol−1 and −13.8 kJ mol−1, respectively, when placed over rings 4–5 (site 6). The solvent–risperidone (1:1) binding energies averaged over the eight sites form an order: 1-propanol (−16.2 kJ mol−1) > acetone (−12.8 kJ mol−1) > toluene (−8.2 kJ mol−1).
When comparing all the eight interaction sites it appears that site 5 is amongst the strongest interacting sites. This site provides a set of polar groups, including carbonyl oxygen and tertiary amine nitrogen, which result in strong binding with a solvent molecule. In order to compare strength of the risperidone–solvent interaction for all the seven solvents, the site 5 was chosen, and the respective binding energies are shown in Fig. 7.
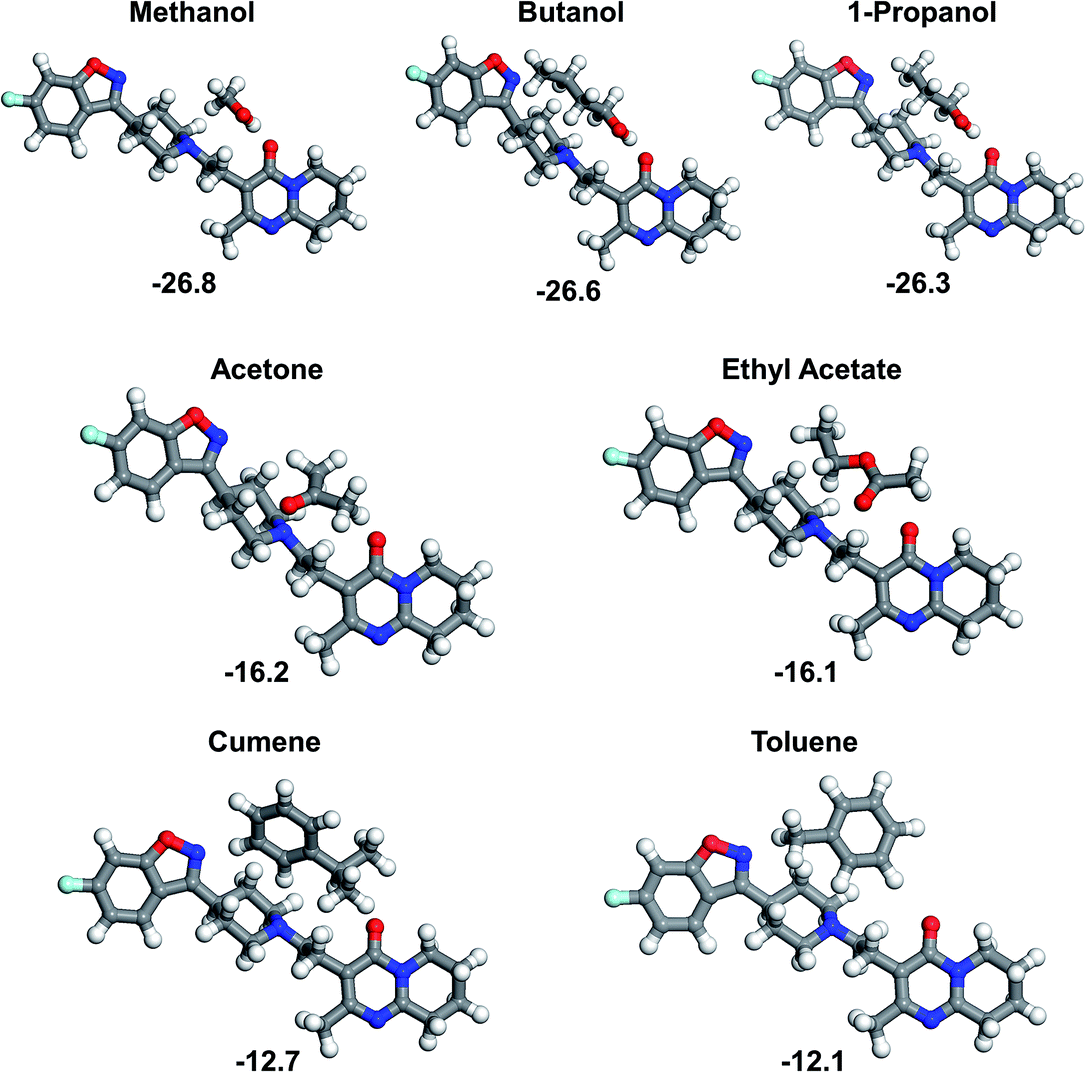 | ||
| Fig. 7 Risperidone–solvent (1:1) associates at site 5, with all the seven solvents used in the experiment (B97-D3/6-31G(d,p) level). Binding energy in kJ mol−1, calculated at B2PLYP-D3/def2-QZVP level. | ||
The binding energies calculated at site 5 form three distinct groups of comparable values: (i) alcohols, yielding very similar and the strongest binding interactions, (ii) acetone and ethyl acetate, with moderate binding, and (iii) cumene and toluene, being relatively weakly bound to the risperidone molecule.
3.3. Solution spectroscopy
N3, C1C2)26 in the pyrimidine ring (ring 4) and also some contribution from v(C1–N3) of the same ring (cf.Fig. 2). Functional groups responsible for this vibration frequency correspond to site 7 as shown in Fig. 2.
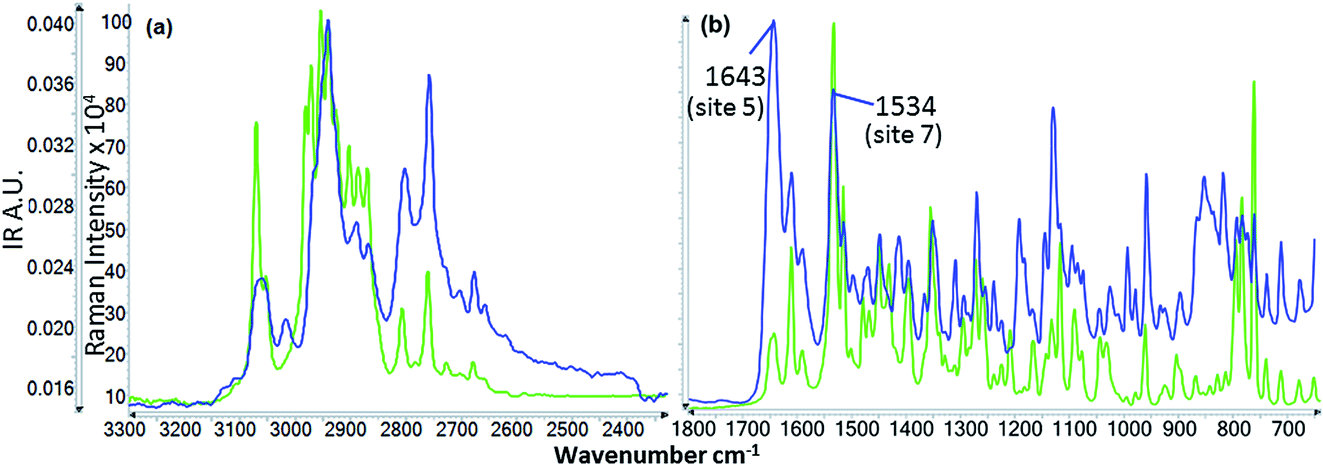 | ||
| Fig. 8 IR (blue) and Raman (green) spectrum of risperidone Form I (solid). | ||
O) is sensitive to hydrogen bonding and other solvent–solute interactions due to the effect of interacting species on the double bond characteristics of the carbonyl bond. The v(CO), has often been used as a probe to study the solvent–solute interaction12,28,29 and generally the stronger the solvent interacts, the lower the frequency at which it absorbs. The shifts of the carbonyl frequency suggest that at site 5 of the risperidone molecule (Fig. 2) the alcohols are strongly bound followed by acetone and ethyl acetate, and the methyl benzenes (toluene and cumene) are relatively weakly interacting at this site.
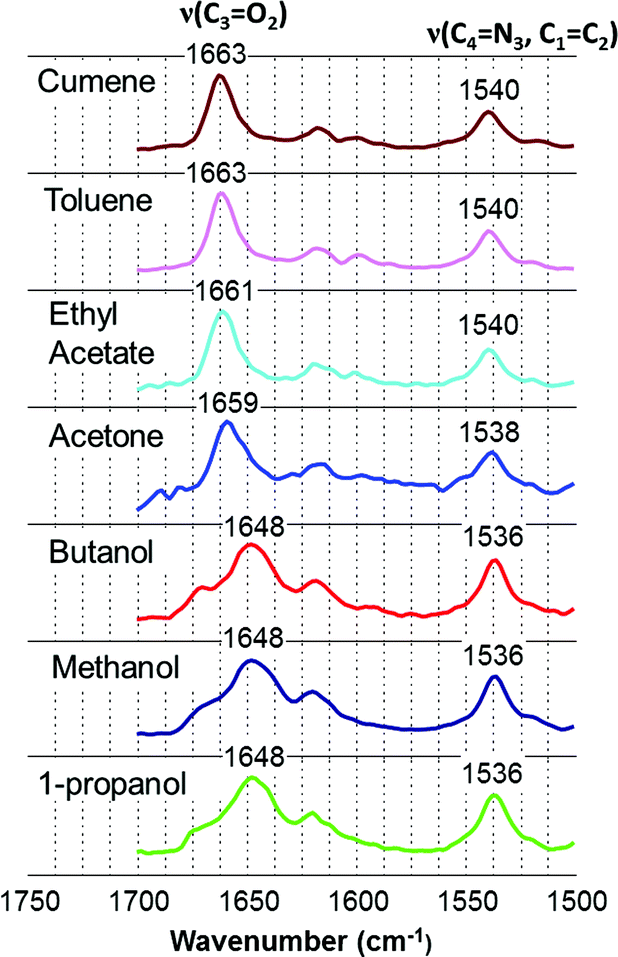 | ||
| Fig. 9 IR spectra of risperidone showing the peak shift observed in different solvents. The solvent spectrum is subtracted from the solution spectra. | ||
Similar to the carbonyl peak, shifts were also observed for the peak (1534 cm−1) corresponding to site 7. This peak at 1534 cm−1 (in solid material) moves to somewhat higher frequency in alcoholic solutions (1536 cm−1) followed by slightly higher frequency in acetone solution (1538 cm−1) and further higher frequency (1540 cm−1) in toluene, cumene and ethyl acetate solutions. The relative shift difference was smaller than that observed for the carbonyl peak which is expected considering a higher force constant for carbonyl stretching frequency over the frequency responsible for the peak at 1534 cm−1 in IR. The concentration effect on the spectra was also analysed for toluene, 1-propanol, methanol and acetone solutions: there was no clear difference in the spectra due to the different concentrations. Details of concentration and the wavenumber accuracy are shown in the ESI† file.
4. Discussion
The induction time experiments reveal that in order to reach the same induction time the required driving force increases in the order: cumene < toluene < acetone < ethyl acetate < methanol < propanol < butanol. We will refer to this order as increasing difficulty of nucleation, and point out that this observation is directly extracted from the experimental work without assumption of a particular theory. This work further shows that if the data are processed within the framework of the classical nucleation theory the obtained interfacial energies increase in a similar but not identical order. Overall, the difference in interfacial energy of risperidone depending on the solvent is rather limited, spanning from 1.58 to 2.25 mJ m−2 for the seven solvents investigated. In the alcohols the value is clearly higher than in the other four solvents, but within these two groups of solvents the differences are relatively small. With respect to the experimental uncertainties we do not see a big discrepancy between the driving force order and the interfacial energy order even though we do want to point out the difference with respect to ethyl acetate in particular. Furthermore, since the nucleation rate in the classical nucleation theory depends on the preexponential factor as well as the nucleation work, the order of nucleation according to the driving force scale doesn't necessarily have to correlate with the interfacial energy order.Risperidone is a relatively large molecule (410.49 g mol−1) that possesses various functional groups of different polarity, serving as sites for intermolecular interactions. Because of this DFT simulation of the complete first solvation shell10 becomes too demanding and we settled for simulating a number of one-to-one molecule solvent–solute pairs. In Fig. 10 the spectroscopic results are compared with the DFT-computed solvent–risperidone binding energies at the carbonyl group (Fig. 10a) site 5 and the pyrimidine ring (Fig. 10b) site 7. Obviously, there is a good correlation in the data from the two methods: with increasing binding energy the carbonyl peak and the pyrimidine peak both shift towards lower frequencies. The results of both methods suggest that the binding to the non-polar solvents cumene and toluene, is quite weak, is somewhat stronger to the aprotic solvents ethyl acetate and acetone, and clearly stronger than the alcohols. Since the carbonyl site shows a relatively larger shift in solution spectra, as compared to the (CN, CC) vibrations of ring 4, the carbonyl group appears a relatively more sensitive probe for investigating the solvent–solute interactions in solution.
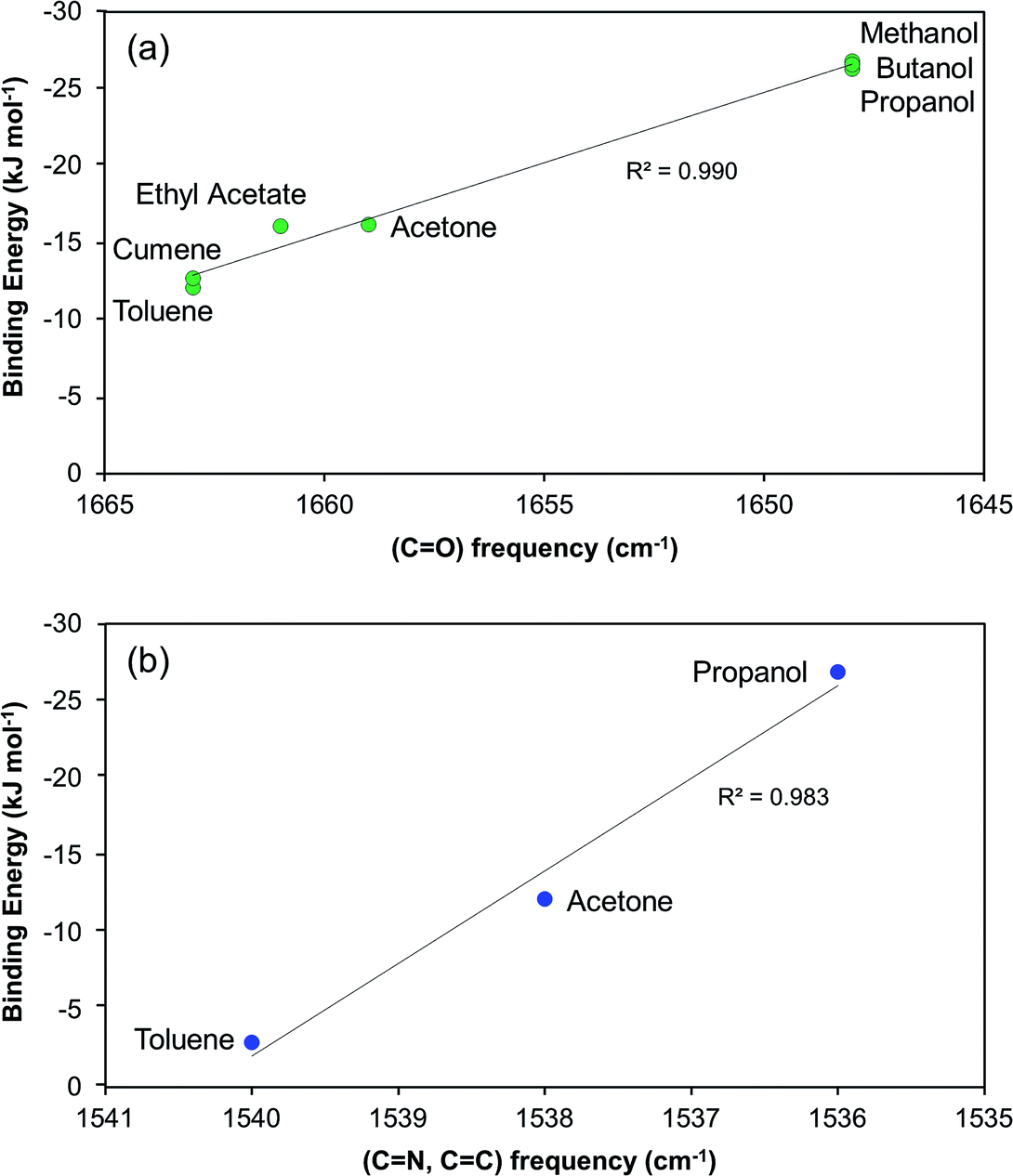 | ||
| Fig. 10 Cross-validation of solvent–risperidone interactions: DFT binding energy vs. FTIR vibrational frequency at (a) carbonyl v(CO) (site 5) for all the 7 solvents and (b) v(CN + CC) in ring 4 (site 7) for selected 3 solvents. | ||
In Fig. 11 we examine the correlation between the driving force required to reach a median induction time of 2000 s and the FTIR carbonyl peak frequency. The overall trend is that the required driving force increases with decreasing peak frequency suggesting that the crystal nucleation of risperidone becomes gradually more difficult the stronger the solvent binds to the carbonyl site. The correlation rather clearly distinguishes between the non-polar solvents, the aprotic solvents and especially the alcohols. It must be noted though that the correlation in Fig. 11 shows strength of solvent–solute interaction only at one site. However a similar correlation could be observed for the shift in the pyrimidine ring vibrations affecting site 7 as well. Unfortunately, in the solution IR spectroscopy only certain sites of interactions can be probed, and the resolution is somewhat limited for solvents bearing similar structural features such as the group of alcohols and the pair of toluene and cumene. This points out certain limitations of FTIR spectroscopy in such work.
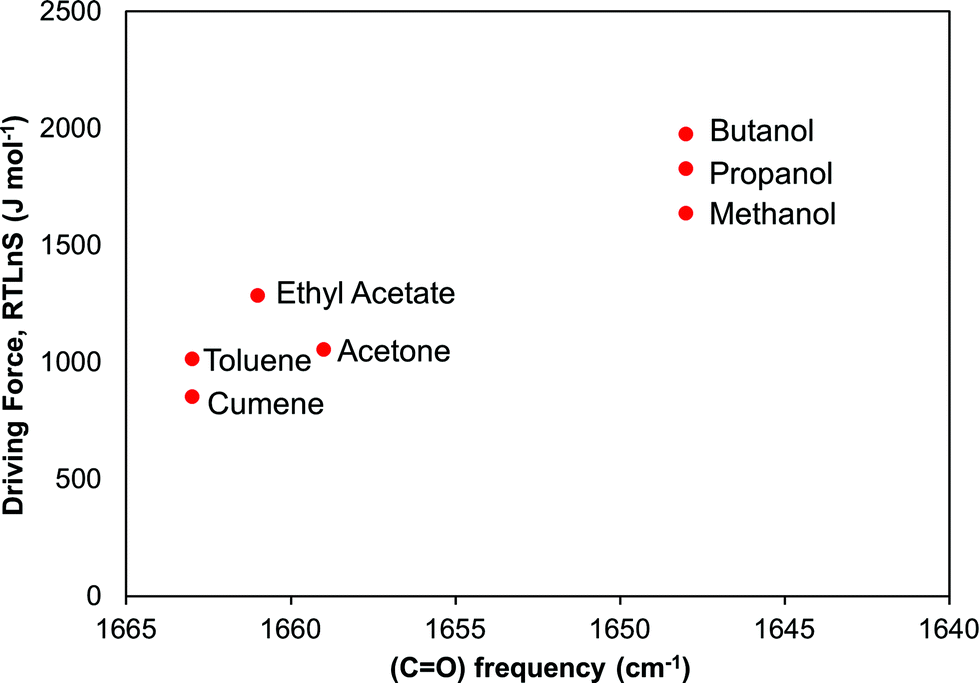 | ||
| Fig. 11 Relationship between driving force required to reach a median induction time of 2000 s and the FTIR vibrational frequency of the risperidone carbonyl group, site 5. | ||
Fig. 12a shows, as expected, that the nucleation driving force correlates nicely with the DFT computed (1:1) binding energy to the carbonyl group at site 5. At the sites where the DFT binding energies are the strongest, i.e. sites 5, 6, and 7, we also observe a correlation between binding energy and the ease of nucleation for the three considered solvents. It is noteworthy that, for five sites out of the eight considered, the binding energies follow the order of difficulty of nucleation. The three sites where there is no such a correlation are those where the solvent–solute interactions are relatively weak, as they feature either relatively non-polar C–H hydrogens (site 1 and 8) or a combined interaction of hydrogen and fluorine (site 2). For the three solvents where we have DFT-computed binding energies to all 8 sites, we have calculated a mean interaction energy, and the correlation to the nucleation driving force is shown in Fig. 12b. Based on these three solvents only the correlation is not overly impressive, since the nucleation driving force required in acetone for 2000 s induction time is almost the same as for toluene. It is noteworthy though that at 3000 s induction time (Table 2), there is a somewhat more clear difference in driving force between toluene and acetone.
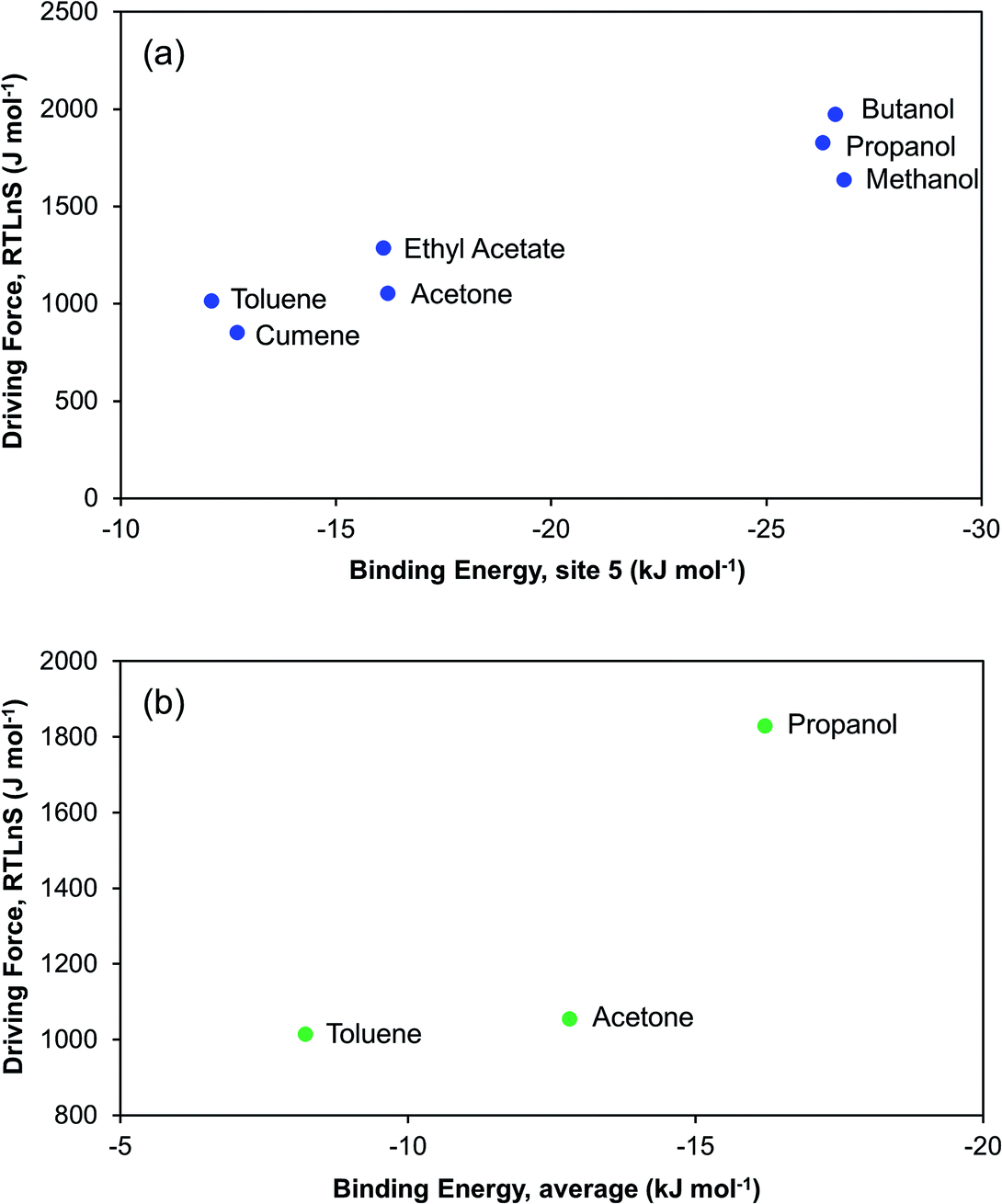 | ||
| Fig. 12 Relationship between driving force at median induction time of 2000 s and DFT binding energy: (a) for seven solvents at site 5, and (b) averaged over eight interaction sites considering three representative solvents. | ||
Since often the nucleation propensity is characterised by the interfacial energy only, for completeness we also show the corresponding correlations in Fig. 13. Fig. 13a shows the correlation between the interfacial energy and the driving force required to induce the same induction time. Overall, the difference between the alcohols and the other solvents is distinguished but the more detailed order differs, and in particular a discrepancy is observed in case of ethyl acetate, where the lowest interfacial energy of 1.58 mJ m−2 is calculated, despite the driving force being somewhat in the mid-range (cf.Fig. 4 and Table 2). The correlations of interfacial energy with carbonyl frequency (Fig. 13b), the DFT calculated binding energies at sites 5 and 7 (Fig. 13c), and the average binding energy (Fig. 13d), are similar to that shown in Fig. 13a. Overall, we observe that the strength of solvent–solute interaction affects the nucleation process, such that the stronger the solvent binds to the solute, the greater the difficulty of nucleation becomes. This very much follows the observations made for salicylic acid,13 further supporting the hypothesis that desolvation of the solvent molecules during the process of formation of crystal nuclei can be a key step in explaining the influence of the solvent on the nucleation process.
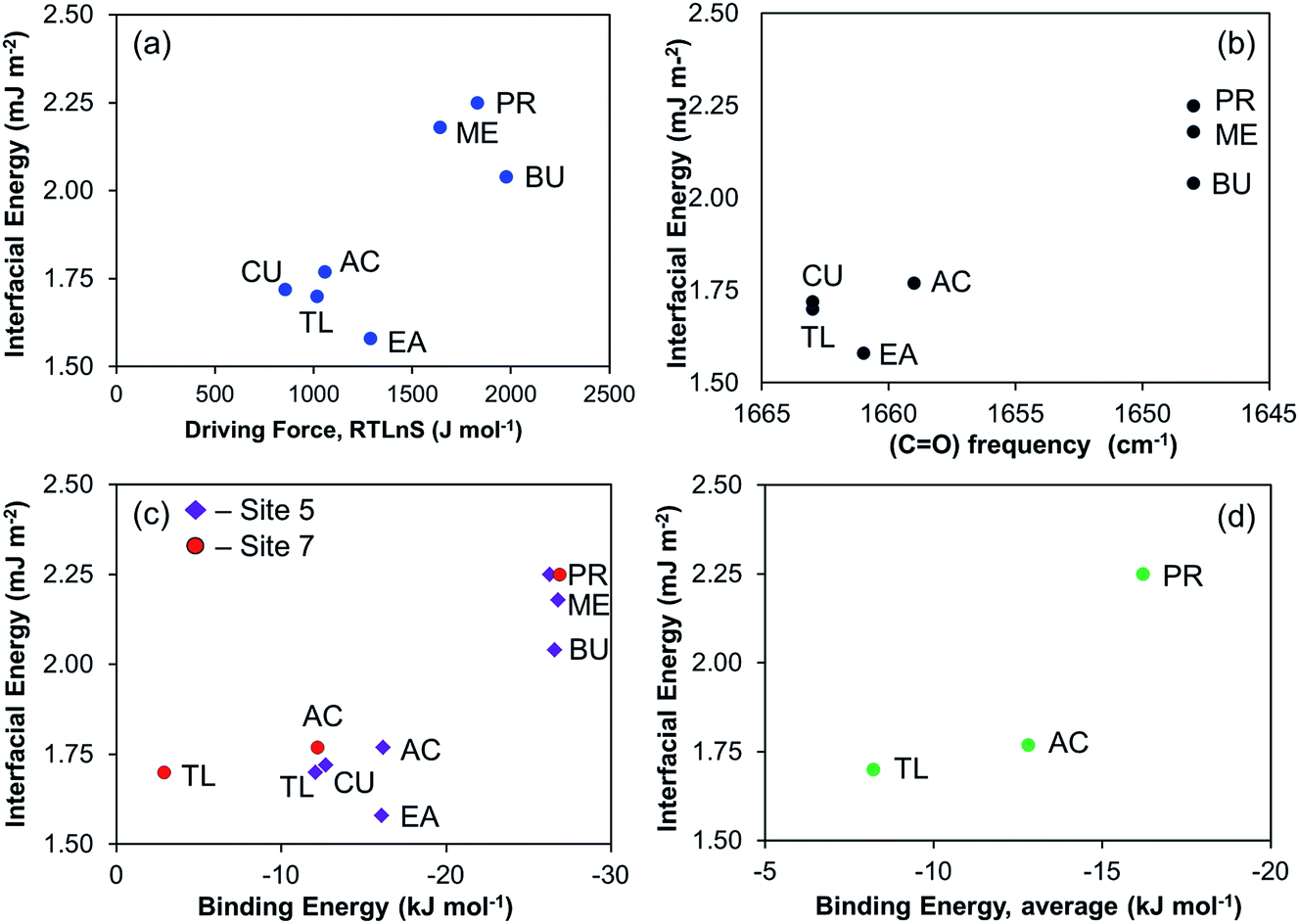 | ||
| Fig. 13 Relationship between interfacial energy and (a) driving force at median induction time of 2000 s, (b) FTIR vibrational frequency of risperidone carbonyl group, (c) DFT binding energy of solvent–solute at site 5 (purple diamonds) and site 7 (red dots), and (d) DFT binding energy of solvent–solute as averaged over eight interaction sites. Cumene (CU), toluene (TL), acetone (AC), ethyl acetate (EA), methanol (ME), propanol (PR), and butanol (BU). | ||
5. Conclusions
The nucleation of risperidone depends on the solvent. To reach the same induction time the required driving force increases in the order: cumene, toluene, acetone, ethyl acetate, methanol, propanol, and butanol. This order overall corresponds to the order of increasing interfacial energy as determined within the classical nucleation theory, but there are deviations in the detailed order. The frequency of the FTIR spectroscopy peaks corresponding to the carbonyl and pyrimidine group vibrations decreases in the polarity order of the three solvent groups. DFT (1:1) solvent–solute binding energies depend significantly on the binding site for all three solvents investigated. For the carbonyl site the binding energy increases essentially in the same order as the order of increasing driving force. A good correlation between spectroscopic data and DFT simulations provides validation for the relevance of these techniques in characterising solvent–solute interactions. The results of this work support the hypothesis that the influence of the solvent on the crystal nucleation is related to the desolvation of the risperidone molecules. The stronger the solvent binds to the risperidone molecule in solution, the more difficult the desolvation and the slower the nucleation becomes.
Acknowledgements
The support of the Science Foundation Ireland (10/IN.1/B3038) is gratefully acknowledged. J. Zeglinski acknowledges the Science Foundation Ireland (SFI) and Higher Education Authority funded Irish Centre for High End Computing (ICHEC) for access to computational facilities, and Å. C. Rasmuson the support of the Swedish Research Council (621-2010-5391).References
- R. A. Granberg, C. Ducreux, S. Gracin and Å. C. Rasmuson, Chem. Eng. Sci., 2001, 56, 2305–2313 CrossRef CAS.
- S. Teychene and B. Biscans, Cryst. Growth Des., 2008, 8, 1133–1139 Search PubMed.
- M. Svärd and Å. C. Rasmuson, Cryst. Growth Des., 2013, 13, 1140–1152 Search PubMed.
- R. J. Davey, G. Dent, R. K. Mughal and S. Parveen, Cryst. Growth Des., 2006, 6, 1788–1796 CAS.
- S. A. Kulkarni, E. S. McGarrity, H. Meekes and J. H. ter Horst, Chem. Commun., 2012, 48, 4983 RSC.
- C. A. Hunter, J. F. McCabe and A. Spitaleri, CrystEngComm, 2012, 14, 7115 RSC.
- R. J. Davey, N. Blagden, S. Righini, H. Alison, M. J. Quayle and S. Fuller, Cryst. Growth Des., 2001, 1, 59–65 CAS.
- C. S. Towler and L. S. Taylor, Cryst. Growth Des., 2007, 7, 633–638 CAS.
- R. J. Davey, S. L. Schroeder and J. H. ter Horst, Angew. Chem., Int. Ed. Engl., 2013, 52, 2166–2179 CrossRef CAS PubMed.
- A. Mattei, X. Mei, A. Miller and T. Li, Cryst. Growth Des., 2013, 13, 3303–3307 CAS.
- M. Lahav and L. Leiserowitz, Cryst. Growth Des., 2006, 6, 619–624 CAS.
- D. Khamar, J. Zeglinski, D. Mealey and Å. C. Rasmuson, J. Am. Chem. Soc., 2014, 136, 11664–11673 CrossRef CAS PubMed.
- S. Lohani, I. V. Nesmelova, R. Suryanarayanan and D. J. W. Grant, Cryst. Growth Des., 2011, 11, 2368–2378 CAS.
- W. Du, Q. Yin, J. Gong, Y. Bao, X. Zhang, X. Sun, S. Ding, C. Xie, M. Zhang and H. Hao, Cryst. Growth Des., 2014, 14, 4519–4525 CAS.
- R. A. Sullivan, R. J. Davey, G. Sadiq, G. Dent, K. R. Back, J. H. ter Horst, D. Toroz and R. B. Hammond, Cryst. Growth Des., 2014, 14, 2689–2696 CAS.
- L. Zhao, C. Zhu, J. Ji, J. Chen and H. H. Teng, Geochim. Cosmochim. Acta, 2013, 106, 192–202 CrossRef CAS PubMed.
- D. Mealey, D. M. Croker and Å. C. Rasmuson, CrystEngComm, 2015 10.1039/C4CE01428F.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al., Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2013, 24 Search PubMed.
- S. Portmann and H. P. Luthi, Chimia, 2000, 54, 766–770 CAS.
- S. Jiang and J. H. ter Horst, Cryst. Growth Des., 2011, 11, 256–261 CAS.
- H. Y. Yang and Å. C. Rasmuson, Cryst. Growth Des., 2013, 13, 4226–4238 CAS.
- A. Alparone, Spectrochim. Acta, Part A, 2011, 81, 631–639 CrossRef CAS PubMed.
- C. Reichardt and T. Welton, Solvents and Solvent Effect in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 425–490 Search PubMed.
- R. A. Nyquist, in Interpreting Infrared, Raman, and Nuclear Magnetic Resonance Spectra, ed. R. A. Nyquist, Academic Press, San Diego, 2001, pp. 165–203 Search PubMed.
- S. I. Nam, E. S. Min, Y. M. Jung and M. S. Lee, Bull. Korean Chem. Soc., 2001, 22, 989–993 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4fd00223g |
| This journal is © The Royal Society of Chemistry 2015 |