
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotentiometric sensing array for monitoring aquatic systems†
Nadezda
Pankratova
a,
Gastón A.
Crespo
a,
Majid Ghahraman
Afshar
a,
Miquel Coll
Crespi
a,
Stéphane
Jeanneret
a,
Thomas
Cherubini
a,
Mary-Lou
Tercier-Waeber
a,
Francesco
Pomati
b and
Eric
Bakker
*a
aUniversity of Geneva, Department of Inorganic and Analytical Chemistry, Quai Ernest-Ansermet 30, CH-1211 Geneva, Switzerland
bEawag: Swiss Federal Institute of Aquatic Science and Technology, Department of Aquatic Ecology, Ueberlandstrasse 133, CH-8600 Duebendorf, Switzerland
First published on 25th March 2015
Abstract
Since aquatic environments are highly heterogeneous and dynamic, there is the need in aquatic ecosystem monitoring to replace traditional approaches based on periodical sampling followed by laboratory analysis with new automated techniques that allow one to obtain monitoring data with high spatial and temporal resolution. We report here on a potentiometric sensing array based on polymeric membrane materials for the continuous monitoring of nutrients and chemical species relevant for the carbon cycle in freshwater ecosystems. The proposed setup operates autonomously, with measurement, calibration, fluidic control and acquisition triggers all integrated into a self-contained instrument. Experimental validation was performed on an automated monitoring platform on lake Greifensee (Switzerland) using potentiometric sensors selective for hydrogen ions, carbonate, calcium, nitrate and ammonium. Results from the field tests were compared with those obtained by traditional laboratory analysis. A linear correlation between calcium and nitrate activities measured with ISEs and relevant concentrations measured in the laboratory was found, with the slopes corresponding to apparent single ion activity coefficients  and
and 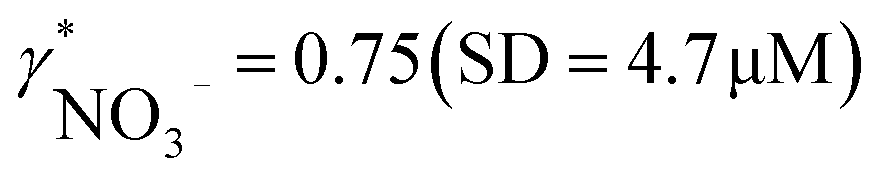 . Good correlation between pH values measured with ISE and CTD probes (SD = 0.2 pH) suggests adequate reliability of the methodology.
. Good correlation between pH values measured with ISE and CTD probes (SD = 0.2 pH) suggests adequate reliability of the methodology.
Environmental impactWe aim to record time dependent concentration data of nutrients and species relevant to the carbon cycle at varying depths in freshwater systems with as little perturbation as possible, and to eventually relate this information with microbiological data obtained on the same system by scanning flow cytometry. An array of potentiometric sensors was developed for this purpose that operates autonomously on a floating platform where lake samples are pumped from specific depths and measured. The sensor array can be calibrated with up to two calibrant solutions at regular intervals. The performance of the device is successfully compared to the benchtop analysis on samples obtained at the same depth. |
1. Introduction
Freshwater ecosystems, and in particular lakes, are threatened worldwide by an interaction of anthropogenic environmental changes, including eutrophication and climate change which strongly affect the physics and chemistry of the aquatic habitats.1 Within aquatic ecosystems, phytoplankton communities are common indicators of environmental changes since they have fast generation times and respond rapidly to variations in both physical and chemical parameters.2 Phytoplankton growth relies on macro-nutrients (nitrate, phosphate, salicylic acid), sunlight and other crucial substances such as vitamin B and trace metals. While sunlight is attenuated as a function of depth, most of the nutrients are supplied from either deeper water or the sediments. As a result, light and concentration gradients in a water column are changing in opposed directions.3 The effect of these natural processes on the growth of phytoplankton is still an open question to the scientific community. Indeed, recent theoretical studies have elucidated the understanding of the dynamic interactions between phytoplankton growth and the resources in a water column.3 However, theoretical models need to be experimentally validated to more fully understand such complex systems, which have implications to questions of climate change, eutrophication and pollution.2High quality monitoring data are in this context crucial for tracing and understanding the mechanisms that drive phytoplankton and lake ecosystem dynamics. The latter requires a coupling of biological data obtained in situ with automated physical and chemical profiling. So far, most monitoring programs only poorly integrate biological data with direct sensing of standard physico-chemical parameters (e.g. temperature, conductivity, pH, oxygen), nutrients (nitrate, ammonium, phosphate) and rarely pigment data like chlorophyll-a.4–6 Current developments include the integration of existing methods into automated, complex and operational sensing systems for a comprehensive and long-term monitoring of both the biology and chemistry of water environments. The combination of a diverse set of sensor techniques will be most valuable for achieving a large chemical, spatial and temporal coverage.7
Over the past decades, potentiometric sensors based on liquid polymer membrane materials have become a promising tool for the realization of in situ sensing devices. In the past years considerable improvement of the detection limit and the discrimination of interfering ions and the development of new molecular receptors along with appropriate theoretical treatments were achieved. This has resulted in a strong foundation for the application of potentiometric sensors in environmental trace analysis and potentiometric biosensing.8 For example, this has allowed one to develop potentiometric sensors for the monitoring of microprofiles in sediments.9,10
We aim here to establish an integrated platform based on a flow potentiometric sensing array for measuring nitrogen and carbon cycle related ionic species involved in phytoplankton growth in freshwaters. The sensing platform was initially validated and automated in house. A self-contained data acquisition instrument, GalvaPot v2 (see ESI† for more details), was developed specifically for field applications. The device allows one to read out up to 12 electrodes and offers simultaneous control of a number of peripherals that include pumps, valves and other actuators. GalvaPot v2 can be programmed by a touch screen interface, without the need for a PC, and the recorded data are stored in internal memory. The potentiometric platform was validated in the field (lake Greifensee) during four days using an automated Eawag monitoring system, allowing for water sampling at different depths and subsequent potentiometric analysis after a delay of a few minutes. The analytical characterization of the proposed system is reported on in detail. The development of this sensing tool builds on a synergy between analytical chemists and limnologists to help stimulate this field and understand yet unresolved environmental processes.
2. Experimental
2.1 Materials and chemicals
Potassium tetrakis[3,5-bis-(trifluoromethyl)phenyl]borate (KTFPB), tridodecylmethylammonium nitrate (TDMAN), bis(2-ethylhexyl) phthalate (DEHP), tetrakis(4-chlorophenyl)borate tetradodecylammonium salt (ETH 500), 2-nitrophenyloctylether (o-NPOE), ammonium ionophore I (nonactin), potassium tetrakis (4-chlorophenyl) borate (KTpClPB), hydrogen ionophore I (tridodecylamine), N,N-dioctyl-3α,12α-bis(4-trifluoroacetylbenzoyloxy)-5β-cholan-24-amide (carbonate ionophore VII), N,N-dicyclohexyl-N′,N′-dioctadecyl-3-oxapentanediamide (calcium ionophore IV, ETH 5234), bis(2-ethylhexyl) adipate (DOA), tridodecylmethylammonium chloride (TDMACl), high molecular weight poly(vinyl chloride) (PVC), tetradodecylammonium chloride (TDDA), 2-nitrophenyl octyl ether (o-NPOE), high molecular weight poly(vinyl chloride) (PVC), tetrahydrofuran (THF), calcium chloride dihydrate, sodium nitrate, sodium bicarbonate, ammonium chloride, potassium chloride, sodium hydroxide standard solution 2 mol L−1, tris(hydroxymethyl)aminomethane (TRIS), nitrilotriacetic acid (NTA) and sulfuric acid 96% were purchased from Sigma-Aldrich and Fluka. Nitric and hydrochloric acids suprapur grade were purchased from Merck. Aqueous solutions were prepared by dissolving the appropriate salts in Milli-Q water (18.2 MΩ cm).2.2 Electrodes and membrane preparation
The commercial pH glass electrode (InLab Mono Pro) was purchased from Mettler Toledo. The cocktail for carbonate-selective membranes was prepared by dissolving 8.3 mg of carbonate ionophore VII, 60 mg of PVC, 2 mg of TDMACl, and 100 µL of DOA in 2 mL of THF. The cocktail for calcium selective membranes contained 15 mmol kg−1 of calcium ionophore IV, 5 mmol kg−1 of ion exchanger KTFPB, 66 mg of PVC and 131 mg of o-NPOE, dissolved in 2 mL of THF. The nitrate ISE-cocktail was composed of 10 mmol kg−1 ion exchanger salt TDMAN, 15 mmol kg−1 inert lipophilic salt ETH 500, 65 mg of PVC and 130 mg of plasticizer DEHP. The cocktail for pH-sensitive PVC based electrodes consisted of 20 mmol kg−1 TDDA, 5 mmol kg−1 KTpClPB, 66 mg of PVC and 132 mg of plasticizer o-NPOE. Ammonium PVC-based membranes were prepared using 3 mmol kg−1 of nonactin, 62 mg PVC and 138 mg of plasticizer o-NPOE, without additional ion exchanger.Each individual cocktail was poured into glass rings (22 mm in diameter) placed on a glass slide and dried overnight at room temperature under a dust-free environment. After THF evaporation a membrane of 200 µm approximate thickness was obtained. Small disks (diameter ca. 8 mm) were punched from the cast films and subsequently conditioned in 1 mM solution of the corresponding salt NaHCO3, CaCl2, NaNO3, NH4Cl or HCl. The membranes were then mounted in Ostec electrode bodies with inner silver–silver chloride elements (Oesch Sensor Technology, Sargans, Switzerland). The inner solution was composed of 1 mM solution of the corresponding salt. In the case chloride ion was not present in the salt, 1 mM NaCl was added to the inner electrolyte to ensure a defined potential at the inner silver–silver chloride element.
2.3 Automated analysis setup
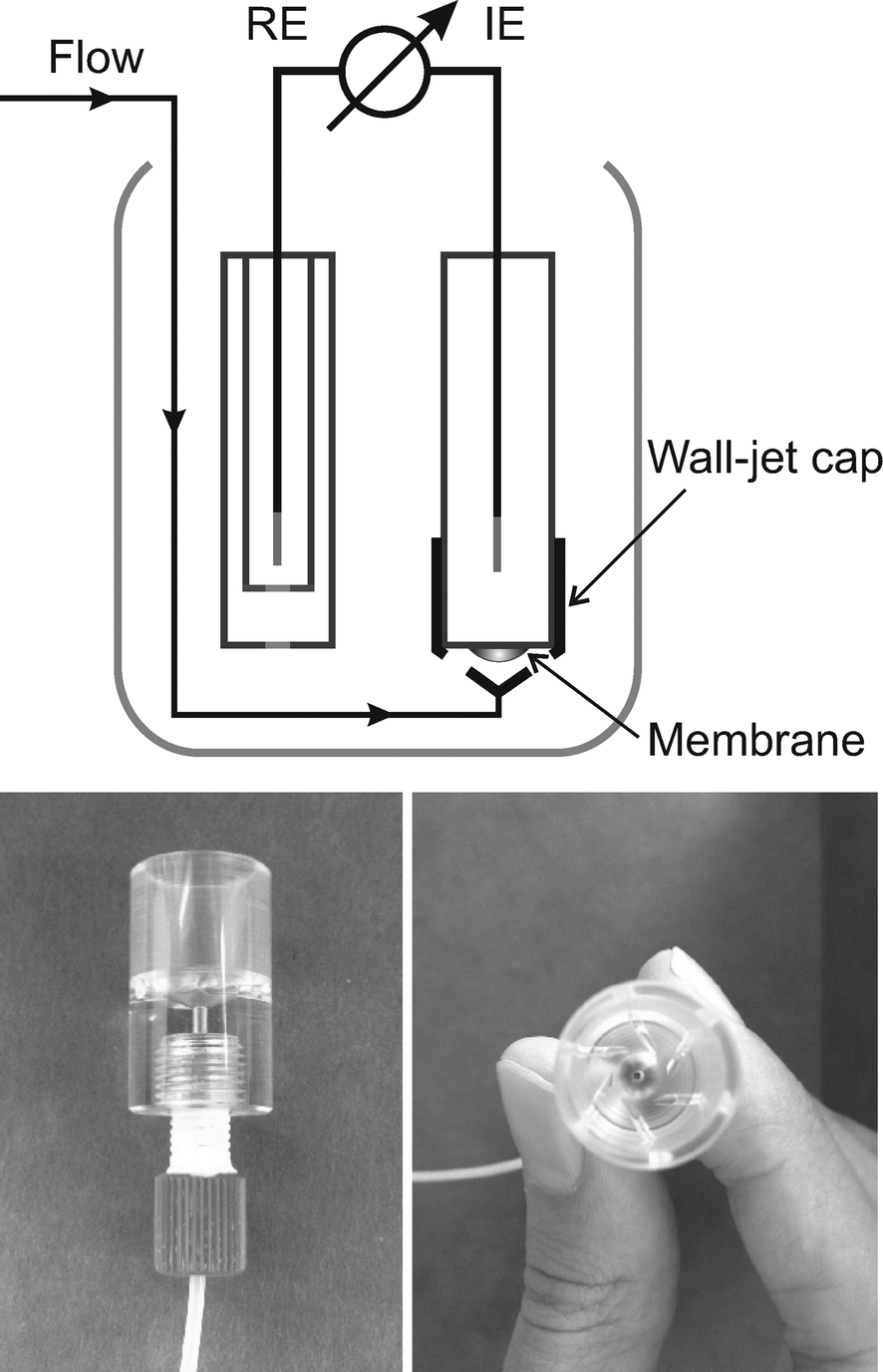 | ||
| Fig. 1 Flow cell design.11 | ||
The calibration of the sensors was performed using two solutions, the compositions of which are given in Table 1. Solutions were prepared in 5 L polyethylene bottles by dissolving corresponding chemicals in Milli-Q-purified water. The setup of the experiment provides for a continuous flow through the cell since stop flow conditions would lead to the penetration of concentrated outer KCl solution from the beaker into the cell containing the ISE. To achieve this, calibration solutions were continuously flushing the membranes whenever measurements of water samples were stopped. For the same reason, another pump was started just before turning off one of main Pumps 1–3, keeping one of the main Pumps 1, 2 or 3 on at all times. Pumps 4 and 5 as well as Pump 6, were turned on throughout the experiment. Calibration solutions were delivered to the flow cell in the same way as the sample after starting corresponding Pumps 1 or 2 and immediate disconnection of Pump 3.
| Chemical | Calibration solution 1 | Calibration solution 2 |
|---|---|---|
| a pH value indicated in Table 1 is approximate. The actual value may vary for different portions of the same calibration solution (prepared on different days) maximum by 0.3 units of pH. | ||
| Tris(hydroxymethyl)aminomethane, mM | 10 | 10 |
| Nitrilotriacetic acid, mM | 3 | 3 |
| Potassium chloride, µM | 130 | 130 |
| Calcium chloride dihydrate, mM | 1 | 1 |
| Sodium bicarbonate, mM | 5 | 5 |
| Sodium nitrate, µM | 100 | 10 |
| Ammonium chloride, µM | 10 | 1 |
| pHa (adjusted using sodium hydroxide/sulfuric acid) | 8.5 | 7.6 |
Both Calibration and Experiment methods were automated using GalvaPot v2. The detailed description of corresponding methods are given in ESI.† Calibration solutions were pumped through the system continuously, excluding the time window taken by Experiment method. Starting Experiment method caused the end of the running Calibration method. Upon finishing Experiment method, Calibration method was resumed. The limited internal memory of the GalvaPot v2 did not provide for an automatic saving of every data point read by the device, so the reading of the potential values was performed over specified time windows, averaged and then stored in internal memory. Every single reading was obtained by averaging the signal on each electrode over 6 s with a 12 Hz data acquisition frequency. 8–10 averaged readings were saved when running Experiment method and 8–10 readings for each calibration solution for the time window of calibration right before the trigger and immediately after the end of the Experiment method. As a consequence of the limited internal memory the collected data was transferred to external memory once a day.
The composition of calibration solutions is indicated in Table 1. Carbonate (CO32−) and ammonium (NH4+) concentrations were recalculated for every particular calibration solution depending on its precise pH value determined using conventional pH meter and standard buffer solutions, in laboratory conditions. pH, nitrate, ammonium and carbonate concentrations chosen for preparing calibration solutions correspond to the relevant concentration ranges in lake Greifensee. The anticipated level of analytes in lake water was determined during preliminary trials of the described setup in the field.
The calcium concentration in the calibration solutions had to be chosen in a lower range than the one expected for calcium levels in lake water. The latter accounts for the fact that the addition of millimolar levels of calcium to a solution containing millimolar levels of total carbonate would cause precipitation of both calcium and carbonate, resulting in an uncertainty of the calibration solution composition affected by temperature changes as well as precipitation kinetics. Therefore, the concentration of calcium in the calibration solutions was buffered using NTA that left approximately 0.5 wt% of total calcium in the uncomplexed form. The free calcium fraction is sensitive to NTA concentration and is highly affected by pH changes. For this reason, an additional calibration of the installed ISEs was performed a few times a week using two calibration solutions containing just 10−3 M and 10−2 M calcium chloride.
2.4. Field measurements
Field measurements of nutrients and chemical species related to carbon cycle were performed at the end of August (18-22.08.2014) on the lake Greifensee, a small eutrophic lake (surface area 8.45 km2, water volume 0.148 km3) located in the canton of Zurich, Switzerland. The automated analysis setup described above was synchronized with the Eawag automated monitoring platform by allowing the GalvaPot v2 to accept an electric signal from the system's controller module (Idronaut Controller Module operating also with a CTD probe with traditional sensors for profiling)2 as a trigger to start potentiometric measurements of the samples taken at different depths: 1, 2.5, 4, 5.5, 7 and 8.5 m.The Eawag platform is equipped with a system to retrieve water from selected depths. Water was brought to the acrylic sampling chamber (volume 250 mL) through an antimicrobial, silver-nanoparticle coated and shaded flexible polyethylene tubing.2 The trigger signal activates the external pump (boat water system pump, Jabsco PAR-Max 1) that flushes the chamber with water sample from the selected depth for 3 min at a flow rate of 4.2 L min−1. After flushing the chamber Experiment method started automatically. The sample collected in the chamber and delivered to the cells using Pump 3 was used for potentiometric measurements. A new profiling started every 4 h: at 00![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :00, 04:00, 08:00, 12:00, 16:00 and 20:00. The total time for potentiometric measurements at one depth was limited to about 16 min due to the Eawag automated monitoring cycle, with the entire cycle taking around 1 h 40 min.
:00, 04:00, 08:00, 12:00, 16:00 and 20:00. The total time for potentiometric measurements at one depth was limited to about 16 min due to the Eawag automated monitoring cycle, with the entire cycle taking around 1 h 40 min.
2.5. Field data validation
3. Results and discussion
3.1 Preliminary laboratory experiments
Considering the high calcium concentration in the lake, the choice of a sensor for calcium determination was straightforward. A PVC based membrane doped with calcium ionophore IV was used for this purpose. The selectivity, stability and detection limits of this type of membrane have been thoroughly studied 18,19 and confirm its applicability at pH 7–9 at the millimolar concentration range for freshwater analysis.
A tridodecylamine doped membrane was chosen for preparing the pH electrode. Membranes of this type have been subject of considerable research and have already been used in a number of applications.20,21 A calibration curve for a PVC based pH electrode is presented in Fig. S6 (see ESI).† Despite the applicability of the PVC based pH sensor for water samples analysis a commercial glass electrode (half-cell, Mettler Toledo) was also implemented to ensure a reliable pH detection. The latter is very important since carbonate speciation is highly affected by pH changes, and providing reliable pH data is vital for the analysis of carbonate and CO2 levels.
Previous studies have shown PVC based ISEs doped with carbonate ionophore VII to be reliable sensors for carbonate detection with exceptionally high selectivity over the key interfering ions, giving detection limit lower than 10−6 M at pH 8.0.22 A strategy has recently been explored that allows one to directly measure CO2 levels by measuring pH electrode against a carbonate electrode without using a traditional reference electrode.23 Since the flow setup described above includes a reference electrode at all times, both pH and carbonate electrodes were measured against the reference electrode while CO2 levels may be inferred by subtraction of the two potentials values based on carbonate species equilibria.23
Nitrate and ammonium levels in freshwater are quite low. Insufficient detection limits of nitrate24 and ammonium25,26 selective PVC based sensors as well as less attractive selectivity might be a limiting factor when implementing these sensors for environmental analysis. Considering the low detection limit of 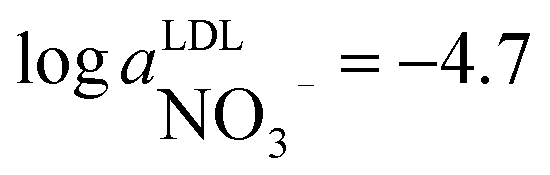 (ESI Fig. S7†) and a rather low nitrate levels in freshwater (10−5 to 10−4 M), monitoring data analysis needed to be performed by a nonlinear regression of the curve given in Fig. S7† with an adjustment of the intercept on the EMF axis according to the experimentally obtained value.
(ESI Fig. S7†) and a rather low nitrate levels in freshwater (10−5 to 10−4 M), monitoring data analysis needed to be performed by a nonlinear regression of the curve given in Fig. S7† with an adjustment of the intercept on the EMF axis according to the experimentally obtained value.
When choosing the composition of the membrane for ammonia sensing we aimed at preparing a membrane with the lowest possible detection limit. Considering the results of preliminary studies (see ESI Fig. S8†), membranes containing nonactin without additional ion exchanger were prepared. The detection limit for the ammonium selective electrode with a 100 µM potassium background corresponds to  , which is not sufficiently low for the chosen application as suggested from the average ammonium level indicated above. However, the actual ammonium concentrations may vary significantly from 10−8 M in the surface layer to few hundred µM in freshwater sediments.5 Therefore, the ammonium sensor was also implemented into the flow analysis setup to allow for an estimation of ammonium level changes when profiling at larger depths.
, which is not sufficiently low for the chosen application as suggested from the average ammonium level indicated above. However, the actual ammonium concentrations may vary significantly from 10−8 M in the surface layer to few hundred µM in freshwater sediments.5 Therefore, the ammonium sensor was also implemented into the flow analysis setup to allow for an estimation of ammonium level changes when profiling at larger depths.
Bracketed calibrations using the proposed flow analysis setup are shown in ESI Fig. S9† with the following observed electrode slopes in mV: 31.4 ± 1 for calcium, 58.3 ± 1 for pH (PVC based electrode), 58.7 ± 1 for pH glass electrode, 31.5 ± 1 for carbonate, 37.3 ± 3 for nitrate and 7.6 ± 2 for ammonium. Single ion activities ai were calculated based on the concentration values indicated in Table 1 according to the common relationship:
| ai = γici | (1) |
As discussed above, a near Nernstian response slope was obtained for calcium, carbonate and pH electrodes, suggesting that they are adequate for quantitative analysis. In contrast, a sub-Nernstian slope for nitrate necessitated the use of a nonlinear calibration curve based on the Nikolskii equation for calculating the nitrate activity in water samples. Indeed, the deviation between linear and non-linear calibrations was important for this ion (5–25% RSD depending on sample activity). The relatively high concentration of potassium deteriorates the limit of detection for the ammonium selective membrane and explains the relatively low slope (see ESI, Fig. S8†). Nonetheless, this electrode was maintained for comparative estimation of ammonium levels in water samples.
We note that a multivariate approach could also be efficiently applied for data analysis when observing nonlinear calibration curves, as these techniques are useful when determining concentrations close to the detection limit.27 In this early work, it was initially preferred to apply an analytical approach based on Nikolskii equation for a rational discussion of the data.
3.2 Field measurements
Measurements on lake Greifensee were performed during a period of 90 h, starting on August 18, 2014 and finishing on August 22, 2014. Fig. 2a shows the obtained potentiometric time trace between Thursday afternoon (12 pm) and Friday morning (10 am) for the five analyzed ions. As discussed above, the experimental arrangement allowed for the measurement of up to eight ISEs. In this experiment the following ISEs were installed: 1 carbonate electrode, 1 glass pH electrode, 2 nitrate electrodes, 2 ammonium electrodes and 2 calcium electrodes. For simplicity and to avoid overlap of the graphs, just 5 traces, one for each type of ISE, are presented in Fig. 2a and b. Every cycle consisted of two 2-point-calibrations performed both at the beginning and at the end of the cycle (four calibration points), six measurements corresponding to six water samples taken from different depths and 6 measurements corresponding to the 1-point calibration between water sample measurements. One of the cycles is separately shown in Fig. 2b. Every cycle (including calibrations) took approximately 2 h to be completed and a new cycle started every 4 h. The time when a particular solution reached the surface of ion selective membrane is indicated with vertical lines in Fig. 2b: water sample measurements are highlighted with solid lines and measurements corresponding to calibration solutions 1 and 2 are indicated with dashed and dotted lines respectively.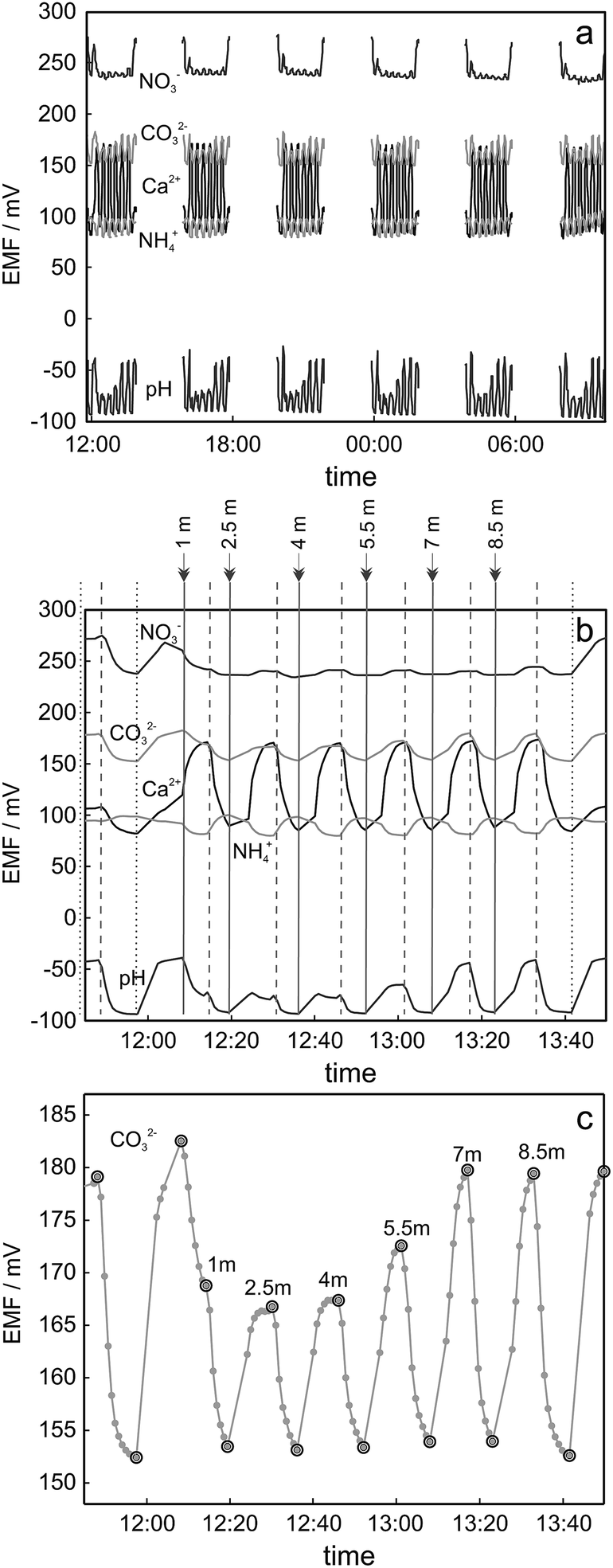 | ||
| Fig. 2 Potentiometric data obtained during filed measurements using proposed automated analysis arrangement (replicates not shown in the figure). (a) Experimental curves obtained during the experiment performed on 21-22.08.2014. (b) Zoom for the first cycle shown in Fig. 5a. Vertical lines indicate the time when corresponding new solution reached the surface of the ISE membrane: calibration solution 1 (dashed line), calibration solution 2 (dotted line) or lake water from the depth specified in the figure (solid line). (c) Zoom for the cycle presented in Fig. 5b for carbonate ISE: grey markers represent the single data points acquired by GalvaPot during field measurements, black circles correspond to the EMF value corresponding to relevant sample/calibration solution. | ||
The collected data points are shown in Fig. 2c. A confirmation of calibration reproducibility within 2 h of measurements is obtained with the injection of a standard solution immediately after measuring each lake sample. The long-term stability of sensor calibration is illustrated in ESI Fig. S10.† The drift of the calibration observed during 90 h of measurements was in the range of 5–15 mV depending on the sensor and confirms the advantage of chosen approach using continuous calibration between the profiles. Considerable changes of pH and carbonate activities were observed as a function of depth. Nitrate levels did not significantly change for the first few meters, but a notable reduction of nitrate activity started to be observed at 8.5 m (see also 3.3). The ammonium level was found to be lower than the detection limit of the electrode (ca. 10 µM with potassium background 100 µM). The ammonium concentration is an indicative parameter of the anoxic region in the lake, which is often present at larger depths9,10 than the ones explored here. The low observed ammonium activity was therefore expected for the conditions used here.
Calcium activity is used as an example of the capability of the established sensing platform. Since the obtained activity is a function of depth and time (f = c (depth, time)), the data are visualized with a 2D surface plot as shown in Fig. 3 and show an increase of calcium activity in the lake with depth and its decrease during the night.
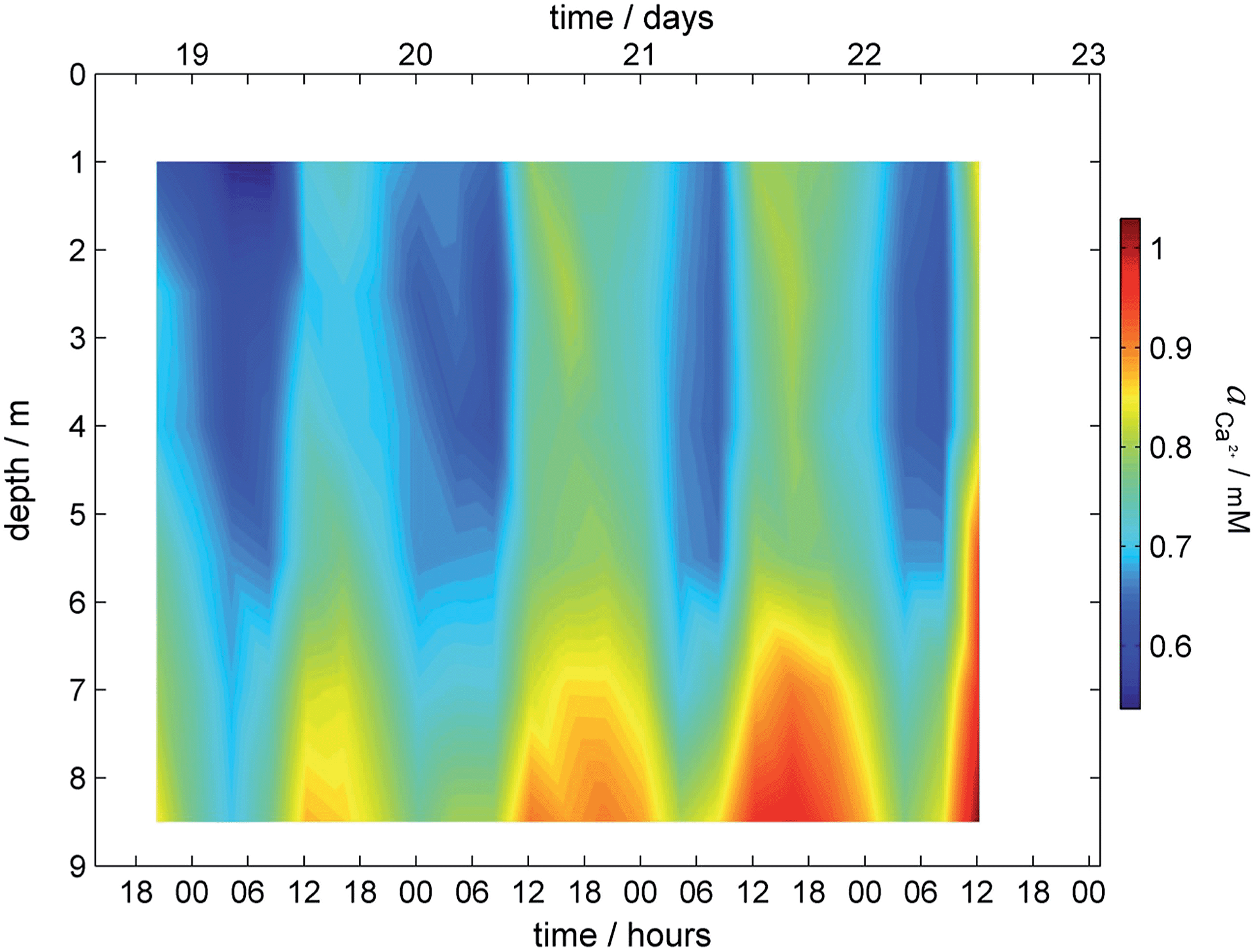 | ||
| Fig. 3 Surface plot for calcium profile obtained during field monitoring on the lake Greifensee (Switzerland) 18-22.08.2014. | ||
3.3 Data validation
To validate the consistency of the approach, laboratory measurements of calcium and nitrate concentrations in lake water were performed. Fig. 4a and b shows the correlation between ion activities determined by potentiometry in the field and the concentrations obtained with laboratory analysis of water samples taken at corresponding depths. A linear correlation is assumed based on the linear relationship between activity and concentration (eqn (1)). The slope of the linear regression corresponds to an apparent single ion activity coefficient γ*i that was estimated as the mean value of experimentally determined single ion activity coefficients , where aexpi is the single ion activity determined by ISE in the field, cexpi is the molar concentration of the ion i obtained by laboratory analysis. The obtained mean values γ*i were found as
, where aexpi is the single ion activity determined by ISE in the field, cexpi is the molar concentration of the ion i obtained by laboratory analysis. The obtained mean values γ*i were found as 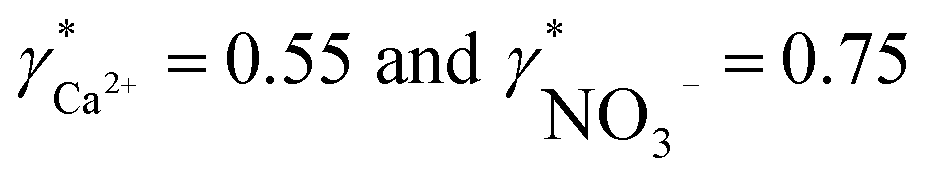 .
.
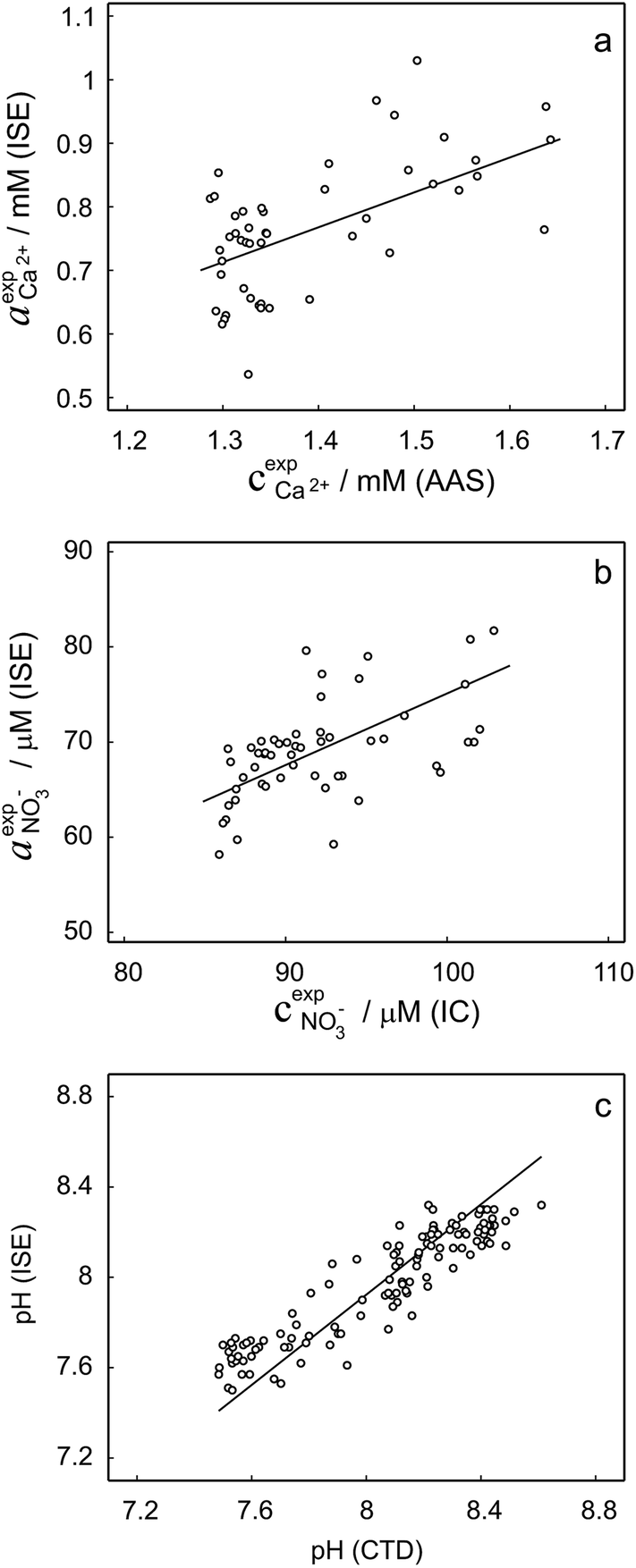 | ||
Fig. 4 (a and b) Correlation between activities measured in the field by ISEs and concentrations obtained within laboratory analysis using atomic absorption spectroscopy (a, calcium) and ion chromatography (b, nitrate). The slopes of linear regressions correspond to apparent single ion activity coefficients being  and and 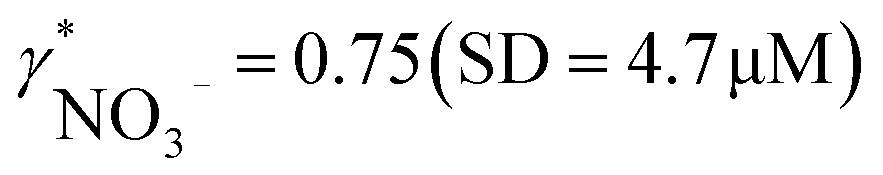 . (c) Correlation between pH measurements with glass electrode implemented in the proposed flow setup and alternative in situ pH measurements performed by EAWAG using CTD probe (SD = 0.2 pH). . (c) Correlation between pH measurements with glass electrode implemented in the proposed flow setup and alternative in situ pH measurements performed by EAWAG using CTD probe (SD = 0.2 pH). | ||
Note that the γ*i value includes not only the thermodynamic contribution from the ion activity coefficients due to the ionic strength of the lake, but also any matrix effects of the natural water sample as well as systematic errors of both field and laboratory measurements. The thermodynamic contribution to the experimental γ*i value was estimated with the single ion activity coefficient γi calculated with the first approximation of the Debye–Hückel theory (see ESI, eqn (2), (3) and (5);† bicarbonate and calcium ions were considered as relevant counter ions as these are the major species in the sample).
The value of the ionic strength was estimated based on conductivity measurements. Electrical conductivity κ is given by the following equation:
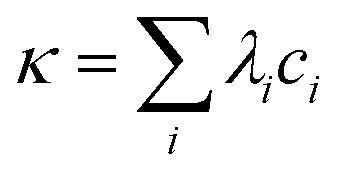 | (2) |
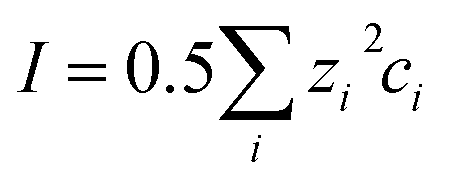 , see ESI eqn (3)†) was calculated assuming calcium bicarbonate as the major contributor to conductivity and ionic strength (λCa2+ = 11.894 mS m2 mol−1, λHCO3− = 4.45 mS m2 mol−1).28 The conductivity of the water samples increased with depth and was found to be in the range 380–470 µS cm−1, giving an ionic strength in the range 5.5–6.8 mM. Based on the simplified Debye–Hückel theory (see ESI, eqn (2) and (5)†) single ion activity coefficients were found as 0.68–0.71 for γCa2+ and 0.91–0.92 for γNO3− considering bicarbonate and calcium as the counter ions accordingly.
, see ESI eqn (3)†) was calculated assuming calcium bicarbonate as the major contributor to conductivity and ionic strength (λCa2+ = 11.894 mS m2 mol−1, λHCO3− = 4.45 mS m2 mol−1).28 The conductivity of the water samples increased with depth and was found to be in the range 380–470 µS cm−1, giving an ionic strength in the range 5.5–6.8 mM. Based on the simplified Debye–Hückel theory (see ESI, eqn (2) and (5)†) single ion activity coefficients were found as 0.68–0.71 for γCa2+ and 0.91–0.92 for γNO3− considering bicarbonate and calcium as the counter ions accordingly.
A comparison of theoretically estimated single ion activity coefficients γi(γCa2+ = 0.68–0.71, γNO3− = 0.91–0.92) with apparent single ion activity coefficients γ*i obtained experimentally  shows γ*i to be somewhat lower than the estimated γi. It should be considered that the procedure for γi calculation presented above allows only a rough estimation of γCa2+ and γNO3− since the complexity of the natural sample media along with relatively high ionic strength goes beyond the first approximation of the Debye–Hückel theory. To a certain extent the difference between estimated (γi) and experimental (γ*i) values may originate from systematic experimental errors deriving from calibration inaccuracy of field measurements and/or differences in sampling procedures. In particular, the samples for laboratory analysis of calcium were acidified, which may result in the release of complexed or adsorbed calcium that is not detected with ISEs.
shows γ*i to be somewhat lower than the estimated γi. It should be considered that the procedure for γi calculation presented above allows only a rough estimation of γCa2+ and γNO3− since the complexity of the natural sample media along with relatively high ionic strength goes beyond the first approximation of the Debye–Hückel theory. To a certain extent the difference between estimated (γi) and experimental (γ*i) values may originate from systematic experimental errors deriving from calibration inaccuracy of field measurements and/or differences in sampling procedures. In particular, the samples for laboratory analysis of calcium were acidified, which may result in the release of complexed or adsorbed calcium that is not detected with ISEs.
Activity and concentration profiles (for field and laboratory data, respectively) are presented in Fig. 5. The scales of the concentration and activity axes correspond to the relationship aexpi = γ*icexpi where apparent single ion activity coefficients γ*i are equal to 0.55 for calcium and 0.75 for nitrate as discussed above. The profiles suggest good correlation between field and laboratory measurements. When comparing field and laboratory results it must be taken into account that owing to the monitoring system arrangement, a manual sampling for laboratory analysis was performed with slightly different time resolution and not exactly at the same location. The latter may result in some discrepancy between field and laboratory measurements, so the data points in Fig. 5 are not expected to match perfectly. Importantly, despite the relatively small changes of the parameters, the tendency of relative changes of calcium and nitrate activities agree with the relevant concentration changes. Fig. 4c shows the correlation between pH values measured using commercial pH glass electrode in the proposed flow arrangement and those obtained by EAWAG using the CTD probe that ran immediately before the ISE profiling. A good correlation between the results obtained using the two different methods confirms that the observed pH data are adequate and applicable in the presented sensing array.
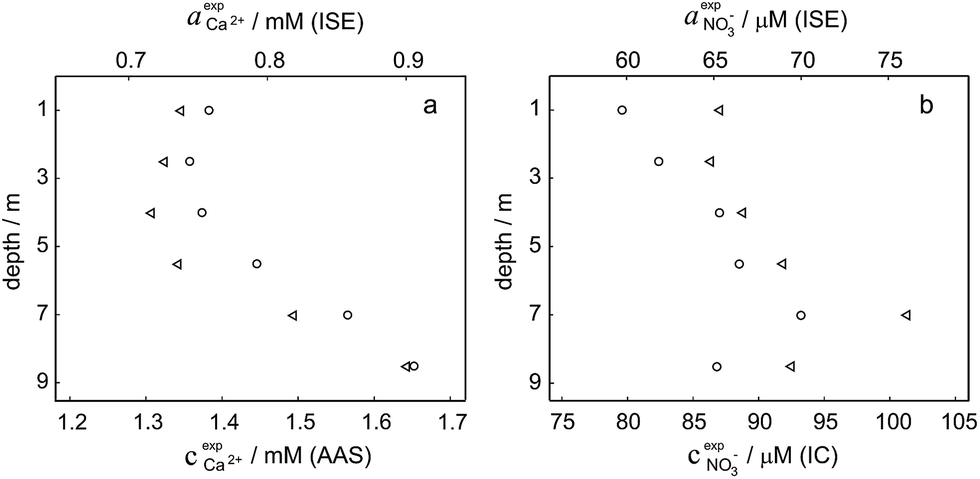 | ||
Fig. 5 Field data versus laboratory analysis. (a) Calcium activity (circles) measured by ISE in the field and calcium concentration (triangles) measured by AAS depending on the depth. (b) Nitrate activity (circles) measured by ISE in the field and nitrate concentration (triangles) measured by IC depending on the depth. The scales of activity and concentration axes are defined by the relationship aexpi = γ*icexpi where γ*i is apparent single ion activity coefficient 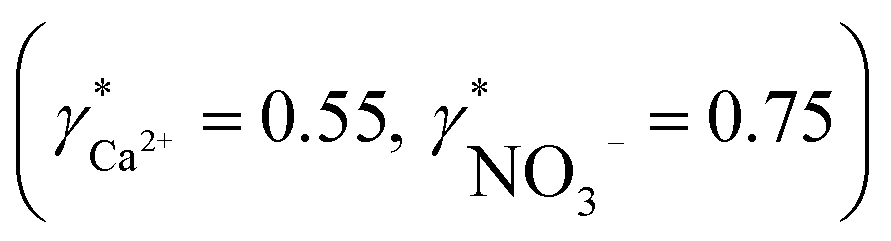 . . | ||
4. Conclusions
The potentiometric sensing array for the detection of nutrients and species relevant to the carbon cycle was successfully deployed for continuous monitoring of pH, calcium, nitrate and carbonate levels at the EAWAG monitoring platform on the lake Greifensee (Switzerland), in parallel with scanning flow cytometer measurements. The proposed setup suggests continuous calibration of potentiometric sensors between the measurements of water samples thus providing more reliable data compared to ISE arrays using relatively infrequent calibration in field conditions. Comparison of field and laboratory measurements confirms the consistency of the setup for field analysis. We believe that the potentiometric sensing array described here may serve as a promising tool for ecological monitoring aimed at tracking and understanding environmental changes in aquatic ecosystems.Acknowledgements
The authors acknowledge financial support by the Swiss National Science Foundation (Sinergia Grant).References
- R. Adrian, C. M. O’Reilly, H. Zagarese, S. B. Baines, D. O. Hessen, W. Keller, D. M. Livingstone, R. Sommaruga, D. Straile, E. Van Donk, G. A. Weyhenmeyer and M. Winder, Limnol. Oceanogr., 2009, 2283–2297 CrossRef.
- F. Pomati, J. Jokela, M. Simona, M. Veronesi and B. W. Ibelings, Environ. Sci. Technol., 2011, 45, 9658–9665 CrossRef CAS PubMed.
- K. Yoshiyama, J. P. Mellard, E. Litchman and C. A. Klausmeier, Am. Nat., 2009, 174, 190–203 CrossRef PubMed.
- H. S. Jacobsen and A. L. Jensen, in Monitoring of Water Quality: The Contribution of Advanced Technologies, ed. F. Colin and P. Quevauviller, Elsevier, The Netherlands, 1998, pp. 89–102 Search PubMed.
- M. Maerki, B. Muller, C. Dinkel and B. Wehrli, Limnol. Oceanogr., 2009, 54, 428–438 CrossRef CAS.
- D. Odermatt, F. Pomati, J. Pitarch, J. Carpenter, M. Kawka, M. Schaepman and A. Wüest, Rem. Sens. Environ., 2012, 126, 232–239 CrossRef PubMed.
- O. Zielinski, J. A. Busch, A. D. Cembella, K. L. Daly, J. Engelbrektsson, A. K. Hannides and H. Schmidt, Ocean Sci., 2009, 5, 329–349 CrossRef CAS PubMed.
- E. Bakker and E. Pretsch, Angew. Chem., Int. Ed., 2007, 46, 5660–5668 CrossRef CAS PubMed.
- D. de Beer, A. Bissett, R. de Wit, H. Jonkers, S. Koehler-Rink, H. Nam, B. H. Kim, G. Eickert and M. Grinstain, Limnol. Oceanogr.: Methods, 2008, 6, 532–541 CrossRef CAS.
- B. Muller, K. Buis, R. Stierli and B. Wehrli, Limnol. Oceanogr., 1998, 43, 1728–1733 CrossRef CAS.
- A. K. Bell-Vlasov, J. Zajda, A. Eldourghamy, E. Malinowska and M. E. Meyerhoff, Anal. Chem., 2014, 86, 4041–4046 CrossRef CAS PubMed.
- S. Jeanneret, G. A. Crespo, M. Ghahraman Afshar and E. Bakker, Sens. Actuators, B, 2015, 207, 631–639 CrossRef CAS PubMed.
- A. Heini, I. Puustinen, M. Tikka, A. Jokiniemi, M. Lepparanta and L. Arvola, Hydrobiologia, 2014, 731, 139–150 CrossRef CAS PubMed.
- T. Sobczynski and T. Joniak, Pol. J. Environ. Stud., 2013, 22, 227–237 CAS.
- O. Farhadian, S. Kolivand, K. M. Mahmoudi, D. E. Ebrahimi and S. N. Mahboobii, Iran. J. Fish. Sci., 2013, 12, 301–319 Search PubMed.
- E. Goransson, Ph.D. Thesis, Swedish University of Agricultural Sciences, 2003.
- K. Hollocher, L. Quintin and D. Ruscitto, in New York State Geological Association Field Trip Guidebook, 74th annual meeting, ed. J. McClelland, and Karabinos, P., Lake George, New York, 2002, pp. C11.11–C11.15 Search PubMed.
- E. Pretsch, Chimia, 2001, 55, 875–878 CAS.
- A. Ceresa, E. Bakker, B. Hattendorf, D. Guenther and E. Pretsch, Anal. Chem., 2001, 73, 343–351 CrossRef CAS.
- S. C. Ma and M. E. Meyerhoff, Mikrochim. Acta, 1990, 1, 197–208 CrossRef CAS.
- C. Espadas-Torre, E. Bakker, S. Barker and M. E. Meyerhoff, Anal. Chem., 1996, 68, 1623–1631 CrossRef CAS.
- Y. S. Choi, L. Lvova, J. H. Shin, S. H. Oh, C. S. Lee, B. H. Kim, G. S. Cha and H. Nam, Anal. Chem., 2002, 74, 2435–2440 CrossRef CAS.
- X. Xie and E. Bakker, Anal. Chem., 2013, 85, 1332–1336 CrossRef CAS PubMed.
- M. A. Arada Pérez, L. P. Marín, J. C. Quintana and M. Yazdani-Pedram, Sens. Actuators, B, 2003, 89, 262–268 CrossRef.
- M. S. Ghauri and J. D. Thomas, Analyst, 1994, 119, 2323–2326 RSC.
- O. G. Davies, G. J. Moody and J. D. R. Thomas, Analyst, 1988, 113, 497–500 RSC.
- J. Gallardo, S. Alegret and M. del Valle, Sens. Actuators, B, 2004, 101, 72–80 CrossRef CAS PubMed.
- Handbook of Chemistry and Physics, ed. D. R. Lide, 72 edn, CRC Press, 1991–1992 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of Galvapot v2 design and methods, extended results and discussions. Table S1, Fig. S1–S10. See DOI: 10.1039/c5em00038f |
| This journal is © The Royal Society of Chemistry 2015 |