
Research highlights: probing adsorbed organic coatings on nanoparticle surfaces
Stacey M.
Louie
* and
John M.
Pettibone
Materials Measurement Science Division, National Institute of Standards and Technology, Gaithersburg, MD 20899, USA. E-mail: stacey.louie@nist.gov
First published on 23rd September 2015
Abstract
Organic coatings adsorbed at a nanoparticle surface, together with the system properties, change the physicochemical properties of the nanoparticle, thereby possibly modifying its behavior and interactions with its surroundings. A detailed understanding of the coating and system properties and their relationship to nanoparticle behavior is therefore of great interest across all fields of nanotechnology, from medical and industrial applications to environmental implications of nanomaterials. Here, we highlight three studies that probe the properties and interactions of a variety of coatings on nanoparticle surfaces. In one study, both surface characterization methods and modeling approaches are applied to assess the persistence of citrate capping agents on gold nanoparticles upon adsorption of thiol ligands in ethanol. The second highlighted study demonstrates a labeling method for spatial mapping of binding sites on proteins adsorbed to colloidal particles, providing information on the composition and orientation of the adsorbed proteins. Finally, we highlight research that characterizes the interaction of proteins with grafted polymer coatings on nanoparticles and relates the structure of the resulting adsorbed layer to observed biological effects. These three studies outline general methodologies applicable for coating characterization.
The behavior of an engineered nanoparticle (NP) in environmental and biological systems is defined in part by adsorbed organic molecules or macromolecules that reside on the NP surface (Lowry et al., Environ. Sci. Technol., 2012, 46, 6893; Philippe and Schaumann, Environ. Sci. Technol., 2014, 48, 8946; Nel et al., Nat. Mater., 2009, 8, 543; Monopoli et al., Nat. Nanotechnol., 2012, 7, 779). A detailed understanding of the adsorbed coatings may be necessary to predict the behavior of NPs, including their attachment at interfaces, adsorptivity, reactivity, biouptake, and toxicity. A research need thus exists for thorough characterization of the properties of these coatings and determination of the most important properties to predict coated NP behavior in applied or natural systems. These properties may include the coating’s chemical composition, physical conformation or structure, and spatial orientation of functional moieties around the NP. Here, three recent articles are highlighted that demonstrate comprehensive or novel characterization methodologies to investigate adsorbed ligands and macromolecules on NP surfaces.
Characterizing the adsorbed layer structure of citrate-stabilized gold NPs upon thiol functionalization
Citrate-stabilized gold nanoparticles (AuNPs) are used ubiquitously in nanomaterial studies, and thiol ligands are often applied to these NPs for the purpose of obtaining a dense, strongly-bound surface coating with a desired chemistry or functionality (Sperling and Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333). Often, the citrate layer is assumed to be readily displaced by thiols, which have higher binding energies to the gold surface. However, for ligand exchange performed in an organic solvent (ethanol), a recent study by Park and Shumaker-Parry demonstrated that citrate molecules on AuNPs show a strong resistance to desorption under aryl- and alkanethiol functionalization (Park and Shumaker-Parry, ACS Nano, 2015, 9, 1665). Although extrapolation to aqueous solutions of electronically similar thiols may not result in similar retained citrate, the experimental and modeling approaches used in the paper demonstrate characterization methods that can be applied in future work using environmental media.Park and Shumaker-Parry began by quantifying the citrate and dodecanethiolate (DDT) moieties present on the surface of citrate-reduced AuNPs (≈40 nm diameter) after functionalization in ethanolic solution. Both attenuated total reflectance infrared (ATR-IR) spectra and X-ray photoelectron spectroscopy (XPS) indicated the persistence of COOH groups and gold–carboxylate (Au–COO−) coordinates after functionalization with dodecanethiol. Notably, ATR-IR bands attributed to surface hydrogen-bonded COOH moieties of the citrate were not significantly diminished upon thiol functionalization, suggesting the role of a stable hydrogen-bonded citrate network on the AuNP surface. The authors also used XPS to quantitatively assess the surface coverage of various aryl and alkane thiols. In brief, the authors found lower than monolayer (ML) surface coverages of the thiols and a nonzero citrate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :alkanethiolate ratio, which provided further evidence of incomplete exchange of citrate by thiol.
:alkanethiolate ratio, which provided further evidence of incomplete exchange of citrate by thiol.
Finally, Park and Shumaker-Parry proposed a model of the surface structure of the mixed thiolate and citrate layer on the AuNP surface, in which the alkanethiolates bind in the vacant surface area unoccupied by the hydrogen-bonded citrate network (Fig. 1). The proposed model and experimental results describing an upper limit of 0.69 ML coverage were consistent for the specific system studied by the authors. Again, the authors emphasized the contribution of the strong hydrogen bonding, as well as possible steric and chelating effects of the citrate layer, to the resistance of citrate to ligand exchange by thiols.
 | ||
| Fig. 1 Structural model of the hydrogen-bonded citrate layer on a AuNP (111) surface (a) and adsorption of alkanethiolates (CH3–C9–S–) at vacant sites (b), resulting in a mixed layer on the AuNP in ethanol solution. Reprinted with permission from Park and Shumaker-Parry, ACS Nano, 2015, 9, 1665. Copyright 2015 American Chemical Society. | ||
In summary, Park and Shumaker-Parry provided direct measurement and a mechanistic explanation for the existence of a mixed citrate and thiolate layer on AuNPs in ethanol. While the characterization approaches used in this study are often used in surface science, wider adoption of this methodology is needed in environmental studies for analogous coated NPs in aqueous systems. Ligand exchange is often used in the preparation of commonly used macromolecule-coated NPs (e.g., coated Au or Ag NPs prepared by polymer adsorption to citrate-stabilized NPs), and possible co-adsorption of ligands may result in a more complex surface chemistry than assumed. Detailed measurement of the adsorbed layer, as demonstrated in the highlighted article, will enable a more complete understanding of the surface properties of the NPs and better predictions of their environmental and biological fate.
Mapping spatial locations of functional motifs of protein coatings on NPs
The behavior of NPs in biological systems is well known to depend on the formation of protein coatings, or coronas, on the NPs (Nel et al., Nat. Mater., 2009, 8, 543; Monopoli et al., Nat. Nanotechnol., 2012, 7, 779; Lynch et al., Adv. Colloid Interface Sci., 2007, 134–135, 167). Properties of interest for the coatings include not only adsorbed mass and composition, but also the conformation and orientation of the proteins, which affect the activity of the folded protein. Therefore, a detailed understanding of the structure of the protein coating will likely be necessary to evaluate the biological pathway of engineered NPs. In a recent article, Kelly et al. demonstrated a methodology incorporating immunogold labeling and characterization by differential centrifugal sedimentation (DCS), transmission electron microscopy (TEM), and scanning TEM (STEM) to identify, quantify, and map the location of epitopes on the protein coating (Kelly et al., Nat. Nanotechnol., 2015, 10, 472).Kelly et al. first demonstrated the labeling method for polystyrene particles (220 nm diameter) coated with transferrin protein (Tf). Two types of immunogold NPs (e.g., 5 nm gold NPs coated with antibody conjugates) were prepared with antibodies specific to two epitopes near the receptor binding site of Tf. Semi-quantitative assessment of the number of target epitopes on the Tf-coated polystyrene was performed by titration with the immunogold NPs and characterization by DCS. In DCS, the sedimentation time of the particles depends on the mass and size of the particle, and hence the number of bound immunogold labels (Fig. 2a). The authors confirmed qualitative consistency between the DCS results and the number of bound immunogold labels to the Tf-coated particles counted in TEM images (Fig. 2b). A relatively small number of epitopes were labeled by the immunogold compared to the estimated number of adsorbed Tf, which was attributed to random orientation of the adsorbed Tf and hence obscuration of epitopes at the particle surface or within the protein coating. Finally, STEM imaging was used to map the three-dimensional spatial orientation of the immunogold labels. Analysis of the inter-particle distances between the immunogold NPs further suggested a random arrangement and adsorption process for Tf on the polystyrene particle (Fig. 2c).
 | ||
| Fig. 2 Schematic of the immunogold labeling method and analysis by DCS (a), in which the net particle density increases with titration of the epitopes from low to high immunogold concentrations and results in reduced sedimentation time of the particles. TEM imaging is also used to count the number of adsorbed labels for titration from low to high concentrations of two immunogold antibody labels (monoclonal (mTf) and polyclonal (pTf)) (b). STEM is used for three-dimensional mapping of the location of the immunogold labels (c). Adapted by permission from Macmillan Publishers Ltd: Nature Nanotechnology, Kelly et al., Nat. Nanotechnol., 2015, 10, 472, copyright 2015. | ||
Complementary analyses were performed to further assess the conformation and binding properties of the adsorbed protein. These methods included tryptophan fluorescence analysis to assess possible denaturation of the Tf, as well as sequential titration of the Tf-coated particles with transferrin receptor (TfR) followed by the immunogold labels to better assess the degree of receptor binding capacity.
In addition to the single-protein (Tf) system, Kelly et al. demonstrated the immunogold labelling approach for a complex protein coating that was formed on carboxylated polystyrene NPs in media containing human plasma. Immunogold labels were prepared for two of the adsorbed proteins, Tf and immunoglobulin G (IgG). Sequential titration with the two immunogold labels indicated a higher abundance of IgG than Tf, consistent with mass spectrometry results for the stripped coating. The authors also noted a wide variance in the number of adsorbed labels per particle for the complex protein mixture compared to the single-protein coating, suggesting that heterogeneity in coating formation across a NP population may need to be considered in realistic biological systems.
This study demonstrated that immunogold labeling can be a useful approach for spatial mapping of epitopes on protein coatings and evaluation of potential biological interactions at the outer surface of the coated NPs. As shown by the authors, the application of this method as part of a suite of complementary coating characterization methods can provide a more holistic understanding of the structure and behavior of NPs transformed by protein adsorption in biological systems.
Probing interactions of proteins with polyethylene glycol coatings on NPs
Polyethylene glycol (PEG) coatings are often applied to NPs for drug delivery applications to change the pharmacokinetics of the NPs (Jokerst et al., Nanomedicine, 2011, 6, 715). One mechanism by which PEG coatings modify biological uptake and circulation is through the reduction of protein adsorption onto the PEGylated NP. We highlight research by Pelaz et al. that probes the adsorption of protein onto PEGylated iron platinum (FePt) NPs and relates the structure of the resulting adsorbed layer to cellular uptake (Pelaz et al., ACS Nano, 2015, 9, 6996).Pelaz et al. used FePt NPs (1.6 nm radius) coated with poly(maleic anhydride-alt-dodecene) (PMA) that was covalently labeled with the fluorescent dye DY-636, enabling hydrodynamic size determination by fluorescence correlation spectroscopy (FCS) as well as fluorescence lifetime studies to evaluate the environment around the fluorophore. Amine-terminated PEGs of three molecular weights were covalently bound to the carboxyl groups of the PMA to achieve a high grafting density of PEG. To distinguish the intrinsic effects of the PEG coating from its effects on the size and surface charge of the NPs, two controls were also assessed: a PMA-coated Fe3O4 NP of comparable size to the PEGylated NPs (but similar surface charge to the non-PEGylated FePt-PMA), and a glucose-coated FePt-PMA NP of comparable charge to the PEGylated NPs (but similar size to the non-PEGylated FePt-PMA) (Fig. 3).
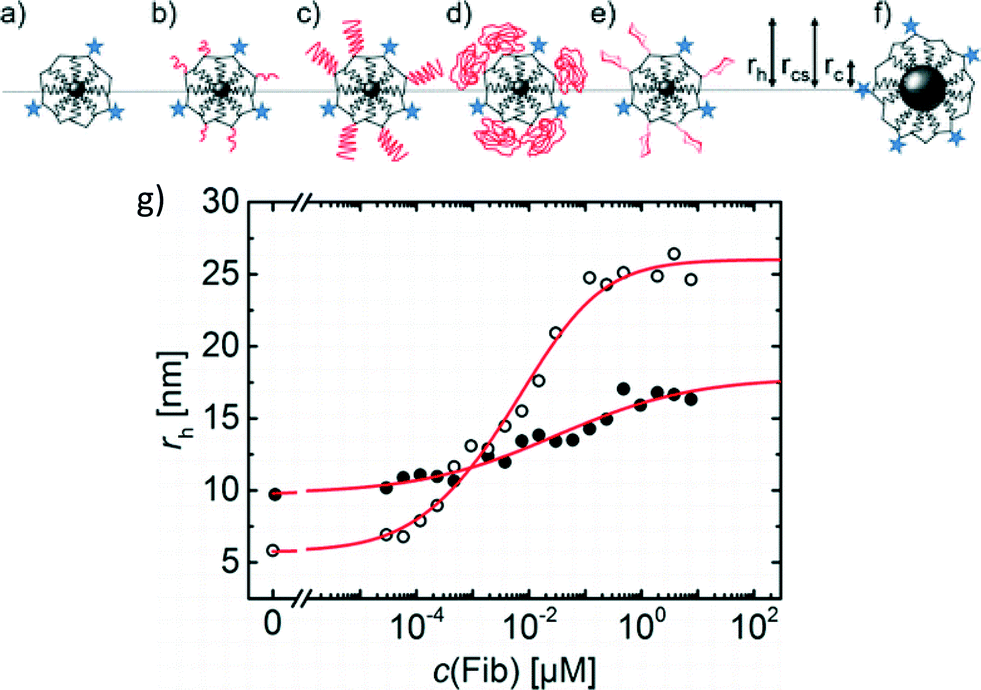 | ||
| Fig. 3 Sketches of the NPs assessed: FePt-PMA (a), FePt-PMA-PEG750 (b), FePt-PMA-PEG5k (c), FePt-PMA-PEG10k (d), FePt-PMA-glucose (e), and Fe3O4-PMA (f). The radius of the core (rc), core with polymer shell (rcs), and hydrodynamic radius (rh) were measured. Blue stars represent the DY-636 fluorophore, molecules drawn in black represent PMA, and molecules drawn in red represent either PEG or glucose. (g) FCS measurements indicated a lower Δrh for FIB adsorption to the PEGylated FePt-PMA (filled circles) than the non-PEGylated FePt-PMA (open circles), similar to results for HSA adsorption. Adapted with permission from Pelaz et al., ACS Nano, 2015, 9, 6996. Copyright 2015 American Chemical Society. | ||
Adsorption of human serum albumin (HSA) to the non-PEGylated and PEGylated NPs and the two controls was assessed. By fitting the increase in hydrodynamic diameter (rh) with increasing HSA concentration to the Hill equation, several adsorption parameters were calculated, including the maximum change in rh at HSA saturation (Δrh) and the binding affinity of the HSA. PEGylation did not prevent adsorption of HSA, but a lower Δrh was observed compared to the non-PEGylated and control NPs. The authors postulated that the lower Δrh could result from either a lower number of protein molecules adsorbed to the PEGylated particles or penetration of HSA into the PEG layer. To distinguish these scenarios, fluorescence lifetime measurements were performed. Adsorption of HSA resulted in a longer fluorescence lifetime of the DY-636 for both the PEGylated and non-PEGylated NPs, suggesting that HSA penetrated the PEG coating and interacted with the underlying DY-636-tagged NP surface. Similar conclusions were also drawn for adsorption of large, rod-like fibrinogen (FIB) proteins, where the lower Δrh for adsorption to the PEGylated NPs compared to the non-PEGylated NPs was attributed to penetration of the FIB into the PEG layer (Fig. 3g).
Uptake of the NPs by 3T3 murine fibroblast cells in medium containing fetal bovine serum proteins was then assessed. Despite adsorption of protein onto all particles, a significant decrease in uptake by the NPs coated with higher molecular weight PEGs was observed compared to the non-PEGylated FePt-PMA and control NPs. This result further suggested that the proteins penetrated into the PEG coating, such that the PEG layer provided a physical barrier between the proteins and the cells and hence could still reduce cellular uptake of the PEGylated NPs.
Pelaz et al. concluded that, even with dense covalent grafting of PEG to the NPs, the high surface curvature of the NPs likely resulted in a PEG layer of insufficient density to prevent protein adsorption. Discernment of protein penetration within the PEG layer provided an explanation for the physical properties (e.g. size) of the PEGylated NPs upon protein adsorption as well as the effects on cellular uptake. More broadly, this study shows that interactions of polymer-coated NPs with other macromolecules in solution may result in layers containing interspersed macromolecules rather than simple displacement or overcoating of the initial coating. Such interactions should be considered in future studies assessing transformations of polymer-coated NPs in complex environmental and biological media containing natural organic matter and proteins.
| This journal is © The Royal Society of Chemistry 2015 |