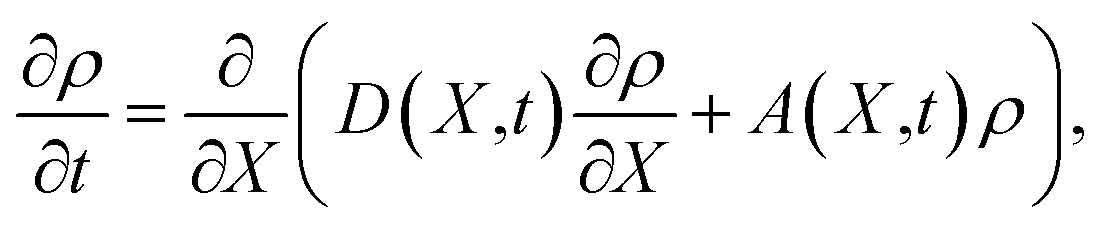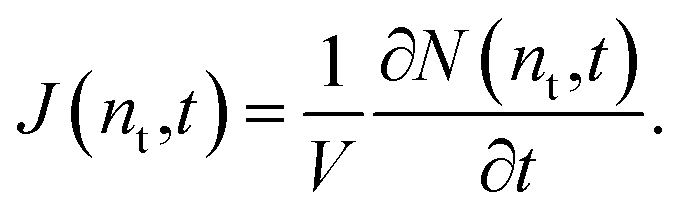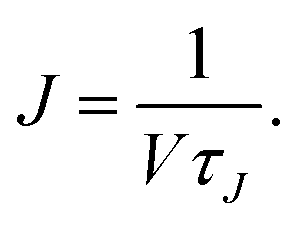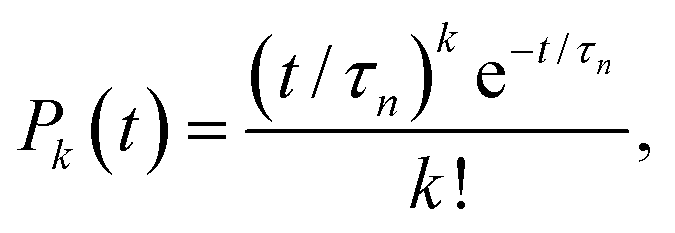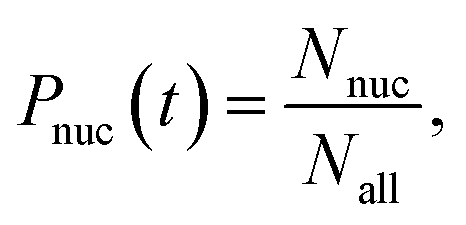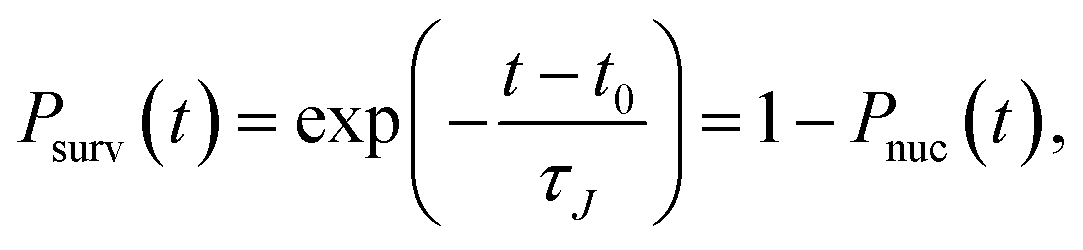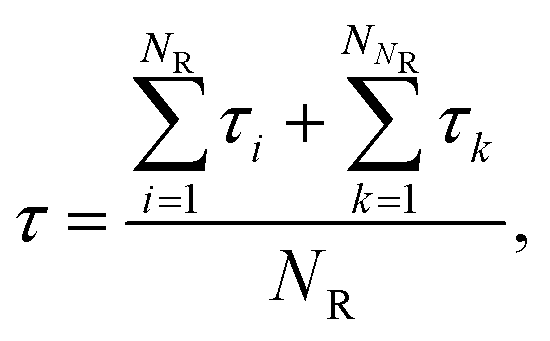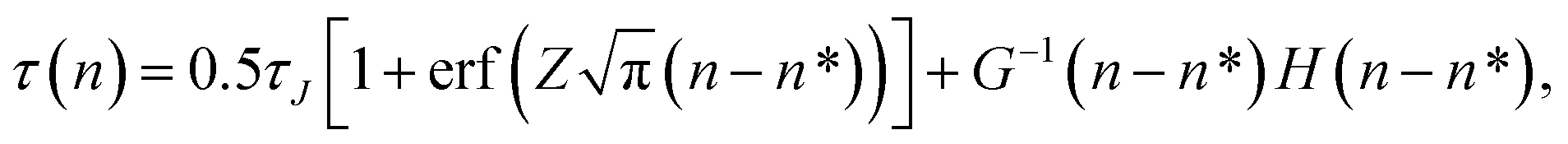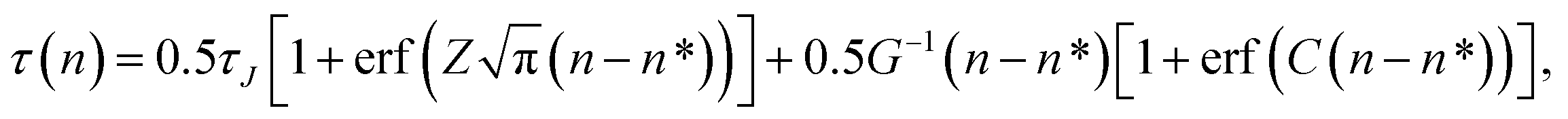DOI:
10.1039/C4FD00219A
(Paper)
Faraday Discuss., 2015,
179, 463-474
Nucleation rate analysis of methane hydrate from molecular dynamics simulations
Received
11th November 2014
, Accepted 6th January 2015
First published on 6th January 2015
Abstract
Clathrate hydrates are solid crystalline structures most commonly formed from solutions that have nucleated to form a mixed solid composed of water and gas. Understanding the mechanism of clathrate hydrate nucleation is essential to grasp the fundamental chemistry of these complex structures and their applications. Molecular dynamics (MD) simulation is an ideal method to study nucleation at the molecular level because the size of the critical nucleus and formation rate occur on the nano scale. Various analysis methods for nucleation have been developed through MD to analyze nucleation. In particular, the mean first-passage time (MFPT) and survival probability (SP) methods have proven to be effective in procuring the nucleation rate and critical nucleus size for monatomic systems. This study assesses the MFPT and SP methods, previously used for monatomic systems, when applied to analyzing clathrate hydrate nucleation. Because clathrate hydrate nucleation is relatively difficult to observe in MD simulations (due to its high free energy barrier), these methods have yet to be applied to clathrate hydrate systems. In this study, we have analyzed the nucleation rate and critical nucleus size of methane hydrate using MFPT and SP methods from data generated by MD simulations at 255 K and 50 MPa. MFPT was modified for clathrate hydrate from the original version by adding the maximum likelihood estimate and growth effect term. The nucleation rates calculated by MFPT and SP methods are within 5%, and the critical nucleus size estimated by the MFPT method was 50% higher, than values obtained through other more rigorous but computationally expensive estimates. These methods can also be extended to the analysis of other clathrate hydrates.
1 Introduction
Clathrate hydrates are ice-like structures in which guest molecules are trapped inside water cages connected by a hydrogen-bonded network.1 The formation of clathrate hydrates of natural gases, also called gas hydrates, is a serious problem in the flow assurance of oil/gas flow lines. Inhibiting and mitigating hydrate formation in flow lines are crucial for the safety and the reduction of operating costs of maintaining flow lines.1,2 Gas hydrates are also abundant in the seafloor and have attracted attention as a potential energy resource.3 Depressurization of these hydrate deposits is projected to be an efficient method to produce natural gas from the hydrates in the sea.4 To depressurize the hydrate reservoir, it is important to induce dissociation to release the gas from the hydrates, which will consequently generate thermodynamically favorable conditions for hydrate reformation. Therefore, understanding hydrate formation is required to develop efficient energy production strategies. At the most fundamental level, the mechanism of hydrate formation must be understood, since the incipient hydrate crystallization phenomenon can be controlled in flow lines during the production of oil/gas and for the gathering of gas from hydrate reservoirs. Moreover, a thorough understanding of the hydrate formation process will improve the efficiency of other challenging hydrate applications such as gas transport,2,3 hydrogen storage,5 and capture/sequestration of carbon dioxide.6
Nucleation is the first stage in hydrate formation, which is generally an activated process where small clusters of the new phase are formed from a super-saturated phase. A free energy barrier exists between the two phases, and small clusters which exceed the critical size located at the peak of the barrier become the nucleus of the new phase.7 Hydrate growth can be observed experimentally,8 however, hydrate nucleation cannot be observed, as it is a molecular level process that involves nuclei size and time scales that are on the nano scale. Molecular dynamics (MD) simulation has proven to be an invaluable tool to observe hydrate nucleation.9–11 There have been numerous studies on hydrate nucleation,12–14 growth,15–17 and stability18–20 by simulations. Previous studies have thoroughly examined the formation mechanisms of various hydrate structures.21–26 Order parameters that characterize the hydrate structures formed in the nucleation process have also been developed.27–31 Nonetheless, the analysis of hydrate nucleation is still in the early stages, and there are remarkably scarce reports in which the nucleation rate and the critical nucleus size are discussed.32,33
Nucleation is a stochastic process and commonly considered to be a “rare event” in molecular simulations.34 Direct molecular simulations typically require long calculation times (100s nanoseconds to microseconds) to observe hydrate nucleation. But more importantly, the critical nucleus size of hydrate formation, which is essential in calculating the nucleation rate, cannot be found beforehand. Although there are many difficulties in the simulations of hydrate nucleation, Walsh et al. succeeded in observing hydrate nucleation in microsecond simulations.12 Recently, Barnes et al. performed a number of similar simulations using high-performance computing and analyzed the nucleation rate and critical nucleus size.35
Nucleation rate analyses using MD simulations have actually been well documented for monatomic systems forming droplets or bubbles through homogeneous and heterogeneous nucleation.36–45 This study is initiated from the assumption that the various methods to analyze the nucleation rate and critical nucleus size may be applicable for other nucleation processes, and therefore we have used these methods to analyze hydrate nucleation.46–49 In this study, we analyzed the simulation results from Barnes et al.35 and calculated the nucleation rate and critical nucleus size of the methane hydrates by implementing methods originally applied to analyze vapor-to-liquid nucleation. This work introduces a methodology for analyzing methane hydrate nucleation but the findings are easily applicable to other complex clathrate hydrate structures.
2 Methods
2.1 Simulation details
Barnes et al. performed 200 MD simulations of methane hydrate nucleation at T = 255 K, P = 50 MPa.35 Simulation cells included 2944 water and 512 methane molecules with a cylindrical water/methane interface. The initial configuration was created by melting 64 unit cells of structure I hydrate at T = 550 K.12 Water and methane models were TIP4P/Ice50 and the unified atom model,51 respectively, and the Lorentz–Berthelot combining rules were used to calculate water–methane interactions. GROMACS 4.5 and 4.6 were used to perform the simulations, using the Verlet leapfrog algorithm for time integration.52 The isobaric–isothermal ensemble was applied, where the pressure was controlled by the Parrinello–Rahman barostat53 with a time constant of 4 ps, and the temperature was controlled by the Nosé–Hoover thermostat54,55 with a time constant of 2 ps. The SETTLE algorithm was used to constrain the bond lengths and angles of water molecules.56 A time step of 2 fs was used, with short-range interactions truncated at 1 nm, and the long-range electrostatic interactions were calculated by the particle mesh Ewald algorithm57,58 with a Fourier spacing of 0.12 nm. Over 90% of the simulations were performed for a minimum of 3 μs.
2.2 Analysis of nucleation
An activated process typified by nucleation is the formation of small embryos of a new phase from an existing metastable phase by overcoming a free energy barrier.7 The rate of which the critical-sized embryos are formed during the nucleation process is the nucleation rate. The nucleation process is considered to be a diffusion process over a barrier in an internal space, which can be described by the Fokker–Planck equation in terms of a variable X that is an internal coordinate or degree of freedom,| | 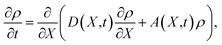 | (1) |
where ρ is the density in the internal space, and D and A are the diffusion and drift coefficients in this space, respectively. In the nucleation process, eqn (1) can be expressed as a function of a variable n, which is the number of molecules constituting a cluster, instead of X. In this case, ρ indicates the cluster size distribution, and eqn (1) can be expressed in the form of a continuity equation in the internal space,| | 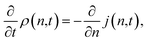 | (2) |
where ρ(n,t) is the number density of clusters containing n monomers at time t and j(n,t) is the formation rate of size n clusters in the system. At steady state, ∂ρ(n,t)/∂t = 0 and j(n,t) is constant, so the nucleation rate at steady state can be defined as J = j/V, where V is the volume of the system. The nucleation rate can also be described by means of integrating eqn (2) with respect to n, ∂N(nt,t)/∂t = j(nt,t)· N(nt,t) is the total number of clusters larger than a threshold size of nt. The nucleation rate obtained by this expression is| | 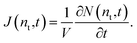 | (3) |
In this equation, the nucleation rate is described by a time derivative of the number of clusters greater than nt per unit volume. This rate should be constant at steady state and independent of nt, so nt must be greater than the critical size. In this work, the nucleation rate was evaluated by this definition based on eqn (3).
2.2.1 Definition of clusters.
The definition of cluster size is essential in the analysis of nucleation. Recent hydrate nucleation studies indicate that amorphous structures are initially formed and these may anneal and crystallize.59,60 Nucleation and growth of hydrates are usually characterized by order parameters that distinguish the phase of water molecules, structure of cages, and coordinates of guest molecules.27–31,61 However, it is difficult to identify clusters that have formed initially using these order parameters due to the complex molecular geometries. Most recently, Barnes et al. developed an order parameter, called the Mutually Coordinated Guest (MCG) order parameter, that identifies guest molecules separated by water clusters consisting of five or six-member rings.62 This order parameter (OP) can estimate the cluster size of methane hydrates sufficiently, so the MCG-1 OP from the MCG algorithm was used as the cluster size n in this work.
2.2.2 Mean first-passage time method (MFPT).
This method is suitable to analyze nucleation in MD simulations. The method makes the simplifying assumption that the free energy barrier is high, and only the height and curvature of the barrier control the nucleation kinetics, and that growth is much faster than nucleation.48,49 MFPT does not require large systems for the simulation, but rather demands numerous replications of small nucleating systems to analyze the statistics of the phenomenon. Full reaction coordinate analysis of methane hydrate nucleation requires enormous calculation time, so MFPT is advantageous in analyzing this event because it only uses the trajectories of direct MD simulations of nucleating systems.
The mean first-passage time in the case of nucleation is defined as the mean time τ(n) that the largest cluster in each system requires to reach or exceed a threshold size n for the first time. If the free energy barrier is high enough, MFPT as a function of the cluster size n can be described by a specific sigmoidal curve
| | 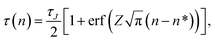 | (4) |
where
τJ is the nucleation time, erf(
x) is the error function, and
Z is the Zeldovich factor. A fitting of the simulation results to
eqn (4) directly yields the nucleation time
τJ, critical nucleus size
n*, and the Zeldovich factor. In the case of a high free energy barrier, the MFPT curve has a clear plateau at the end of the sigmoidal shape, which indicates the nucleation time
τJ. The critical nucleus size is considered to be the size at time
τJ/2 in the MFPT curve because the probability of the transition at the top of the barrier is 50%. The nucleation rate
J is calculated from the nucleation time
τJ as
| | 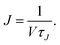 | (5) |
2.2.3 Survival probability.
The formation of a large enough post-critical cluster in the presence of a high free energy barrier is a random event, which follows the Poisson distribution| | 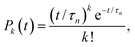 | (6) |
where Pk(t) is the probability that k clusters larger than n appear at time t, and τn is the average time when these clusters appear. In the case of k = 0, eqn (6) shows the survival probability (SP) P0(t),8,47,49i.e., the probability that there are no clusters larger than n after a time t in the system, to be
On the other hand, the nucleation probability Pnuc(t) is given by
| | 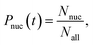 | (8) |
where
Nnuc is the number of systems in which nucleation is observed and
Nall is the number of all simulation systems. From
eqn (7) and
(8), the SP
Psurv(
t) becomes
| | 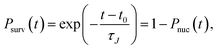 | (9) |
where
t0 is the fastest time a cluster takes to reach the threshold size
n among all simulations. If the free energy barrier is high, these probabilities do not depend on the threshold size of cluster
n. A fitting of the simulation results to
eqn (9) yields the nucleation time
τJ, and the nucleation rate can be obtained with
eqn (5).
3 Results and discussion
Nucleation and growth of methane hydrate was observed in 46 out of the 200 replications of the MD simulation trajectories. We analyze the nucleation rate from this set using the MFPT and SP, and the critical nucleus size is also calculated from MFPT.
3.1 Mean first-passage time
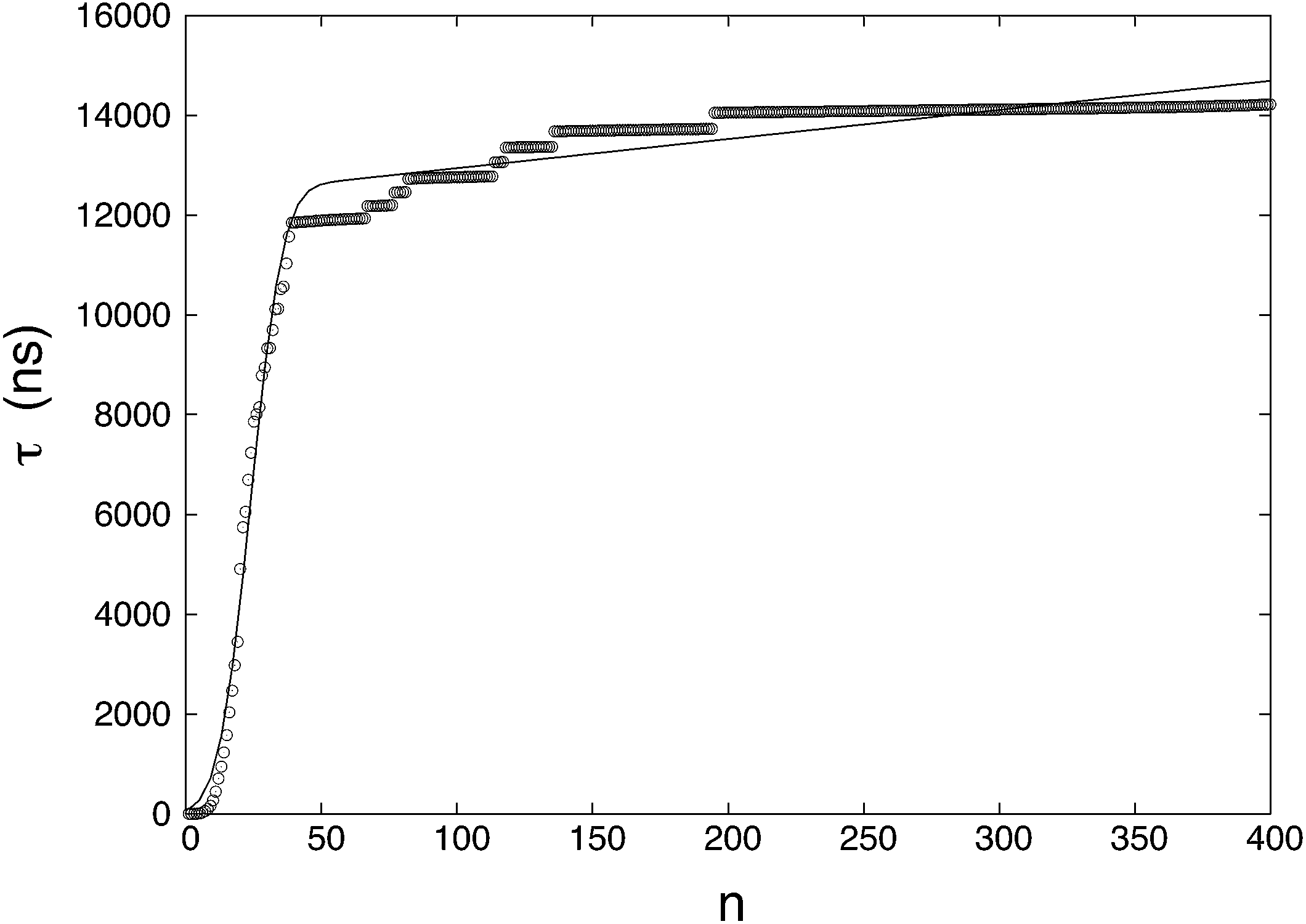
The MFPT of each cluster size n (up to n = 400, corresponding to near complete solidification of the system) was calculated and plotted in Fig. 1. The statistics for small n are better than those for large n. This is mainly due to the observation time for small clusters being relatively short (even though this “short” time consumed massive computational resources) compared to those needed to fully observe complete nucleation. The ratio of nucleated to non-nucleated systems most likely influences the calculation of MFPT, so τ is calculated by a supplemental equation, which is based on the maximum likelihood estimate,32,63| | 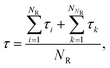 | (10) |
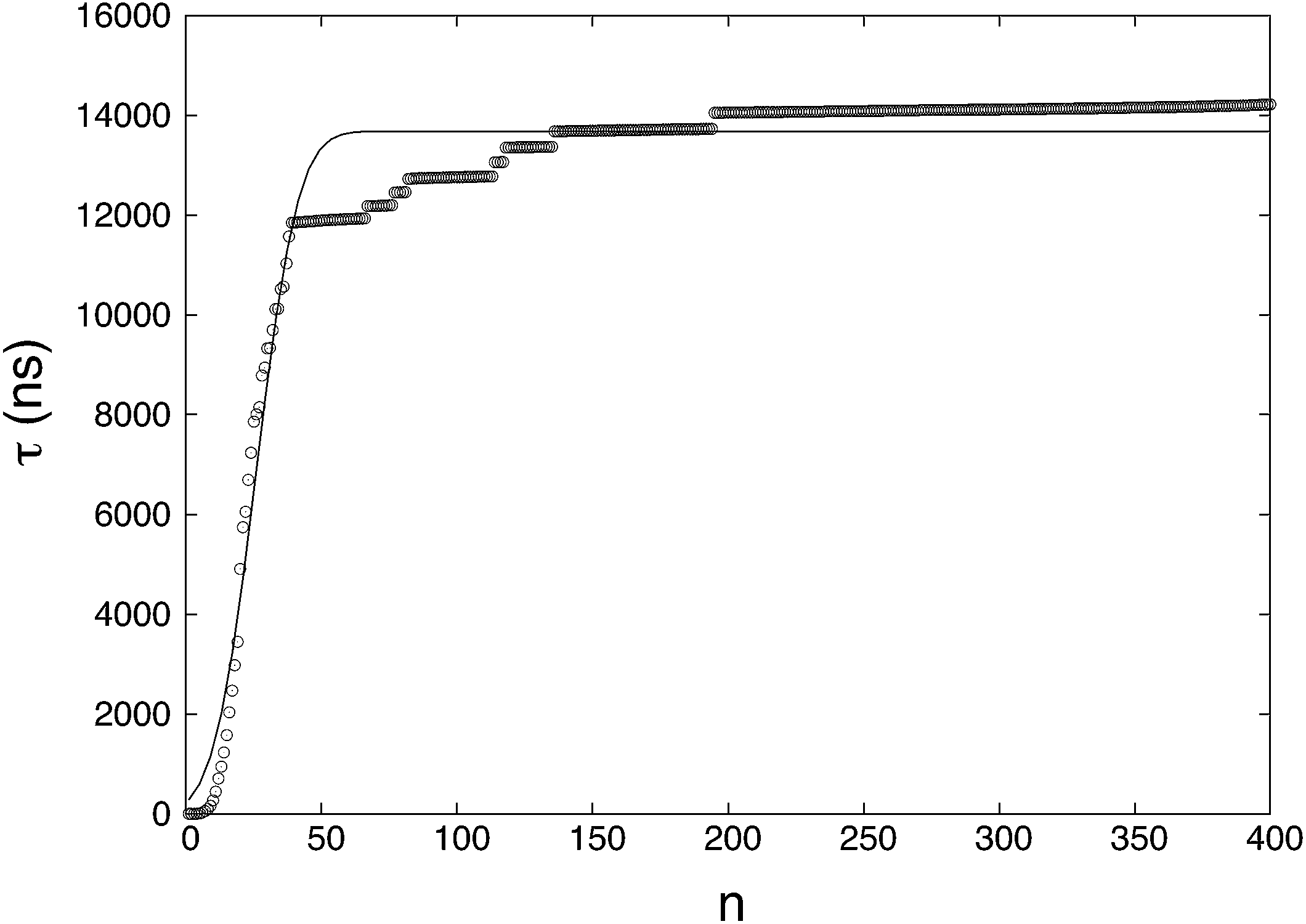
where NR (= 46) is the number of reacted (clusters reaching or exceeding a particular size n) trajectories and NNR (= 154) is the number of remaining trajectories. τi is the nucleation time, and τk is the total simulation time for non-nucleating trajectories. The nucleation time τJ and critical nucleus size n* are obtained by fitting the data to eqn (4). The nucleation rate J is calculated by plugging the obtained values into eqn (5) with V = 84.9 nm3 (estimated by considering the volume of the aqueous phase in a non-nucleating trajectory − volume of bulk methane phase subtracted from total volume). The nucleation rate from Fig. 1 is J = 8.61 × 1023 cm−3 s−1, and the critical nucleus size is n* = 25.9.
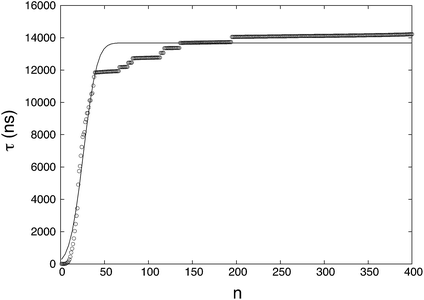 |
| | Fig. 1 The MFPT curve obtained from 200 nucleation trajectories. τ is the MFPT and n is the threshold size of a cluster. The symbols are mean first-passage times calculated from the results of the simulations and the solid line is a fitting of the MFPT data. | |
The fitting of the MFPT curve from eqn (4) is poor in the range of 40 ≤ n ≤ 120. Under the thermodynamic conditions considered, the free energy barrier is relatively high, so it is difficult to observe nucleation (only about a quarter of replications nucleated). In systems where nucleation solely occurs, the plateau is expected to appear right after the sigmoidal curve.48 Compared to nucleation, the time scale of growth is commonly much shorter, but in our case, there seems to be an overlap of the time scales in the nucleation and growth processes, which can affect the MFPT results. Therefore, a modification to the MFPT curve is applied as suggested by Yi et al. by adding an additional term to eqn (4) to account for finite growth rates of post-critical clusters, with64
| | 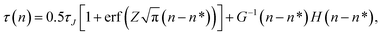 | (11) |
where
G is the growth rate and
H(
X) is the Heaviside function. The Heaviside function becomes effective when a cluster size exceeds the critical nucleus size,
i.e., when growth occurs after nucleation. The smooth approximation of the Heaviside function can also be presented by an error function, transforming
eqn (11) into
| | 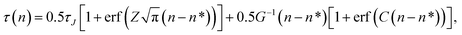 | (12) |
where
C is required to be a large positive number.
Fig. 2 shows the fitting given by
eqn (12). The plateau in
Fig. 1 becomes a positive slope that incorporates the growth contribution that is absent in
Fig. 1. The newly calculated nucleation rate is
J = 9.43 × 10
23 cm
−3 s
−1, and the critical nucleus size becomes
n* = 23.8. The variation in the nucleation rate is greater than that of the critical nucleus size without growth.
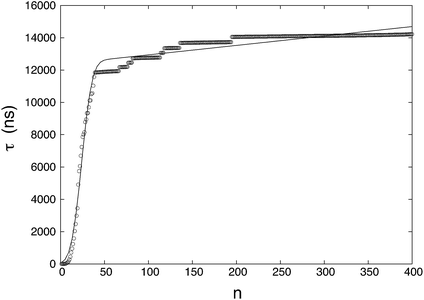 |
| | Fig. 2 The MFPT curve considering nucleation and growth simultaneously. The symbols are mean first-passage times calculated from the results of the simulations and the solid line is a fitting of MFPT data points. | |
To analyze how the number of replications influences the results when using the MFPT method, a sensitivity analysis is conducted. Subsets of replications are randomly taken from the total of 200 and the nucleation times are estimated by the maximum likelihood method. The MFPT curve is generated from the averages of the subsets. Table 1 shows the averaged results of MFPT for different numbers of subsets. The results show that the differences in both the nucleation rate and critical nucleus are within 20%, which is an insignificant variation for typical nucleation studies.46,48,49 Despite the variation, a greater number of replications are expected to produce more accurate results.
Table 1 The MFPT results calculated from the various number of simulations. Nall is the number of total simulation, J is the nucleation rate, and n* is the critical nucleus size
|
N
all
|
20 |
50 |
100 |
150 |
200 |
|
J (1023 cm−3 s−1) |
8.19 |
10.28 |
10.53 |
8.49 |
9.43 |
|
n* |
21.8 |
24.9 |
23.6 |
23.1 |
23.8 |
As shown in Table 1, from all 200 replications, the nucleation rate and critical nucleus size are estimated as 9.43 × 1023 cm−3 s−1 and n* = 23.8, respectively. Nucleation in the monatomic systems can be observed relatively quickly, so the MFPT curve has a clear plateau right after the sigmoidal shape.48,49 On the other hand, hydrate nucleation required a long calculation time due to a high free energy barrier, so nucleation may not be observed for many systems within the limited calculation time. Furthermore, hydrate formation is a complex process and growth just after the nucleation process tend to occur at the same time scale of nucleation. Therefore, MFPT for methane hydrate was modified from its original function using the maximum likelihood estimate by adding the growth term. The nucleation rate result is within 5% of that obtained by Barnes et al.,35 though the critical nucleus size is larger by 50%. The critical nucleus size obtained by Barnes et al. was estimated from a pb histogram test and this seems to have a higher variance.35,65–67 However, MFPT is a function of the nucleus size n only, so there are limitations in fully capturing the complexities of hydrate nucleation. The source of the discrepancy in the critical nucleus size also may be from the aforementioned comparable nucleation and growth time scales as well as the diffusion kinetics depending on the non-classical shape of the free energy curve.35 Although further studies are required to clarify the discrepancy in the nucleus size, the agreement in the nucleation rate confirms the effectiveness of the MFPT analysis method for a qualitative estimate without further free energy computations.
3.2 Survival probability
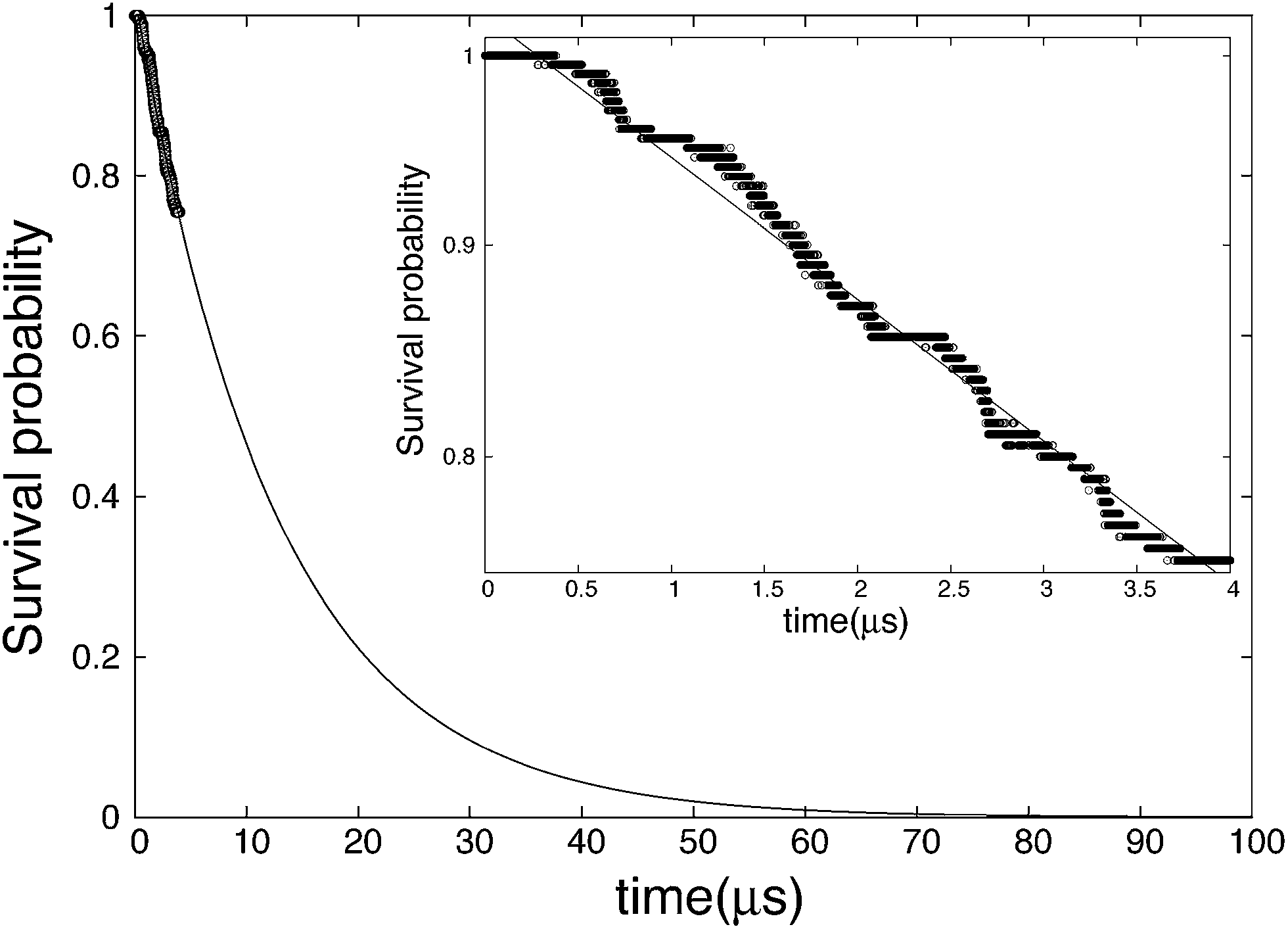
The survival probability is calculated by eqn (9) and the results are shown in Fig. 3. Unlike completely nucleated systems (nt = 400) that had 46 out of 200 occurrences, nt = 40 had 49, and the range of the survival probability is within 0.76 ≤ Psurv(t) ≤ 1.00. The systems that nucleated are found to grow and not dissociate. If the threshold size is increased to nt = 80, the number of nuclei converges to 46. In other words, three replications did not have a nucleus that reached 80 in size within the simulation time. The variation in the threshold size is analyzed and is found to have little effect on the results of SP. The fitting line in Fig. 3 illustrates SP at a longer time scale, and presents the asymptotic tendency at infinite time. Based on Fig. 3, one can see that if the simulations of this study are performed to around 70 μs, the SP will become close to zero, meaning nearly all systems will have likely nucleated. The inset in Fig. 3 portrays a single logarithmic transformation from the original SP plot, and the nucleation rate is calculated from the slope of this graph. The nucleation rate is J = 9.31 × 1023 cm−3 s−1, which is close to that obtained by MFPT.
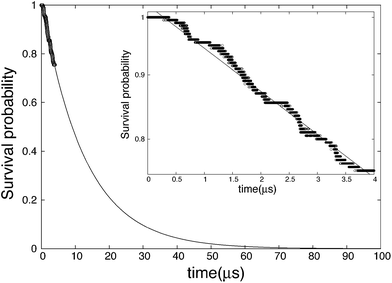 |
| | Fig. 3 Survival probability in the case of threshold nt = 40. Points are calculated from the results of the simulations and the solid line is the fitting of the data points. The inset is a single logarithmic graph in the range of SP calculated from the results of the simulations (0.76 ≤ Psurv(t) ≤ 1.00). | |
SP is commonly independent of the threshold cluster size in the case that the free energy barrier is high. We verified this by changing the threshold cluster size, and Table 2 contains the results. The difference among the values is around 5%, which is remarkable considering the deviation in typical nucleation studies. The nucleation rate will slightly decrease with an increasing threshold size because the number of nucleated systems decreases. The rate at nt = 20, however, was the smallest. The reason for this is from the fact that 20 is around the critical nucleus size obtained by Barnes et al. and MFPT, hence the inconsistency.49 Therefore, when using the SP method, the threshold size should be set by an adequately larger number than the critical nucleus size (nt ≥ 40 was sufficient in this study). On the other hand, if the threshold size is too large, the analysis range may spill over to the growth stage rather than staying purely in the nucleation stage, which is the primary target.
Table 2 The SP of the various threshold cluster sizes. nt is the threshold size, Nnuc is the number of nucleated systems, and J is the nucleation rate
|
n
t
|
10 |
20 |
40 |
60 |
80 |
100 |
|
N
nuc
|
54 |
49 |
49 |
47 |
46 |
46 |
|
J (1023 cm−3 s−1) |
9.52 |
8.98 |
9.31 |
9.25 |
9.10 |
9.10 |
The nucleation rate calculated by Walsh et al. is from the maximum likelihood estimate.31 They performed the six replications at T = 250 K, P = 50 MPa and the induction times were verified by analyzing the evolution of the global F461 order parameter as well as the appearance of cages larger than seven through the MD trajectories. The nucleation rate from Walsh et al. is around Jsim = 5.00 × 1024 cm−3 s−1. Their results are one order of magnitude greater than our results from MFPT and SP. This difference may be attributed from the small number of samples and the definition of induction time. On the other hand, the nucleation rate calculated by Barnes et al. is from the maximum likelihood estimate based on prior knowledge of the critical nucleus size.32,35 The nucleation rate from Barnes et al. is Jsim = 9.07 × 1023 cm−3 s−1, which is very close to our results from MFPT and SP. The biggest advantage of this study compared to previous studies is that the MFPT and SP methods can generate the nucleation rate directly from the MD trajectories.
4 Conclusion
The nucleation of methane hydrate is a phenomenon that is complicated to analyze at the molecular level due to a high free energy barrier. Recently, this process has been observed by molecular dynamics simulations using high performance computing. In this study, we analyzed the nucleation rate and critical nucleus size of methane hydrate using MFPT and SP, and assessed the applicability of these methods for methane hydrate nucleation analysis. In this study, MFPT was modified from its original function using the maximum likelihood estimate and adding the growth term. The nucleation rates obtained by MFPT and SP are in good agreement (within 20%) and these results are also close to the rate Barnes et al. estimated using direct calculations based on the maximum likelihood estimate. MFPT and SP are convenient methods to calculate the methane hydrate nucleation rate since they only require simulation trajectories. The critical nucleus size was also calculated by MFPT, which was found to be larger than that of Barnes et al. based on the pb histogram test. This difference is likely to come from the non-classical shape of the free energy surface and the methane hydrate system having comparable nucleation and growth time scales, which will influence the critical nucleus size generated from MFPT. However, a modification for growth for MFPT slightly improves the fit of the simple fitting function. Though the MFPT and SP methods are only applied for the estimation of the nucleation rate and critical nucleus size of methane hydrate, the methods can easily be extended to the analysis of other clathrate hydrates.
Acknowledgements
This work was supported (in part) by MEXT Grant-in-Aid for the ”Program for Leading Graduate Schools”. This project was partially funded by the U.S. National Science Foundation (CHE-1125235). B.C.K. and G.T.B. thank the NREL Directors Fellowship Program for Funding. High-performance computing resources were provided by the National Renewable Energy Laboratory, Golden Energy Computing Organization (Colorado School of Mines), and Sandia National Laboratories.
References
-
E. D. Sloan Jr and C. Koh, Clathrate Hydrates of Natural Gases, CRC press, Boca Raton, 2007 Search PubMed.
- E. D. Sloan, Nature, 2003, 426, 353–363 CrossRef CAS PubMed.
- R. Boswell, Science, 2009, 325, 957–958 CrossRef CAS PubMed.
- M. R. Walsh, S. H. Hancock, S. J. Wilson, S. L. Patil, G. J. Moridis, R. Boswell, T. S. Collett, C. A. Koh and E. D. Sloan, Energy Econ., 2009, 31, 815–823 CrossRef PubMed.
- L. J. Florusse, C. J. Peters, J. Schoonman, K. C. Hester, C. A. Koh, S. F. Dec, K. N. Marsh and E. D. Sloan, Science, 2004, 306, 469–471 CrossRef CAS PubMed.
- Y. Park, D.-Y. Kim, J.-W. Lee, D.-G. Huh, K.-P. Park, J. Lee and H. Lee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12690–12694 CrossRef CAS PubMed.
- D. Reguera, J. Rubı and A. Pérez-Madrid, Phys. A, 1998, 259, 10–23 CrossRef CAS.
- R. Ohmura, M. Ogawa, K. Yasuoka and Y. H. Mori, J. Phys. Chem. B, 2003, 107, 5289–5293 CrossRef CAS.
- C. P. Ribeiro Jr and P. L. Lage, Chem. Eng. Sci., 2008, 63, 2007–2034 CrossRef PubMed.
- J. A. Ripmeester and S. Alavi, ChemPhysChem, 2010, 11, 978–980 CrossRef CAS PubMed.
- B. C. Barnes and A. K. Sum, Curr. Opin. Chem. Eng., 2013, 2, 1–7 CrossRef PubMed.
- M. R. Walsh, C. A. Koh, E. D. Sloan, A. K. Sum and D. T. Wu, Science, 2009, 326, 1095–1098 CrossRef CAS PubMed.
- S. Sarupria and P. G. Debenedetti, J. Phys. Chem. Lett., 2012, 3, 2942–2947 CrossRef CAS.
- N. J. English, M. Lauricella and S. Meloni, J. Chem. Phys., 2014, 140, 204714 CrossRef PubMed.
- S. Liang and P. G. Kusalik, Chem. Phys. Lett., 2010, 494, 123–133 CrossRef CAS PubMed.
- D. Yuhara, M. Hiratsuka, D. Takaiwa and K. Yasuoka, Mol. Simul., 2014, 1–5 CrossRef PubMed.
- D. Bai, B. Liu, G. Chen, X. Zhang and W. Wang, AIChE J., 2013, 59, 2621–2629 CrossRef CAS PubMed.
- N. J. English and G. M. Phelan, J. Chem. Phys., 2009, 131, 074704 CrossRef PubMed.
- S. A. Bagherzadeh, P. Englezos, S. Alavi and J. A. Ripmeester, J. Phys. Chem. B, 2012, 116, 3188–3197 CrossRef CAS PubMed.
- M. Conde and C. Vega, J. Chem. Phys., 2013, 138, 056101 CrossRef CAS PubMed.
- M. Matsumoto, J. Phys. Chem. Lett., 2010, 1, 1552–1556 CrossRef CAS.
- L. C. Jacobson and V. Molinero, J. Am. Chem. Soc., 2011, 133, 6458–6463 CrossRef CAS PubMed.
- A. H. Nguyen, L. C. Jacobson and V. Molinero, J. Phys. Chem. C, 2012, 116, 19828–19838 CAS.
- F. Jimenez-Angeles and A. Firoozabadi, J. Phys. Chem. C, 2014, 118, 11310–11318 CAS.
- F. Jimenez-Angeles and A. Firoozabadi, J. Phys. Chem. C, 2014, 118, 26041–26048 CAS.
- G. S. Smirnov and V. V. Stegailov, J. Chem. Phys., 2012, 136, 044523 CrossRef PubMed.
- R. W. Hawtin, D. Quigley and P. M. Rodger, Phys. Chem. Chem. Phys., 2008, 10, 4853–4864 RSC.
- M. R. Walsh, J. D. Rainey, P. G. Lafond, D.-H. Park, G. T. Beckham, M. D. Jones, K.-H. Lee, C. A. Koh, E. D. Sloan and D. T. Wu,
et al.
, Phys. Chem. Chem. Phys., 2011, 13, 19951–19959 RSC.
- L. C. Jacobson, W. Hujo and V. Molinero, J. Phys. Chem. B, 2009, 113, 10298–10307 CrossRef CAS PubMed.
- M. Matsumoto, A. Baba and I. Ohmine, J. Chem. Phys., 2007, 127, 134504 CrossRef CAS PubMed.
- S. N. Chakraborty, E. M. Grzelak, B. C. Barnes, D. T. Wu and A. K. Sum, J. Phys. Chem. C, 2012, 116, 20040–20046 CAS.
- M. R. Walsh, G. T. Beckham, C. A. Koh, E. D. Sloan, D. T. Wu and A. K. Sum, J. Phys. Chem. C, 2011, 115, 21241–21248 CAS.
- B. C. Knott, V. Molinero, M. F. Doherty and B. Peters, J. Am. Chem. Soc., 2012, 134, 19544–19547 CrossRef CAS PubMed.
-
D. Frenkel and B. Smit, Understanding Molecular Simulation, 2nd edn, Academic, San Diego, 2002 Search PubMed.
- B. C. Barnes, B. C. Knott, G. T. Beckham, D. T. Wu and A. K. Sum, J. Phys. Chem. B, 2014, 118, 13236–13243 CrossRef CAS PubMed.
- K. Yasuoka and M. Matsumoto, J. Chem. Phys., 1998, 109, 8463–8470 CrossRef CAS PubMed.
- T. Kinjo, K. Ohguchi, K. Yasuoka and M. Matsumoto, Comput. Mater. Sci., 1999, 14, 138–141 CrossRef CAS.
- P. R. Ten Wolde and D. Frenkel, J. Chem. Phys., 1998, 109, 9901–9918 CrossRef CAS PubMed.
- K. Yasuoka and X. C. Zeng, J. Chem. Phys., 2007, 126, 124320 CrossRef PubMed.
- H. Matsubara, T. Koishi, T. Ebisuzaki and K. Yasuoka, J. Chem. Phys., 2007, 127, 214507 CrossRef PubMed.
- B. R. Novak, E. J. Maginn and M. J. McCready, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 085413 CrossRef.
- J. Wedekind, D. Reguera and R. Strey, J. Chem. Phys., 2006, 125, 214505 CrossRef PubMed.
- J. Wedekind and D. Reguera, J. Phys. Chem. B, 2008, 112, 11060–11063 CrossRef CAS PubMed.
- D. Suh and K. Yasuoka, J. Phys. Chem. B, 2011, 115, 10631–10645 CrossRef CAS PubMed.
- D. Suh and K. Yasuoka, J. Phys. Chem. B, 2012, 116, 14637–14649 CrossRef CAS PubMed.
- K. Yasuoka and M. Matsumoto, J. Chem. Phys., 1998, 109, 8451 CrossRef CAS PubMed.
- M. Sekine, K. Yasuoka, T. Kinjo and M. Matsumoto, Fluid Dynam. Res., 2008, 40, 597–605 CrossRef PubMed.
- J. Wedekind, R. Strey and D. Reguera, J. Chem. Phys., 2007, 126, 134103 CrossRef PubMed.
- G. Chkonia, J. Wölk, R. Strey, J. Wedekind and D. Reguera, J. Chem. Phys., 2009, 130, 064505 CrossRef PubMed.
- J. Abascal, E. Sanz, R. G. Fernández and C. Vega, J. Chem. Phys., 2005, 122, 234511 CrossRef CAS PubMed.
- W. L. Jorgensen, J. D. Madura and C. J. Swenson, J. Am. Chem. Soc., 1984, 106, 6638–6646 CrossRef CAS.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511 CrossRef PubMed.
- W. Hoover, Phys. Rev. A, 1985, 31, 1695 CrossRef.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS PubMed.
- U. Essmann, L. Perera, M. Berkowitz, T. Darden, H. Lee and L. Pedersen, J. Chem. Phys., 1995, 103, 8577 CrossRef CAS PubMed.
- L. C. Jacobson, W. Hujo and V. Molinero, J. Phys. Chem. B, 2010, 114, 13796–13807 CrossRef CAS PubMed.
- L. C. Jacobson, W. Hujo and V. Molinero, J. Am. Chem. Soc., 2010, 132, 11806–11811 CrossRef CAS PubMed.
- C. Moon, R. Hawtin and P. M. Rodger, Faraday Discuss., 2007, 136, 367–382 RSC.
- B. C. Barnes, G. T. Beckham, D. T. Wu and A. K. Sum, J. Chem. Phys., 2014, 140, 164506 CrossRef PubMed.
-
D. D. Dunlop and A. C. Tamhane, Statistics and Data Analysis: From Elementary to Intermediate, Prentice Hall, 2000 Search PubMed.
- P. Yi, C. R. Locker and G. C. Rutledge, Macromolecules, 2013, 46, 4723–4733 CrossRef CAS.
- R. Du, V. S. Pande, A. Y. Grosberg, T. Tanaka and E. S. Shakhnovich, J. Chem. Phys., 1998, 108, 334–350 CrossRef CAS PubMed.
- P. L. Geissler, C. Dellago and D. Chandler, J. Phys. Chem. B, 1999, 103, 3706–3710 CrossRef CAS.
- P. G. Bolhuis, C. Dellago and D. Chandler, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5877–5882 CrossRef CAS PubMed.
Footnote |
| † Present address: Weapons and Materials Research Directorate, U.S. Army Research Laboratory, Aberdeen Proving Ground, Maryland, USA. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a