 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
X-ray diffraction in temporally and spatially resolved biomolecular science†
John R.
Helliwell
*a,
Alice
Brink
ab,
Surasak
Kaenket
a,
Victoria Laurina
Starkey
a and
Simon W. M.
Tanley
a
aSchool of Chemistry, University of Manchester M13 9PL, UK. E-mail: john.helliwell@manchester.ac.uk; Tel: +44 (0)1612754970
bDepartment of Chemistry, University of the Free State, Bloemfontein 9300, South Africa. E-mail: alice.brink@gmail.com
First published on 21st January 2015
Abstract
Time-resolved Laue protein crystallography at the European Synchrotron Radiation Facility (ESRF) opened up the field of sub-nanosecond protein crystal structure analyses. There are a limited number of such time-resolved studies in the literature. Why is this? The X-ray laser now gives us femtosecond (fs) duration pulses, typically 10 fs up to ∼50 fs. Their use is attractive for the fastest time-resolved protein crystallography studies. It has been proposed that single molecules could even be studied with the advantage of being able to measure X-ray diffraction from a ‘crystal lattice free’ single molecule, with or without temporal resolved structural changes. This is altogether very challenging R&D. So as to assist this effort we have undertaken studies of metal clusters that bind to proteins, both ‘fresh’ and after repeated X-ray irradiation to assess their X-ray-photo-dynamics, namely Ta6Br12, K2PtI6 and K2PtBr6 bound to a test protein, hen egg white lysozyme. These metal complexes have the major advantage of being very recognisable shapes (pseudo spherical or octahedral) and thereby offer a start to (probably very difficult) single molecule electron density map interpretations, both static and dynamic. A further approach is to investigate the X-ray laser beam diffraction strength of a well scattering nano-cluster; an example from nature being the iron containing ferritin. Electron crystallography and single particle electron microscopy imaging offers alternatives to X-ray structural studies; our structural studies of crustacyanin, a 320 kDa protein carotenoid complex, can be extended either by electron based techniques or with the X-ray laser representing a fascinating range of options. General outlook remarks concerning X-ray, electron and neutron macromolecular crystallography as well as ‘NMR crystallography’ conclude the article.
1 Introduction
The advent of intense synchrotron X-ray sources has allowed time-resolved diffraction in general, and macromolecular crystallography in particular, to have wide applicability to study time dependent phenomena at the molecular level; as spearhead time-resolved Laue protein crystallography at the ESRF (http://www.esrf.eu/UsersAndScience/Experiments/SoftMatter/ID09B) opened up the field of sub-nanosecond crystal structure analyses.1 Biomolecular science ‘fast time-resolved’ examples include carbon monoxy myoglobin and phospho yellow protein (PYP).2 More complex cases include following the enzyme reaction of hydroxymethylbilane synthase in the crystal which is slower by nature.3 There are a limited number of such time-resolved studies in the literature. Why is this? Firstly, crystal lattice interactions can block the necessary structural changes for a given biochemical reaction to proceed. Secondly, crystal size determines a sample's scattering strength and thereby the required exposure time, clearly increasing as a sample gets smaller. The latter obviously can be at odds with the intrinsic time resolution required to monitor a given molecular structural change; different measuring protocols try and ease past this challenge such as the Hadamard measuring sequence4 or the simpler approach of crystal to crystal averaging at equivalent time-slices.3 Meanwhile the X-ray laser now gives us femtosecond (fs) duration pulses, typically 10 fs up to ∼50 fs.5 Their use is attractive for the fastest time-resolved protein crystallography studies. It has been proposed that single molecules could even be studied,6 which would free us of the crystal lattice restrictions referred to above. Again though, the sample scattering strength along with the current X-ray laser maximum photons per pulse achievable from the source currently restrict the samples to crystalline arrays albeit micron to submicron (‘nano’) i.e. small crystal sizes.7 In considering these challenges and the clear advantage of being able to measure X-ray diffraction from a single molecule, with or without temporal resolved structural changes, we have studied in detail a class of metal complexes, the platinum hexahalides, as well as the tantalum bromide cluster when bound to hen egg-white lysozyme (HEWL) as a test protein crystal including their ‘X-ray-photo-dynamics.8,9 These metal clusters have the major advantage of being very recognisable shapes (octahedral and pseudo spherical). So, where we can expect difficult to interpret electron density maps, and their time-resolved changes in biochemical reactions, these ‘marker well-shaped complexes’ will offer a start to electron density map interpretations.9 A further approach is to investigate the X-ray laser beam diffraction strength of a well scattering nano-cluster; an example from nature being the iron containing ferritin (which has 2000 iron atoms in ferrihydrite form within a protein multi-subunit shell).10 Another example would be, in a bionanotechnology approach, an encapsulated metal core within a virus protein shell.2 Understanding the X-ray-photo-dynamics properties of the platinum hexahalides and of the tantalum bromide cluster (Ta6Br12) using X-ray crystallography
We have studied in detail a class of metal complexes, the platinum hexahalides, as well as the tantalum bromide cluster when bound to hen egg-white lysozyme (HEWL) as a test protein crystal.8,9 Our interest first arose from the use of protein powder X-ray diffraction to exploit the wavelength dependent dispersive differences for phasing of extracted powder diffraction Bragg reflection amplitudes. The larger delta f ‘ signal arising from hexabromide was attractive.8 The bromine K edge at 0.92 Å X-ray wavelength was very conveniently placed for two wavelength measurements involving ‘on edge’ and a remote X-ray wavelength. For most electron dense clusters of this type however, platinum hexaiodide is more attractive. In ref. 9 we reported the binding behaviour to histidine in HEWL as a test protein. In this paper we report the stability of the platinum hexahalides to prolonged X-ray irradiation. The platinum hexabromide and its photochemical response to UV light has been studied11,12 but not the platinum hexaiodide to our knowledge. These metal clusters have the distinct advantage of being particularly recognizable shapes (octahedral and pseudo spherical). So, where we can expect difficult to interpret electron density maps, and their time-resolved changes in biochemical reactions, these ‘marker well-shaped complexes’ will offer a start to electron density map interpretations.9 The break-up of platinum hexabromide, even when cryocooled, upon prolonged X-ray irradiation, akin to the UV study,11 was reported previously.13 Here we report the HEWL with bound PtBr6 at room temperature also upon prolonged X-ray irradiation.2.1 PtBr6, PtI6 and Ta6Br12 heavy atom clusters bound to a protein: their X-ray-photo-dynamics
In the case of ‘HEWL Ta6Br12 PtBr6’ the protein was co-crystallised with 2 mM Ta6Br12, forming beautiful green coloured crystals (Fig. 1a), of the usual tetragonal HEWL crystal morphology. After being soaked in PtBr6, the crystals turned from green to the orange colour of the PtBr6 (Fig. 1b). Fig. 1c shows the distinctive colour of the ‘HEWL soaked in K2PtI6’ crystals.
 | ||
| Fig. 1 Lysozyme crystals cocrystallized with Ta6Br12 (a. top) and soaked with K2PtBr6 (b. middle); the orange of PtBr6 beats the green of Ta6Br12 and finally (c. bottom) HEWL crystals soaked in K2PtI6. | ||
The crystal structures were solved using PHASER_MR16 and then rigid body and restrained refinement with CCP4i REFMAC5,17 using the previously reported lysozyme structure 2W1Y as the molecular search model.18 The use of PHASER_MR was probably not required as 2W1Y is relatively isomorphous with these crystals. Model building, adjustment and refinement were carried out respectively using the COOT molecular graphics programme19 and REFMAC5 in CCP4i. The crystallographic and molecular model refinement parameters are summarized in the ESI† footnotes. All figures were produced using COOT.19
The experimental conditions for preparing the ‘HEWL PtBr6 room temperature’ crystal sample were basically the same as described above namely a native hen egg white lysozyme protein crystal was soaked in 10 mM K2PtBr6 for 24 hours. A crystal was selected to be smaller than the X-ray beam size (approx. 0.3 mm) but not too small to give too weak a diffraction on an APEXII microfocus sealed tube X-ray source. The crystal was mounted ‘wet’ in a glass capillary and thereafter remained held at room temperature, approximately 20° C. X-ray diffraction data were recorded over an angular range of rotation of the crystal of 210° by which time, i.e. X-ray dose (approximately 2 MGy), the diffraction pattern had noticeably deteriorated. Data sets were grouped according to 30° sweeps making seven data sets in all. The high symmetry of the tetragonal lysozyme space group (P43212) yielded a reasonably good data set completeness for each 30° sweep of approximately 70%.
2.1.3.1 Cryo experiments. K2PtBr6 and Ta6Br12 bound in the same crystal allow a direct comparison, side by side as it were, of the two metal complexes. As reported in ref. 13 a break up of PtBr6 (Fig. 2), occurs versus a stability of Ta6Br12 throughout. The other sensitive marker of X-ray irradiation damage is the splitting of the protein disulphides20 and such an effect was shown for two of the disulphides by a 7th repeat X-ray diffraction data set.
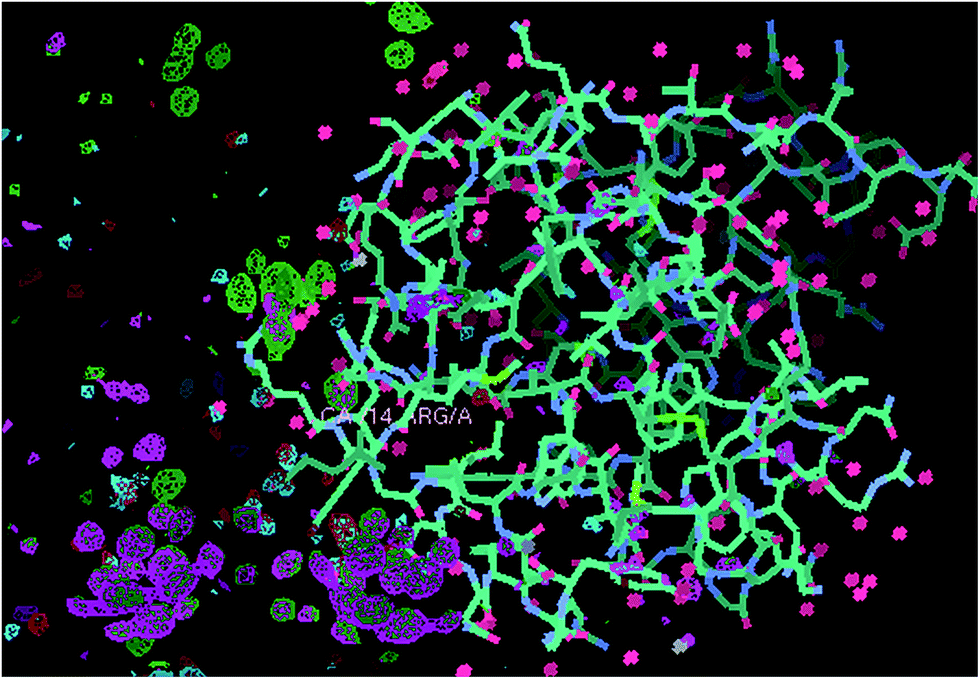 | ||
| Fig. 2 Comparison of the XRD stability behaviour of Ta6Br12, which is stable, versus PtBr6 and its clear break up as evidenced by X-ray data set 0 (electron density difference map contoured at 3 sigma green) and data set 6 (electron density difference map contoured at 3 sigma lilac). The nearest amino acid residue to the PtBr6 complex, which is Arg 14, is labelled; note the nice octahedral electron density for the first X-ray diffraction data set (green) and the essentially absent lilac density for it at the 6th data set. At the lower portion of the figure the equivalent green and lilac electron densities are for the Ta6Br12 moieties, there are two of them, and their stabilities to the X-rays, which is very good, are essentially identical. The protein model is 2W1Y and the maps are each rigid body 2W1Y model refinement Fo-Fc ‘omit’ (i.e. neither Ta6Br12 or PtBr6 are included in the model) electron density difference maps. | ||
The extension of the cryocrystallographic studies to include the multiple X-ray data set irradiation stability of the iodo complex (i.e. K2PtI6) bound to lysozyme is a natural extension and complements the aim of using these platinum octahedral complexes to facilitate interpreting difficult electron density maps. The first X-ray diffraction data set electron density (green in Fig. 3) and the somewhat diminished (lilac in Fig. 3) electron density for PtI6 at the 6th XRD data set are not as severe as the PtBr6 break up upon X-ray irradiation. There is a PtI3 moiety, directly bound to one of the His15 imidazole nitrogens, which is stable versus X-ray dose (i.e. the green and lilac electron densities in Fig. 3 are identical).
 | ||
| Fig. 3 Comparison of the XRD stability behaviour of PtI6 and PtI3X; comparison of data set 0 (electron density difference map contoured at 3 sigma green) and data set 6 (electron density difference map contoured at 3 sigma lilac). The nearest amino acid residue to both moieties, His15, is labelled; note the nice octahedral electron density for PtI6 (right, green; viewed down the three fold axis of symmetry of the octahedron). The first X-ray diffraction data set electron density (green) and the somewhat diminished lilac electron density for PtI6 at the 6th XRD data set are not as severe as the PtBr6 break up upon X-ray irradiation. The PtI3X moiety, directly bound to one of the His15 imidazole nitrogens, is stable versus X-ray dose (i.e. the green and lilac electron densities are identical). The refined structure of the HEWL PtI6 data set 0 PDB code is 4owc (ref. 9). The protein model shown here is 2W1Y and the maps are each rigid body 2W1Y model refinement Fo-Fc ‘omit’ (i.e. neither PtI6 or the PtI3X are included in the model) electron density difference maps. | ||
2.1.3.2 Room temperature X-ray crystallographic study of hen egg white lysozyme bound PtBr6 and relation to its chemical kinetics. The X-ray-photo-dynamics behaviour of PtBr6 at room temperature when bound to hen egg white lysozyme and the stages of its stepwise removal of bromine atoms are shown in Fig. 4 which compares the metal complex at successive stages of X-ray irradiation, namely data sets 1, 3, 5 and 7 (data sets 2, 4 and 6 are not shown), i.e. with cumulatively increasing X-ray dose to the same crystal. The occupancies of Pt and Br are approximately 0.3; the bound water that would be expected (see Fig. 5) to complete the octahedral arrangement around the platinum centre at 0.3 occupancy is not visible in Fig. 4b–d; this is due to the limited diffraction resolution of 2.2 Å and a diffraction data completeness of ∼70% in each case.
 | ||
| Fig. 4 The X-ray-photo-dynamics behaviour of PtBr6 at room temperature when bound to hen egg white lysozyme and the stages of its stepwise removal of bromine atoms evidenced by a comparison of (a) data set 1, (b) data set 3, (c) data set 5 and (d) data set 7. Data sets 1 through to 7 are with cumulatively increasing X-ray dose to the same crystal. The nearest amino acid residue to the PtBr6 complex is Arg 14, which is on the right hand side of the platinum complex and labelled. The protein model is the restrained refined ‘HEWL_PtBr6 RT data set 1’ and the electron density difference maps in blue are each rigid body 2W1Y model refinement 2Fo-Fc ‘omit’ maps, i.e. PtBr6 is not included in the model (1.4 rms contour level). The occupancies of Pt and Br are approximately 0.3; the bound water that would be expected (see Fig. 5) to complete the octahedral arrangement around the platinum centre at 0.3 occupancy is not visible in Fig. 4b–d; this is due to the limited diffraction resolution of 2.2 Å and diffraction data completeness of ∼70% in each case. | ||
 | ||
| Fig. 5 Glebov et al.11 reported results as follows:- “Photochemistry of the PtBr62−complex in aqueous solution was studied by steady state and laser flash photolysis (308 nm). The multistep photo-aquation of the complex occurs in the nano- and micro-second time intervals without the formation of intermediate platinum(III) complexes.” | ||
The chemical kinetics is basically that of a unimolecular reaction (i.e. break up) of the platinum hexabromide upon X-ray illumination. Since the X-ray illumination is at a continuous level, simply chosen to allow reasonable strength X–ray diffraction data to be recorded, we cannot determine an absolute rate constant for the break up. We note though that at room temperature the platinum hexabromide break up was more prompt than at cryo temperature in the lysozyme + PtBr6 + Ta6Br12 study, since the room temperature X-ray diffraction data had to be subdivided into smaller angular portions than that for the cryo data. A future experiment could be undertaken whereby a larger X-ray intensity would be used to illuminate a crystal sample e.g. from an intense synchrotron undulator X-ray beam, which would form an X-ray flash photolysis approach, and then the irradiated sample be plunged frozen. This would be repeated with several samples each at different time lapse intervals after X-ray irradiation. Neutron macromolecular crystallography diffraction data sets would then be recorded for each time lapse plunge freeze sample; the neutron beam would then allow the molecular state of PtBr6 to be probed without further radiation damage. Of course it would be entirely possible that the break up state would be identical in each neutron derived crystal structure, i.e. if the damage in an intense X-ray beam was ‘instantaneous’. Glebov et al.11 in their UV-vis studies indeed undertook flash photolysis and ‘steady state illumination’ experiments and kinetics analyses and from which we quote:-“The spectral curves obtained by steady state irradiation and pulse photolysis coincide, indicating that the first photoaquation step occurred within the time <50 ns”.
3 A short summary of the UV photochemistry of PtBr62−
The inorganic photophysics of K2PtBr6 in aqueous solution under steady state and laser flash photolysis (308 nm) has also been reported previously11 including a reaction mechanism proposal taking place on a timescale of nanoseconds or shorter involving charge transfer to an excited d–d state for lengthening of the Pt–Br bond distance and subsequent departure of a bromine atom. It was also described that successive steps of bromine removal and take up of a water molecule is possible.Glebov et al.11 reported their experimental results as follows:-
“Photochemistry of the PtBr 6 2− complex in aqueous solution was studied by steady state and laser flash photolysis (308 nm). The multistep photo-aquation of the complex occurs in the nano- and micro-second time intervals without the formation of intermediate platinum(III) complexes.” See their figure reproduced as Fig. 5 here. They conclude that “Thus, experiments with pico- or femto-second time resolution are necessary to perform for detailed investigation of the processes that occur upon the excitation of the PtBr62−complex”.
Our protein single crystal results are consistent with these models showing the successive steps in the removal of bromine atoms in the room temperature PtBr6 experiment and crystal structure analyses (Fig. 4a–d).
4 Specific conclusions
Ta6Br12 is relatively the most stable under repeated X-ray irradiation (Fig. 2) with PtI6 only showing some small changes (Fig. 3). Relatively less stable as a heavy atom complex is PtBr6 (Fig. 2 and 4).The study by Glebov et al.11,12 of PtBr6 shows that ‘with and without UV-vis light flash’ leads to photo dissociation of the platinum hexabromide (Fig. 5). The cryo ‘HEWL Ta6Br12 PtBr6’ study (Fig. 2) and the room temperature XRD study of ‘HEWL with PtBr6’ (Fig. 4a–d) concurs with the UV study, all showing a light induced break-up of PtBr6.11 Whilst the UV study of platinum hexaiodide seems not to have been undertaken, the HEWL PtI6 behaviour shows that it is relatively more stable under X-ray irradiation than PtBr6 and suggests therefore that the UV stability of PtI6 would likewise be more stable.
The respective behaviours of the temporal and structural stabilities of these heavy atom clusters are clearly of interest for a number of reasons. With the femtosecond XRD ‘diffract before destroy’ method5 the X-ray sensitivity of PtBr6 is not relevant. However its irradiation by UV light before X-ray laser diffraction would prepare an altered oxidation state11 which may be useful. The stability of Ta6Br12 and of PtI6 upon prolonged X-ray irradiation is a good property when synchrotron or home lab X-ray sources are used for biological crystal structure determination.
5 General outlook
Whilst our article title is specifically on X-ray diffraction, a more general question is whether there will be a surge of electron based diffraction techniques for temporally and spatially resolved molecular science. Electrons interact much more strongly with atoms than do X-rays. Electron crystallography and single particle electron microscopy (EM) imaging offer alternatives to the trend with X-ray sources such as the X-ray laser for the study of ever smaller samples. Our X-ray crystal structural studies of the lobster carapace coloration protein beta-crustacyanin21 have been extended to determine the layout at around 30 Å resolution of the alpha-crustacyanin complex, a 320 kDa complex of eight beta-crustacyanins,22 using negative stain single particle EM and small angle X-ray scattering along with ‘rigid body fitting’ of the beta-crustacyanin crystal structure. This structural study of the alpha-crustacyanin complex can be extended either by electron based techniques or with the X-ray laser representing a fascinating range of options.23 The technique of electron single crystal crystallography has very recently gained a major impetus with successful 3D structural analysis at 2 Å resolution, using molecular replacement, of hen egg white lysozyme as a test protein and using two individual microcrystals.24 It may not be possible to make de novo, high resolution protein structure determinations unless accurate isomorphous derivative intensity differences can be measured.25 However molecular replacement is now the predominant technique in protein crystallography not least due to the fact that the protein data bank contains more and more of the protein folds seen in nature (the PDB now contains over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 entries). Thus alpha-crustacyanin might yet be solvable with electron (microcrystal) crystallography and using the beta-crustacyanin X-ray crystal structure determined with a xenon heavy atom derivative and softer X-rays from the Daresbury SRS. Clearly there are important general implications for biological crystallography from the electron technique results of ref. 24 using microcrystals of proteins and complexes of biological macromolecules. Besides, static structure determination dynamic studies based on electron crystallography from a microcrystal sample might be easier than from a larger crystal, i.e. preserving crystallinity and so on. Again though, like the comment regarding the challenge for electron based techniques of measuring accurate isomorphous heavy atom derivative intensity differences, Bragg reflection intensity changes from temporal structural sequences can be smaller than those induced by a heavy atom and thereby even more difficult to measure precisely. Nevertheless further development of electron crystallography may well open up new options in this fascinating field; Zewail and Thomas have championed electron based techniques for a general realisation of 4D, space and time, structural studies26a and the mapping of atomic motions with ultrabright electrons and the fundamental limits in space-time resolution are discussed by Dwayne Miller and coauthors in ref. 26b.
000 entries). Thus alpha-crustacyanin might yet be solvable with electron (microcrystal) crystallography and using the beta-crustacyanin X-ray crystal structure determined with a xenon heavy atom derivative and softer X-rays from the Daresbury SRS. Clearly there are important general implications for biological crystallography from the electron technique results of ref. 24 using microcrystals of proteins and complexes of biological macromolecules. Besides, static structure determination dynamic studies based on electron crystallography from a microcrystal sample might be easier than from a larger crystal, i.e. preserving crystallinity and so on. Again though, like the comment regarding the challenge for electron based techniques of measuring accurate isomorphous heavy atom derivative intensity differences, Bragg reflection intensity changes from temporal structural sequences can be smaller than those induced by a heavy atom and thereby even more difficult to measure precisely. Nevertheless further development of electron crystallography may well open up new options in this fascinating field; Zewail and Thomas have championed electron based techniques for a general realisation of 4D, space and time, structural studies26a and the mapping of atomic motions with ultrabright electrons and the fundamental limits in space-time resolution are discussed by Dwayne Miller and coauthors in ref. 26b.
A regular challenge for X-ray crystal structure analysis of biological macromolecules is the determination of the protonation states of ionisable amino acid side chains of aspartic acid, glutamic acid and histidine. Likewise the orientation of bound water molecules or their state as hydroxyl or hydronium ions is even more challenging. Thus a growing development at neutron sources is the provision of instruments for neutron macromolecular crystallography notably in Japan at JAERI and JPARC, at the Institut Laue Langevin in Grenoble and at Oak Ridge National Lab, USA for both the research reactor and the SNS. Likewise a variety of macromolecular crystallographic software analysis packages now provide macromolecular refinement with neutron and X-ray scattering factors. Given the low flux of neutron beams at first sight it would seem that studies of structural and temporal dynamics using neutron macromolecular crystallography would not be possible. Is this the case? In fact ‘no’, it is possible with sample freezing to study structural intermediates albeit on a timescale set by the freezing time of the crystal sample.27 This area of neutron macromolecular crystallography is expected to grow; a very recent example is ref. 28. One such ‘next experiment’, as mentioned above, is suggested from consideration of the chemical kinetics of the X-ray induced break up of PtBr6.
Diffraction based techniques have limits. These have been highlighted in the recent book on NMR crystallography,29 which I reviewed.30 Quoting from Chapter 1 of this book:- “A key advantage is the case of host–guest complexes where the guest is mobile and obviously thereby disordered and its visibility absent or limited in the diffraction-derived structural analyses…(thus)…diffraction-based information forms an important background to investigations and where NMR is essential to delineate motional effects'…NMR measurements are ‘in addition to the relaxation times information which extends down to the microseconds region’. …There are two major areas in which complementary information (to diffraction) is desirable: Molecular-level mobility and purely spatial, i.e. static, disorder’.” Chapter 27 of the book31 is devoted to structural biology and includes NMR measurements by Martin and Zilm of a protein, ubiquitin (consisting of 76 amino acids and which has a molecular mass of about 8.5 kDa), in three different sample states “of microcrystals, nanocrystals and lyophilized (i.e. amorphous)”. The solid state NMR spectra for the nanocrystals of ubiquitin were equivalent to the spectra from the larger crystals; the difference was in the amorphous solid state ubiquitin spectra. As macromolecular crystallographers we tend not to worry too much about the sample state details and yet the properties of metallic nanoparticles, for example, are known to be different from bulk solids. This is an interesting further question for debate as diffraction methods take macromolecular crystallography into the nanocrystal sample size domain.
Acknowledgements
We are grateful to the University of Manchester for general support, to the EPSRC for a PhD studentship to SWMT, to the Thailand Government for studentship support to SK, to the SASOL Young Academic Collaboration Initiative for Dr Alice Brink, to the School of Chemistry for crystallization and computing facilities and to the Faculty of Life Sciences for X-ray diffraction facilities. We are very grateful to Dr Colin Levy and Dr Pat Bryant for their excellent stewardship of the Manchester Institute of Biotechnology (‘MIB’) X-ray Facility.Notes and references
- Time–Resolved Diffraction, ed. J. R. Helliwell and P. M. Rentzepis, Oxford University Press, 1997 Search PubMed; Time-resolved Macromolecular Crystallography, ed. D. W. J. Cruickshank, J. R. Helliwell and L.N. Johnson, Oxford Science Publications, 1992 Search PubMed.
- Z. Ren, D. Bourgeois, J. R. Helliwell, K. Moffat, V. Srajer and B. L. Stoddard, J. Synchrotron Radiat., 1999, 6, 891–917 CrossRef CAS.
- J. R. Helliwell, Y. P. Nieh, J. Raftery, A. Cassetta, J. Habash, P. D. Carr, T. Ursby, M. Wulff, A. W. Thompson, A. C. Niemann and A. Hädener, J. Chem. Soc., Faraday Trans., 1998, 94, 2615–2622 RSC.
- B. A. Yorke, G. S. Beddard, R. L. Owen and A. R. Pearson, Nat. Methods, 2014, 11(11), 1131–1134 CrossRef CAS PubMed.
- J. C. H. Spence and H. N. Chapman, Biology with free-electron X-ray lasers, Philos. Trans. R. Soc., B, 2014, 369, 1647 Search PubMed.
- R. Neutze, R. Wouts, D. van der Spoel, E. Weckert and J. Hajdu, Nature, 2000, 406, 752–757 CrossRef CAS PubMed.
- J. R. Helliwell, Science, 2013, 339, 146–147 CrossRef CAS PubMed.
- J. R. Helliwell, A. M. T. Bell, P. Bryant, S. J. Fisher, J. Habash, M. Helliwell, I. Margiolaki, S. Kaenket, Y. Watier, J. Wright and S. Yalamanchilli, Z. Krist., 2010, 225, 570–575 CrossRef CAS.
- S. W. M. Tanley, L.-V. Starkey, L. Lamplough, S. Kaenket and J. R. Helliwell, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2014, 1132–1134 CAS.
- J. M. A. Smith, J. R. Helliwell and M. Z. Papiz, Inorg. Chim. Acta, 1985, 106, 193–196 CrossRef CAS.
- E. M. Glebov, V. F. Plyusnin, V. P. Grivin, A. B. Venediktov and S. V. Korenev, Russ. Chem. Bull., Int. Ed., 2007, 56, 2357–2363 CrossRef CAS.
- V. Balzani and V. Carassiti, J. Phys. Chem., 1968, 72, 383–388 CrossRef CAS.
- J. R. Helliwell, S. Kaenket, C. Levy, P. Bryant, L. Lamplough, J. Raftery, L.-V. Starkey, E. Kotsiliti, I. Margiolaki, A. Fitch and J. P. Wright, Reported at 7th Radiation Damage ‘RD7’ Conference, held Diamond Light Source, Harwell Science and Innovation Campus, Oxfordshire, UK, 2012 Search PubMed.
- T. L. Blundell, L. N. Johnson, Protein Crystallography, Academic Press, New York, 1976, pp. 66–77 Search PubMed.
- Bruker, PROTEUM2. Version 2, 2006, Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., 2011, D67, 355–367 CrossRef PubMed.
- M. Cianci, J. R. Helliwell and A. Suzuki, Acta Crystallogr., 2008, D64, 1196–1209 Search PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., 2004, D60, 2126–2132 CrossRef CAS PubMed.
- J. R. Helliwell, J. Cryst. Growth, 1988, 90, 259–272 CrossRef CAS.
- M. Cianci, P. J. Rizkallah, A. Olczak, J. Raftery, N. E. Chayen, P. F. Zagalsky and J. R. Helliwell, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9795–9800 CrossRef CAS PubMed.
- N. H. Rhys, M.-C. Wang, T. A. Jowitt, J. R. Helliwell, J. G. Grossmann and C. Baldock, J. Synchrotron Radiat., 2011, 18, 79–83 CAS.
- J. R. Helliwell, Crystallogr. Rev., 2010, 16, 231–242 CrossRef CAS.
- B. L. Nannenga, D. Shi, A. G. W. Leslie and T. Gonen, Nat. Methods, 2014, 11(9), 927–930 CrossRef CAS PubMed.
- M. E. Dumont, J. W. Wiggins and S. B. Hayward, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2947–2951 CrossRef CAS.
- (a) A. Zewail and J. M. Thomas, 4D Electron Microscopy: Imaging in Space and Time, Imperial College Press, London, 2010 Search PubMed; (b) S. Manz, A. Casandruc, D. Zhang, Y. Zhong, R. A. Loch, A. Marx, T. Hasegawa, L. C. Liu, S. Bayesteh, H. Delsim-Hashemi, M. Hoffmann, M. Felber, M. Hachmann, F. Mayet, J. Hirscht, S. Keskin, M. Hada, S. W. Epp, K. Flöttmann and R. J. Dwayne Miller, Faraday Discuss. 10.1039/c4fd00204k.
- M. P. Blakeley, A. J. Kalb (Gilboa), J. R. Helliwell and D. A. A. Myles, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16405–16410 CrossRef CAS PubMed.
- C. M. Casadei, A. Gumiero, C. L. Metcalfe, E. J. Murphy, J. Basran, M. G. Concilio, S. C. M. Teixeira, T. E. Schrader, A. J. Fielding, A. Ostermann, M. P. Blakeley, E. L. Raven and P. C. E. Moody, Science, 2014, 345(6193), 193–197 CrossRef CAS PubMed.
- NMR Crystallography, ed. R. K. Harris, R. E. Wasylishen and M. J. Duer, John Wiley & Sons, Ltd., Chichester, 2009 Search PubMed.
- J. R. Helliwell, Crystallogr. Rev., 2014, 18, 241–245 CrossRef.
- D. Middleton, in NMR Crystallography, ed. R. K. Harris, R. E. Wasylishen and M. J. Duer, John Wiley & Sons, Ltd., Chichester, 2009, ch. 27, and see specifically CrossRef CAS; R. W. Martin and K. W. Zilm, J. Magn. Reson., 2003, 165, 162–174 CrossRef CAS.
- J. R. Helliwell and S. W. M. Tanley, Acta Crystallogr., 2013, D69, 121–125 Search PubMed.
- R. L. Owen, E. Rudino-Pinera and E. F. Garman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4912–4917 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. 2–4 were calculated using Refmac5 (main text ref. 17) using hen egg white lysozyme, coordinates PDB and code 2W1Y (main text ref. 18). The CCP4 mtz format processed structure factor amplitudes ‘X-ray radiation damaged’ diffraction data files used to make Fig. 2–4, displayed with COOT (main text ref. 19), are provided as ESI files to this paper. The cumulative absorbed X-ray dose by the time of the final data sets in the cryo cases was approximately 10 MGy and in the room temperature case approximately 2 MGy; uncertainties in these numbers arise from the X-ray generator flux at the sample being an estimate and the chemical composition of the protein crystal irradiated in each case is also somewhat approximate due to the inherent uncertainty in the bound atom occupancies and the final chemical composition of the protein crystal solvent channels. A detailed examination of such X-ray dose estimates can be found in main text ref. 32. Nevertheless the cumulative X-ray dose given is a good guide to the dose absorbed by the crystal samples. An absorbed X-ray dose of 30 MGy was recommended by Owen et al. 2006 (main text ref. 33) as an appropriate dose limit for typical macromolecular crystallography experiments conducted with the sample held at cryo temperatures i.e. typically 100 K; the cryo data collection experiments reported here are well within that absorbed X-ray dose limit as is also evident by the good quality of the protein electron density even at the final repeat X-ray diffraction data set with the exception of some disulphide bridge splitting. Fig. 2–4: HEWL crystal space group in each case was P43212; the electron density maps displayed in Fig. 2–4 were respectively calculated at 1.7 Å, 1.6 Å and 2.2 Å. The PDB codes for the previously published ‘HEWL PtBr6’ and ‘HEWL PtI6’ X-ray crystal structures determined at 100 K (ref. 9) are respectively 4OWH and 4OWC. The PDB code for the ‘HEWL Ta6Br12 PtBr6’ crystal structure determined at 100 K described for the first time here is 4R6C. Full details of the XRD data sets and molecular model parameters for these metal atom complexes bound to the HEWL protein can be found in these PDB files. See DOI: 10.1039/c4fd00166d |
| This journal is © The Royal Society of Chemistry 2015 |