
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysicochemical and ion-binding properties of highly aliphatic humic substances extracted from deep sedimentary groundwater†
Takumi
Saito
*ab,
Motoki
Terashima
c,
Noboru
Aoyagi
d,
Seiya
Nagao
e,
Nobuhide
Fujitake
f and
Toshihiko
Ohnuki
b
aNuclear Professional School, School of Engineering, The University of Tokyo, 2-22 Shirakata Shirane, Tokai-mura, Ibaraki, 319-1188, Japan. E-mail: saito.takumi@jaea.go.jp; takumi.saito@nuclear.jp; Fax: +81-29-282-5927; Tel: +81-29-284-3518
bAdvanced Science Research Center, Japan Atomic Energy Agency (JAEA), 2-4 Shirakata, Tokai-mura, Ibaraki, 319-1195, Japan
cRadioactive Waste Processing and Disposal Research Department, JAEA, 4-33 Muramatsu, Tokai-mura, Ibaraki, 319-1194, Japan
dNuclear Science and Engineering Center, JAEA, 2-4 Shirakata, Tokai-mura, Ibaraki, 319-1195, Japan
eInstitute of Natural and Environmental Technology, Kanazawa University, Wake, Nomi, Ishikawa 923-1224, Japan
fGraduate School of Agricultural Science, Kobe University, Rokkodai1, Kobe 657-8501, Japan
First published on 2nd July 2015
Abstract
Humic substances (HSs) are ubiquitous in various aquatic systems and play important roles in many geochemical processes. There is increasing evidence of the presence of HSs in deep groundwater; nevertheless, their ion binding properties are largely unknown. In this study we investigated the physicochemical and ion-binding properties of humic and fulvic acids extracted from deep sedimentary groundwater. The binding isotherms of protons (H+) and copper (Cu2+) were measured by potentiometry and fitted to the NICA-Donnan model, and the obtained parameters were compared with the generic parameters of the model, which are the average parameters for HSs from surface environments. The deep groundwater HSs were different from surface HSs, having high aliphaticities, high sulfur contents, and small molecular sizes. Their amounts of acidic functional groups were comparable to or slightly larger than those of surface HSs; however, the magnitude of Cu2+ binding to the deep groundwater HSs was smaller. The NICA-Donnan model attributed this to the binding of Cu2+ to chemically homogeneous low affinity sites, which presumably consist of carboxylic groups, via mono-dentate coordination at relatively low pH. The binding mode tended to shift to multi-dentate coordination with carboxylic groups and more heterogeneous alcoholic/phenolic groups at higher pH. X-ray absorption spectroscopy also revealed that Cu2+ binds to O/N containing functional groups and to a lesser extent S containing functional groups as its divalent from. This study shows the particularity of the deep groundwater HSs in terms of their physicochemical and ion-binding properties, compared with surface HSs.
Environmental impactFor future use of deep underground space it is necessary to monitor and protect the quality of deep groundwater. Development of mechanistic models that can describe reactions of pollutants with components in groundwater is mandatory, as is the case for surface water systems. Humic substances (HSs) play important roles in the speciation of metal ions; nevertheless, the details of ion binding to deep groundwater HSs are largely unknown. This study reveals the particularity of the physicochemical and ion-binding properties of the HSs extracted from sedimentary groundwater by comparing them to those of surface HSs. |
1. Introduction
Humic substances (HSs) are a class of natural organic matter resulting from the degradation and condensation of animal, plant and microbial remains, and are ubiquitous in various environments: surface and ground water, ocean, soil, and the atmosphere.1,2 A HS is not a molecular entity with a distinct structure, but should be considered as a group of organic molecules with certain physicochemical properties in common.3 Based on the solubility to water at different pH values, they are operationally divided into humic acid (HA), which is soluble at pH > 2, fulvic acid (FA), which is soluble at both acidic and alkaline pH, and insoluble humin. HSs play important roles in various environmentally-relevant processes; they determine structures of micro aggregates in soils,3 stabilize metastable minerals,4 catalyze redox reactions,5 and capture inorganic and organic contaminants.6–8 Protons and metal ions readily bind to the functional groups of HSs,8,9 mostly carboxylic and phenolic groups and less significantly amine and sulfur-containing groups, and alter their reactivity, bioavailability, and mobility.7,10Ion binding to HSs has been an active topic of research for decades.11–13 The particularity of HSs as ligands lies in their chemical heterogeneity and polyelectrolyte nature.9,13 The former is manifested in the distribution of the affinity constant of a HS for a given metal ion due to the diversity of the environments surrounding its functional groups. The latter originates from negative charges located on its carbon backbone, which creates a negative electrostatic potential that attracts cations and excludes anions.14,15 Recent mechanistic models for ion binding to HSs such as the NICA-Donnan model9,11 and the Model VI and its successor12,16 take these aspects into account and can successfully describe the binding of various cations over a wide range of conditions. These models have been well tested for HSs extracted from different surface environments.12,17,18 There are certain similarities in the obtained model parameters, once HAs and FAs are separately discussed.17 Thus, so-called “generic” parameters have been proposed for these two groups of HSs12,17,18 and are widely used to estimate the level of metal binding to HSs for which specific model parameters are unavailable.19,20
There is increasing evidence showing the presence of HSs in deep underground environments either as dissolved forms in pore water or bound to host rocks.21–29 Deep underground environments are rather different from surface aquatic systems, as manifested by slow groundwater flow, which leads to prolonged interaction between rocks and dissolved/suspended components, low oxygen concentration, and no direct energy input from the sun. It is likely that HSs in deep underground environments are different from their counterparts in surface environments with respect to their structures and ion binding properties.21,23 Underground HSs may originate from surface waters transported by downward recharge, dissolution of sedimentary organic materials, or in situ production from the remains of microorganisms or algae in connate water, and have experienced long-term diagenesis. Ratios of dissolved and bound HSs in deep underground environments are different from site to site, and they may have rather different properties even in a given geological setting.30
The uniqueness of deep underground HSs has been pointed out by several researchers.21,23–26,29,31,32 Schäfer et al.23 studied possible sources of FAs in the Gorleben aquifer, based on isotopic data and C and S X-ray absorption near-edge spectroscopy (XANES). They reported that FAs derived from the deep brine groundwater at around −216 m below ground level (bgl) had similar carbon backbone structures to those of FA in the corresponding shallow recharge groundwater and that HAs and FAs originated from lignite in Miocene sediments were highly aromatic. Alberts et al.32 reported that the properties of HA and FA extracted from groundwater at −30 to −70 m bgl were different from those of their counterparts from surface water, but copper binding to them was similar. Courdouan et al.30 studied the binding of trivalent metal ions to extracts of organic matter from pore water and rocks in the Opalinus clay (OPA) and the Callovo-Oxfordian formations and found stronger binding of curium to pore-water organic matter from OPA. Although some properties of deep underground HSs such as elemental composition, 13C NMR carbon distribution, optical properties, and molecular-size distribution have been reported,21,24–26 their ion binding properties over a wide range of conditions remain largely unknown.31–33 This is particularly the case for deep groundwater HSs, as large-scale extraction of HSs from deep groundwater is limited.
Deep groundwater HSs can be extracted from pumped groundwater, using boreholes from the surface. Some countries are operating underground research laboratories (URLs) for feasibility tests of geological disposal of nuclear wastes, where large amounts of groundwater samples are available with less contamination or alteration.34–37 This makes URLs appropriate places for extraction of deep groundwater HSs.
Considering the future uses of deep underground space by mankind such as geological disposal of nuclear wastes and potential deterioration of groundwater quality, the ion binding properties of deep groundwater HSs are to be studied and the applicability of the aforementioned mechanistic models is to be tested, as is the case for surface HSs. Thus, the objective of this study is to reveal the physicochemical and ion-binding properties of HA and FA isolated from sedimentary groundwater at the Horonobe URL of the Japan Atomic Energy Agency (JAEA).35 The physicochemical properties of the Horonobe HSs, which are denoted as HHSs hereafter, were compared with those of surface HAs and FAs to discuss their structural differences. Binding isotherms of protons (H+) and copper (Cu2+) were measured over a wide range of conditions by potentiometric titration and fitted to the NICA-Donnan model.9,11 The results were compared to the model calculations with the generic parameters proposed by Milne et al.,18 which capture average trends of ion binding to HSs from surface environments. The oxidation state and local coordination environment of Cu2+ bound to the HA fraction of the HHSs were also assessed by X-ray absorption spectroscopy (XAS). Copper was chosen as a representative divalent metal ion in this study to examine the general metal binding properties of the HHSs, as it can be easily quantified by an ion selective electrode (ISE) and its binding to surface HSs has been well studied.18,38,39 Copper is also an essential trace element for organisms at low concentration and becomes toxic at elevated concentrations.40,41 It could be introduced to deep groundwater systems by exploitation of underground space, as it is an important constituent of various materials used in modern industries. Thus, studies on the speciation Cu2+ in the presence of groundwater HSs are relevant for its fate in deep groundwater environments. Although the HA and FA from a single groundwater source are examined in this study, the outcomes can be applied or be a good starting point to estimate the degree of metal binding to HSs in sedimentary groundwater similar to this study.
2. Materials and methods
2.1. Materials
For all experiments, Milli-Q grade pure water and analytical-grade chemicals purchased from Wako Pure Chemical Industries were used, unless otherwise noted.HA and FA were extracted from groundwater collected at the −250 m gallery of the Horonobe URL located in the northern part of Hokkaido Prefecture, Japan. The geology and geochemistry of the site are described elsewhere.35,42 The −250 m gallery is located at the boundary of the Pliocene Koetoi and the Miocene Wakkanai formations, which are composed of diatomaceous and siliceous mudstones, respectively. Groundwater at the sampling location is a weakly alkaline Na+/HCO3− type with relatively high total organic carbon (TOC) and Cl− levels. Groundwater after filtration and acidification was passed through a column packed with DAX-8 resins (Supelite DAX-8, Sigma-Aldrich). Separation and extraction of HA and FA fractions from the loaded resins and subsequent purification was performed in a laboratory on the surface, according to the protocol recommended by the International Humic Substances Society (IHSS).43 In total 6.6 g of HA and 3.5 g of FA were obtained by treating approximately 6000 L of the groundwater. These values correspond to approximately 8.5 and 4.5% of the TOC in the groundwater, respectively. Hereafter, the HA and FA fractions are denoted as HHA and HFA, respectively.
2.2. Characterization of HSs
Elemental compositions and 13C NMR carbon distributions of the HHSs were evaluated in the same manner as a previous report.33 Carbon, H and N contents were determined using an elemental analyzer (Yanagimoto, MT-6), and that of S was analyzed by ion chromatography after conversion to SO42−. Ash contents were also determined by combustion at 550 °C. For 13C NMR a 50 mg HS sample was dissolved in a mixture of 0.02 mL of 10 M NaOD (99.9% deuteration, Sigma-Aldrich) and 0.4 mL of D2O (99.9% deuteration, Sigma-Aldrich) solution. As a reference material for the chemical shifts, 0.02 mL of a 1.0% solution of sodium 3-trimethylsilylpropionate-2,2,3,3-D4 (TMSP; 98% deuteration, French Atomic Energy Commission, CEA) was added into the mixture. The test solution was then passed through a glass-fiber filter with a pore size of 0.7 μm, and placed in a 5 mm diameter spin tube. 13C NMR spectra were recorded by a Bruker AVANCE 500 spectrometer operating at 125.77 MHz. The inverse gated decoupling technique was applied for the measurement with a pulse width of 45° and acquisition time of 0.839 s. A total repetition time of 2.5 s was applied to permit relaxation of all the spins, and 4000–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans were accumulated. Chemical shift assignments were made using data reported by Wilson44 and Fujitake and Kawahigashi.45
000 scans were accumulated. Chemical shift assignments were made using data reported by Wilson44 and Fujitake and Kawahigashi.45
The UV/Vis absorption spectra of the HHS as well as those of the standard or reference HSs from the international humic substances society (IHSS) and the Japanese humic substance society (JHSS) and purified Aldrich HA (PAHA)46 were measured in this study by a UV/Vis spectrometer (UV-3100, Shimadzu). The samples were prepared as 50 mg L−1 HS solutions in 0.01 M NaHCO3 buffer.47 The size distributions of HHA and HFA were determined by flow-field flow fractionation (Fl-FFF) with 1 kDa polyethersulfone membrane (AF2000, Postnova), according to Lukman et al.48 The electron accepting capacities (EAC) of the HHS and the standard HSs from the IHSS and JHSS were determined by the mediator electrochemical reduction (MER) in a similar way to Aeschbacher et al.,49 using diquat dibromide monohydrate (99.5%, Supelco) as a mediator. The details of the MER measurement are given in the ESI.†
2.3. Potentiometric titration
Potentiometric titration of HHA and HFA was performed using the Wallingford titration system.50 HHS solutions were prepared by dissolving the freeze-dried samples in alkaline solutions at around pH 9 and kept stirred overnight.51 The pH and Cu2+ activities were measured by a glass electrode (Metrohm, 6.0150.100) and a Cu ISE (Methrom, 6.0502.140), combined with a 3 M KCl Ag/AgCl reference electrode (Metrohm, 6.0733.100) in an electrolyte bridge (0.1 M NaClO4, Schott B511). All titrations were performed in a thermostated vessel under a slight over-pressure of moisturized Ar and continuous stirring. The glass electrode was calibrated by titrating HClO4 solutions with 0.1 M NaOH. The Cu ISE was calibrated by titrating 0.36 mM Cu(ClO4)2 solution with 0.02 M ethylenediamine solution (Aldrich). The electrode calibrations were performed at the same salt levels as those in the subsequent sample titrations.Proton binding isotherms of the HHSs were obtained by acid–base titration, as described elsewhere.46 30 mL of a 1 g L−1 HHA or HFA solution was first titrated to pH 4 and stirred for 1 hour; then three-sets of forward and backward titrations were performed. The salt concentration of the solution was increased by adding a 1 M NaClO4 solution (Merck) between the different sets of titrations. At every point of the titration the reading of the glass electrode was recorded when the drift became less than 0.1 mV min−1 or after 30 min. The relative positions of the charge (−q)–pH curves of the HHSs at the different salt levels were determined from the amounts of base and acid titrants necessary to back-titrate H+ released in the pH-stat salt titration. The absolute position of the curves was then determined by optimizing the initial negative charge, q0, at the beginning of the titration in the NICA-Donnan fitting,11 as described in the ESI.† The results of the forward base titration were used in the subsequent fitting, as the hysteresis between the forward and backward titrations at a given salt level was small. The uncertainty in the determination of the HHS charge, q, was estimated to be less than 0.1 meq g−1, using a typical standard deviation of the glass electrode calibration (0.05 pH unit).
Copper binding isotherms to the HHSs were measured by pH-stat titration at three pH levels (pH 4, 6, and 8) and 0.1 M NaClO4.38 At pH 4 the additional titration of 0.01 M NaClO4 was performed. The Cu2+ titration to HFA at pH 8 failed most likely because of poor pH buffering (see the discussion below). 30 mL of a 1 g L−1 HHA or HFA solution was first titrated to pH 4 and stirred for 1 hour, and then to a desired pH and equilibrated within 0.2 mV (0.003 pH unit) for 30 min. After equilibration a 0.1 M Cu(ClO4)2 solution or 10−3 M Cu(ClO4)2 solution in 0.1 M NaClO4 was added. The pH of the sample solution was back-titrated to the original value and kept within 0.2 mV for 20 min by addition of the acid and base titrants. Readings of the electrodes were recorded after their drifts became less than 0.1 mV min−1 or after 20 min. At each titration point the solution was checked for the formation of Cu(OH)2(s) (logKsp = −19.32 (ref. 52)) and the Cu2+ binding amount ([Cu2+]bound) was calculated by subtracting the sum of the concentrations of free Cu2+ and its hydrolysis products from the total concentration, using the hydrolysis constants of Cu2+.52 The magnitude of the uncertainty in log[Cu2+]bound was estimated to be no more than 0.2, using a typical error of the Cu ISE calibration (0.06 as the logarithm of Cu2+ activity, logaCu).
The obtained H+ and Cu2+ binding isotherms to HHA and HFA were fit to the NICA-Donnan model,9,11 using an in-house MATLAB® program. The details of the model as well as the fitting procedure are given in the ESI.† First, the maximum density of H+ binding sites, Qmaxj,H of the site j (j = 1 and 2 for the low-affinity and high-affinity sites, respectively), the median values of the affinity constants of the site j for H+, ![[K with combining tilde]](https://www.rsc.org/images/entities/i_char_004b_0303.gif) j,H, the apparent heterogeneity parameters of the site j, mj, the Donnan parameter, b, and q0 were optimized, using the charge/pH curves. Then, the median values of the affinity constants of the site j for Cu2+, j,Cu, the ion-specific non-ideality parameters of the site j for H+ and Cu2+, nj,H, nj,Cu, and the heterogeneity parameters of the site j, pj, which correspond to the reciprocal of the width of the affinity distribution, were optimized by fitting to the Cu2+ binding isotherms, while nj,H × pj. was kept equal to mj.11 The lower and upper boundaries were set to 0 and 1 for the mj, nj,i and pj parameters.
j,H, the apparent heterogeneity parameters of the site j, mj, the Donnan parameter, b, and q0 were optimized, using the charge/pH curves. Then, the median values of the affinity constants of the site j for Cu2+, j,Cu, the ion-specific non-ideality parameters of the site j for H+ and Cu2+, nj,H, nj,Cu, and the heterogeneity parameters of the site j, pj, which correspond to the reciprocal of the width of the affinity distribution, were optimized by fitting to the Cu2+ binding isotherms, while nj,H × pj. was kept equal to mj.11 The lower and upper boundaries were set to 0 and 1 for the mj, nj,i and pj parameters.
2.4. XAS analysis
Copper K-edge XANES and extended X-ray absorption fine structure (EXAFS) analyses of Cu2+ reacted with HHA and PAHA were carried out at the BL-27B in the KEK Photon Factory (Tsukuba, Japan). The HA samples were dissolved in alkaline solutions at 4.0 g L−1 and stirred overnight under an Ar atmosphere. After adjusting the pH to 4 or 7 with 0.1 or 0.01 M HCl and NaOH, a 10 mM CuCl2 solution was added to achieve a Cu2+ loading of 80 mmol Cu per kg HA, and pH was re-adjusted to the original values. These values correspond to the Cu2+ binding amounts of 13.3 (log[Cu2+]bound = −1.88) and 310 (log[Cu2+]bound = −0.51) mmol kg−1, respectively, according to the NICA-Donnan model calculation with the optimized parameters for HHA. After equilibration for two days, the samples were freeze-dried, mixed with boron nitride (BN), and pressed into pellets, which were covered by a Kapton® tape. Reference solid compounds (CuIIO, CuIICl2, CuICl, and CuISCN) were dispersed in BN and pressed into pellets. In addition a 0.02 M Cu2+ solution with 0.04 M L(+)-tartrate at pH 7 was also measured as a reference.Copper K-edge X-ray absorption spectra (XAS) were measured in fluorescence mode at 148 K using a liquid N2 cryostat equipped with Kapton® windows (CoolSpek, UNISOKU) for the HA samples and in transmission mode at room temperature for the reference materials. A Si(111) double crystal monochromator was detuned by about 50% to reject higher harmonic intensity. Reduction and theoretical fitting of the obtained XAS data was performed by the Athena and Artemis software packages53 and FEFF 6.54 The details of the data reduction and fitting are given in the ESI.†
3. Results and discussion
3.1. Physicochemical properties of the HHSs
The elemental compositions and 13C NMR distributions of HHA and HFA are given in Table 1 and the more detailed physicochemical properties of HHA and HFA are summarized in Table S1 in the ESI† together with those of the IHSS and JHSS HSs and PAHA. These standard or reference HSs are isolated from various surface environments, ranging from soils (EHA from the Elliot soil, IHA and IFA from the Inogashira volcanic ash soil, DHA and DFA from the Dando forest soil), peats (PHA from the Pahokee peat), oxidation products of lignite (Leonardite HA, LHA), rivers (SRHA and SRFA from the Suwannee River), and lakes (NHA and NFA from the Nordic lake and BFA from the Biwako lake). The elemental compositions of the HHSs are characterized by their low oxygen and ash contents and high hydrogen and sulfur contents, compared to HAs and FAs from surface environments. The relatively small O/C and large H/C ratios of the HHSs can be seen in the van Krevelen plot (Fig. 1).55 In Fig. S1 of the ESI† a similar plot with broader data from the literature is also shown. The locations of the standard and commercial surface HAs and FAs depend on their types and origins (Fig. 1). The former are characterized by relatively high O/C ratios; the latter exhibit a wide range of H/C ratios with constant and relatively small O/C ratios. The aquatic HA and FA are close to each other except for BFA. Groundwater HSs tend to have small O/C ratios, as is shown in Fig. S1.† Some groundwater HAs as well as marine HSs show relatively large H/C ratios. Compared with HSs from various environments, HHA and HFA are different in terms of the O/C and H/C ratios as is shown in Fig. 1. The large H/C values of the HHSs indicate the abundance of saturated carbons, which is in line with their small aromaticities estimated from the 13C NMR C distribution (Tables 1 and S1 in the ESI†). The highly aliphatic nature of organic matter in deep underground environments has been pointed out by several researchers.26,29,31 Pettersson et al.26 reported an even higher H/C value (H/C = 1.7) for HA extracted from granitic groundwater at −280 m bgl. Claret et al.31 also reported the highly aliphatic nature of FAs extracted from argillite of Meuse and Opalinus shale and discussed their origin as oceanic sediments at high burial temperature.| Elemental compositiona (%) | 13C NMRb (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C | H | N | O | S | Ash | CI | CII | CIII | CIV | CV | |
| a Ash free basis. b CI: carbonyl C (190–220 ppm), CII: carboxyl C (165–190 ppm), CIII: aromatic C (110–165 ppm), CIV: methoxyl and carbohydrate C (48–110 ppm), CV: aliphatic C (5–48 ppm). c Not detected. | |||||||||||
| HHA | 62.29 | 6.40 | 3.36 | 25.44 | 2.51 | N.D.c | 3.0 | 12.7 | 26.4 | 17.9 | 40.6 |
| HFA | 60.23 | 6.84 | 2.06 | 29.00 | 1.87 | N.D.c | 4.2 | 13.9 | 21.4 | 15.5 | 45.6 |
 | ||
| Fig. 1 van Krevelen plot of the HHSs, the IHSS and JHSS reference and standard HSs, and PAHA. The types of HS and origins are designated by different symbols and colors. | ||
The densities of oxygen-containing carboxylic and phenolic functional groups of the HHSs determined by conventional end-point acid–base titration in Table S1† are comparable to those of the surface HSs. This means that the oxygen depletion indicated by the small O/C ratios of the HHSs occurs in functional groups other than carboxylic and phenolic-type groups, as described by Thurman.56 The UV/Vis optical properties of the HHSs are characterized by relatively small A250/210 and A350/280 ratios (Table S1†). This points to the presence of small conjugated systems in the HHSs48 and again in accordance with their low aromaticities.
The size distributions of the HHSs measured by Fl-FFF are compared to those of the IHSS and JHSS standard HSs and PAHA in Fig. 2. For the surface HSs the sizes of the HAs are larger than those of the FAs. The size distributions of the HAs are largely overlapping, although their shapes are somewhat different from each other; EHA, IHA, and LHA possess multiple peaks. The size distributions of the FAs are all mono-modal, and the peak locations are different, depending on their sources. It seems that the JHSS FAs (BFA, DFA, and IFA) are somewhat smaller than the IHSS FAs (SRFA and NFA). The size distributions of HHA and HFA are mono-modal with the modal sizes of 0.6 and 0.3 nm, respectively, which are appreciably smaller than those of the surface HSs. Relatively small sizes of deep underground organic matter have been reported in the literature.57,58 Bouby et al.58 reported that organic matter in the Gorleben groundwater had a modal size of 1 nm. Saito et al.57 compared the size distribution of organic matter in granitic and sedimentary groundwater by Fl-FFF. The sedimentary groundwater was taken from a borehole at the same depth in the Horonobe URL as in this study and exhibited a mono-modal size distribution with a peak around 2 nm. This may indicate that HA and FA fractions may account for only a part of the dissolved organic matter in this groundwater.
 | ||
| Fig. 2 Fractograms of the HHSs, the IHSS and JHSS reference or standard HSs, and PAHA by Fl-FFF, using 5 mM Tris buffer as effluent. Fractionated HSs were measured by a UV/Vis detector at 255 nm. The fractograms of HAs are shown in (a) and those of FAs in (b). | ||
The EAC of a HS corresponds to the number of electrons transferred to the HS from the mediator, normalized by the mass of the HS, and represents its redox capacity. For the HHSs, the EAC values are relatively small compared with those of the IHSS and JHSS HSs (Fig. S2 in the ESI†). As in Aeschbacher et al.,49 we found a linear relationship between the EAC and the aromaticities of the HSs investigated (Fig. S2†). This is because the concentration of quinone moieties, that are predominantly responsible for redox reactions in HSs, tends to be proportional to the amount of aromatic carbon.42 Thus, the HHSs with low aromaticities have small redox capacities compared with the surface HSs.
In summary the HHSs can be viewed as relatively small organic matter with abundant aliphatic carbons and sulfurs. The densities of acidic functional groups are comparable to those of the surface HSs. The differences between HHA and HFA are small. The cluster analysis (Fig. S3 in the ESI†) performed for the physicochemical properties compiled in Table S1† clearly indicates that they are different from the IHSS and JHSS HSs. BFA is an exception, being clustered into the same group as the HHSs. This may indicate the presence of similar formation processes among them.
3.2. H+ and Cu2+ binding isotherms to the HHSs
The charge/pH curves of HHA and HFA at different salt levels are presented in Fig. 3. The negative charge (−q) of the HHSs increases with pH and salt concentration, as is usually seen for surface HSs.17 The maximum negative charge of HFA is larger than that of HHA, which predominantly arises from greater deprotonation at acidic pH (pH < 6). This further suggests that HFA possesses more acidic functional groups, mostly carboxylic groups, than HHA. In Fig. 3 the charge/pH curves of the HHSs are compared to those calculated by the NICA-Donnan model with the generic parameters derived for surface HSs (Table 2). At acidic pH the slopes of the curves are larger for the HHSs. At neutral and alkaline pH this trend is reversed, although the differences are small. The slope of a charge/pH curve of a HS reflects the width of the corresponding affinity distribution of its functional groups. A smaller slope means a wider distribution and larger chemical heterogeneity.9 Note that at 6 < pH < 9 the negative charges of the HHSs hardly change, indicating that the number of the acidic functional groups having corresponding pKa is small. Thus, these comparisons imply that the negative charges of the HHSs largely originate from H+ dissociation from chemically homogeneous low-affinity sites, which should mainly consist of carboxylic groups located on the aliphatic chains of the HHSs, considering their elemental compositions and 13C-NMR carbon distributions (Table 1). The salt dependence of the charge/pH curves is also different between the HHSs and the model calculation. Relatively large salt effects are observed at neutral to alkaline pH for the HHSs, but at acidic pH in the model calculation with the generic parameters.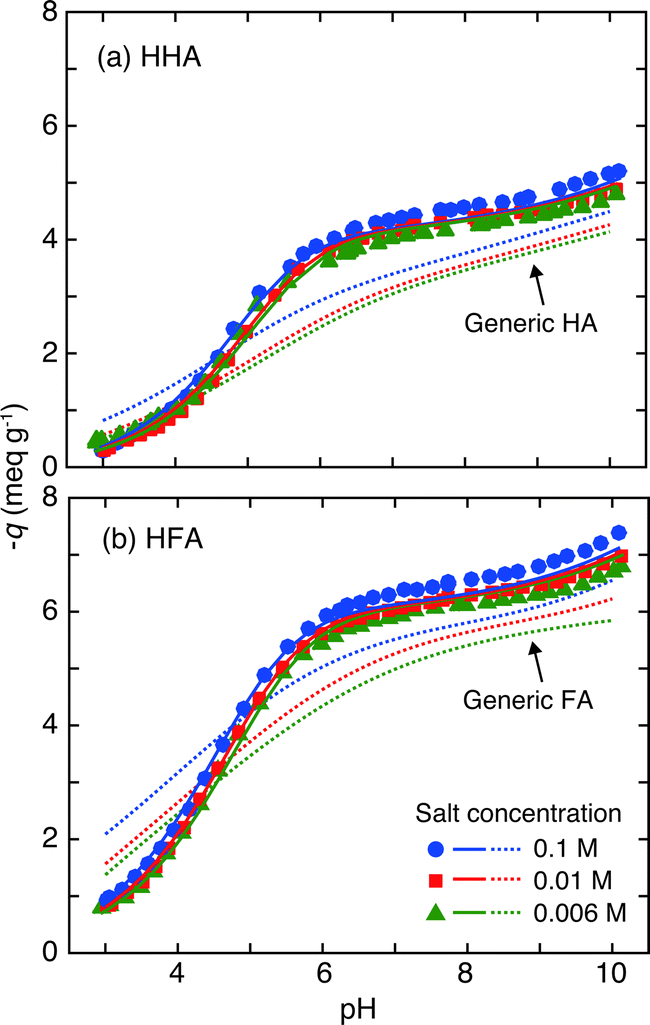 | ||
| Fig. 3 Charge–pH curves (symbols) of HHA (a) and HFA (b) at the three salt concentrations. The solid curves represent the results of fitting to the NICA-Donnan model for the HHSs and the dotted curves correspond to the calculation of the NICA-Donnan model with the generic parameters in Table 2.18 The negative charges (−q) are plotted in the ordinates. | ||
| HHA | HFA | GHA | GFA | |
|---|---|---|---|---|
| a The values in italic are constrained in physically meaningful ranges of the corresponding parameters (see the text of the ESI for details). b The correlation coefficients of the fitting for H+ and Cu2+. | ||||
| q 0 (eq kg−1) | −0.64 | −0.53 | — | — |
| b | 0.81 | 0.87 | 0.49 | 0.57 |
| Q max1,H (eq kg−1) | 4.38 | 5.64 | 3.15 | 5.88 |
| p 1 | 1 | 1 | 0.62 | 0.59 |
| Q max2,H (eq kg−1) | 4.44 | 4.09 | 2.55 | 1.86 |
| p 2 | 0.36 | 0.27 | 0.41 | 0.70 |
| log1,H |
3.74 | 3.63 | 2.93 | 2.34 |
| n 1,H | 0.82 | 1 | 0.81 | 0.66 |
| log2,H |
10.62 | 10.48 | 8.00 | 8.60 |
| n 2,H | 1 | 1 | 0.63 | 0.76 |
| log1,Cu |
1.32 | 1.16 | 2.23 | 0.26 |
| n 1,Cu | 1 | 1 | 0.56 | 0.53 |
| logK2,Cu |
14.43 | 15.05 | 6.85 | 8.26 |
| n 2,Cu | 0.28 | 0.29 | 0.34 | 0.36 |
| R 2 (H+)b | 0.9970 | 0.9975 | — | — |
| R 2 (Cu2+)b | 0.9951 | 0.9876 | — | — |
The Cu2+ binding isotherms to HHA and HFA are presented in Fig. 4. The isotherms are similar between them. The binding amounts of Cu2+ to the HHSs increases with the Cu2+ concentration and pH, as is expected for metal binding to surface HSs.18,38 The observed pH dependency of Cu2+ binding is the result of diminished H+ competition to the functional groups of the HHSs with an increase of pH. The Cu2+ binding amounts also slightly decrease with an increase of salt concentration due to screening of the negative electrostatic potential of the HHSs. In Fig. 4 the Cu2+ binding isotherms calculated by the NICA-Donnan model with the generic parameters given in Table 2 are presented for comparison. The binding amounts of Cu2+ to the HHSs are smaller than those of the model calculations regardless of pH and salt concentration. Weak binding of europium is also reported for HA and FA extracted from groundwater collected at −495 to 550 m bgl through a surface borehole in the Horonobe URL.33 At pH 4 the slopes of the isotherms to the HHSs are close to 1 in the log–log plot, which are larger than those of the model calculations with the generic parameters at the same pH. Interestingly, the differences in the slopes became smaller at higher pH, and at pH 8 for HHA it becomes similar to the slope of the calculated Cu2+ isotherms. The slope of a metal-binding isotherm of HS in a log–log plot is determined by the combination of the chemical heterogeneity of sites and the ion-specific non-ideality such as stoichiometry of the binding.9 The slope close to 1 in the isotherms of the HHSs at pH 4 together with the relatively large slopes of their charge–pH curves at acidic pH (Fig. 3) indicate Cu2+ binding to relatively homogeneous sites via mono-dentate coordination. At higher pH it seems that the binding mode tends to shift to coordination with greater denticity such as bi-dentate coordination.
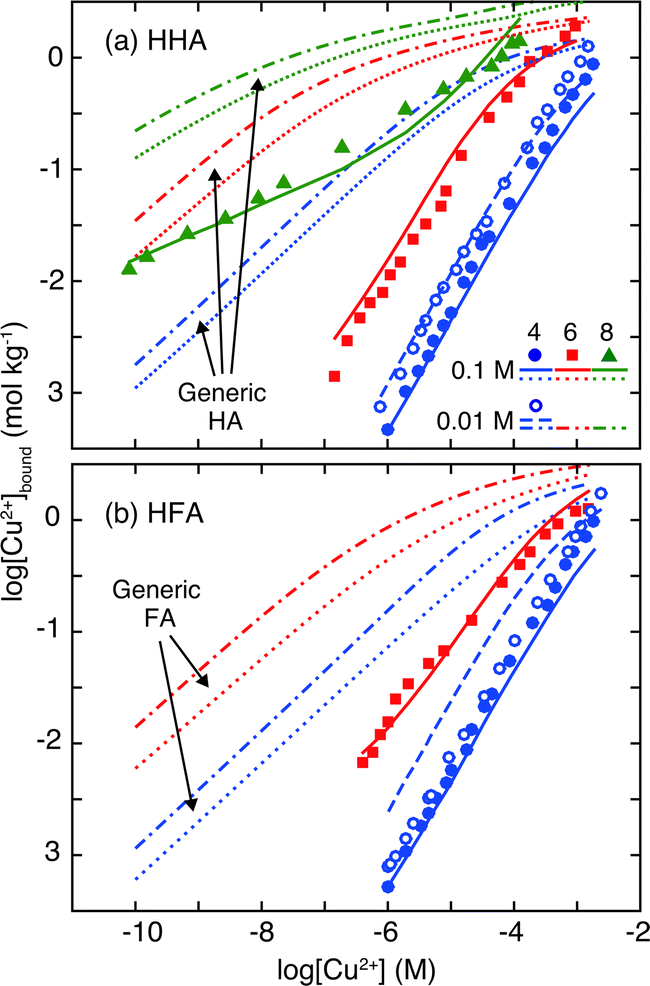 | ||
| Fig. 4 Copper binding isotherms to HHA (a) at pH 4, 6, and 8 and to HFA (b) at pH 4 and 6, measured in the presence of 0.1 M NaClO4. For pH 4, the results with 0.01 M NaClO4 are also presented. The solid and dashed curves correspond to the results of the NICA-Donnan fitting at 0.1 and 0.01 M NaClO4, respectively. The dotted and chained curves correspond to the calculation of the NICA-Donnan model with the generic parameters in Table 2 at 0.1 and 0.01 M salt concentrations.18 | ||
3.3. NICA-Donnan modeling
The results of the NICA-Donnan fitting to the H+ and Cu2+ isotherms to HHA and HFA are presented in Fig. 3 and 4 and the optimized parameters are given in Table 2 together with Milne's generic parameters.17 The 95% confidence intervals and the correlation matrices of the optimized parameters are given in Tables S2–S4 in the ESI.† Note that some parameters associated with the high-affinity sites, namely Qmax2,H, log2,H, and log2,Cu suffered from large errors. This is because that these parameters were not well fitted to the model due to the limited experimental conditions for H+ and Cu2+ binding to the HHSs in alkaline pH (pH ≤ 10 for the charge–pH curves and pH ≤ 8 or 6 for the Cu2+ isotherms). In order to unequivocally determine these parameters, potentiometric titration in non-aqueous media would be necessary.
For H+ binding the model successfully reproduces the charge–pH curves, especially at pH < 6. At neutral to alkaline pH the model somewhat underestimated the magnitude of the salt effect. The electrostatic part of the NICA-Donnan model (eqn (S4) and (S5) in the ESI†) assumes a relatively simple functional form for the so-called Donnan volume, which depends only on the salt concentration.14 Although the Donnan model is relatively simple with only one adjustable parameter and advantageous over other more sophisticated but complex models, its potential flaw for small FAs has been recognized.59 Considering the sizes of HHA and HFA, which are smaller than the Debye length of the solutions (1 and 3 nm for 0.1 and 0.01 M NaClO4), electrostatic potential calculation by the rigid-sphere or ion-permeable sphere model would be more realistic.14 The discrepancy observed at pH > 6 may also indicate the presence of a pH-dependent conformational change in the HHSs. Such conformational changes are common for linear aliphatic polyelectrolytes such as polymethacrylic acid.60 For HHA, Cu2+ binding is well reproduced by the model over a wide range of conditions, using the single set of the parameters. For HFA the model overestimated the salt effect and failed to describe the Cu2+ binding at 0.01 M NaClO4 and pH 4. It is likely that the model mishandles the electrostatic potential of HFA.
The optimized NICA-Donnan parameters for HHA and HFA are more or less similar to each other except for Qmax1,H, which is larger for HFA. The maximum densities of H+ binding sites are similar between the high (j = 1) and low (j = 2) affinity sites in HHA. For HFA the density of the latter group was smaller by 1.5 meq g−1. The obtained parameters can be compared to those of surface HSs with various origins and the generic parameters in Table 2.18 The values of Qmax1,H of the HHSs are in the ranges reported for the surface HSs, while those of Qmax2,H are larger.17 The median affinity constants of H+ of the HHS are larger than those of most of the surface HSs and Milne's generic parameters. The heterogeneity parameter, pj, and ion-specific non-ideality parameter, nj,H are also relatively large for the HHSs. This is especially the case for the low affinity carboxylic-type sites, reflecting the large slopes of their charge/pH curves (Fig. 3) at pH < 6. The Donnan parameters, b, are also larger for the HHSs. Considering the large aliphaticity of the HHSs as discussed in the physicochemical characterization, the log2,H values of HHA (10.62) and HFA (10.48), which are larger than the corresponding values of the generic HA (8.60) and FA (8.00), may indicate a larger contribution of alcoholic hydroxyl groups to the sites of the HHSs than those of surface HSs, although the presence of phenolic groups with large pKa cannot be entirely neglected as the HHSs still contain a certain amount of aromatic carbons (Table 1). This can also explain the weak H+ buffering by the HHSs at neutral to alkaline pH (Fig. 3).
The NICA-Donnan parameters of Cu2+ binding to HHSs are rather different from those of the generic parameters derived by Milne et al. (Table 1).18 For the low-affinity sites log1,Cu is larger for HHA and smaller for HFA than the corresponding generic parameters; whereas n1,H for both HHA and HFA are 1 and larger than the corresponding generic parameters (0.56 for GHA and 0.53 for GFA). For the high-affinity sites log2,Cu of HHA and HFA are larger than those of the generic parameters, and n2,H are smaller. Thus, the Cu2+ binding to the HHSs are characterized by n1,Cu = 1 for the low-affinity sites and large log2,Cu and small n2,Cu values for the high-affinity sites. A ratio of the parameter nj,i of a metal ion and proton is a good indicator of the underlying denticity of the complexation reaction (ni,j/nj,H close to 1 for mono dentate binding and 0.5 for bi-dentate binding). Thus, the optimized NICA-Donnan parameters for the HHSs suggest the mono-dentate nature of Cu2+ binding to the chemically homogeneous low-affinity sites at acidic pH. Considering the comparable or larger densities of the sites, Qmax1,H, of the HHS to/than those of surface HSs, this is most likely caused by sparsely distributed carboxylic groups on the aliphatic backbones of the HHSs, with which it is hard to form bi-dentate coordination with Cu2+. This can also explain the weak Cu2+ binding to the HHSs (Fig. 4). The relatively small n2,Cu/n2,H values for the HHSs together with the relatively small p2 values indicate that the more heterogeneous phenolic/alcoholic-type groups are involved in the binding of Cu2+ at neutral to alkaline pH via multi-dentate coordination. It is of interest to compare the NICA-Donnan parameters for the HHSs to those optimized for other groundwater HSs. Marang et al.61 reported the NICA-Donnan parameters for the binding of divalent and trivalent metal ions including Cu2+ to HA extracted from deep groundwater (−139 m bgl) of the Gorleben aquifer in Germany. The NICA-Donnan parameters of Cu2+ for this HA are more like those of GHA than those of HHA, suggesting a diversity of the ion binding properties of groundwater HSs. Further research is needed to relate it to the origin and genesis of groundwater HSs.
3.4. Oxidation state and chemical environment of Cu2+ bound to HHA
The oxidation state of Cu and its coordination environment bound to HHA was assessed by XAS. The XANES spectra of Cu2+ bound to HHA and PAHA are compared to those of the reference compounds in Fig. 5. The CuI reference materials exhibit characteristic pre-edge features (8.985 keV for CuSCN and 8.988 keV for CuCl) originating from 1s → 4p transitions.62 The CuII compounds, on the other hand, are characterized by intense white lines around 8.997 to 9.000 keV due to 1s → continuum transitions.62 The XANES spectra of Cu2+ bound to HHA and PAHA at pH 4 and 7 resemble those of CuII reference materials, especially Cu-tartrate. This suggests that reduction of Cu2+ bound to HHA and PAHA is negligible, although the magnified plot of the edge regions reveals a slight increase at 8.985 keV and a decrease at 8.998 keV for HHA (Fig. S4 in ESI†). Fulda et al.62 reported reduction of Cu2+ upon binding to a soil HA under anoxic conditions by XAS. It has also been reported that SRFA rapidly reduces Cu2+ even under oxic conditions.63 Virtually no reduction of Cu2+ by HHA may agree with its low EAC obtained by MER (Fig. S2†).49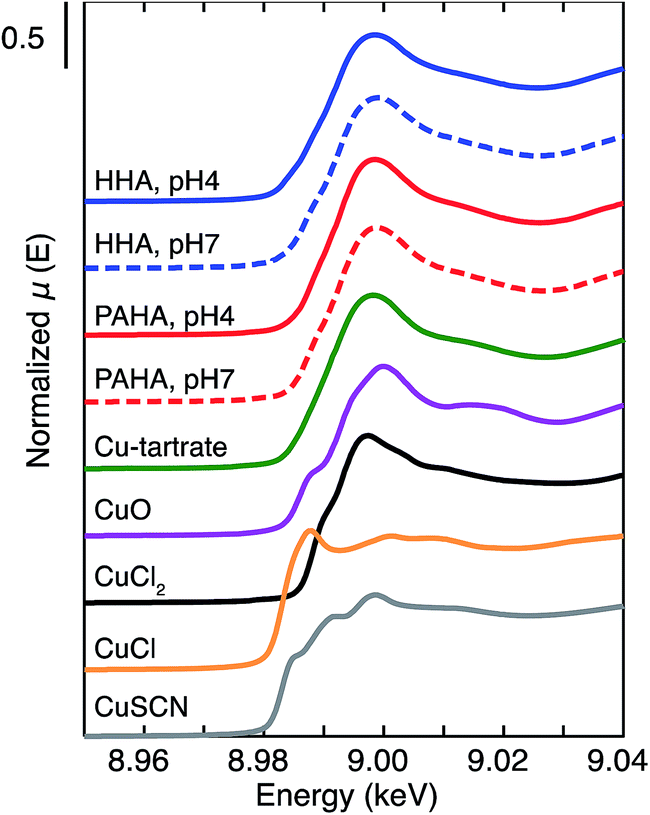 | ||
| Fig. 5 XANES spectra of Cu2+ bound to HHA and PAHA at pH 4 and 7 together with those of the CuII and CuI reference materials. | ||
The k3-weighted EXAFS spectra of Cu2+ bound to HHA and PAHA at pH 4 and 7 (Fig. 6(a)) exhibit systematic differences between the two HAs at 5.7, 6.5, and 7.4 Å−1. In Fig. S4 of the ESI† the same spectra are shown as an overlapped plot. The magnitudes of the corresponding Fourier transforms (Fig. 6(b) and S5(b) in the ESI†) are also different between them. For both HAs the Fourier transformed magnitudes are dominated by the intense peaks around 1.5 Å, which correspond to the scattering from the nearest O (and, to a lesser extent, N). For HHA additional peaks are noticeable around 2 Å, although their magnitudes are small. These peaks most likely originate from the Cu–S path as suggested by others.49,64 This assignment agrees with the high S content of HHA (Table 1). Quantitative modeling of the first coordination sphere of Cu2+ (Fig. 5(b) and Table 3) shows that approximately four O exist at 1.93 Å for PAHA, which corresponds to the Jahn–Teller distorted coordination geometry around Cu2+. For HHA at pH 4 the coordination number (CN) of O in the first shell is decreased and a small but non-negligible number of S (CN = 0.4 ± 0.2) is found at 2.35 Å. At pH 7 the presence of S in the second shell is inconclusive. Note that this does not necessarily mean bi-dentate coordination of Cu2+ with O/N and S in HHA. Considering the results of the NICA-Donnan fitting (Fig. 4 and Table 2), it seems more likely that the two independent mono-dentate sites exist in HHA, as an EXAFS spectrum is a one-dimensional representation of the coexisting coordination environments of a target element. The S containing functional group could be the thioacetic group, R–COSH, which then should be a part of the low affinity sites in the NICA-Donnan modeling, as the pKa value of thioacetic acid (CH3COSH, pKa = 3.33) is close to that of acetic acid (pKa = 4.76) and smaller than that of methanethiol (CH3SH, pKa = 10.33).65 It is also noteworthy to mention that the contribution of S to Cu2+ binding to HHA might be underestimated, compared with that in the original groundwater, as the functional groups containing reduced S may be oxidized during the extraction and storage. For more detailed discussion, S speciation in the HHSs should be determined by soft X-ray absorption spectroscopy.23
 | ||
| Fig. 6 k 3-weighted Cu K-edge EXAFS spectra (a) and the corresponding Fourier transform magnitude (b) of Cu2+ bound to HHA and PAHA at pH 4 and 7. The Cu2+ loading was 80 mmol Cu per kg HA. Solid curves correspond to the experimental results and dashed curves in the Fourier transform magnitude plots to the results of theoretical fitting of the first coordination sphere of Cu2+ (see the text for the details). | ||
| First shell (Cu–O/N) | Second shell (Cu–S) | ΔE0 (eV) | |||||
|---|---|---|---|---|---|---|---|
| R (Å) | CNa | σ 2 (103 Å2) | R (Å) | CN | σ 2 (103 Å2) | ||
| a Coordination number. b Debye–Waller factor. The Debye–Waller factor of the Cu–S shell was set to be equal to that of the Cu–O/H shell. | |||||||
| HHA | |||||||
| pH 4 | 1.95 ± 0.01 | 2.7 ± 0.6 | 3 ± 1 | 2.35 ± 0.04 | 0.4 ± 0.2 | 3 ± 1 | 1.53 ± 2.36 |
| pH 7 | 1.95 ± 0.02 | 3.1 ± 0.9 | 4 ± 3 | 2.35 ± 0.16 | 0.1 ± 0.3 | 4 ± 3 | 1.52 ± 3.51 |
|
|||||||
| PAHA | |||||||
| pH 4 | 1.94 ± 0.01 | 3.7 ± 0.8 | 5 ± 2 | — | — | — | 0.71 ± 2.80 |
| pH 7 | 1.93 ± 0.01 | 3.6 ± 0.5 | 4 ± 1 | — | — | — | 1.19 ± 2.07 |
4. Conclusion
The present study aims to reveal physicochemical and ion binding properties of the HHSs isolated from deep groundwater in a sedimentary rock formation. It is found that the HHSs are rather different from surface HSs, characterized by high aliphaticity and S contents and relatively small sizes. Proton and Cu2+ binding was studied by potentiometric titration and fit to the NICA-Donnan model for comparison to their binding to surface HSs. The results clearly indicate distinctive binding behaviors of H+ and Cu2+ to the HHSs, likely caused by the unique chemical nature of their functional groups compared with those of surface HSs. These ions predominantly bind to chemically homogeneous carboxylic groups of the HHSs and to a lesser extent S containing groups by mono-dentate coordination at low pH, which can explain the larger slopes of their isotherms and the smaller magnitude of Cu2+ binding than those of surface HSs. At neutral to alkaline pH H+ dissociation from the HHSs is small, likely because the majority of the high affinity sites consist of alcoholic OH groups with larger pKa. At the same pH range the slope of the Cu2+ isotherm to HHA becomes smaller and is close to the model calculations with the generic parameters, suggesting that the binding mode could change to bi-dentate coordination with both carboxylic and alcoholic/phenolic groups. The XAS analyses further indicate that Cu2+ binds to HHA largely as CuIIvia O/N containing functional groups and to a lesser extent S containing functional groups. The generality of the results obtained in this study should be examined in the future by performing similar investigations for HSs isolated from various groundwater samples and generic parameter sets applicable for groundwater HSs should be developed. The effects of co-existing metal ions, which are present in deep underground water at relatively high concentrations, on ion binding to the HHSs are of great interest as they can alter the electrostatic properties and secondary structures of the HHSs.Acknowledgements
The authors (T. S.) would like to thank Mr Lukman Steven for his assistance in the potentiometric titration and Dr Masayuki Watanabe for the UV/Vis measurements. This research was partly supported by the Ministry of Economy, Trade and Industry (METI), Japan, and “Grant-in-Aid for Young Scientists (B)” (Grant No. 25820446), the Japan Society for the Promotion of Science. This work was carried out under the approval of the KEK-PF (2012G723 and 2012G527).References
- G. R. Aiken, D. M. McKnight, R. L. Wershaw and P. MacCarthy, Humic Substances in Soil, Sediment and Water, Wiley, New York, 1985 Search PubMed.
- C. Baduel, M. E. Monge, D. Voisin, J. L. Jaffrezo, C. George, I. El Haddad, N. Marchand and B. D'Anna, Environ. Sci. Technol., 2011, 45, 5238–5244 CrossRef CAS PubMed.
- F. J. Stevenson, in Humic Substances in Soil, Sediments and Water, ed. G. R. Aiken, D. M. Mcknight, R. L. Wershaw and P. MacCarthy, John Wily & Sons, New York, 1985, pp. 13–52 Search PubMed.
- A. M. Jones, R. N. Collins, J. Rose and T. D. Waite, Geochim. Cosmochim. Acta, 2009, 73, 4409–4422 CrossRef CAS PubMed.
- J. Jiang and A. Kappler, Environ. Sci. Technol., 2008, 42, 3563–3569 CrossRef CAS.
- G. Bronner and K. U. Goss, Environ. Sci. Technol., 2011, 45, 1307–1312 CrossRef CAS PubMed.
- J. Buffle, Complexation Reactions in Aquatic Systems: An Analytical Approach, Ellis Horwood, Chichester, 1985 Search PubMed.
- E. Tipping, Cation binding by humic substances, Cambridge Univeristy Press, Cambridge, 2002 Search PubMed.
- L. K. Koopal, T. Saito, J. P. Pinheiro and W. H. van Riemsdijk, Colloids Surf., A, 2005, 265, 40–54 CrossRef CAS PubMed.
- J. F. McCarthy and J. M. Zachara, Environ. Sci. Technol., 1989, 23, 496–502 CAS.
- D. G. Kinniburgh, W. H. van Riemsdijk, L. K. Koopal, M. Borkovec, M. F. Benedetti and M. J. Avena, Colloids Surf., A, 1999, 151, 147–166 CrossRef CAS.
- E. Tipping, Aquat. Geochem., 1998, 4, 3–48 CrossRef CAS.
- J. A. Marinsky and J. H. Ephraim, Environ. Sci. Technol., 1986, 20, 349–354 CrossRef CAS PubMed.
- T. Saito, S. Nagasaki, S. Tanaka and L. K. Koopal, Colloids Surf., A, 2005, 265, 104–113 CrossRef CAS PubMed.
- G. S. Manning, J. Phys. Chem., 1969, 51, 924–934 CrossRef CAS PubMed.
- E. Tipping, S. Lofts and J. E. Sonke, Environ. Chem., 2011, 8, 225–235 CrossRef CAS.
- C. J. Milne, D. G. Kinniburgh and E. Tipping, Environ. Sci. Technol., 2001, 35, 2049–2059 CrossRef CAS.
- C. J. Milne, D. G. Kinniburgh, W. H. van Riemsdijk and E. Tipping, Environ. Sci. Technol., 2003, 37, 958–971 CrossRef CAS.
- I. A. M. Ahmed, J. H. Taylor, M. Bieroza, H. Zhang and W. Davison, Water Res., 2014, 67, 276–291 CrossRef CAS PubMed.
- J. Xiong, L. K. Koopal, W. F. Tan, L. C. Fang, M. X. Wang, W. Zhao, F. Liu, J. Zhang and L. P. Weng, Environ. Sci. Technol., 2013, 47, 11634–11642 CrossRef CAS PubMed.
- K. Kovács, A. Gáspár, C. Sajgó, P. Schmitt-Kopplin and E. Tombácz, Geochem. J., 2012, 46, 211–224 CrossRef.
- K. Longnecker and E. B. Kujawinski, Geochim. Cosmochim. Acta, 2011, 75, 2752–2761 CrossRef CAS PubMed.
- T. Schäfer, G. Buckau, R. Artinger, J. I. Kim, S. Geyer, M. Wolf, W. F. Bleam, S. Wirick and C. Jacobsen, Org. Geochem., 2005, 36, 567–582 CrossRef PubMed.
- R. Artinger, G. Buckau, S. Geyer, P. Fritz, M. Wolf and J. I. Kim, Appl. Geochem., 2000, 15, 97–116 CrossRef CAS.
- C. Gron, L. Wassenaar and M. Krog, Environ. Int., 1996, 22, 519–534 CrossRef.
- C. Pettersson, J. Ephraim and B. Allard, Org. Geochem., 1994, 21, 443–451 CrossRef CAS.
- A. Courdouan, I. Christl, S. Meylan, P. Wersin and R. Kretzschmar, Appl. Geochem., 2007, 22, 1537–1548 CrossRef CAS PubMed.
- A. Courdouan, I. Christl, S. Meylan, P. Wersin and R. Kretzschmar, Appl. Geochem., 2007, 22, 2926–2939 CrossRef CAS PubMed.
- L. Grasset, J. Brevet, T. Schafer, F. Claret, E. C. Gaucher, A. Albrecht and A. Ambles, Org. Geochem., 2010, 41, 221–233 CrossRef CAS PubMed.
- A. Courdouan, I. Christl, T. Rabung, P. Wersin and R. Kretzschmar, Environ. Sci. Technol., 2008, 42, 5985–5991 CrossRef CAS.
- F. Claret, T. Schafer, T. Rabung, M. Wolf, A. Bauer and G. Buckau, Appl. Geochem., 2005, 20, 1158–1168 CrossRef CAS PubMed.
- J. J. Alberts, Z. Filip and N. Hertkorn, J. Contam. Hydrol., 1992, 11, 317–330 CrossRef CAS.
- M. Terashima, S. Nagao, T. Iwatsuki, N. Fujitake, Y. Seida, K. Iijima and H. Yoshikawa, J. Nucl. Sci. Technol., 2012, 49, 804–815 CrossRef CAS PubMed.
- T. Iwatsuki, R. Furue, H. Mie, S. Ioka and T. Mizuno, Appl. Geochem., 2005, 20, 2283–2302 CrossRef CAS PubMed.
- K. Hama, T. Kunimaru, R. Metcalfe and J. Martin, Phys. Chem. Earth, 2007, 32, 170–180 CrossRef PubMed.
- S. Kurosawa, S. C. James, M. Yui and M. Ibaraki, J. Colloid Interface Sci., 2006, 298, 467–475 CrossRef CAS PubMed.
- Y. Tachi, K. Yotsuji, Y. Seida and M. Yui, Geochim. Cosmochim. Acta, 2011, 75, 6742–6759 CrossRef CAS PubMed.
- T. Saito, S. Nagasaki, S. Tanaka and L. K. Koopal, Radiochim. Acta, 2004, 92, 567–574 CrossRef CAS.
- J. W. J. van Schaik, D. B. Kleja and J. P. Gustafsson, Geochim. Cosmochim. Acta, 2010, 74, 1391–1406 CrossRef CAS PubMed.
- L. M. Gaetke and C. K. Chow, Toxicology, 2003, 189, 147–163 CrossRef CAS.
- J. T. Rubino and K. J. Franz, J. Inorg. Biochem., 2012, 107, 129–143 CrossRef CAS PubMed.
- H. Kurikami, R. Takeuchi and S. Yabuuchi, Phys. Chem. Earth, 2008, 33, S37–S44 CrossRef PubMed.
- R. S. Swift, in Methods of soil analysis. Part 3-chemical methods, ed. D. L. Sparks, A. L. Page, P. A. Helmke, R. H. Loeppert, P. Soltanpour, M. A. Tabatabai, C. Johnston and M. E. Sumner, Soil Science Society of America, Madison, 1996, p. 1011 Search PubMed.
- M. A. Wilson, Eur. J. Soil Sci., 1981, 32, 167–187 CrossRef CAS PubMed.
- N. Fujitake and M. Kawahigashi, Soil Sci. Plant Nutr., 1999, 45, 359–366 CrossRef CAS PubMed.
- T. Saito, L. K. Koopal, W. H. van Riemsdijk, S. Nagasaki and S. Tanaka, Langmuir, 2004, 20, 689–700 CrossRef CAS.
- F. J. Stevenson, Humus Chemistry, John Wiley & Sons, New York, 1982 Search PubMed.
- S. Lukman, T. Saito, N. Aoyagi, T. Kimura and S. Nagasaki, Geochim. Cosmochim. Acta, 2012, 88, 199–215 CrossRef CAS PubMed.
- M. Aeschbacher, M. Sander and R. P. Schwarzenbach, Environ. Sci. Technol., 2010, 44, 87–93 CrossRef CAS PubMed.
- D. G. Kinniburgh, C. J. Milne and P. Venema, Soil Sci. Soc. Am. J., 1995, 59, 417–422 CrossRef CAS.
- C. J. Milne, D. G. Kinniburgh, J. C. M. de Wit, W. H. van Riemsdijk and L. K. Koopal, J. Colloid Interface Sci., 1995, 175, 448–460 CrossRef CAS.
- W. L. Lindsay, Chemical equilibria in soils, John Wiley & Sons, New York, 1979 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- S. I. Zabinsky, J. J. Rehr, A. Ankudinov, R. C. Albers and M. J. Eller, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 2995–3009 CrossRef CAS.
- J. A. Rice and P. MacCarthy, Org. Geochem., 1991, 17, 635–648 CrossRef CAS.
- E. M. Thurman, in Humic Substances in Soil, Sediments and Water, ed. G. R. Aiken, D. M. Mcknight, R. L. Wershaw and P. MacCarthy, John Wily & Sons, New York, 1985 Search PubMed.
- T. Saito, T. Hamamoto, T. MIzuno, T. Iwatsuki and S. Tanaka, J. Anal. At. Spectrom., 2015, 30, 1229–1236 RSC.
- M. Bouby, N. Finck and H. Geckeis, Mineral. Mag., 2012, 76, 2709–2721 CrossRef CAS PubMed.
- M. F. Benedetti, W. H. van Riemsdijk and L. K. Koopal, Environ. Sci. Technol., 1996, 30, 1805–1813 CrossRef CAS.
- R. D. Porasso, J. C. Benegas and M. A. G. T. van den Hoop, J. Phys. Chem. B, 1999, 103, 2361–2365 CrossRef CAS.
- L. Marang, P. E. Reiller, S. Eidner, M. U. Kumke and M. F. Benedetti, Environ. Sci. Technol., 2008, 42, 5094–5098 CrossRef CAS.
- B. Fulda, A. Voegelin, F. Maurer, I. Christl and R. Kretzschmar, Environ. Sci. Technol., 2013, 47, 10903–10911 CrossRef CAS PubMed.
- A. N. Pham, A. L. Rose and T. D. Waite, J. Phys. Chem. A, 2012, 116, 6590–6599 CrossRef CAS PubMed.
- A. Manceau and A. Matynia, Geochim. Cosmochim. Acta, 2010, 74, 2556–2580 CrossRef CAS PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, Taylor & Francis, Boca Raton, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5em00176e |
| This journal is © The Royal Society of Chemistry 2015 |