
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microbial composition of purified waters and implications for regrowth control in municipal water systems†
Caitlin R.
Proctor
,
Marc A.
Edwards
and
Amy
Pruden
*
Via Department of Civil and Environmental Engineering, 418 Durham Hall, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: apruden@vt.edu; Tel: +540 231 3980
First published on 25th August 2015
Abstract
The limits of water treatment to control microbial regrowth were examined using highly purified waters. Measurable microbial genetic material was detected in the product water in a survey of thirteen laboratory pure water systems. Illumina 16S rRNA gene amplicon sequencing revealed surprisingly diverse microbial assemblages, confirmed to be active in bioassays, with no direct relationship to quality or maintenance of the systems. With storage under both light and dark conditions, a 2-log increase in bacterial genetic markers was observed within 10 days, indicating viable oligotrophic communities despite rigorous treatment steps. With growth, microbial communities shifted concurrent with enrichment of Proteobacteria groups capable of nitrogen fixation (Bradyrhizobium) and H2 oxidation (Comamonadaceae). This study has implications not only for laboratory studies, which rely on highly purified waters, but also for municipal drinking water, which depends on treatment to reduce nutrients sufficiently to limit downstream regrowth of microorganisms.
Water impactHighly purified waters are depended upon for a variety of laboratory, industrial, and other applications. The same general principle of disinfection alongside removal of nutrients, such as organic carbon and nitrogen, to prevent downstream regrowth is applied in municipal water systems to protect public health. Here we survey the microbial assemblage composition of thirteen laboratory-grade water purification systems and identify which microbes are associated with regrowth. We observed a wide diversity of DNA sequences, with a 2-log increase in total bacterial gene markers in less than 10 days. This study highlights the practical limits of nutrient limitation as a means of microbial control and indicates that additional measures are also needed to deliver high quality drinking water, especially when pathogen re-growth is a concern. |
1 Introduction
Water purification systems are core infrastructure in research labs and for many industrial applications, with production scales varying from 1–2 to tens of thousands of L per day. These systems employ a range of treatment approaches to achieve a high standard of water quality suitable for the target application.1–4The American Society for Testing and Materials International (ASTM) classifies three types of highly purified water based on specific attributes and use of the produced water – “ultra-pure”, “reagent grade”, and “bio-application grade” water. “Ultra-pure” water is defined for use in industrial applications and is characterized by a wide range of physical, chemical, and biological parameters, depending on the specific use. For example minimum resistivity ranges from 0.5–18.2 MΩ cm2. Since impurities (i.e. ionic compounds) conduct electricity through water, resistivity is considered to be directly proportional to the purity of the water. “Reagent grade” water is commonly used in a variety of laboratories, with minimum resistivity of 18.0 MΩ cm1. “Bio-application grade” water is intended for use in clinical, pharmaceutical, or biomedical applications, and has more stringent standards with respect to colony forming units (CFUs) and total organic carbon (TOC).3 ASTM standards for both reagent grade and bio-application water advise against any storage of produced water and dictate periodic monitoring of relevant water quality parameters in addition to in-line measurements.
Treatment processes for purified water can vary, but several technologies are commonly employed individually, or in combination, to meet the specific standards of each application.1,5 Reverse osmosis (RO) uses pressure to pass water through a membrane that generally allows water molecules, but not ions, to permeate. Ion-exchange resins (IER) have an affinity for dissolved ions, removing them from the aqueous phase and replacing them with H+ and OH−. Distillation acts through boiling the water and condensing the steam to generate water with very low dissolved salts and depletion of other constituents with a higher boiling point than water itself. Ultraviolet (UV) irradiation kills or inhibits bacteria by damaging DNA and thus its ability to replicate. UV can also degrade organic carbon in low-pressure drinking water scenarios6 or destroy it to less than 5 ppb in pure water applications7 thus indirectly limiting subsequent heterotrophic microbial growth. Activated carbon filters take advantage of the vast surface area of activated carbon and its affinity for organic and non-polar chemical impurities to remove them from the water. The high surface area also makes activated carbon an ideal attachment substrate for microbial biofilms, which can in turn degrade residual organic carbon and remove other constituents, thus improving overall biostability of the water.8–11 A variety of materials and pore-sizes can be employed in filtration to remove particles by sieving and other mechanisms, with ultrafiltration removing particles larger than 0.1–0.001 μm. Recirculation is also sometimes used to limit regrowth, but few studies have specifically examined this process.12 Regular disinfection of pure water systems is beneficial for reducing bacterial concentrations in product water, but levels have been observed to increase back to pre-disinfection levels within three weeks.13
Survival and regrowth of bacteria is a concern in highly purified water systems just as it is in the treatment and distribution of municipal drinking water.14 In highly purified water systems, the concern may be even greater as even minute levels of microbial cells can be detrimental to intended uses, such as rinsing of electrical components.15 In the scientific community, consistent and high water quality is crucial for conducting reproducible and comparable experiments across laboratories.16 One logical approach for limiting microbial regrowth in any water system is to minimize the availability of nutrients available for growth.17 In drinking water systems, removal of assimilable organic carbon (AOC), or carbon that is readily available to bacteria for growth,11,18 has gained attention as a means of limiting regrowth in continuously flowing water distribution systems. Reduction of AOC below 10 μg L−1 has been cited as a critical threshold for microbial control in drinking water distribution systems with little or no disinfectant,19 and levels less than 100 ug L−1 have been recommended to control growth of bacteria with moderate levels of disinfectant.20,21
Remarkably, despite the stringency of the treatment methods applied and the extreme oligotrophic conditions achieved, highly purified water systems can be host to significant microbial growth,15,22,23 and even pathogens like Pseudomonas aeriginosa.24 In particular, IERs25 and activated carbon9 can provide suitable biofilm attachment substrate and access to organic matter. A diverse range of bacteria have been observed in highly purified water systems using both culture-based and molecular-based tools.26–29 However, little is known about potential for microbial growth in laboratory grade water,22,30 and the few studies that have attempted to fully characterize the microbial communities observed are limited to industrial application systems.26–29 Identification of the bacteria in highly purified water has traditionally used culture-based techniques,26,29,31 which are particularly limited for oligotrophic microbes that are characteristic of these low-nutrient environments. Culturability with heterotrophic plate count (HPC) methods may represent as little as 0.001% of flow cytometry total cell counts for potable drinking water.32 Molecular methods, which capture both the cultured and uncultured fractions of bacteria, have been used in only a limited number of the studies26–30 and, to the authors' knowledge, next-generation DNA sequencing has not been reported for deep profiling of the microbial community composition of highly purified water systems.
The purpose of this study was to survey the microbial assemblages inhabiting a range of laboratory-grade water systems using Illumina sequencing to deeply profile 16S rRNA gene amplicons and to determine the effect of storage on microbial communities. In addition to providing insight into the microbial ecology of these extremely oligotrophic systems, the results also serve as a reference point to the practical limits of water quality that can reasonably be attained via nutrient limitations in water systems, with and without storage. The systems analyzed in this study employ the highest standards of treatment, and thus represent a best-case scenario for all oligotrophic waters. The results have important implications in light of certain emerging advanced water systems that employ costly reverse osmosis and UV treatments.
2 Methods
Two studies were undertaken to characterize the bacterial communities that colonize laboratory grade waters. First, a survey was conducted with laboratory grade water systems representing a range of treatment and maintenance approaches housed in several laboratories across the Virginia Tech campus. Second, an experiment was conducted to gain insight into the biostability of a subset of waters by tracking bulk water bacterial growth during storage.2.1 Survey of water purification systems
Thirteen laboratory grade water purification systems were included in this study. Information about age and maintenance history of the systems was obtained from lab users (Table 1).| System code name | Flow rate (L min−1) | In-line resistivity reading (MΩ cm) | Components of system/feed watera | Frequency of maintenance/time since last maintenance | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pre-filter | RO | DI | IER | GAC | UF | UV | ||||
| a RO = reverse osmosis; DI = de-ionized; IER = ion-exchange resin; GAC = granular activated carbon; UF = ultrafiltration; UV = sterilization with UV light. | ||||||||||
| Collected 12.3.12 | ||||||||||
| A-1 | 0.96 | 18.2 | X | X | X | X | 1.5 years | |||
| A-2 | 1.16 | 18.2 | X | X | 2 months | |||||
| A-3 | 1.13 | 18.0 | X | X | X | 2 months | ||||
| A-4 | 1.61 | 18.2 | X | X | After malfunction | |||||
| A-5 | 1.62 | N/A | X | X | 6 months | |||||
| A-6 | 0.82 | 18.3 | X | X | 6 months | |||||
| Collected 12.4.12 | ||||||||||
| B-1 | 1.89 | 18.2 | X | X | 1 month | |||||
| B-2 | 1.76 | 18.07 | X | 4 months | ||||||
| B-3 | 1.10 | 18.2 | X | X | X | 6 months | ||||
| B-4 | 1.01 | 19.2 | X | X | X | 6 months | ||||
| Collected 12.5.12 | ||||||||||
| C-1 | 2.25 | 17.7 | X | X | 5.5 years | |||||
| C-2 | 0.29 | 18.3 | X | 2 years | ||||||
| C-3 | 1.67 | 18.32 | X | X | X | X | 6 months | |||
Systems were sampled using pre-sterilized 1 L high-density polyethylene (HDPE) Nalgene bottles with polypropylene caps, which had previously been soaked and rinsed in reagent grade water for more than 1 month. Two consecutive 1 L samples were collected from each system using the highest flow conditions possible. In order to capture the maximum possible microbial contamination, water was not intentionally flushed before sampling.
After sample collection, an additional 60 mL was collected for adenosine triphosphate (ATP) analysis. Samples were stabilized on site by filtering to capture cellular contents using a Quench-Gone LuminUltra (NB, Canada) syringe filter. Cells were lysed to release and preserve ATP for analysis by filtering 1 mL of UltraLyse (LuminUltra) through the syringe. Stabilized samples were maintained on ice until further analysis.
Water flow rates were determined at the time of sample collection by recording the time required to fill containers of pre-determined volume. Water samples were immediately placed on ice in a cooler. Upon return to lab, all samples were maintained at 4 °C until filtration, which was carried out within 12 hours of sample collection.
Blanks consisted of 1 L of water sterilized by autoclaving under standard conditions. Trip blanks and field blanks consisted of 1 L of laboratory grade water (Barnstead; system C-3, Table 1) stored in the same type of container as the samples. This system was selected based on extensive experience with the system suggesting optimal performance and convenient access to an autoclave to minimize contamination. Field blanks were opened at each site for an equivalent duration of sample collection while trip blanks remained closed. Filter blanks were not exposed to water and were analyzed as a quality control to monitor any potential background sources of contamination from the filter, DNA extraction procedure, and laboratory manipulation.
2.2 Time series study
Two experiments were conducted to determine the effects of storage on microbial composition of laboratory grade water. The first one was conducted from 1/30/2013–2/9/2013 [Time Study 1] and the second was conducted from 5/31/2013–7/1/2013 [Time Study 2]. Time Study 1 (9 days) was carried out under exposure to ambient light in order to account for possible phototrophic effects, whereas Time Study 2 was carried out over a longer time frame (32 days) in a closed cabinet shielded from light in order to exclude phototrophy. Sacrificial samples were collected after 0, 1, 2, 3, 6, and 9 days in Time Study 1, and were collected after 0, 1, 3, 7, 14, 21, and 32 days in Time Study 2. Both were conducted in a temperature-controlled laboratory at room temperature, 20 °C.In each experiment, nanopure water (Barnstead; system C-3, Table 1) was aliquotted into a glass Pyrex 10 L media storage bottle with screw cap that had both been acid washed and sterilized via autoclaving. Water was thoroughly mixed via manual shaking then distributed into six or seven (respectively for Time Study 1 and 2) Pyrex 1 L media storage bottles with screw caps that had been acid washed and sterilized via baking at 550 °C for 4 hours (glass bottles) or autoclaving (caps). Approximately 1 L was transferred into each storage bottle under sterile conditions and was subsequently tightly capped. Time 0 samples were taken immediately after distribution of all waters.
2.3 ATP and AMP quantification
ATP provides an indicator of viable biomass activity levels, while adenosine monophosphate (AMP) is an indicator of cell stress. ATP and AMP concentrations, and their ratios, were measured using a LuminUltra® Quench-Gone™ Aqueous Test Kit (LuminUltra). Preserved samples were analyzed according to manufacturer protocol within 12 hours to determine ATP, AMP, and the ATP:AMP index.2.4 Sample concentration and DNA extraction
For each sampling event, the entire liter was sacrificed for filtration. Each sample event included a filter blank sample (analysis of the filters only). Time zero samples were collected immediately after transfer into the 1 L incubation bottles. Each storage bottle was shaken vigorously by hand in the same fashion prior to sample concentration.Samples were concentrated onto 0.22 μm pore-size sterile mixed cellulose ester filters (Millipore, Billerica, MA) by vacuum filtration using sterile technique. The filter was folded and torn using sterile tweezers and transferred to a Lysing Matrix A tube provided in the FastDNA® SPIN Kit (MP Biomedicals, Solon, OH). DNA extraction was conducted according to manufacturer instructions.
2.5 Quantitative polymerase chain reaction (q-PCR)
All DNA samples were analyzed with quantitative polymerase chain reaction (q-PCR), which was applied to quantify bacterial 16S rRNA genes as an indicator of the level of total bacteria.33 Briefly, the primers BACT1369F: CGGTGAATACGTTCYCGG and Prok: GGWTACCTTGTTACGACTT were used with a denaturation step of 98 °C for 2 minutes and 40 cycles with 98 °C for 5 s and 55 °C for 5 s. Blank qPCR reactions and calibration curves spanning seven orders of magnitude were included in every run. The calculated limit of quantification was 5 copies per mL based on the lowest point on the curve and assuming a 2 L sample volume for DNA extraction. Q-PCR was carried out using a CFX96™ Realtime system (Bio-Rad, Hercules, CA). Q-PCR assays were previously validated for drinking water samples in terms of specificity and limit of quantification.34 Previous tests (data not shown) indicated that a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 dilution was appropriate for dilution of potential inhibitors and consistent quantification of highly purified water samples.
:10 dilution was appropriate for dilution of potential inhibitors and consistent quantification of highly purified water samples.
2.6 Illumina sequencing of 16S rRNA gene amplicons
Illumina amplicon sequencing was applied to a subset of samples to characterize the compositions of the microbial assemblages of the water systems. Bacterial and Archaeal 16S rRNA genes were amplified with barcoded primers 515F/806R35 using published protocols.36 In order to normalize depth of reads/sample, 20 ng of DNA of each amplification product were mixed according to quantification using the Qubit® ds DNA HS Assay Kit (Invitrogen™) and Qubit® 2.0 Fluorometer. Combined PCR products were cleaned using QIAGEN PCR Purification Kit. Sequencing was performed on an Illumina Miseq® benchtop sequencer using paired-end 250 bp protocol by the Virginia Bioinformatics Institute (Blacksburg, VA).2.7 Statistical methods and data analysis
Statistical analysis for quantitative measures was performed using JMP (SAS, Cary, NC) and R (http://www.r-project.org/). In order to appropriately compare blanks to samples, all q-PCR data was normalized to two liters, assuming that the volume of the samples was common to that of both the water and filter blanks. Given that data were not normally distributed, non-parametric tests including the Mann–Whitney U Test (Willcox) and the Kruskal Wallis Test were used to compare means of groups for q-PCR data. Least-squared regression was applied to determine correlations. Significance was set at α = 0.05.Sequence reads were contigued using PAired-eND Assembler for DNA Sequences (PandaSeq).37 QIIME (Quantitative Insights Into Microbial Ecology) was used as a pipeline for sequence analysis. Operational Taxonomical Units (OTUs) were assigned using uclust38 based on 97% similarity to the Greengenes database.39 Weighted and unweighted Unifrac40 distance was computed between all samples using an equal sampling depth of 11000 sequences per sample. Unweighted Unifrac distances are constructed based on which unique OTUs are present, whereas weighted Unifrac distance also takes into consideration the abundance of each OTU. A smaller distance indicates that communities are more similar and composed of more closely related taxonomical OTUs. These distances were employed for multidimensional scaling (MDS) and analysis of similarity (ANOSIM) as implemented in Primer-E software (Plymouth, United Kingdom). ANOSIM produces global R values which range from 0 to 1, with 1 indicating that samples within the group are more similar to each other than any samples outside the group.41 Bootstrapped jackknife trees were produced in QIIME using Unifrac distances.
3 Results
3.1 Survey of water purification systems
The treatment, maintenance, and operating conditions of the thirteen laboratory grade systems included in this survey are described in Table 1. All samples were collected within a three day period in December 2012 [mean outdoor temperatures 51–57 °F (11–14 °C)].The systems represented a range of treatment and maintenance conditions, ages of systems, and quality of feed water. Yet, similar in-line resistivity readings were noted across most of the systems (mean 18.24 MΩ cm, 95% CI [18.02–18.46 MΩ cm], outliers C-1, B-4, A-3; N = 12). Quantification of 16S rRNA genes suggested measurable levels of bacteria (Fig. 1) even when systems had final UV treatments designed to remove organic carbon and disinfect the water at the point of use. Although the average for all blanks together was lower than that of samples (p = 0.03, Wilcox), that of particular blank types exposed to water did not vary significantly from the samples (for trip blanks – p = 0.0572, for field blanks – p = 0.9; Wilcox). The average across all samples and across field blanks were nearly equal. Notably, samples were capped immediately after sampling and remained closed until analyzed, whereas field blanks were opened as much as 6 times in a day. Trip blanks remained tightly capped throughout the sampling day. Both field and trip blanks were originally collected at the same time from the same system (C-3), autoclaved prior to the experiment, and subject to the same holding times and temperature shifts during sampling. Filter blanks (filter only – no contact with water) yielded significantly lower concentrations of 16S rRNA when compared to all other samples, which were exposed to either 1 L (blanks) or 2 L (all sample locations) of laboratory grade water (p = 0.0026, Wilcox).
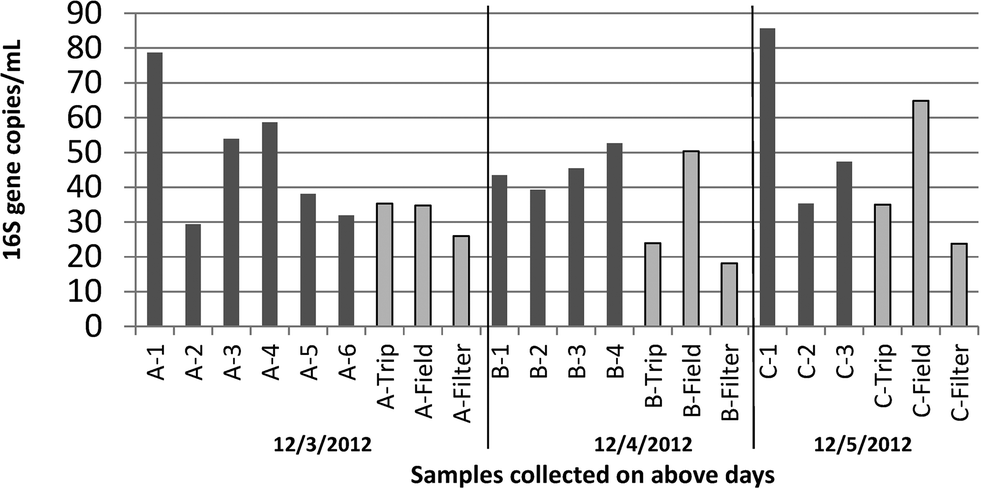 | ||
| Fig. 1 Concentration of 16S rRNA genes [gene copies per mL] in 2 L samples of pure water collected from a range of systems over a three day period and the corresponding blanks for each day. Trip blanks and field blanks consisted of 1 L of autoclaved pure water collected from system C-3 and subject to equivalent storage conditions during sampling. Field blanks were opened at each site, trip blanks were not. Filter blanks were not exposed to any water. For each bar, n = 1, as average of q-PCR analytical triplicates. | ||
Most samples were characterized by very low levels of ATP, in the range of <0.5 pg mL−1, which assay manufacturers describe as indicative of “good” microbial control for drinking water (Fig. 2A). However, three samples, all collected on the same day, were in the range of 0.5–10 pg mL−1 which is indicative of “preventative measures needed”. As all of the high values were collected on the same day, it is possible that this could be due to systematic error in ATP measurements on that particular day. In contrast to the ATP data, sampling days were not significantly different for q-PCR data (p = 0.86, Kruskal Wallis). The AMP Index was above 3.0 for all samples, which assay manufacturers describe as “lethal stress” (Fig. 2B). Neither ATP nor AMP correlated with 16S rRNA gene measurements (p > 0.05, least squared regression).
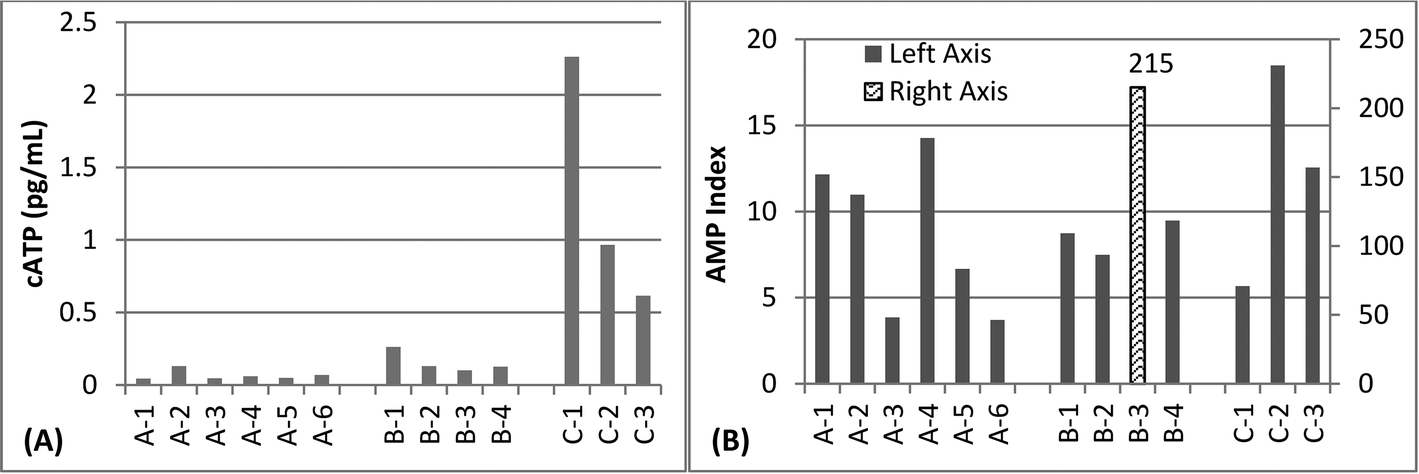 | ||
| Fig. 2 A) Concentration of adenosine tri-phosphate (ATP) in various pure water systems (n = 1 for each bar). B) AMP Index, the ratio between measured ATP and adenosine mono-phosphate (AMP) in various pure water systems (n = 1 for each bar). | ||
3.2 Effect of storage on levels of bacterial gene markers
A 2-log increase in 16S rRNA genes was observed within about 10 days in both the Time Study 1 (10 days) and Time Study 2 (32 days) storage experiments (Fig. 3). In Time Study 2, the concentration of 16S rRNA genes stabilized within ±1-log by the final 2 weeks of the experiment.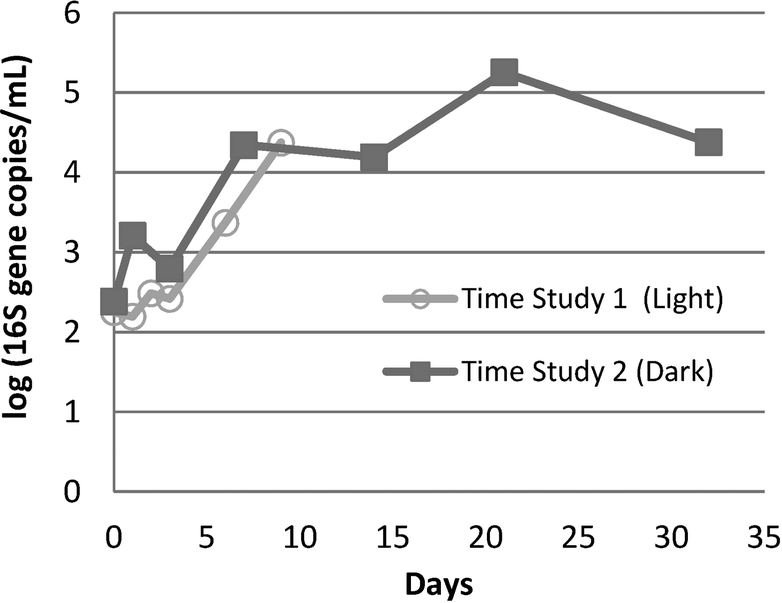 | ||
| Fig. 3 Log (16S rRNA gene copies) detected by q-PCR in two time storage studies of nano-pure water from the same system (C-3). In both studies, water was stored in sterilized 1 L glass containers at room temperature after homogenization of all samples for each study. All growth conditions were similar between Time Study 1 and Time Study 2, with the exception of light exposure and the time frame/season. Time Study 1 was conducted in winter and Time Study 2 was conducted in spring (n = 1 for each time point). | ||
3.3 Comparison of the microbial assemblages
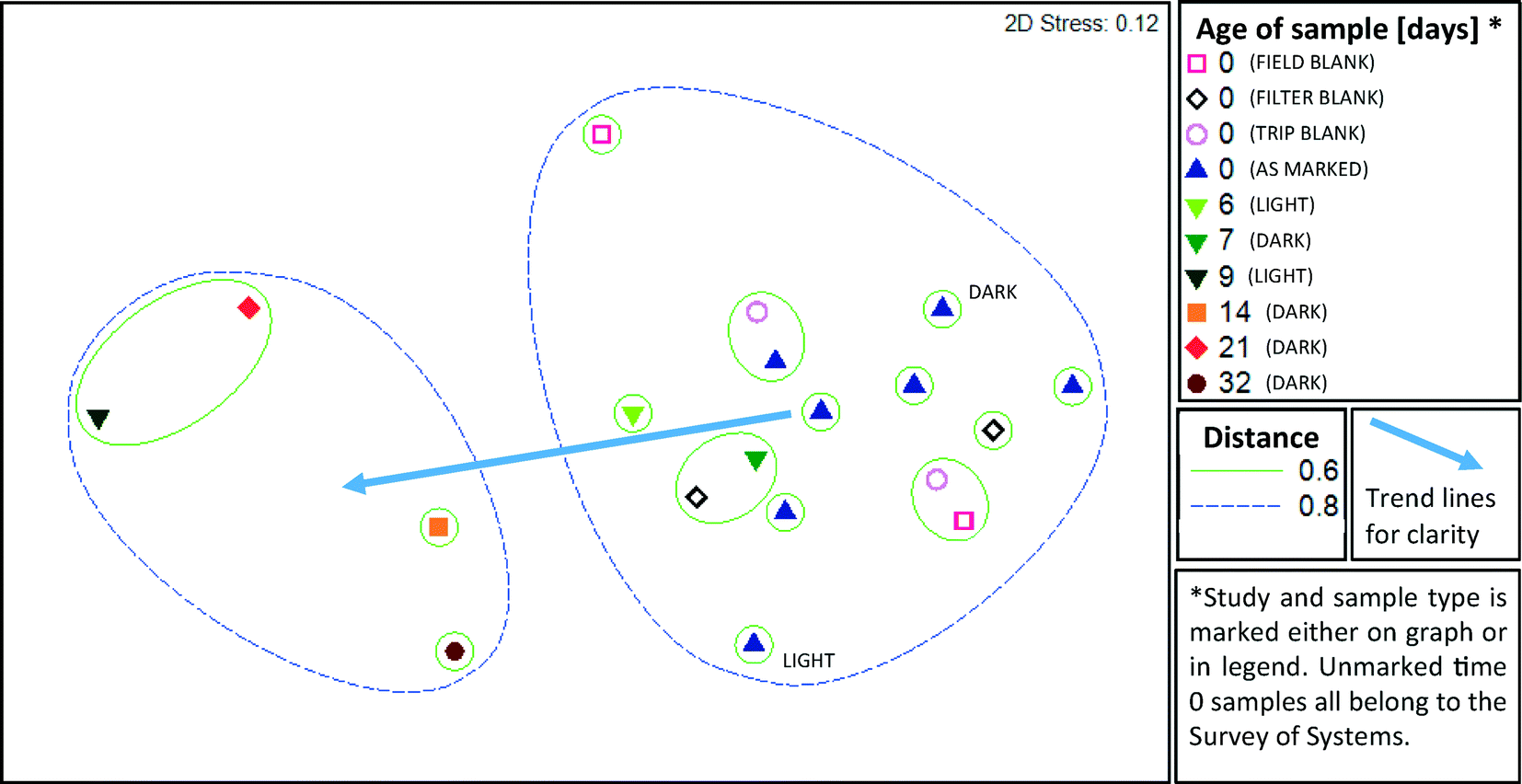
A cross section of samples (n = 19) were selected for microbial profiling by Illumina sequencing of 16S rRNA gene amplicons. From the field survey, 5 of 13 water purification systems (A-1, A-2, A-5, C-1, and C-3) and all three types of blanks from the two days encompassed by those samples were subject to amplicon sequencing. From Time Study 1, samples from day 0, 6 and 9 were selected. From Time Study 2, samples from day 0, 7, 14, 21 and 32 were selected.Samples from all studies were pooled together for ANOSIM analysis, which demonstrated that the storage time was a significant factor driving the microbial community structure (R = 0.646, p = 0.001, ANOSIM). The strongest difference was observed between samples aged 0–1 days and those aged more than 8 days (R = 0.836, p = 0.001, ANOSIM). A two dimensional MDS plot (Fig. 4) illustrates the shift in composition of the microbial assemblages that took place as the water aged. The microbial composition did not cluster based on the kind of water purification system that the field samples were collected from, i.e. distinct clusters were not apparent. Blanks clustered closely with the samples that were not subject to storage and none of the three types of blanks (trip, field, and filter) could be differentiated from the samples (R = −0.012, p = 0.51, ANOSIM). A distinct cluster was apparent between samples aged six and seven days in Time Study 1 and Time Study 2, respectively. This suggests that the composition of microbial assemblages converge as water ages, regardless of the source of the water and despite differences in experimental set-up (i.e., shielding from light in Time Study 2).
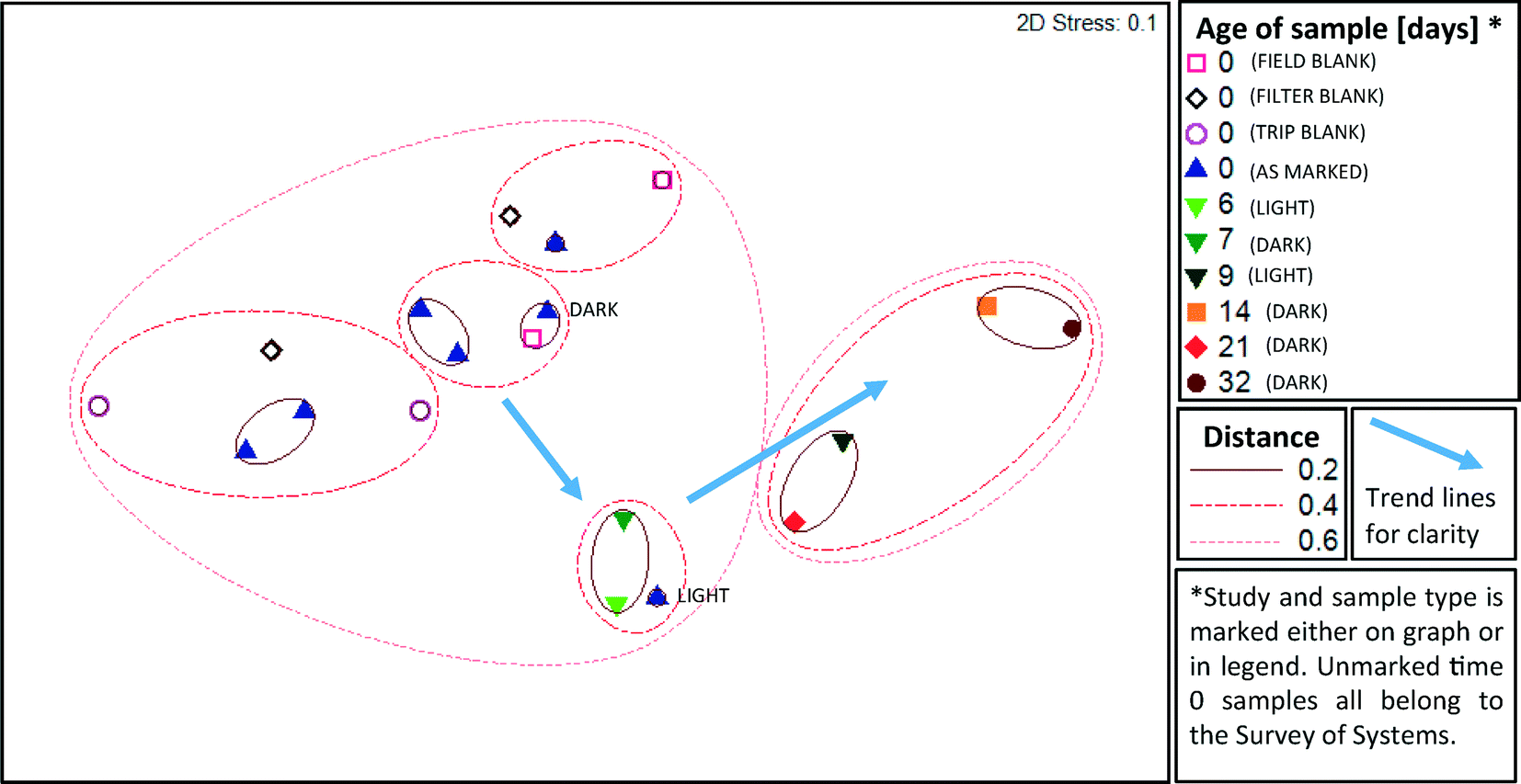 | ||
| Fig. 4 Comparison of microbial assemblage composition in highly purified water and their shifts during storage according to multi-dimensional scaling (MDS) of weighted Unifrac distance matrices. Select samples from two distinct time studies in both light (Time Study 1) and dark (Time Study 2) conditions are represented. Additional time 0 samples from five additional systems and blanks are from the field survey of pure water systems. A smaller distance between samples indicates greater similarity, i.e. samples within a circle marked 0.2 are more similar than those in a circle marked 0.6. The relative abundance of unique OTUs is taken into consideration in this weighted analysis. | ||
An unweighted Unifrac analysis, which does not take into consideration the relative abundance of each new OTU, produced similar trends, although clustering was generally weaker when subject to MDS and ANOSIM analysis. As with the weighted analysis discussed above, age of sample was a significant factor driving the kinds of microbes detected (R = 0.55, p = 0.002, ANOSIM), and all three types of blanks were not distinct from samples also aged 0 days (R = −0.012, p = 0.75, ANOSIM). Based on MDS (Fig. 5) analysis, clustering distances were greater than with weighted Unifrac analysis, indicating that abundant species, rather than rare species, were particularly important in defining community differences. Jack-knife clustering (Fig. S1 and S2†) also indicated greater distinction as a function of water age with weighted, rather than unweighted, analysis, further indicating that abundance and growth were a critical factor in the differences observed among the microbial assemblages.
 | ||
| Fig. 5 Multi-dimensional scaling (MDS) of an unweighted Unifrac distance analysis. A smaller distance between samples indicates greater similarity. The abundance of unique OTUs is not taken into consideration in this unweighted analysis. Select samples from two distinct time studies under both light (Time Study 1) and dark (Time Study 2) conditions are represented. Additional time 0 samples from five additional systems and blanks are from the field survey of pure water systems. A smaller distance between samples indicates greater similarity, i.e. samples within a circle marked 0.2 are more similar than those in a circle marked 0.6. | ||
3.4 Composition of the microbial assemblages
Analysis of the 16S rRNA gene amplicon sequences indicated clear shifts in the overall compositions of the microbial assemblages during water storage (Fig. 6). Through taxonomic analysis of the DNA sequences, it was possible to identify which groups of bacteria were associated with the genetic material detected and estimate how the populations shifted during water storage. In particular, Proteobacteria, especially Alpha Proteobacteria and Beta Proteobacteria, tended to dominate with greater storage time.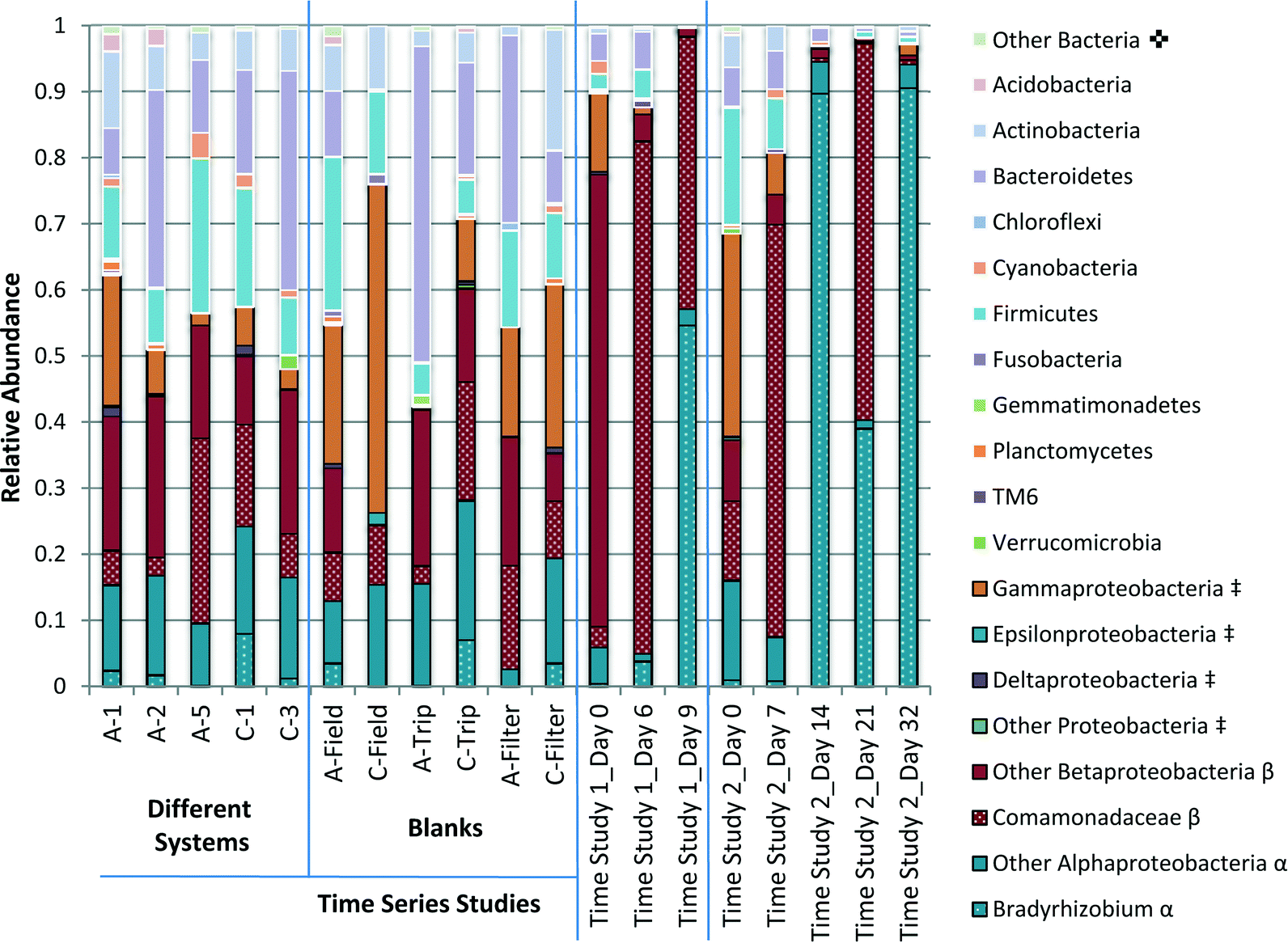 | ||
Fig. 6 Comparison of the relative abundance of the phyla detected in pure water samples. Select samples from two distinct time studies in both light (Time Study 1) and dark (Time Study 2) conditions are represented, as well as samples from several pure water systems and blanks collected during that survey campaign. Taxa separated by phylum unless otherwise marked.  Other bacteria includes all phyla that contributed to less than 1% of all samples. ‡Proteobacteria subdivided into classes (Alpha-, Beta-, Gamma-, Delta-, Epsilon-Proteobacteria and other). β BetaProteobacteria further divided into the family Comamonadaceae and other. α AlphaProteobacteria further divided into the genus Bradyrhizobium and other. Other bacteria includes all phyla that contributed to less than 1% of all samples. ‡Proteobacteria subdivided into classes (Alpha-, Beta-, Gamma-, Delta-, Epsilon-Proteobacteria and other). β BetaProteobacteria further divided into the family Comamonadaceae and other. α AlphaProteobacteria further divided into the genus Bradyrhizobium and other. | ||
The phyla with the highest abundance across most samples included Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria. Actinobacteria, Firmicutes and Bacteroidetes were all detected in greater relative abundance in the samples that were not subject to storage, including samples collected from the field survey of water systems and blank samples, than samples with greater storage time. Overall, a surprising diversity was suggested, even in filter-blank samples that were not exposed to water.
Of Actinobacteria, Mycobacteria 16S rRNA gene sequences were found in all samples, and were highest in relative abundance in the systems A-5 (3.1%), C-1 (3.8%), and Time Study 2 day 0 (2.7%). In Time Study 2, the relative prevalence of Mycobacteria appeared to decrease with time, with day 7 (1.9% of amplicons) to a low on day 21 (0.08% of amplicons). Prevalence of Mycobacteria also decreased with storage time in Time Study 1. Within Firmicutes, both Clostridia and Bacilli were detected among the amplicon sequences. The most common taxa detected within Bacteroidetes was Chitinophagaceae.
The candidate phylum TM6 was ubiquitous to all samples, including filter blanks, although on average it made up only 0.2% of the amplicon pool across samples. It was at highest concentration on day 6 of the Time Study 1 (subject to light exposure) (1.2% of amplicons).
Cyanobacteria were found in all samples and were in highest relative abundance (3.8% of amplicons) in the A-5 system. In Time Study 1, they were found in highest abundance in the Time 0 sample. Clade MLE1-12 was identified in 17 of 19 samples, including blanks. Of the phylum Chloroflexi, the greatest relative abundance of phototrophic OTUs (1.2%) was found in the Filter Blank_12.3 sample.
Nitrifying bacteria were sporadically found in low relative abundances. Nitrospira was found with the greatest relative abundance in Field Blank_12.3 (0.6%) and Nitrosomonadaceae were found in greatest abundance in Time Study 2, day 0 (1.2%).
Proteobacteria were detected in greater relative abundance in samples with greater storage time. Apha- Beta- and Gamma-Proteobacteria were the most prevalent classes. Gamma Proteobacteria encompass many pathogens including Legionella, which was detected in this study at the genus level in two samples with only 1 OTU per sample. Gamma Proteobacteria became a less significant class with greater storage time. The relative dominance of Alpha- and Beta-Proteobacteria in relation to each other varies over time (Fig. 6).
Alpha Proteobacteria detected in laboratory grade water systems was dominated by the genus Bradyrhizobium within the family Bradyrhizobiaceae and the class Rhizobiales. Bradyrhizobium accounted for up to 90% of OTUs detected in samples collected at day 14 and day 32 of Time Study 2, as well as 55% of OTUs detected in samples collected on day 9 of Time Study 1.
Among Beta Proteobacteria, the order Burkholderiales dominated and was highly variable. Within this order, the Ralstonia genus within the Oxalobacteraceae family and an unidentified genus in the Comamonadaceae family dominated. The Comamonadaceae family dominated in samples allowed to stagnate for longer periods of time, accounting for 60% and 57% of OTUs detected in samples collected on day 7 and 21 of Time Study 2, and 75% and 41% of OTUs detected in samples collected on day 6 and 9 of Time Study 1. Ralstonia accounted for 60% of OTUs detected in the initial sample for Time Study 1.
4 Discussion
4.1 Comparison of microbial assemblage composition of various water purification systems
All of the systems analyzed in this study were used for similar applications and all were advertised to provide Type 1 reagent grade water or better. Resistivity readings were generally above 18.0 MΩ cm (Table 1), indicating acceptable quality according to standard criteria. ATP readings also indicated reasonable water quality and that surviving cells were under “lethal stress,” suggesting that the biomass that was present was not initially thriving. This is expected, as nutrient limitation and ultrafiltration in highly purified water treatment systems are likely to place high stress on any surviving bacteria.Despite these positive indications of water quality, 16S rRNA genes were still detected in all samples. As DNA detection methods cannot differentiate between live and dead cells, detection of 16S rRNA does not necessarily indicate that systems were contaminated with live bacteria. Autoclaved water samples still yielded detectable signal, possibly as a result of intact DNA released from killed cells. All samples exposed to water yielded higher concentrations of 16S rRNA genes than filter blanks, suggesting that DNA contamination persists in many types of laboratory grade waters and that the source of all DNA contamination was neither the filter itself nor the filtering and DNA extraction process. Field blanks, which were opened throughout the day at each sampling location, yielded 16S rRNA gene concentrations that were higher than trip blanks, which were not opened throughout the day, but similar to that of the samples. Given that system samples were capped immediately after collection, this suggests that the process of opening the bottles for sampling contributes to bacterial contamination. All detected concentrations of DNA were considerably lower than that of the local municipal tap water that fed the systems, which was previously reported to range from 102 to 106 gene copies per mL, with an average of about 104 gene copies per mL, using the same quantification methods.34 Thus, purification methods did succeed in reducing bacterial contamination compared with the source tap water, which contained disinfectant residual of ~2 mg L−1 chloramine. These samples were taken during an unseasonably warm winter, which is noted here since season commonly has an effect on drinking water.42 However, there is no evidence that season had a major influence in the present study.
The lack of clustering of microbial communities by any particular identifier amongst samples from different systems indicates that the particular treatment train in a laboratory grade water system is not a fundamental factor driving microbial community composition. Rather, the community may be dictated by the common source water. It is possible that the entire distribution system is to an extent governed by filtration at the drinking water treatment plant43 or the disinfectant used,44 as reported by others. The similar background chemistry of the water is also a likely factor shaping the microbial assemblages.45 It is also possible that all the systems analyzed provided a similar level of stress as indicated by the similar resistivity readings, thus selecting for similar communities.
In addition, microbial assemblage compositions of the different systems did not differ greatly from blanks, which were either autoclaved (field and trip blanks) or not exposed to water (filter blanks). Thus, the DNA sequences detected may also represent microbes ubiquitous to the “sterile” environment, and thus a bias to consider in the profiling of microbial communities from samples with relatively low DNA yields. Much of the detected community diversity in blanks and samples prior to incubation could also be an artifact of DNA extraction kits, as explored by Salter et al.46 However, while it is true that some or most of the DNA amplicons detected across this study may have represented non-living microbes or contamination, some portion must have been viable based on the responsive growth observed when the waters were incubated over time in Time Study 1 and Time Study 2. Live bacteria are also a clear possibility in highly purified water systems, as others have observed total coliforms at the effluent of a 10000 L per day purification system reached 27 or more CFU per 100 mL.31
4.2 Comparison with other potable waters and putative functional capabilities
The predominant phyla detected were similar to those in drinking water systems in the U.S. as reported based on sampling of 17 drinking water distribution systems,45 and drinking water in China,47 though the exact compositions and relative abundances differ. Proteobacteria are metabolically diverse and dominated in both of these prior drinking water studies (35% and 47% respectively), as well as the present study (minimum in a sample 42%). However, in the prior drinking water studies, Cyanobacteria was a major contributor, comprising 29% and 11% of DNA sequences across all samples in each study, respectively. While Cyanobacteria were also found in the present study, it was in lower relative abundance (maximum 3.8% in one sample).The OTUs identified were also similar to those reported in other highly purified waters and reagents. Both Ralstonia and Bradyrhizobium were isolated from several industrial ultra pure water systems,28,29 and Bradyrhizobium was isolated from a pharmaceutical water.27 Bradyrhizobium, Chitinophagaceae, and Comomonadaceae were also found in contamination from the laboratory and reagents in DNA extraction kits.46 While their ubiquity across these low-biomass systems could be attributed to DNA extraction bias, their growth indicates that these taxa thrive in the oligotrophic drinking water environment.
Actinobacteria are Gram positive and play an important role in carbon recycling. Thus it is not surprising that Mycobacteria, extremely slow-growing oligotrophic bacteria commonly found in drinking water,45,48 were ubiquitous in these highly purified water samples. Some mycobacteria from drinking water are associated with disease,49 but the resolution of the methodology applied in this study did not allow for identification of pathogenic species.
Firmicutes are known to produce endospores, which may account for their survival through rigorous treatment processes. Primarily anaerobes (i.e., Clostridia and Bacilli) were detected. The family Chitinophagaceae within Bacteriodetes has been identified as surviving within free living amoeba in drinking water.50 This, along with the presence of other taxa that are known to infect amoebae in drinking water (including Bacillus, Ralstonia, Mycobacterium, Lactococcus, and Legionella)50 may indicate that amoeba play an important role in the survival and growth of bacteria in highly purified water.
The phylum TM6 is proposed as a symbiont of an unknown organism and it has been recovered from sinks in hospitals and several other drinking water related biofilms.51 It was also a frequently detected phylum based on RNA analysis of both bulk water and biofilms in a drinking water system in Germany.52 Further investigation into the phylum may be of importance to controlling oligotrophic bacteria.
Cyanobacteria are generally thought to be phototrophic bacteria, but have also been detected in municipal drinking water samples shielded from light.45 The relative abundance of Cyanobacteria decreased with storage time. Of the other phototrophic phyla, Chloroflexi, some of which were reported to be anaerobic,53 and Chlorobi, were found only sporadically and were not detected in Time Study 1, which was exposed to ambient light during storage. Thus phototrophy did not likely contribute measurably to the observed growth. The clade MLE1-12 was nearly ubiquitous and has also been identified in drinking water distribution systems45 and pharmaceutical wastewater,54 both of which are typically not exposed to light. Thus, the clade may not truly be phototrophic, although it is a member of the Cyanobacteria phylum.
The presence of ammonia oxidizers, nitrifiers and denitrifiers suggests that the nitrogen cycle may play an important role in nutrient-limited purified water environment. Besides the previously mentioned Nitrospira, a nitrite oxidizer, and Nitrosomonadaceae, a group of ammonia oxidizers, some species of the genus Ralstonia are associated with opportunistic pathogens and denitrification.55 DNA of ammonia oxidizers could also be an artifact of the use of chloramination for secondary disinfection in source tap water.
Proteobacteria appeared to be the primary drivers of growth in both time studies. These were able to proliferate in extremely oligotrophic environments, perhaps due to the phylum's wide variety of available metabolisms. Those that most effectively proliferated include the Bradyrhizobium genus and the Comamonadaceae family. Their roles in nitrogen fixation and H2 oxidation may play an important role in oligotrophic bacterial growth. Bradyrhizobium is commonly associated with nitrogen fixation in soils, and has previously been found in several ultra-pure water systems.26–28 It is also associated with free living amoeba in drinking water.50 The Comamondaceae family is associated with H2 oxidation.56
4.3 Implications for municipal water treatment and delivery
The increase in concentration of 16S rRNA genes collected over time from previously sterilized glass containers is suggestive of regrowth. The experiment was intended to identify the minimum possible proliferation likely in storage situations. As this study implemented pre-sterilized and baked labware, aseptic sample collection techniques, and focus on the bulk water rather than biofilm, it is likely that bacterial proliferation is even higher under typical storage conditions where such precautions are not taken. Similar growth occurred under both light and dark conditions, indicating that phototrophic effects are not likely the driving factor.This study may have implications for use of laboratory grade water as controls in laboratories. Although laboratory grade water used directly after production will only cause a minimal q-PCR increase, storage of the same water for as little as 48 hours may give as much as a 2–3 log increase in 16S rRNA genes detected and may not be adequate for comparison to experimental samples, especially if samples have inherently low DNA concentrations (i.e., drinking water experiments).
The kinds of microbes detected and their relative abundances were most profoundly affected by stagnation times. As differences in microbial assemblage compositions were more pronounced when abundance was taken into account (weighted), this may indicate that certain subsets of the bacteria present in the systems were especially prone to survive and thrive in the bulk of laboratory grade water. Samples collected on days six and seven from the two independent time experiments clustered closely together, indicating that the bacteria subject to re-growth in both experiments may have had similarly slow growth times, even with a difference in incubation conditions (light and dark).
Results from the storage tests also have important implications for nutrient limitations as a strategy for the control of bacterial regrowth in municipal waters. Under conditions engineered to minimize all nutrients including nitrogen, phosphorus, potassium and organic matter, including UV destruction of TOC (typically 2 ppb) and sterilization, the lowest level of bacterial growth achievable in bulk water after 10 days was 3 log 16S rRNA gene copies per mL. Assuming 5 16S rRNA gene copies per bacterial cell,57 the number of cells is estimated to be in the range of 2–3 logs per mL. Such stringent treatment approaches are not generally practical for municipal water systems, and even if implemented it is extremely difficult to maintain such low levels of nutrient levels in the distribution system, and even more so in building plumbing. At the end of drinking water distribution lines, and especially within buildings, stagnation cannot be avoided. Water age also increases when water-saving devices are used, further contributing to water quality issues.58 Stagnation of drinking water has previously been linked with changes in bacterial quantification and community composition in drinking water distributions systems.34,59 Stagnation of drinking water in Switzerland overnight resulted in a 2–3 fold increase in cell concentrations measured by flow-cytometry, and a change in microbial composition according to denaturing gradient gel electrophoresis.59 Even in systems providing a chloramine disinfectant residual, stagnation in the home resulted in significant increases in concentrations of genes of several organisms of concern.34 Stagnation in distillation systems in hospitals supported growth of the opportunistic pathogen Pseudomonas aeruginosa.24 This study reaffirms that total prevention of growth as water ages in a distribution system and in buildings is not plausible, as it occurs even with minimal supply of nutrients and maximized cell stress. Thus, nutrient limitation as a sole strategy for microbial control in distributed drinking water as it ages will have limited effectiveness, especially considering accumulation/concentration of nutrients and biomass in biofilms in ultrapure and potable water systems.10,18,22,25,32,60
5 Conclusions
Surveys of the water purification systems resulted in detection of a surprising array of bacterial 16S rRNA gene sequences. A portion of bacteria were alive and active, growing up to two logs during storage of as little as ten days, even under sterile set-up and dark incubation conditions. A shift in the microbial assemblage composition after about one week indicated that the Proteobacteria phylum was a key player in the regrowth occurring in this extremely oligotrophic environment. Nitrogen fixing (Bradyrhizobium) and H2 oxidizing (Comamonadaceae) bacteria were particularly dominant in highly purified water allowed to grow in storage for extended time periods.Acknowledgements
This study was funded by the US National Science Foundation (NSF) [CBET award 1336650], The Alfred P. Sloan Foundation – Microbiology of the Built Environment Program, and the Virginia Tech Institute for Critical Technology and Applied Science (ICTAS). The authors also thank the Lisa Belden laboratory at Virginia Tech for assistance in DNA sequence analysis. The findings do not represent the views of the sponsors.References
- ASTM-D1193, Specification for Reagent Water, ASTM International, 2011.
- ASTM-D5127, Standard Guide for Ultra-Pure Water Used in the Electronics and Semiconductor Industries, ASTM International, 2013.
- ASTM-D5196, Standard Guide for Bio-Applications Grade Water, ASTM International, 2013.
- M. R. Freije, ASHRAE J., 2012, 54, 100–101 Search PubMed.
- NIH Office of Research Facilities, Laboratory Water: It's Importance and Uses. National Institutes of Health, Division of Technical Resources, 2013 Search PubMed.
- M. J. Lehtola, I. T. Miettinen, T. Vartiainen, P. Rantakokko, A. Hirvonen and P. J. Martikainen, Water Res., 2003, 37, 1064–1070 CrossRef CAS.
- Thermo Scientific, Barnstead Nanopure™ TOC-UV ultrapure water systems Operation Manual, Dubuque, Iowa, 2009 Search PubMed.
- C. C. Chien, C. M. Kao, C. W. Chen, C. D. Dong and C. Y. Wu, Chemosphere, 2008, 71, 1786–1793 CrossRef CAS PubMed.
- S. Velten, M. Boller, O. Koster, J. Helbing, H. U. Weilenmann and F. Hammes, Water Res., 2011, 45, 6347–6354 CrossRef CAS PubMed.
- P. Servais, G. Billen and P. Bouillot, J. Environ. Eng., 1994, 120, 888–899 CrossRef CAS.
- C. C. Chien, C. M. Kao, C. D. Dong, T. Y. Chen and J. Y. Chen, Desalination, 2007, 202, 318–325 CrossRef CAS PubMed.
- W. V. Kayser, K. C. Hickman, W. W. Bond, M. S. Favero and L. A. Carson, Appl. Microbiol., 1975, 30, 704–706 CAS.
- U.S. Filter, Preventing microbial contamination in analytical grade water with regular water system sanitation, Siemens, 2001 Search PubMed.
- A. K. Rathod, S. Diwakar, A. Mundrigi and E. Herbig, Filtr. Sep., 2013, 50, 27–29 CrossRef.
- A. Gough, R. W. Attwell, D. F. D. Hardy and R. Caldwell, Solid State Technol., 1986, 29, 139–142 CAS.
- A. J. Semiao, O. Habimana, H. Cao, R. Heffernan, A. Safari and E. Casey, Water Res., 2013, 47, 2909–2920 CrossRef CAS PubMed.
- M. J. Lehtola, I. T. Miettinen, T. Vartiainen and P. J. Martikainen, Water Res., 2002, 36, 3681–3690 CrossRef CAS.
- I. C. Escobar, A. A. Randall and J. S. Taylor, Environ. Sci. Technol., 2001, 35, 3442–3447 CrossRef CAS.
- D. van der Kooij, J. – Am. Water Works Assoc., 1992, 84, 57–65 CAS.
- M. W. LeChevallier, N. J. Welch and D. B. Smith, Appl. Environ. Microbiol., 1996, 62, 2201–2211 CAS.
- A. K. Camper, B. Ellis, P. Butterfield, B. Anderson, P. Huck, C. Volk and M. LeChevallier, Investigation of the Biological Stability of Water in Treatment Plants and Distribution Systems, ed. AwwaRF, Denver, CO, 2000 Search PubMed.
- G. A. McFeters, S. C. Broadaway, B. H. Pyle and Y. Egozy, Appl. Environ. Microbiol., 1993, 59, 1410–1415 CAS.
- R. A. Governal, M. T. Yahya, C. P. Gerba and F. Shadman, J. Ind. Microbiol., 1991, 8, 223–228 CrossRef.
- M. S. Favero, L. A. Carson, W. W. Bond and N. J. Petersen, Science, 1971, 173, 836–838 CAS.
- H.-C. Flemming, Water Res., 1987, 21, 745–756 CrossRef CAS.
- C. L. Chen, W. T. Liu, M. L. Chong, M. T. Wong, S. L. Ong, H. Seah and W. J. Ng, Appl. Microbiol. Biotechnol., 2004, 63, 466–473 CrossRef CAS PubMed.
- M. Kawai, E. Matsutera, H. Kanda, N. Yamaguchi, K. Tani and M. Nasu, Appl. Environ. Microbiol., 2002, 68, 699–704 CrossRef CAS.
- L. A. Kulakov, M. B. McAlister, K. L. Ogden, M. J. Larkin and J. F. O'Hanlon, Appl. Environ. Microbiol., 2002, 68, 1548–1555 CrossRef CAS.
- V. Bohus, E. M. Tóth, A. J. Székely, J. Makk, K. Baranyi, G. Patek, J. Schunk and K. Márialigeti, Water Res., 2010, 44, 6124–6132 CrossRef CAS PubMed.
- N. Matsuda, W. Agui, T. Tougou, H. Sakai, K. Ogino and A. Masahiko, Colloids Surf., B, 1996, 5, 279–289 CrossRef CAS.
- V. T. Penna, S. A. Martins and P. G. Mazzola, BMC Public Health, 2002, 2, 13 CrossRef.
- F. Hammes, M. Berney, Y. Wang, M. Vital, O. Koster and T. Egli, Water Res., 2008, 42, 269–277 CrossRef CAS PubMed.
- M. T. Suzuki, L. T. Taylor and E. F. DeLong, Appl. Environ. Microbiol., 2000, 66, 4605–4614 CrossRef CAS.
- H. Wang, M. Edwards, J. O. Falkinham and A. Pruden, Appl. Environ. Microbiol., 2012, 78, 6285–6294 CrossRef CAS PubMed.
- J. G. Caporaso, C. L. Lauber, W. A. Walters, D. Berg-Lyons, J. Huntley, N. Fierer, S. M. Owens, J. Betley, L. Fraser, M. Bauer, N. Gormley, J. A. Gilbert, G. Smith and R. Knight, ISME J., 2012, 6, 1621–1624 CrossRef CAS PubMed.
- J. G. Caporaso, C. L. Lauber, W. A. Walters, D. Berg-Lyons, C. A. Lozupone, P. J. Turnbaugh, N. Fierer and R. Knight, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(Suppl 1), 4516–4522 CrossRef CAS PubMed.
- A. P. Masella, A. K. Bartram, J. M. Truszkowski, D. G. Brown and J. D. Neufeld, BMC Bioinf., 2012, 13, 31 CrossRef CAS PubMed.
- R. C. Edgar, Bioinformatics, 2010, 26, 2460–2461 CrossRef CAS PubMed.
- T. Z. DeSantis, P. Hugenholtz, N. Larsen, M. Rojas, E. L. Brodie, K. Keller, T. Huber, D. Dalevi, P. Hu and G. L. Andersen, Appl. Environ. Microbiol., 2006, 72, 5069–5072 CrossRef CAS PubMed.
- C. Lozupone and R. Knight, Appl. Environ. Microbiol., 2005, 71, 8228–8235 CrossRef CAS PubMed.
- K. R. Clarke and R. M. Warwick, Change in marine communities: an approach to statistical analysis and interpretation, 2nd edn, PRIMER-E Ltd., Plymouth, United Kingdom, 2001 Search PubMed.
- A. J. Pinto, J. Schroeder, M. Lunn, W. Sloan and L. Raskin, mBio, 2014, 5, e01135-14 CrossRef PubMed.
- A. J. Pinto, C. Xi and L. Raskin, Environ. Sci. Technol., 2012, 46, 8851–8859 CrossRef CAS PubMed.
- V. Gomez-Alvarez, R. P. Revetta and J. W. Santo Domingo, Appl. Environ. Microbiol., 2012, 78, 6095–6102 CrossRef CAS PubMed.
- E. P. Holinger, K. A. Ross, C. E. Robertson, M. J. Stevens, J. K. Harris and N. R. Pace, Water Res., 2014, 49, 225–235 CrossRef CAS PubMed.
- S. J. Salter, M. J. Cox, E. M. Turek, S. T. Calus, W. O. Cookson, M. F. Moffatt, P. Turner, J. Parkhill, N. J. Loman and A. W. Walker, BMC Biol., 2014, 12, 87 CrossRef PubMed.
- W. Lin, Z. Yu, H. Zhang and I. P. Thompson, Water Res., 2014, 52, 218–230 CrossRef CAS PubMed.
- R. Liu, Z. Yu, H. Zhang, M. Yang, B. Shi and X. Liu, Can. J. Microbiol., 2012, 58, 261–270 CrossRef CAS PubMed.
- J. O. Falkinham, 3rd, Emerging Infect. Dis., 2011, 17, 419–424 CrossRef PubMed.
- V. Delafont, A. Brouke, D. Bouchon, L. Moulin and Y. Hechard, Water Res., 2013, 47, 6958–6965 CrossRef CAS PubMed.
- J. S. McLean, M. J. Lombardo, J. H. Badger, A. Edlund, M. Novotny, J. Yee-Greenbaum, N. Vyahhi, A. P. Hall, Y. Yang, C. L. Dupont, M. G. Ziegler, H. Chitsaz, A. E. Allen, S. Yooseph, G. Tesler, P. A. Pevzner, R. M. Friedman, K. H. Nealson, J. C. Venter and R. S. Lasken, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2390–2399 CrossRef CAS PubMed.
- K. Henne, L. Kahlisch, I. Brettar and M. G. Hofle, Appl. Environ. Microbiol., 2012, 78, 3530–3538 CrossRef CAS PubMed.
- T. Yamada, Y. Sekiguchi, S. Hanada, H. Imachi, A. Ohashi, H. Harada and Y. Kamagata, Int. J. Syst. Evol. Microbiol., 2006, 56, 1331–1340 CrossRef CAS PubMed.
- T. M. LaPara, C. H. Nakatsu, L. Pantea and J. E. Alleman, Appl. Environ. Microbiol., 2000, 66, 3951–3959 CrossRef CAS.
- M. P. Ryan, J. T. Pembroke and C. C. Adley, Eur. J. Clin. Microbiol. Infect. Dis., 2011, 30, 1245–1247 CrossRef CAS PubMed.
- A. Willems, J. De Ley, M. Gillis and K. Kersters, Int. J. Syst. Bacteriol., 1991, 41, 445–450 CrossRef.
- J. A. Klappenbach, J. M. Dunbar and T. M. Schmidt, Appl. Environ. Microbiol., 2000, 66, 1328–1333 CrossRef CAS.
- C. Nguyen, C. Elfland and M. Edwards, Water Res., 2012, 46, 611–621 CrossRef CAS PubMed.
- K. Lautenschlager, N. Boon, Y. Wang, T. Egli and F. Hammes, Water Res., 2010, 44, 4868–4877 CrossRef CAS PubMed.
- K. Williams, A. Pruden, J. Falkinham and M. Edwards, Pathogens, 2015, 4(2), 355–372 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ew00134j |
| This journal is © The Royal Society of Chemistry 2015 |