
Chelation technology: a promising green approach for resource management and waste minimization
Garima
Chauhan
,
K. K.
Pant
and
K. D. P.
Nigam
*
Department of Chemical Engineering, Indian Institute of Technology Delhi, New Delhi, India 110016. E-mail: nigamkdp@gmail.com; Fax: +91-11-26591020; Tel: +91-11-26591020
First published on 13th November 2014
Abstract
Green chemical engineering recognises the concept of developing innovative environmentally benign technologies to protect human health and ecosystems. In order to explore this concept for minimizing industrial waste and for reducing the environmental impact of hazardous chemicals, new greener approaches need to be adopted for the extraction of heavy metals from industrial waste. In this review, a range of conventional processes and new green approaches employed for metal extraction are discussed in brief. Chelation technology, a modern research trend, has shown its potential to develop sustainable technology for metal extraction from various metal-contaminated sites. However, the interaction mechanism of ligands with metals and the ecotoxicological risk associated with the increased bioavailability of heavy metals due to the formation of metal–chelant complexes is still not sufficiently explicated in the literature. Therefore, a need was felt to provide a comprehensive state-of–the-art review of all aspects associated with chelation technology to promote this process as a green chemical engineering approach. This article elucidates the mechanism and thermodynamics associated with metal–ligand complexation in order to have a better understanding of the metal extraction process. The effects of various process parameters on the formation and stability of complexes have been elaborately discussed with respect to optimizing the chelation efficiency. The non-biodegradable attribute of ligands is another important aspect which is currently of concern. Therefore, biotechnological approaches and computational tools have been assessed in this review to illustrate the possibility of ligand degradation, which will help the readers to look for new environmentally safe mobilizing agents. In addition, emerging trends and opportunities in the field of chelation technology have been summarized and the diverse applicability of chelation technology in metal extraction from contaminated sites has also been reviewed.
 Garima Chauhan | Ms Garima Chauhan graduated from the University of Rajasthan (India) with a bachelor's degree (B.Tech.) in Biotechnology in 2006. She obtained her master's degree (M.Tech.) from Malaviya National Institute of Technology (India) in 2009. Currently, she is pursuing her PhD at the Indian Institute of Technology Delhi. Her dissertation work explores the concept of chelation technology to develop an ecofriendly process for the extraction of heavy metals from multimetallic spent catalysts obtained from refineries and fertilizer industries. Her current research interests include waste management, green engineering, and organometallic and computational chemistry. |
 K. K. Pant | Dr K. K. Pant is currently Professor in the chemical engineering department at the Indian Institute of Technology (IIT) Delhi. He has published more than 90 papers in various peer reviewed international research journals with more than 2800 citations (h-index 27). He is currently actively engaged in the research areas of application of heterogeneous catalysis for a cleaner and greener environment, catalytic hydrocarbon conversion processes, hydrogen fuel generation from renewable energy sources, and biomass utilization. He is a reviewer for several international journals and also the member of several chemical engineering professional societies. |
 K. D. P. Nigam | Prof. Krishna Nigam has been on the faculty of the Department of Chemical Engineering, Indian Institute of Technology Delhi, from 1976 and has served in various capacities. He has authored more than 135 research publications, which have received more than 2500 citations in peer reviewed international journals, and has an h-index of 29. His research work has been widely acclaimed and cited in prestigious journals such as Proceedings of Royal Society London, Journal of Fluid Mechanics, Perry's Hand Book etc. He has served on the editorial board of many journals, e.g. Chemical Engineering Research and Design, Chemical Engineering and Processing: Process Intensification, and Education for Chemical Engineers, and guest editor for special issues of Chemical Engineering Science. In recognition of his contributions to chemical engineering, Industrial & Engineering Chemistry Research has brought out a Festschrift issue in his honor (51(4), 1435–2178 (2012)). He has been awarded the HUMBOLDT RESEARCH AWARD of Germany for the year 2011, a rare distinction making him the first Indian chemical engineer recipient of the prestigious award since its inception in 1982. |
Environmental impactHeavy metals are present in industrial waste in significant amounts and may become a threat to ecosystems and human health. Chelation–dechelation technology was conceived by our research group for the extraction of heavy metals from spent catalysts. Nearly “closed-loop” green technology has also been developed in order to extract metals from multimetallic spent catalysts using biodegradable chelating agents. The present review article explores all the aspects of this technology to promote the chelation process as a green chemical engineering approach. This manuscript covers the interaction mechanism of ligands with metals, ecotoxicological risks associated with increased bioavailability of heavy metals and technical applicability of chelation technology in different research areas. Emerging trends and opportunities have also been suggested to carve out new territory for green chelation technology. |
1. Introduction
Sustainability is the primary concern for process industries in the present competitive era, where energy resources are being consumed at much faster pace than ever. Brundtland communicated the most widely adapted definition for sustainability, which suggests that “Sustainable development is development which meets the needs of the present without compromising the ability of future generations to meet their own needs”.1 The necessity of energy resources sprang up to prominence in recent decades due to rapid urban and industrial growth. It is projected that world energy consumption will grow by 56%, from 132 quadrillion kilocalories (kcal) in 2010 to 207 quadrillion kcal in 2040 (ref. 2). A strong conjunction of energy utilization with industrial growth can also be substantiated by the data reported in the IEO2013 (ref. 2) survey, which indicates that the industrial sector is the largest energy consuming sector, utilising 52% of global delivered energy in 2010. This increasing rate of energy dependence is pushing the world towards resource scarcity, price inflation and degraded ecosystems. Therefore, innovative processes are required within an eco-friendly framework that should recognise the global necessity of resources (metals) and significantly reduce hazardous emissions to the environment, without compromising the economics of the process.Although numerous efforts are being made worldwide to call for sustainable industrial processes in order to conserve resources, relatively little has emerged on a practical level so far. The reason behind this lack of practical applicability may be due to the complexity of process industries, where it is difficult to manage economics, ecology and resource needs, all at the same time. Green chemical engineering recognises the concept of developing innovative environmentally benign technologies in order to overcome this complexity and to develop new trends in sustainable process development. Several green research efforts3–9 are being made in process industries to conserve resources and minimize the possibility of pollution.
In the context of conserving resources, heavy metals are one of the major natural resources which are of deep concern. The impossibility of having economic and industrial development without a concomitant increase in resource consumption has now stepped up the necessity of metals, which are considered as ‘keys of industrialization’. Heavy metals are extensively used in the preparation of various catalysts in the petrochemical and fertilizer industries in the form of metal oxides and metal sulphides. The recovery of these metals from industrial waste is an essential issue to consider for industrial and economic benefits. The increasing demand for metals in industrial activity makes them an ineluctable contributor in process industries; however, nor can the negative aspects of heavy metals be disregarded. Heavy metals are bioaccumulative in the ecosystem and their removal from the soil–plant–atmosphere continuum is a tedious process. Waste generated from industrial and manufacturing processes can be considered a substantive source of release of heavy metals into the environment. Waste water treatment sludge, distillation bottom sludge and spent catalysts generated due to organic chemical manufacturing have been classified as potential hazardous waste by the environmental protection agency (EPA), USA.10 Spent catalysts from petroleum refining dual process reactors (spent hydrotreating catalyst (EPA-K171) and spent hydrorefining catalyst (EPA-K172)) have also been listed as hazardous waste due to the presence of toxic metals.10 Wastewater treatment sludge from circuit board manufacturing operations (EPA-F006) and spent pickle liquor generated by the iron and steel industries (EPA – K061, K062) are also considered major sources of environmental pollution.10
In this review, a range of conventional processes (basic inspiring technologies) and green approaches (new modern trends) employed for metal extraction are discussed in brief. The positive and negative aspects of each process have been briefly reviewed in order to understand the current and future process design requirements. Chelation technology, one of the modern approaches, has been of increasing interest to environmental sciences in recent years for the extraction of heavy metals from soil11,12 and industrial waste.13–15 Therefore, the core of this article is a deep review of what has been done in the field of chelation technology so far and what can be done in the near future to promote this technology as a green chemical engineering approach (zero-waste technology). Efforts have been made in the present review to summarize the important aspects associated with metal–chelate complex formation including its chemistry and the interaction mechanism with metals. Ecotoxicological issues associated with conventional chelating agents as well possibilities for degradation of the chelating agents in the environment have also been reviewed in detail. The diverse applications of chelating agents in various research fields have been summed up, with metal extraction from soil and industrial waste using chelation technology being emphasized in particular. In addition, the pertinence of new computational tools in the field of chelation technology has also been considered to illustrate the future possibility of designing new biodegradable chelating agents.
2. Available methods for metal extraction from contaminated sites
2.1. Conventional approaches
Numerous approaches have been reported in the literature for the recovery of heavy metals from contaminated sites. Adsorption has been considered a good alternative for the efficient extraction of metals from industrial waste water.16,17 Membrane separation,18 electrotreatment,19 photocatalytic processes20 and absorption by aquatic plants21,22 have also been employed to remove metals and other pollutants from water. Toxic metals may also be present in soil, and may exhibit chemical interactions, mobility and potential toxicity. Therefore, various techniques such as vitrification, excavation, removal of contaminated soil layers, phytoremediation23,24 and soil washing using surfactant, acids, alkalis and chelating agents25,26 have been investigated for soil remediation. Spent catalysts generated from refineries and fertilizer industries are also considered potential sources of heavy metals and therefore research on metal extraction from spent catalysts is being carried out by researchers. Various pyrometallurgical processes such as calcination followed by leaching27,28 or roasting at high temperatures29,30 have been employed for the extraction of heavy metals from spent catalysts. Hydrometallurgical approaches (leaching,31,32 solvent extraction33,34 and biological methods35,36) have also been reported to recover metals from spent catalysts, soil and other types of industrial waste. Table 1 summarizes the different studies performed to extract heavy metals from contaminated sites using hydro- and pyrometallurgical approaches, along with the positive and negative aspects of each process. Experiments were performed under batch mode for all the references listed in Table 1.| References | Spent catalyst | Metals | Reagent/micro-organism | Reaction conditions | Metal recovery | Advantages | Limitations |
|---|---|---|---|---|---|---|---|
| Bio-leaching | |||||||
| Bhardwaj et al.37 | Spent hydrotreatment catalyst | Fe, Ni, Mo, Al | Acidophilic thermophile, Acidianus brierleyi | Spent medium leaching, 1% w/v pulp density | Nearly 100% Fe, Ni and Mo, 67% Al | No harmful effects to the environment. High extraction efficiency for heavy metals. Low energy requirements. | Longer leaching time. Microorganisms can not withstand at high temperature. Dependency on several atmospheric conditions. Sensitive to any contamination possibility. |
| Gerayeli et al.38 | Spent refinery catalysts | Al, Mo, Ni | Acidianus brierleyi | pH 1.6, pulp density 0.6% (w/v), inoculation 4% (v/v) and elemental sulfur concentration 4 g L−1 | 35% Al, 83% Mo, 69% Ni | ||
| Amiri et al.39 | Spent refinery catalyst | Al, Mo, Ni | Aspergillus niger | Particle size 150–212 μm, sucrose 93.8 g L−1, pulp density 3% w/v, pH 7 | 99.5 ± 0.4% Mo, 45.8 ± 1.2% Ni, 13.9 ± 0.1% Al | ||
| Amiri et al.40 | Tungsten rich spent hydro-cracking catalyst | Ni, Mo, Fe, W, Al | Penicillium simplicissimum | Particle size < 150 μm, pulp density 3% w/v, 30 °C, shaking speed 120 rpm, 14 days incubation time. (Two step bioleaching.) | 100% W, 100% Fe, 92.7% Mo, 66.43% Ni, 25% Al | ||
| Belochini et al.41 | Spent refinery catalysts | Ni, V, Mo | Mixed culture of three strains of Fe/S oxidizing bacteria | 21 days cultivation time, 30 °C and 175 rpm shaking speed, catalyst concentration 10 g L−1, modified 9K medium | 83 ± 4% Ni, 90 ± 5% V, 30–40% Mo | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| Acid leaching | |||||||
| Banda et al.42 | Spent hydro-processing catalyst | Mo, Co | Hydrochloric acid (HCl) | Acid conc.: 3 mol L−1, reaction temp.: 90 °C, reaction time: 60 min, particle size: 250 μm, pulp density: 5% (w/v) | 97% and 94% of Mo and Co | High metal recovery. Better recirculation possibilities (Mishra et al., 2010;32 Zeng & Cheng, 2010).33 Shortest process time. | Usage of hazardous chemicals. May generate corrosive environment. Requires specific material of construction to avoid corrosion. May liberate toxic gases to environment. High energy requirement. High operational cost. Not applicable to highly contaminated substrates (Asghari et al., 2013).185 |
| Mazurek43 | Spent vanadium catalyst | V, K, Fe | Oxalic acid | Acid conc.: 2%, reaction temp.: 50 °C, reaction time: 4 h, particle size: 180–250 μm, pulp density: 4% (w/v) | 91% V, 92% K, 63% Fe | ||
| Parhi et al.44 | Spent refinery catalyst | Ni, Al, Fe | Hydrochloric acid | Acid conc.: 1 M, reaction temp.: 50 °C, reaction time: 120 min, particle size: 50–71 μm, pulp density: 0.2% (w/v) | 99.9% Ni, 1% Al | ||
| Al-Sheeha et al.45 | Spent hydro-processing catalyst | Mo, V, Ni | Organic vs. inorganic acids | Acid conc.: 10%, reaction temp.: 50 °C, reaction time: 6 h, particle size < 500 μm, pulp density: 2.5% (w/v) | Organic acids gave better leaching than inorganic acids | ||
| Barik et al.46 | Spent hydrodesulphurization catalyst | Co, Mo, Al, S | Sulphuric acid along with different oxidants | Acid conc.: 0.5 M, reaction temp.: 50 °C, reaction time: 120 min, particle size: 51–70 μm, pulp density: 1% (w/v), [H2O2] as oxidant: five times stoichiometric amount | 99.87% Mo, 96.25% Co, 11.03% Al | ||
|
|||||||
| Alkali leaching | |||||||
| Huang et al.47 | Spent hydro-desulphurization catalyst | Mo, Ni, Al | NH3·H2O, Na2CO3, NaOH under normal pressure and autoclave conditions | Temperature 160 °C, NaOH dosage: 1.2 times the stoichiometric amount, solid/liquid ratio: 1:4, retention time: 120 min, stirring rate: 300 rpm |
NH3, Na2CO3: <85% Mo; NaOH: <93% Mo; NaOH auto-claving: >96% Mo with 0.2% Al leaching | High extraction efficiency. Short reaction time. | Usage of hazardous chemicals. Large quantity of chemicals needed. May pose harmful effects to human health and the environment. Leaching of support materials along with desired metal affects the extraction efficiency. |
| Al-Sheeha et al.45 | Spent hydro-processing catalyst | Mo, V, Ni | Ammonium hydroxide (NH4OH), ammonium carbonate ((NH4)2CO3), ammonium persulphate ((NH4)2S2O8) | Reagent concentration: 1 molar solution, reaction temperature: 50 °C | NH4OH: 79.4% Mo, 65.4% V, 22.4% Ni, 2.9% Al; (NH4)2CO3: 86.8% Mo, 70.6% V, 25.4% Ni, 0.3% Al; (NH4)2S2O8: 46.7% Mo, 83.7% V, 82.7% Ni, 17.7% Al | ||
| Katsiapi et al.48 | Co–Mn hydroxide precipitates | Co | Ammonical leaching | NH3/(NH4)2CO3: 200 g/200 g, solid to liquid ratio: 10% | 93% Co with less than 0.05% Mn leaching | ||
| Mazurek et al.49 | Spent vanadium catalyst | V, K, Fe | Urea solution | Particle size: 180–250 μm, reaction time: 60 min, reaction temperature: 20 °C, reagent concentration: 40% urea solution, solid to liquid ratio: 10% | 78% V, 90% K, 29% Fe | ||
|
|||||||
| Pyrometallurgical methods | |||||||
| Al-Sheeha et al.45 | Spent hydro-processing catalyst | Mo, V, Ni | Soda ash roasting using NaOH or Na2CO3 | Reaction temperature: 550 °C and 700 °C | More than 97% Mo and V; Ni and Al remain as residue with Na2CO3, while Al leached out in the case of NaOH and affected efficiency | Extraction of highly pure alumina which can be reused as support material. Short reaction time. | High energy requirements and high cost. Difficult to operate and liberates toxic gases to the environment during thermal treatment. May cause major changes in catalyst properties (surface area, phase change) due to sintering. Not an economical approach for extraction of metals from less concentrated raw materials of complex composition. |
| Chen et al.50 | Spent hydro-desulphurization catalyst | Ni, Co, Mo, V, Al2O3 | Roasting | Roasting at 750 °C, roasting time: 30 min, Na2O:Al2O3 = 1.2 |
97.2% Al, 95.8% V, 98.9% Mo | ||
| Kar et al.30 | Spent hydro-refining catalysts | Mo, Ni, S, Co, Fe | Salt roasting using NaCl | Roasting temperature: 900 °C, salt conc.: 20 wt.%, roasting time: 60 min | More than 90% Mo | ||
|
|||||||
| Chlorination | |||||||
| Upadhyay et al.51 | Spent automotive catalyst | Pt, Pd, Rh | Chlorination using electro-generated chlorine | HCl (6.0 mol L−1), current density: 714 A m−2, temperature: 363 K, pulp density: 20 g L−1, agitation speed: 700 rpm | 71% Pt, 68% Pd, 60% Rh | Significant extraction efficiency. Short reaction time. | Large amount of HCl needed. HCl and generated chlorine are toxic. |
| Ojeda et al.52 | Alluvial material | Au | Chlorination + pyro-metallurgical process | 873 K and 5400 s, flow rate: 1.67 × 10−3 m3, molar fraction of chlorine equal to one | 98.73% recovery | High extraction efficiency. Short reaction time. | High temperature requirement. Chemicals needed for reaction are hazardous to handle. May liberate toxic gases to the ecosystem. |
All these processes have shown potential in removing metals from industrial waste. However, the use of hazardous chemicals, the possibility of secondary pollution by these chemicals, lower compatibility with biological processes in the environment and high process costs restrict their use on large scales. Thus, the health and environmental issues associated with these traditional methods are drawing the attention of researchers to look for a sustainable alternative for metal recovery from industrial waste.
2.2. Recent trends
Globally, there is an urgent need to look for new green reagents for metal extraction. This excogitation is now assisting researchers to move towards ionic liquids (ILs), novel resins and polymeric composites, chelating agents and other green extractants. ILs have been efficiently used in the extraction of lanthanides and actinides. Recently, 1-methylimidazole (1-MIM) and 2-methylimidazole (2-MIM) with imidazolium-based ILs were employed for the solvent extraction of lanthanides and yttrium.53 Quaternary phosphonium and ammonium ILs, Cyphos® IL101 (Cytec) and Aliquat® 336 (Henkel), were also demonstrated to remove Pt from aqueous solution by replacing chloride anions with functionalized aromatic anions.54 Liquid phase micro extraction with phosphonium- and ammonium-based ILs, bearing sulphur functionalities in the form of thiosalicylate and 2-(methylthiobenzoate) anions, were also investigated for the extraction of metal ions (Ag, Cu, Hg and Pt) from industrial and communal waste water.55 A wide range of applications of ILs, covering distinct research areas (catalysis, electrolytes and solvents), are currently at an exploratory phase to employ them in industrial processes. However, some ILs may severely contaminate the ecosystem in the case of accidental leakage or wastewater disposal. The search for suitable extractants and the need to understand the complexity of an original aqueous solution due to the interference of added chemicals are other problems associated with this approach.56A new term, “green adsorption”, has also been introduced recently to describe metal extraction from waste water by utilising low-cost, ecofriendly materials originating from agricultural sources, by-products (fruits, vegetables, food), residues and wastes.57 Several authors have employed natural bentonite and zeolites,58,59 rice/peanut husks and fly ash,59–61 banana and orange peel,62 and anaerobic granular sludges63–65 for metal extraction. In spite of having economic potential, these adsorbents are not preferable due to their low adsorption capacities and therefore, chemical modification of these materials is required to improve the performance.66–69 Rice hulls, chemically modified using ethylenediamine, were investigated as an adsorbent for Cr3+ metal removal70 and it was observed that chemical modification of the rice hulls enhanced their adsorption capacity. Fig. 1 shows the structural configuration of some green extractants that have been used in the literature for metal removal.
 | ||
| Fig. 1 Newly synthesized green extractants for metal removal from contaminated sites: (A) DEHPA (di(2-ethylhexyl)phosphoric acid, (B) Aliquat-336 (N-methyl-N,N,N-trioctyl-octan-1-ammonium chloride), (C) Cyanex 272 (bis(2,4,4-trimethylpentyl)phosphinic acid), (D) Cyanex 301 (bis(2,4,4-trimethylpentyl)dithiophosphinic acid), (E) 1-N-butyl-3-methylimidazolium salt, (F) LIX-84-IC (2-hydroxy-5-nonyl-acetophenone oxime), (G) tributyl phosphate, (H) Acorga M5640 (active substance: 2-hydroxy-5-nonylbenzaldehyde oxime), (I) Amberlite resin 400, (J) Dowex M-4195 resin. | ||
Some authors also employed modified biopolymer adsorbents derived from chitin, chitosan and starch for metal removal from contaminated sites.6 Repo et al.71 investigated the adsorption properties of chitosans, modified using different chelating agents, in aqueous solutions of Co2+ and Ni2+ ions. The results indicated that the adsorption efficiency of ethylenediaminetetraacetic acid (EDTA)-modified chitosan was 99.2% for Co2+ and 99.5% for Ni2+, while diethylenetriaminepentaacetic acid (DTPA)-modified chitosan removed 96.7% of Co2+ and 93.6% of Ni2+ under similar reaction conditions, which could be explained on the basis of the crosslinked structure of DTPA-chitosan and lower surface coverage of DTPA compared to EDTA. In a similar manner, Wang et al.72 applied the adsorption process to extract Pb2+ and Cu2+ in a micro-polluted water source using PS–EDTA resin. It was illustrated that under optimum reaction conditions, the PS–EDTA surface adsorbs Cu2+ much better than Pb2+ and seems to be a better adsorbent for the removal of both ions from aqueous solutions. Dzulkefly et al.73 used a chelating resin, Amberlite IRC-718, for the extraction of Ni2+ from spent catalyst samples, obtained from the hydrogenation of palm and palm kernel oils. The oil content was removed from the catalyst by Soxhlet extraction using n-hexane. The Amberlite resin showed more than 90% extraction efficiency for Ni2+, with greater than 90% purity with regard to other metals, Mg and Ca, in the catalyst samples. Kolodynska74 studied the effect of a weak base chelating resin (Dowex M4195) as an ion exchanger for Cu2+, Zn2+ and Ni2+ as well as for Cd2+ and Pb2+ sorption in the presence of complexing agents, glutamicdiacetic acid (GLDA) and ethylenediaminedisuccinic acid (EDDS). It was observed that at low pH there are some deprotonated nitrogen atoms capable of interacting directly with metal ions. Dowex M4195 was reported to have a higher sorption capacity with GLDA than with EDDS. These green adsorbents offer certain advantages such as low-cost, easy operating conditions and a wide pH range; however, low selectivity and waste generation are the major drawbacks that inhibit its applicability on large scales.
Membrane filtration,75–77 electrodialysis78,79 and photocatalysis80,81 are also being considered as green approaches for metal extraction from industrial effluent. These methods require regeneration of the membranes after certain intervals, which increase the operational cost and energy requirement of the process. Nevertheless, high separation selectivity makes these processes feasible for metal removal. Photocatalysts consume photons from the UV-near visible region and may induce the degradation of organic pollutants as well as the recovery of metals in one-pot systems. Traces of the target compounds can also be identified using this method. However, metal removal using photocatalysis processes is still a new research area and therefore cannot be judged at this stage.
It is believed that technical applicability, operational simplicity and economic feasibility are the key factors in selecting the most suitable technology for metal extraction.6 Chelation technology, which has evolved over the last century for use in metal detoxification, has also emerged as a green chemical engineering approach for metal extraction from contaminated sites (soil, waste water, spent catalysts) in recent years. Several studies have been conducted to evaluate the performance of chelation agents as adsorbents, extractants, coating materials to prepare modified composites, etc. This article reviews the technical applicability of chelation technology for metal removal from contaminated sites and also illustrates recent developments in this particular research area.
3. Chelation technology: a path to green economy
‘Chelation’ is defined as the formation of stable metal–ligand complexes which are soluble in water. Although the ‘chelation’ term defines the mobilization of metal ions from contaminated sites using multidentate ligands (chelators) as reagents, some authors have assumed hydration of the metal ion as an initial step (as the reaction takes place in aqueous solution), followed by the multidentate ligand tending to displace water molecules from the metal–water complex in order to form a metal–ligand complex.82,83 In this article, chelation is demonstrated as the process of ligand replacement of monodentate ligands in metal complexes by multidentate ones. The metal ions present in these complexes do not exhibit the chemical activity of uncomplexed ions.84Chelating agents may be either organic or inorganic compounds. Polyphosphates are the best known inorganic chelating agents; however, these are hydrolytically unstable at high temperature and pH. Among the organic chelating agents, aminopolycarboxylates are frequently used chelants and accounted for the largest share (39%) in the world-wide consumption of chelating agents in 2012.85 These aminopolycarboxylates, e.g. EDTA, nitrilotriacetic acid (NTA) and DTPA, have stronger binding capacities than polyphosphates and better sequestering abilities than hydrolytic acid-type chelating agents.86,87 A new discipline, ‘greener chelating agents’, is also gaining the attention of researchers; such agents contribute 15% of the overall world-wide consumption of aminopolycarboxylate chelating agents88 and the demand is expected to increase up to 21% by 2018 due to the replacement of conventional chelating agents by a new range of biodegradable chelating agents. EDDS, GLDA, iminodisuccinic acid (IDSA), etc. are examples of biodegradable green chelating agents which may significantly share the chelating agents market in forthcoming years to avoid the environmental risks associated with non-biodegradable chelators. Fig. 2 shows the molecular arrangement of some newly synthesized biodegradable chelating agents.
 | ||
| Fig. 2 Molecular arrangements of some newly synthesized biodegradable chelating agents: (A) N-2-acetamidoiminodiacetic acid (ADA), (B) ethylenediglutamic acid (EDGA), (C) ethylenediamine dimalonic acid (EDDM), (D) N,N-bis(carboxymethyl)-L-glutamic acid, tetrasodium salt (GLDA), (E) iminodisuccinic acid (IDS), (F) ethylenediamine disuccinic acid (EDDS), (G) methyl glycine diacetic acid, trisodium salt (MGDA), (H) N-bis[2-(1,2-dicarboxymethoxy)ethyl]glycine (BCA3), (I) N-bis[2-(1,2-dicarboxyethoxy)ethyl] aspartic acid (BCEEAA), (J) 2,6-pyridinedicarboxylic acid (DPA). | ||
The key factors driving chelating agent growth in the market include strict environmental legislation, the rising demand for biodegradable chelating agents and the diverse applications of chelating agents. It has been forecasted by Global Industrial Analysts (GIA) that the global market for chelating agents is expected to reach 438 thousands metric tonnes by 2017.89
The extraction of metals with chelating agents does not require high temperatures and the chelating agents can be reused after the extraction of the metals. Due to its recoverable nature, this process is more economical than any other process. This process is ecofriendly due to the easy recovery of the chelating agent used for the process, the non-corrosive environment and the fact that no hazardous byproducts are liberated during the process. The high efficiency of metal extraction and moderate thermodynamic stabilities of the metal complexes make this technology more favorable than any other technology for metal recovery.
3.1. Metal–chelate complex formation
The chelation concept is based on coordinative incorporation of a metal ion into a heterocyclic ring structure. Metal ions in solution always make bonds with ligands and form complex ions. Alfred Werner proposed the concept of transition metal complex formation in 1893 (ref. 90) and solved the puzzling aspect of having higher valences than is requisite for transition-metal compounds. This work suggests the existence of a secondary valence in addition to a “primary” valence of appropriately charged counter ions for transition metal complexes.91 The word ‘chelate’ was introduced in 1920 for caliperlike ligands that function as two associating units and hold the central metal ion in order to make ring structures.92 The coordination environment around the metal ions is demonstrated by Werner's theory according to which metal ions are coordinated by neutral or negatively charged ligands, whose arrangement around the centre depends on the oxidation state of metal.93The metal–chelate complex formation mechanism refers to the step-by-step analysis of reactions involved in the conversion of reactants to products. The general reaction profile for a metal (M)–ligand (L) interaction can be given as eqn (1):
 | (1) |
It can be seen in eqn (1) that there are two transition states where the ligand substitution may take place. A schematic representation of eqn (1) is given in Fig. 3, which suggests that the metal and ligand interact due to the energetic collision of the two molecules. Energetic collisions between molecules cause stretching and bending of the interatomic bonds, which makes them more susceptible to cleavage. Distortion of the bonds can also allow their associated electron clouds to interact with other reactants, which may lead to the formation of new bonds. There are different transition states and intermediate stages between the reactant conversion and product formation. An intermediate occurs at the local energy minimum whereas the transition state always occurs at an energy maximum point and cannot be isolated.
 | ||
| Fig. 3 Metal–ligand interaction – general mechanism. W defines width related to entropy – narrow for −ΔS, wide for +ΔS. | ||
The mechanism of metal–ligand complex (ML) formation can also be demonstrated in terms of a rapid pre-equilibrium. The reaction is carried out in an aqueous phase and the water molecules are replaced by another ligand in the coordination sphere of the metal. The reaction mechanism can be divided into two steps:
(1) Metal–water complexation (formation of an outer sphere complex [M(H2O)]a+
(2) Metal–chelate complexation (replacement of water molecules by the ligand).
The reaction mechanism of metal–chelate complexation in an aqueous system is shown in Fig. 4, which depicts that initially the metal–complex contains water molecules as ligands and then the other ligand is substituting the first one in the rate determining step. Thus, the metal–ligand complex formation reaction can be considered a ligand substitution reaction.
 | ||
| Fig. 4 Schematic representation of the reaction mechanism for metal-complex formation in an aqueous system. | ||
The reaction for the ligand substitution can be given as eqn (2):
| [MLnX]Z+ + Y− ⇌ [MLnY]Z+ + X− | (2) |
Different mechanisms (associative, dissociative and interchange) for the ligand substitution reaction are shown in Fig. 5.
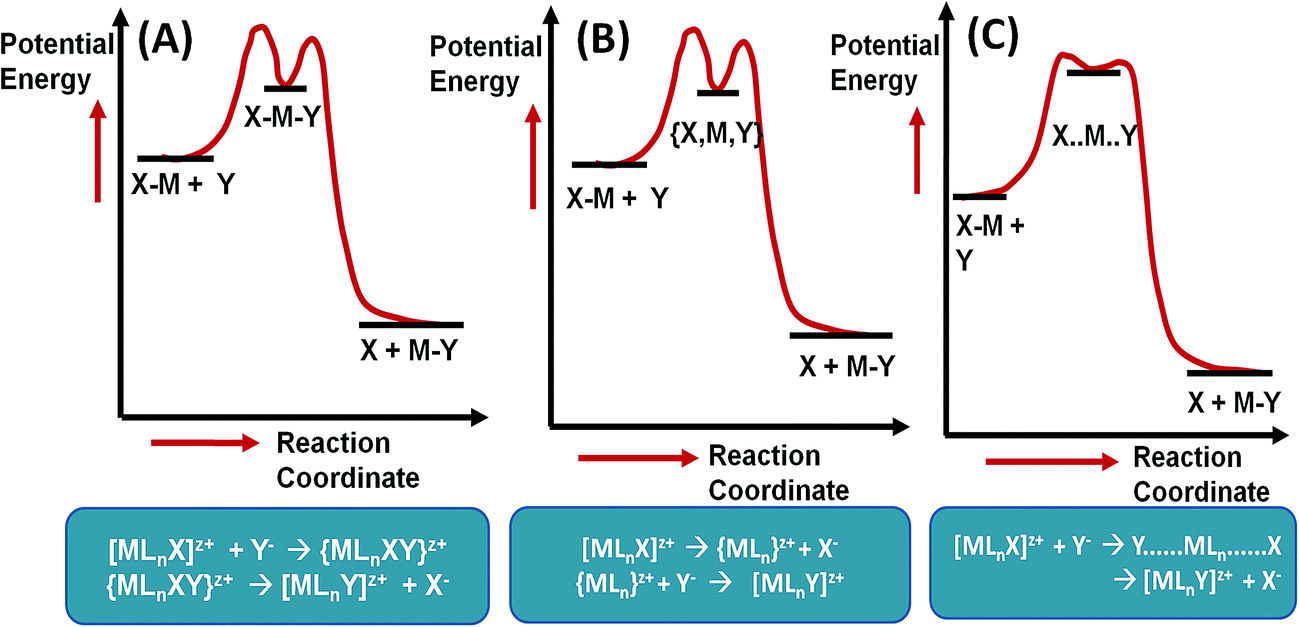 | ||
| Fig. 5 Ligand substitution mechanisms – (a) associative, (b) dissociative, (c) interchange. | ||
Reedjick94 studied the applicability of heavy metals Pt2+ and Ru2+ as anticancer drugs when coordinated with an appropriate ligand. By investigating the reaction kinetics it was observed that the ligand exchange reaction depends on both the metal and the coordinated ligand. Square-planar Pt2+ compounds exchange their ligands via an associative (SN2) process, where the incoming ligand coordinates as a new ligand and the original ligands dissociates. The associative mechanism takes place in just one step and well-defined intermediates are not observed. Octahedral Ru2+ coordination compounds, on the other hand, tend to lose a ligand first (to generate a five-coordinate intermediate), after which the new ligand comes in, and thus follow a dissociative (SN1) mechanism.
The metal extraction processes using a chelating agent is a heterogeneous process because of the transportation of chelated metal ions from the catalyst to the aqueous solution. Thus, it can also be explained as a ‘solid–liquid’ extraction process. The final product of the extraction process does not depend on whether the reaction takes place at the interface or in the aqueous phase. However, it depends on the electron-count of the metal complex undergoing the ligand substitution. The contribution of mass transfer rate to the total rate of the process must be taken into consideration. The process can be considered a ‘diffusion controlled’ process at a low rate of phase mixing, whereas a ‘kinetic region’ can be achieved by increasing the mixing and lowering the particle size.15,95 Thus, the rate of individual chemical stages of the process can be seen as in the region of ‘mixed kinetics’ if the rates of mass transfer and chemical reactions are comparable in magnitude.
3.2. The chelate effect
The formation constant for any chelation process can be calculated using the thermodynamic relationship given in eqn (3):| ΔG° = −RTln(K) = ΔH° − T(ΔS°) | (3) |
Enthalpy (ΔH) contributes to the thermodynamics of complexation process in the form of ligand repulsion, distortion and crystal field stabilization energy96 whereas the entropy (ΔS) includes all probability factors that control the stability of the complex. Schwarzenbach97 delineated the combined effect of these two terms (enthalpy and entropy) as the ‘chelate effect’, which refers to the enhanced stability of a metal–ligand complex due to the chelation process. It was concluded in Schwarzenbach’s work on EDTA versus the corresponding iminodiacetate that the chelating species can be stabilized with a favourable entropy change.97 Adamson98 investigated the effect of standard thermodynamic states and concluded that asymmetry in the choice of the standard thermodynamic states causes the chelate effect.
The enthalpy change in a chelation reaction was assumed to be non-significant until the Myres99 investigation, which suggested that the enthalpy difference in an aqueous solution of ligands is a significant contributor to the Gibbs free energy (ΔG°). The significant entropic effect of a ligand solution and of a coordinated metal ion solution was also studied. An increase in translational entropy was reported when the molecules were released in solution during the chelation process. The entropy of the solution of the monodentate and chelated cations did not differ much whereas the enthalpy deviated from the ideal solution by more than 4.8 kcal.99
Chung et al.100 illustrated an example of chelation for octahedral complexes and suggested that the entropy change of a chelation reaction is a combination of different entropy forms. It was deduced that the solvation entropy and internal rotation entropy give a negative contribution to the overall energy. The reason behind the negative magnitude of solvation entropy is the difference in solvation entropies of the ligands and the different standard states employed. The solvation factor in organic solvents will be less significant than in aqueous solution; therefore, the entropy change of chelation in the gas phase or in an organic solvent will be larger than that in an aqueous solution. It was also suggested that the internal rotation of free ligands and complexes containing monodentate ligands are lost during the chelation reaction, and therefore, a decrease in magnitude of the entropy can be observed with increasing size of the chelate ring.
The temperature dependence of the association constants of metal chelates and complexes was investigated by identifying the ligand-dependent correlations of partial molar entropy of association (ΔSr) of the complexes and chelates with the standard-state entropies (ΔS°) of the aqueous cations at 298 K and 1 bar.101 Quantum chemical calculations were also performed to study the chelate effect of the metal complex formation reaction in aqueous systems.102 It was concluded that the rotational and vibrational entropy contributions are as important as the translational entropy contribution to the total entropy of the reaction. The ΔH° value of the reaction also provides a significant contribution to the ΔG° in complex formation reactions, and therefore, the strong stability of chelate complexes can be considered a combined effect of the enthalpy and various forms of entropy of the reaction.
3.3. Factors affecting the metal–chelate complexation
The formation of a metal–chelate complex may be considered a heterogeneous chemical reaction that depends on certain process parameters, such as reaction pH, molar concentration of chelating agents, reaction temperature, various competing equations in the aqueous phase, etc. This section briefly discusses the effects of various important factors on the metal–ligand complexation process that play a dominant role in increasing the extraction efficiency.| Mn+ + nHL ⇌ MLn + nH+ | (4) |
The extraction constant (Kex) for the above metal–ligand (MLn) complexation reaction can be given as eqn (5):103
| Kex = [MLn][H+]n/[Mn+][HL]n | (5) |
| Kex = E([H+]n/[HL]n), | (6) |
| logE = logKex + npH + nlog[HL]n | (7) |
Thus, it can be shown from eqn (7) that the curve between logE (distribution coefficient) and pH of the solution should be a straight line with slope n, which indicates that if the molar concentration of chelating agent [HL] is constant, then the extraction of heavy metals will be completely dependent on reaction pH.
Several authors have studied the effect of reaction pH on chelant-assisted metal extraction from industrial waste, soil and other contaminated sites. Tandy et al.104 studied the effect of solution pH on the efficiency of various biodegradable and conventional chelating agents for metal extraction from non-calcareous soils, which is shown in Fig. 6. EDTA was found to be the most effective chelating agent over the entire pH range for Cu, Zn and Pb as shown in Fig. 6. EDDS can be observed to be better metal extractant for Cu and Zn than EDTA at pH > 6 and low stoichiometric ratios of chelating agent to metal. NTA also showed comparable extraction of Zn to EDDS at neutral pH; however, NTA was not found to be an effective chelating agent for Cu extraction. IDSA and methylglycinediacetic acid (MGDA) gave better Cu extraction efficiencies than EDTA at neutral pH when the ratio of chelating agent to metal was equimolar. A stronger complexation of Pb with EDTA than with other chelating agents resulted in the highest extraction efficiency of EDTA for Pb metal; however, EDDS also showed a similar Pb extraction efficiency to EDTA for pH > 7.
 | ||
| Fig. 6 Effect of reaction pH on extraction efficiency of various chelating agents for Cu (blue markers), Zn (red markers) and Pb (green markers).104 | ||
Begum et al.105 demonstrated the effect of different extraction variables (reaction pH, chelating agent to metal ratio, stability constants of metal/chelating agent and solid phase distribution of metals) on metal extraction from artificially contaminated soils. EDDS showed the best extraction efficiencies among the biodegradable chelating agents for the extraction of Cu, Pb, and Zn at pH = 7. It was also observed that N,N-bis(carboxymethyl)-L-glutamic acid (GLDA) performed better than the other employed chelating agents for the extraction of Zn, Cd and Cu at pH = 4, while EDTA illustrated the highest efficiency for lead removal. The average extraction efficiency at neutral pH was observed to be either lower than or comparable to that under acidic or alkaline reaction conditions. Acidic pH conditions may increase the concurrent release of the other metal ions and the exchange of H+ ions from functional groups present on the soil surface,106 whereas alkaline pH conditions may be responsible for increasing the reactive species Ln− in aqueous solution and giving a higher rate of formation of the soluble coordination compounds,107 which could be possible reasons for the improved extraction efficiencies at acidic and alkaline pH.
Chatreewongsin108 studied metal extraction from soil samples using EDTA chelation in a microwave system and concluded that the amount of extracted Cu was dependent upon the reaction pH for a classic chelation technique. A higher degree of dissociation of EDTA molecules was observed at alkaline pH, which could be related to the protonation stages of EDTA species (HL3− and L4−) in alkaline medium. Soil surfaces may also contain a large number of negative surface charges at alkaline pH; thus, Cu–EDTA complexes possessing negative charges are preferable in aqueous solutions. Goel et al.13 extracted Ni from the spent catalyst of a fertilizer industry using EDTA and illustrated that the extraction efficiency of EDTA increased with an increase in pH, up to a value of 10; however, above pH = 10, the extraction efficiency starts to decrease. The authors did not explain the reason associated with the decrease in extraction efficiency with increasing in pH. The existence of anionic complexes of MLn+1− in the solution or hydrolysis of the metals may be possible reasons for this observation. Fangueiro et al.109 suggested that solution pH may affect the concentrations of aqueous metal species (and consequently the stability of metal chelates), the solubility of chelating agents, trace metal sorption/desorption, ion exchange phenomena, and the re-adsorption mechanisms of newly formed metal–chelant complexes, and thus strongly affect the performance of chelating agents for extracting metals from contaminated sites. Chauhan et al.15 investigated the performance of the biodegradable chelating agent EDDS to extract Ni from spent catalyst and concluded that EDDS requires a narrower pH range for the chelation–dechelation process than EDTA due to the lower stability constants of the Ni–EDDS complex. It has also been reported that EDDS can extract a higher amount of heavy metals at neutral pH than EDTA.
Thus, the effective pH range for extraction of a particular element may differ for different ligands, which can be explained on the basis of the percentage distribution of the protonation stages of the ligands.96 Curves representing the percentage (%) distribution of various protonation stages for some ligands (3,6-dioxaoctane-1,2,4,5,7,8-hexacarboxylic acid (TDS), EDTA, EDDS, and DTPA) have been reproduced here for the reader's ready reference and are shown in Fig. 7(A)–(D).
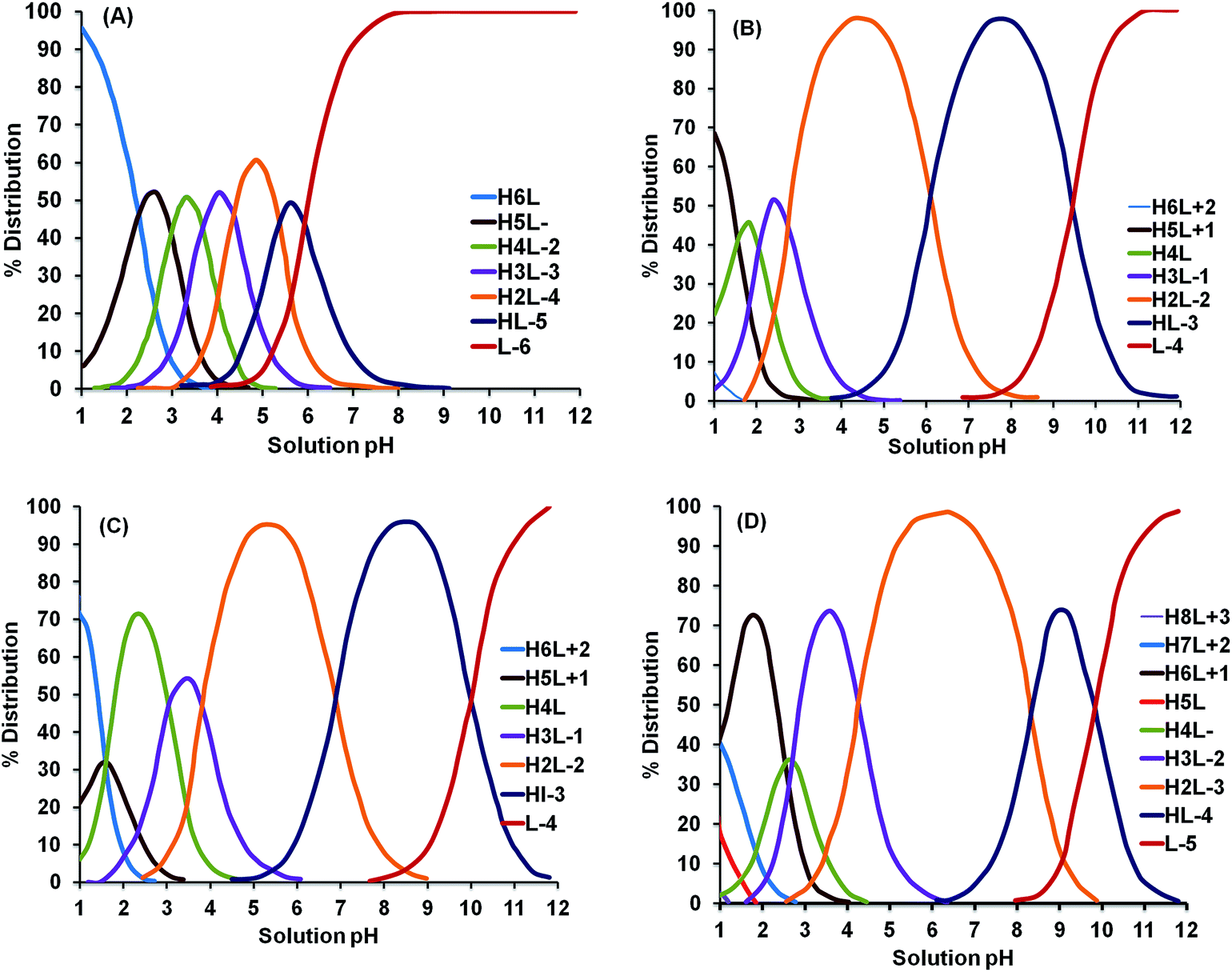 | ||
| Fig. 7 Percentage distribution of protonation stages of various chelating agents: (A) TDS, (B) EDTA, (C) EDDS, (D) DTPA.96 | ||
It can be elucidated that each ligand has a certain pH range for different protonation stages. TDS has more acidic species than EDTA, EDDS and DTPA, whereas DTPA has a wider pH range for the protonated H2L−2 form. HL−3 is predominant for the pH range of 7 to 9 in the case of EDDS, while a wider pH range (pH = 6 to pH = 10) can be observed in the case of EDTA for the same protonation stage. This deviation in the % distribution of the various protonation stages strongly affects the effective pH range for a ligand. It can also be seen from Fig. 7 that the fully protonated H6Ln+ form predominates at the most acidic pH values (pH = 1–2), and as the reaction moves towards alkaline pH, hydrogen availability for displacement decreases and deprotonated forms are prevalent; therefore, after a certain increase in pH value, no further improvement in extraction efficiency can be observed.9 It should also be kept in consideration that the favorable pH range for the efficient extraction of heavy metals may correspond to the precipitation range of the corresponding chelate, which can be determined from either the stability constants15 or the distribution constants of the complexes.103
E and log[HL]. The relationship between pH and the concentration of the reagent has also been explicated in the literature and is given in eqn (8):103| pH50 = (−1/n)logKex − log[HL] | (8) |
Eqn (8) suggests that as the molar concentration of the ligand [HL] increases by one order of magnitude, pH50 decreases by one unit. Here, pH50 indicates the value of pH corresponding to 50% extraction of a metal ion when the distribution coefficient is unity.103,107 The effect of ligand concentration on metal extraction efficiency has been investigated elaborately in the literature.13,74,104,105,110–112 Jiang et al., 2011 (ref. 26) varied the chelating agent concentration from 0.1 g L−1 to 0.4 g L−1 in the extraction of Cu and Ni from artificially contaminated soil and illustrated a significant effect of the molar concentration of chelating agent on extraction efficiency. The metal extraction efficiency improved with an increase in reagent concentration; however, above a certain amount (when the dose of chelating agent is enough for the soil requirement), the extraction efficiency becomes nearly constant. Some authors have also reported the molar concentration of chelating agent in terms of the stoichiometric ratio of chelating agent to metal (MR). Kim et al.113 studied the effect of various factors (solution to soil ratio, presence of major metal cations in samples and stoichiometric ratio of chelating agent to metal ions) responsible for metal extraction from Pb-contaminated soils and observed a high dependence of extraction efficiency on the molar concentration of chelating agent. The extraction efficiency was also found to be dependent on the properties of the soil samples and therefore, at a given stoichiometric ratio, the extraction efficiency varied for different soil samples, as shown in Fig. 8. However, if a sufficiently large amount of EDTA was applied, most of the Pb could be extracted.
 | ||
| Fig. 8 Effect of chelating agent (EDTA) to metal stoichiometric ratio (MR) on Pb extraction from different soil samples.113 | ||
Similar observations were reported for Cu, Pb and Zn extraction using various biodegradable chelating agents104 and the data have been reproduced in Fig. 9. It can be observed from Fig. 9 that as the concentration of chelating agent in the solution increases, the metal extraction efficiency increases. Cu extraction was enhanced significantly for all investigated chelating agents at an equimolar ratio between chelating agent and metal. EDTA showed a significant improvement in Cu extraction at higher chelating agent to metal ratios, which was explained using speciation calculations. These calculations suggest that at a high MR, most of the EDTA was present in the uncomplexed form and therefore, the probability of metal–ligand complex formation increases. On the other hand, 90% of the total EDDS is always accounted for as uncomplexed EDDS and therefore, enough free EDDS is available to extract further Cu even at low MR. Therefore, the Cu extraction efficiency of EDDS is nearly comparable at low and high MR values, while a significant difference in Cu extraction was observed using the other chelation agents.104 When the MR increased (to 10:1), EDTA was observed to be more effective than the other chelating agents for all three metals at neutral pH.
 | ||
| Fig. 9 Effect of chelating agent to metal stoichiometric ratio (MR) on metal extraction using different chelating agents.104 | ||
Research has also been carried out to investigate the effect of the liquid to solid (L/S) ratio on metal extraction from contaminated sites using chelating agents. The effect of L/S was investigated for Co and Mo extraction from spent catalyst at constant MR and the results suggested an increase in extraction efficiency with an increase in L/S9 to a certain extent. An increase in L/S ratio corresponds to a decrease in the concentration of the chelating agent in the aqueous solution, and therefore, beyond the optimum (required) L/S ratio for efficient mixing, the extraction efficiency starts to decrease. However, if the L/S ratio is too low to prepare a chelating agent solution and to minimize the external resistance by performing efficient mixing of particles, then the extraction efficiency will be less; therefore, the optimization of L/S is also requisite to achieve high extraction efficiency. Chauhan et al.112 designed experiments and statistical analysis to optimize the process parameters for a metal extraction process using different chelating agents (EDTA, DTPA, EDDS, NTA). The authors illustrated a significant mutual interaction effect of MR and L/S on Ni extraction. Kim et al.113 varied L/S from 3 to 10 for lead extraction from contaminated soils and concluded that L/S did not have any effect on Pb extraction from the soil. The possible reason for this observation could be the presence of an excess amount of chelating agent, of which only a small fraction was effectively utilized to extract Pb from the soil. The remaining amount of chelating agent may be freely available in the solution or may form complexes with other metal cations; therefore, no effective increase in extraction efficiency can be observed beyond a certain L/S value when the concentration of reagent is enough for the metal extraction. Manouchehri et al.114 also stressed that L/S cannot be considered an indicator to ensure maximum metal extraction efficiency; however, this ratio must be investigated with respect to all the extractable metal ions present in the contaminated sites.
It is recommended that the molar concentration of chelating agents should be greater than that of the target metal species present in contaminated site to minimize the competing effect of other undesirable ions.115,116 However, some reports also suggest that after a certain extent, the extraction efficiency either becomes unaltered or starts to decrease with an increase in concentration, which could be related to the reagent concentration being higher than the requirements of the contaminated site for metal extraction.12 Some other limitations may also arise due to a higher concentration of chelating agent; for example, the reagent is difficult to remove and its residual traces may reduce the accuracy of the determination. Using a lower concentration of ligand and high L/S ratio may be beneficial in order to prevent clogging of the solid particles during leaching and to provide better mixing.117 However, the generation of a large amount of wastewater after the extraction process may increase the treatment cost and possibility of pollution, which is not desirable. Therefore an optimized value of the concentration of chelating agent and L/S ratio are necessary for effective extraction.
Fig. 10 shows the extraction of Ni using EDTA from spent catalyst for different reaction times and reaction temperatures under atmospheric and autogeneous reaction conditions. It can be seen from Fig. 10 that 95% Ni recovery was achieved under hydrothermal conditions in an autoclave at reaction temperatures of 150 °C, over a 4 h reaction time.14 High temperature may also cause an autogeneous pressure build up in the reactor. Extraction experiments were performed under atmospheric reflux conditions at a reaction temperature of 90 °C and a significant difference in extraction efficiency was observed15 when the results were compared with those of the autogeneous reaction conditions with similar process parameters,14 as shown in Fig. 10.
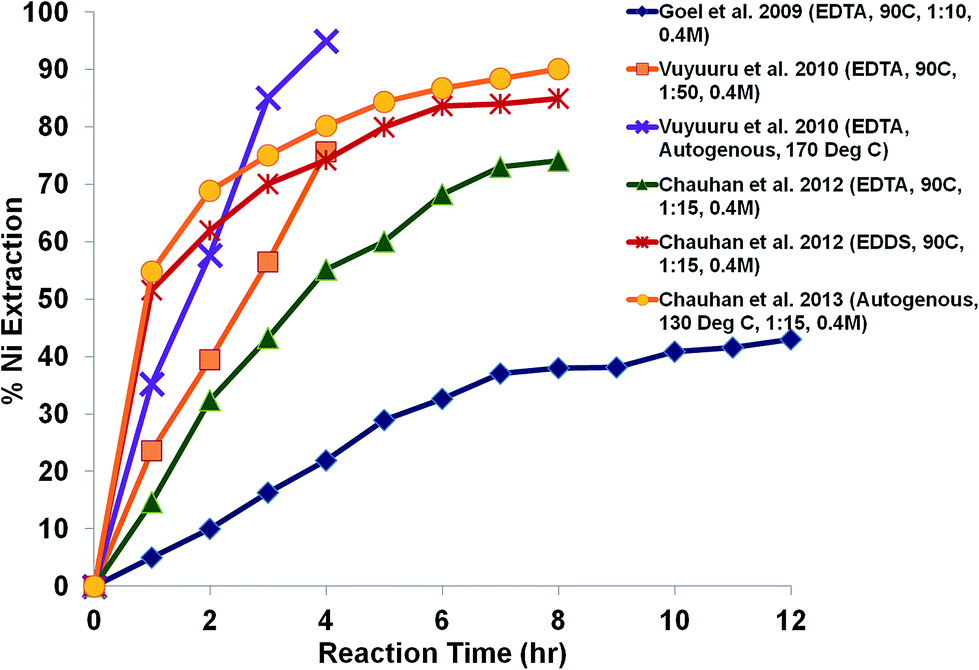 | ||
| Fig. 10 Effect of reaction temperature on Ni extraction from spent catalyst. | ||
This could be due to the autogeneous pressure in the autoclave and the better mixing of particles, which is necessary for an effective extraction process. A higher extraction efficiency was observed with EDDS at a low reaction temperature, which suggested superiority of EDDS over EDTA at low temperature. An increase in the vessel pressure up to 2.7 atm at a temperature of 140 °C during the chelation process for metal extraction has been described in the literature,9,14 which favours the rate of forward reaction, and hence the metal extraction efficiency improves. The extraction process under similar reaction conditions was performed under atmospheric reflux conditions at a reaction temperature 100 °C, and about 58% Mo was extracted, which was nearly 12% less than the extraction efficiency obtained under autogeneous conditions.9 Hong et al.118 also investigated pressure-assisted Pb extraction from soil samples and concluded that soil particles may break into smaller fragments due to high pressure and therefore, the smaller particle size enhances the metal extraction efficiency by controlling the internal diffusion. Therefore, autogeneous reaction conditions are preferable for the chelation process.
Moreover, the metal extraction process using chelation technology could be adopted in a practical industrial plant where the process has to be carried out in stirred autoclaves. Therefore, chelating agent-assisted metal extraction under autogeneous reaction conditions is considered a feasible and efficient means of metal extraction on industrial platforms. However, very high reaction temperatures can adversely affect the process economics, in terms of consumption of steam utilities and equipment costs.
 | (9) |
[MLi] is the concentration of metal–ligand complex including [MLn], [M(OH)j] is the concentration of all hydroxylated components, and [MXq] is the concentration of complexes formed with extraneous complexing agents.103
In et al., 2008 (ref. 119) employed the solvent extraction process with the chelating agent salen-(NEt2)2 to extract Cu2+, Mn2+ and Zn2+ from water samples and investigated the effect of concomitant ions on the metal extraction process. It was illustrated that Mg2+ interfered with the extraction of Mn2+ and the extraction of Mn2+ decreased; however, no significant effects were observed on the extraction of Cu2+ and Zn2+. It was also reported that if Mg2+ coexists in the solution at a very high concentration (>150000 times that of Mn2+), then Mn2+ cannot be determined due to strong interference. Manouchehri et al.114 illustrated the significant effect of the concentration of chelating agent and type of solid matrix on the extraction and reactivity of various metals (Pb, Cu, Cd, Al, Fe, Ca and Mg). Different competitive reactions were seen for the major elements (Al, Ca, Fe and Mg) towards the chelating agent EDTA, depending on the soil Ca content. Calcareous soils preferred the formation of Ca–EDTA due to the abundance of Ca in the soil, while the formation of Al– and Fe–EDTA complexes dominates in non-calcareous soils. The relatively weak bonding strength of Ca to certain soil fractions during the concurrent extraction process in calcareous soils may cause preferable Ca dissolution and displacement of other metals with longer reaction times. Tandy et al.104 observed a strong competitive reaction between target metals and Ca as an important factor for extraction using EDTA, but not with EDDS, which results in a decrease in extraction efficiency for EDTA compared to EDDS. Valverde et al.120 showed that Fe may be neglected during speciation of EDDS in soil samples, whereas a contradictory result was also reported104 which suggested that Fe–EDTA decreases with an increase in reaction pH due to Ca interference, while Fe–EDDS was observed as a relevant species at neutral pH.
Species in aqueous solution exist in formation–dissociation equilibrium, and consequently, displacement reactions of metal ion or ligand by another reactant may occur; therefore, metal–ligand complexation depends on the displacement equilibrium constants when a chelating agent is added to a solution containing two or more metal cations. It is suggested that a ligand must overcome competing metal precipitation, surface complexation and precipitation of solid particles to achieve an effective extraction of metals from the contaminated site.114 Therefore, a sufficient molar concentration of chelating agent in the solution is also desirable to combine with the target metal and other ions which can displace the target metal. The chelating agent should have different stability constants for the two metals, if the selective reaction of one metal is required in presence of the other.121
3.4. Factors affecting the stability of a metal–ligand complex
The metal–ligand complex process can be represented as shown in eqn (10) and (11):| Mn+ + nL− ⇌ MLn; βn = [MLn]/[Mn+][L−] | (10) |
| [MLn] = MLn(0); PML = [MLn]0/[MLn] | (11) |
| Kex = (βnPMLKHLn)/PHLn | (12) |
| logKex = log(βnPML) − nlog(PHL/KHL) | (13) |
Eqn (13) suggests that the stability constant of the complex and the acid dissociation constant of the reagent are interconnected. Consequently, the distribution constants of the reagent and of the complex are also connected.103 Thus, the stability of the metal–ligand complex depends primarily on the ligand properties. Ligands are classified on the basis of their charge, size, identity of the coordinating atom(s), and number of electrons donated to the metal ion (denticity). Ligands can be further characterized into monodentate, bidentate and polydentate, depending on the number of donor atoms present in the ligands, which allow the attachment of two or more donor atoms to the same metal ion simultaneously and produce one or more rings. This ring formation is the major characteristic of chelation, distinguishing it from monodentate metal coordination by the increasing stability of the resultant chelate complex; therefore, the stability constants of chelating agents for an equal number of similar coordinated donor atoms are from one to several orders of magnitude greater than those of monodentate complexes. Five and six-membered rings are considered the most stable in the chelation reaction. The formation of three membered ring structures is prohibited due to the coordination angles on the metal atoms. On the other hand, rings that are more than seven membered are also not desirable due to an unconvincing probability of ring closure. Thus, the size and number of rings, substituents on the rings and the nature of the metal and donor atoms are the common factors which affect the stability of a metal–ion complex. The formation of each additional ring by the same ligand provides extra stability to the metal–ligand complex due to the displacement of coordinated solvent molecules. DTPA is a more stable chelating agent than EDTA due to presence of more donor atoms (8 binding sites) than EDTA (6 binding sites); however, NTA is considered relatively biodegradable and forms a less stable complex due to the availability of only 4 binding sites for complexation.112 The larger the complex formation constant, the more stable is the species. Steric hindrance due to the presence of bulky groups attached to a donor atom also contributes to the stability of a chelating agent. This interaction may cause mutual repulsion between the ligands, and hence the stability of the metal–ligand bond decreases. EDDS has less stability than EDTA (in spite of having the same number of binding sites) in complexes due to the presence of two six membered rings in EDDS, which causes steric hindrance and weakens the metal–ligand bond.121 The biodegradability properties of a chelating agent also depend on the stability constants of the ligands. Aminopolycarboxylates that form complexes with relatively low stability constants (e.g., IDSA, NTA, EDDS) are readily degradable, whereas those forming stronger complexes (e.g., EDTA, DTPA) are relatively resistant to biodegradation. Complex formation constants of selected metal complexes are presented in Table 2 for the reader's reference.
:1 metal–ligand complexes, at 25 °C and with an ionic strength of 0.1 M. Values are expressed as logK135
Thus, assessment of the most suitable and ecofriendly alternative should be performed very carefully by considering all the above described factors, such as pH range, molar concentration of reagent and element, liquid to solid ratio required for efficient metal extraction process, other competing reactions in the aqueous phase, solubility and stability of the metal complex in different reaction conditions, protonation, and stability constants.
4. Environmental scrutiny of chelating agents
Chelating agents have been utilized widely in various domestic and industrial applications for many years and have been recently gaining attention in the field of metal extraction from various contaminated sites for minimizing environmental pollution. A high resistance to biodegradability is requisite to attain stability of the metal–chelate complex during industrial processes; however, non-biodegradable chelating agents may have certain deleterious effects on the ecosystem. Chelating agents may perturb the natural speciation of metals and may have a dissolution effect on heavy metals adsorbed in sediments.136,137 Chelating agents contain nearly 10% nitrogen which may eventually become present in aquatic systems, and thus chelating agents may have a significant effect on the eutrophication process.138 Literature suggests that there is a relatively high concentration of EDTA, the major chelating agent used in various industrial and domestic applications, in surface water and drinking water due to its high persistency in the ecosystem.139,140 According to publications by the European Aminocarboxylates Committee141 and the European Chemical Bureau,142 the predicted no-effect concentration in the aquatic environment (PNECaqua) of EDTA is 2.2 mg l−1, based on the no-effect concentration (NOEC) of EDTA for Daphnia Magna (22 mg l−1). The guideline value for EDTA concentration in drinking water is 0.6 mg l−1, as published by World Health Organization.143Stringent environmental regulations are coercing industries across the globe to increase the utilization and production of biodegradable chelating agents. The biodegradation of these conventional chelating agents (EDTA, DTPA, organophosphonates) was investigated using various isolated bacterial strains;144,145 still, these compounds do not satisfy the criteria for biodegradability.146 Thus, the environmental concerns associated with non-biodegradable chelating agents have been the predominant factor that has stimulated the demand for more ecologically-viable chelants. Biodegradable chelating agents are likely to capture a significant share of the chelating agents market in coming years, owing to the potential health and environmental hazards associated with the use of nonbiodegradable organic chelating compounds. Sykora et al.147 suggested that the biodegradability of a complexing agent depends on the functional group available on the ligands and the number of nitrogen atoms present in the ligands. NTA contains a single nitrogen atom in the molecule and therefore, it is subject to biodegradation. Another important observation suggested that the resistance to biodegradability increases with the number of tertiary amino groups (as in EDTA and DTPA). Optical isomerization is also considered as an important factor for biodegradability, e.g. the [S,S]-EDDS stereoisomer is subject to easy degradation, whereas the [R,R] isomer remains intact and the [R,S] isomer degrades very slowly and incompletely.83,148
In 1993, the Organisation for Economic Co-operation and Development (OECD) guidelines for the testing of chemicals described internationally used standard test methods to investigate the biodegradability of chemicals.149 A brief description of the biodegradability tests have been given in Table 3.
| Major categories | Sub-categories | Basis of testing | Test duration |
|---|---|---|---|
| Ultimate biodegradability screening tests | Modified Sturm test/OECD 301B | By measuring the analytical parameters of mineralisation (CO2 evolution or O2 consumption) | 4 weeks (batch tests) |
| Modified MITI test/OECD 301C | |||
| Closed bottle test/OECD 301D | |||
| Manometric respirometry test/OECD 301F | |||
| BODIS (biodegradability of insoluble substances) test, ISO 10708 | |||
| DOC die-away test/OECD 301A | By determining the extent of ultimate biodegradation by removal of dissolved organic carbon (DOC) | ||
| Modified OECD screening test/OECD 301E | |||
| Inherent biodegradability screening tests | Modified Zahn–Wellens test/OECD 302B | By measuring carbon removal, concentrated sludge inoculums are used to obtain high bacterial concentrations | 28 days |
| Continuous activated sludge (CAS) test | Coupled units test/OECD 303A | By carbon removal (DOC) measurement using activated sludge | 3 h, continuous mode of experiment |
It is vital to mention that different biodegradation tests (coupled units test, Zahn–Wellens test, MITI test, AFNOR test, Sturm test, OECD screening test and closed bottle test) reported in the literature were carried out on EDTA by Gerike and Fischer151 and the results suggest that the most frequently used chelating agent EDTA is not biodegradable at all. The European Union risk assessment report also confirms the non-biodegradability of EDTA.152
Studies of more readily biodegradable complexing agents have been extensively performed in the last few years. Biotechnologically produced EDDS has been an important reagent to investigate for its biodegradability, instead of being a structural isomer of EDTA. MGDA showed a good stability over a wide pH range and also satisfied the OECD criteria.146 Boroweic et al.153 illustrated that GLDA as a chelating agent is biodegradable and non-toxic for ecosystems. Lanham et al.154 investigated ethylenediamine-N,N-diglutaric acid (EDDG), whereas ethylenediamine-N,N-dimalonic acid (EDDM) was synthesized by Aoki and Hara155 as a biodegradable chelating agent. Recently, pyridine-2,6-dicarboxilic acid (PDA) has also been established as a biodegradable ligand able to efficiently chelate metals in a ligand to metal ratio of 2:1.156 Some other biodegradable chelating agents, such as N-bis[2-(carboxymethoxy)ethyl]glycine (BCA3), N-bis[2-(1,2-dicarboxyethoxy)ethyl]glycine (BCA5), N-bis[2-(1,2,dicarboxyethoxy)ethyl]aspartic acid (BCA6), N-bis[2-(methylcarboxymethoxy)ethyl]glycine (MBCA3) and IDSA, have also been investigated in the literature.96
Several new methodologies have been investigated in past few decades to find out new ways of degrading chelating agents. Advanced oxidation processes (AOPs) have been considered a promising technology for the degradation of recalcitrant organic pollutants. These processes apply combinations of radiation, oxidants (ozone, hydrogen peroxide and molecular oxygen) and catalysts for degrading target compounds.157 EDTA was effectively eliminated from bleaching effluent with an ozone dose of 1400 mg l−1.158 UV radiation was employed for the degradation of Fe and Cu complexes of NTA, EDTA, DTPA, and EDDS, whereas Mn2+ complexes were observed to be more recalcitrant to UV-induced degradation than other metal–EDTA or metal–DTPA complexes.159
The biodegradation of chelating agents using pure bacterial cultures is also considered a beneficial approach in order to minimize metal mobilization into the ecosystem. Several microorganisms have been reported to degrade chelating agents under aerobic conditions.160,161Agrobacterium sp.,162 the bacterial strain DSM 9103 (ref. 163) and the bacterial strain BNC1 (ref. 160) were considered as the most efficient enrichment cultures for the degradation of EDTA; however, no bacterial strain was found in the literature to be able to degrade DTPA.157 It has also been mentioned that the bacterial culture that is responsible for the degradation of NTA cannot degrade EDTA; however, the EDTA-degrading bacterial strain BNC1 can grow on both EDTA and NTA.164 The biochemistry of NTA degradation suggested iminodiacetate (IDA) as the major degradation product, which can be obtained by the action of either the NTA monooxygenase of Chelatobacter heintzii (C. Heintzii)165 or the EDTA monooxygenases of BNC1 and DSM 9103.166,167 IDA is transformed to glycine and glyoxylate by a membrane-bound IDA dehydrogenase in C. Heintzii.168 New genes were also identified with the determined N-terminal amino acid sequence, which were overexpressed in Escherichia coli to investigate the IDA oxidase activity.169
The degradation of EDTA is reported to be carried out by the EDTA monooxygenases of strains BNC1 and DSM 9103, which oxidize EDTA to ethylenediaminetriacetate (ED3A) and then to ethylenediaminediacetate (EDDA),163,166,167 and IDA oxidase oxidizes EDDA to ethylenediamine (ED). The bacterium BNC1 may further metabolize ED to obtain the nitrogen present in it.
The degradability of ligands is not only affected by their chemical structure, but may also be influenced by the metal ions present in the complex, which has been demonstrated in the literature for NTA, oxalate, citrate and EDTA.170,171 Kluner et al.164 studied the metabolism of EDTA and its metal chelates using whole cells of the strain BNC1 and the cell-free extracts prepared from it and observed that metal–EDTA chelates with relatively low thermodynamic stability constants (K), such as Ba(105.5), Mg(106.4), and Ca–EDTA (108.4), were readily degraded by growing or resting cells of the strain BNC1; however, metal–EDTA chelates with stability constants above 1012, such as Fe(III)–, Co–, Cd–, Pb–, Ni–, and Cu–EDTA, could not be metabolized by whole cells. This observation demonstrated the fact that the degradability should decrease with increasing stability constants of metal complexes. It was also illustrated that slow metal exchange kinetics plays a significant role in the biodegradation of ligands because of the strict speciation requirements.164
Computer simulations have also been an appropriate means of studying degradation pathways by computing the C–N bond dissociation energies. Chen et al.83 employed molecular simulation (density functional theory at the B3LYP/6-31G* level) to define the dissociation energies of metal–EDTA and metal–EDDS complexes to investigate the superior biodegradability of EDDS compared to EDTA. Fig. 11 gives a collection of proposed and identified degradation pathways of EDTA. Many of the primary and secondary degradation products of EDTA may form soluble complexes with metal ions, such as ED3A, EDDA, and IDA, whereas some of the degradation products are expected to be even more recalcitrant to degradation than EDTA or DTPA themselves, such as 2-ketopiperazine-1,4-diacetic acid (KPDA), 2-ketopiperazine-1/4-acetic acids (KPMAs), and ketopiperazine.172
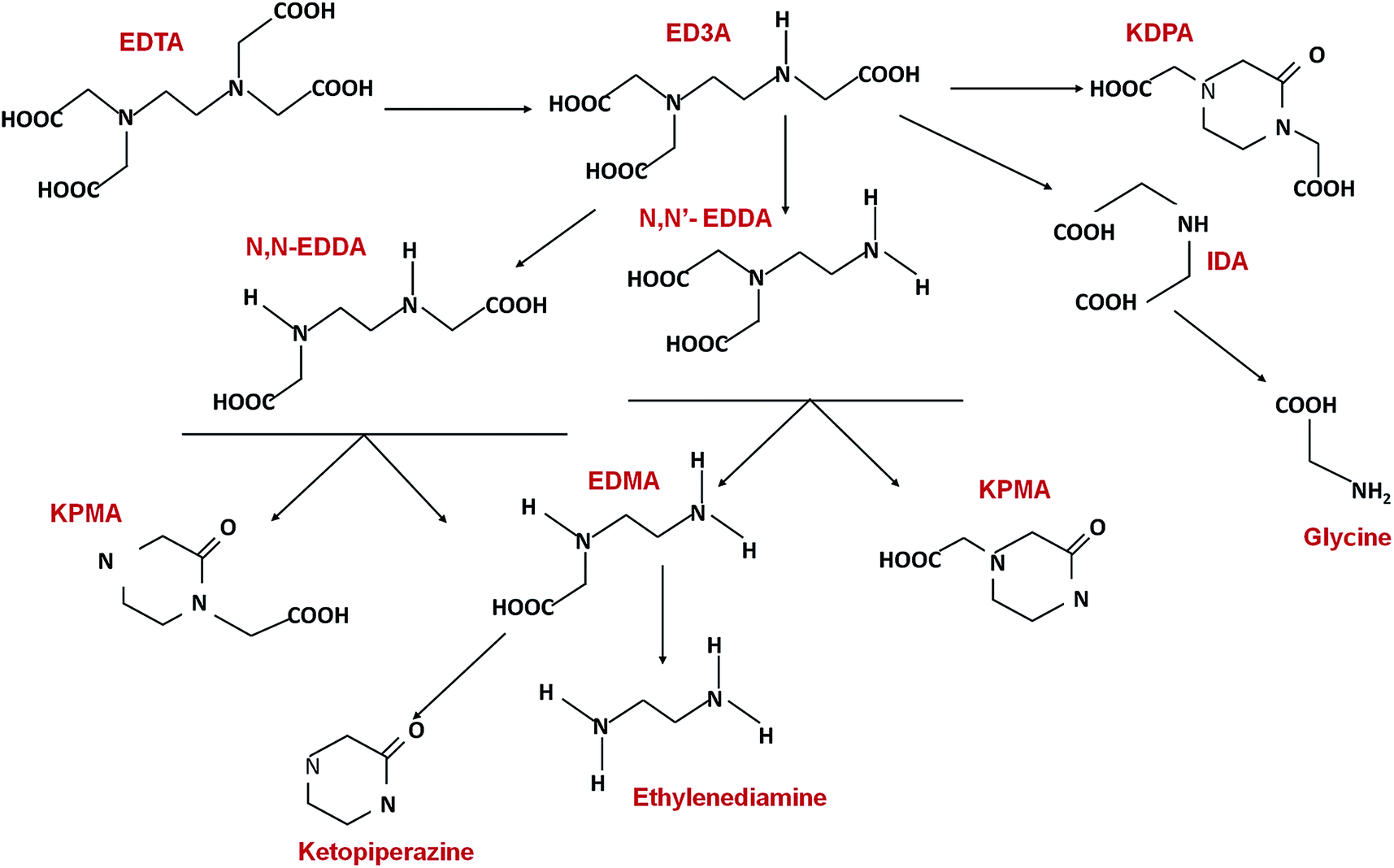 | ||
| Fig. 11 Identified degradation products and pathways of EDTA.135 | ||
The degradation of EDDS was also predicted by calculating the dissociation energy of all C–N bonds present in EDDS.83N-(2-Aminoethyl)aspartic acid (AEAA) was obtained as the first catabolism product of EDDS. The anticipated mechanism and energy profile of [S,S]-EDDS degradation featured N–H bond formation and C–H bond cleavage due to intramolecular transfer of a hydrogen atom from the preferential carbon atom to a nitrogen atom.
Therefore, it can be concluded from the above section that the more stable a metal–ligand complex is, the less possibility there is for it to degrade. The various methods which are available for degrading chelating agents are still not enough to provide an efficient solution to develop an ecofriendly alternative; however, research can move towards new directions to find a better alternative.
5. Applications of chelation technology for metal extraction from contaminated sites
The global market is witnessing an increasing demand for chelating agents and the emergence of many new chelating products. Developed economies capture the largest share of the chelating agent market, as stated by the new market research report on chelating agents.173 Asian markets are also expanding at a growth rate of over 4% annually and thus there is wide applicability for chelating agents in developing countries too. It is reported that more than 70% of the global demand for chelating agents comes from domestic applications, water treatment and pulp & paper industries; however, chelating agents are now finding increasing scope in various applications in order to minimize pollution of ecosystems. The literature available on the interaction of chelating agents with the environment suggests a wide range of applications in medicines, microbial growth, soil remediation and metal extraction from waste materials. The first study on the effect of EDTA on microbial growth and the bioavailability of metal complexes was published in 1950.174 In 1951, the Fe(III)–EDTA complex was investigated as an iron fertilizer for plants.175 Lysine, glyoxaline-4-5-dicarboxylic acid and histidine were also reported in 1953 as chelating agents for the preparation of Keilin horse-heart succinoxidase.176Fig. 12 shows the continuously increasing number of publications every year based on the application of chelating agents in various areas.177 It can be seen from Fig. 12 that chelating agents have been used in medicines for nearly six decades and it is still one of most important disciplines in metal detoxification. The application of the chelation concept in reactions (adsorption and complexation) was published for the first time in 1955 and since then chelating agents have been constantly employed in reactions, which is self explanatory with the increasing number of publications, as shown in Fig. 12. In the 1960s, research was mainly applied to the application of chelating agents in soil, plant and water samples. | ||
| Fig. 12 Cumulative number of publications over the last six decades on the application of chelating agents in various areas (metal extraction from water includes natural water resources and waste water; reactions include adsorption, complexation, surface reactions; environment includes biodegradability and ecotoxicological effects; soil remediation includes phytoextraction and metal mobilization).177 (Database: Scopus; accessed in July 2014.) | ||
Soil-related research expanded its area to soil remediation (extraction of metals and phytoremediation) and the biodegradability aspects of chelating agents until the 1990s. In the 1990s, the applications of chelating agents in ground water, waste water and the surface reactions that occur with natural ions were also investigated. Since then, the versatility of chelating agents has stimulated the continuing research on the uses and environmental effects of chelating agents. The chelation concept has also been employed, in recent years, to metal extraction from industrial waste (spent catalysts) and from waste electrical and electronic equipment (WEEE). The wide range of leading research areas accompanying the applications of chelation agents are shown in Fig. 13, which demonstrates that the highest numbers of publications were featured in the research area of biochemistry, genetics and molecular biology with 27% of the total number of publications on “chelating agents”.177
 | ||
| Fig. 13 Leading research areas in terms of the number of research publications on ‘chelating agents’177 (database: Scopus; accessed in July 2014). | ||
Chelating agents have already proven their applicability in medicines. Nevertheless, extensive research is being carried out on metal detoxification using chelating agents. Chemical engineers are now moving towards research on chelating agents by applying their metal complexation properties in various applications, such as recovery of heavy metals from waste materials, phytoremediation, and many more. The biodegradability issue associated with classical chelating agents has also been a matter of concern for chemical engineers and chemists.
5.1. Chelating agents used for metal extraction from spent catalysts
The applicability of chelating agents in metal extraction from spent catalysts has recently been flourishing as a wide research area for industrial, economical and ecological benefits. Heavy metals are extensively used for the preparation of various catalysts in the petrochemical and fertilizer industries. The market demand for hydrotreatment catalysts is estimated to have an annual growth rate of 4.4%.178 These catalysts start to lose their activity after a certain number of process cycles due to the deposition of metal sulphides and coke on the catalyst pores. At this stage, they need to be replaced with fresh catalysts and are considered as “spent catalysts”. These spent catalysts contain a significant amount of heavy metals and may be a substantial source of release of heavy metals into the environment, if not disposed of in a proper manner. Spent catalysts obtained from the petroleum industry contain 4–12% Mo, 15–30% Al, 1–5% Ni, 0–4% Co, 5–10% S, 1–5% Si, and 0–0.5% V. Disposal of spent catalysts requires compliance with stringent environmental regulations. The avoidance of (heavy) metal release from spent catalysts to the environment is one important motivating factor. Another one is the exploitation of those waste products with respect to metal recycling from an economic point of view. The reuse of spent catalysts after regeneration is prudent and therefore, an environmentally benign means of safe disposal of spent catalysts at the end of the catalyst life should be opted for. It has been reported that the recycling of metal scraps is an eco-efficient process that results in 33% less energy consumption and 60% less pollutant generation than in the production of fresh fuel.179 The large amount of valuable metals present in spent catalysts has also attracted the attention of researchers to explore various new methods for metal extraction in order to develop efficient processing.Numerous hydrometallurgical and pyrometallurgical approaches have been adopted for the recovery of heavy metals from spent catalysts. Pyrometallurgical processes involve smelting of the spent catalyst to metal alloys and slag in a gaseous environment or recovering the metals in the liquid environment, preceded by roasting or wet oxidation of the spent catalyst.180 However, these methods are energy intensive due to high temperature requirements and may liberate toxic gases into the environment. Several hydrometallurgical approaches, such as leaching,181 solvent extraction182,183 and biological methods184,185 have also been reported for the recovery of metals from spent catalysts, soil and other industrial waste that involve the use of aqueous chemistry, and recovery occurs at temperatures lower than the boiling point of the aqueous media. Combinations of pyro- and hydro-metallurgical processes have also been employed for metal recovery from spent catalysts due to the complex nature of spent catalysts.186,187 The chlorination process was also adopted for metal recovery; however, the corrosive nature and toxicity of chlorine gas or any mixture of it inhibits the operation.188,189 All these processes have shown potential in removing metals from industrial waste; however, the use of hazardous chemicals, possibility of secondary pollution by these chemicals, lower tolerance of biological processes in the environment and high process costs restrict their use on large scales. Therefore, the search for an efficient and ecofriendly technology to extract metals from spent catalysts is needed.
The extraction of metals using chelating agents does not require high temperatures and the chelating agents can be reused after the extraction of metals which make it more economical than any other process. Goel et al.13 used spent catalyst from the fertilizer industry for the recovery of Ni using EDTA as a chelating agent. 96% Ni was recovered in the form of NiSO4 with an EDTA concentration of 0.8 M, L/S = 50, reaction time = 10 h, particle size of 100 μm, 700 rpm stirring speed, temperature of 100 °C, and a pH of 10. However, the very high L/S in this study is of great concern due to the large space requirement and increasing possibility of a large amount of waste water generation. The recovered EDTA was reused in successive experiments with more than 73% Ni recovery over four cycles. The authors suggested that interference of competing sulfur ions in the recycled extracted EDTA may be the reason for the decreasing extraction efficiency with recycled EDTA solution. Various reaction parameters, such as molar concentration of chelating agent, solid to liquid ratio, stirring speed, particle size, reaction pH, reaction temperature, reaction time, etc., contribute to the metal extraction efficiency and therefore the effect of each parameter must be studied in order to optimize the extraction efficiency. The non-biodegradability issue associated with EDTA is the major constraint of using it on a large scale. Therefore, a biodegradable chelating agent [S,S]-EDDS was employed for the extraction of Ni from spent catalyst in a batch mode under atmospheric reflux conditions, which recovered 84% Ni in one cycle under optimum reaction conditions.15 The effectiveness of [S,S]-EDDS was also compared with the traditional chelating agent EDTA and it was concluded that [S,S]-EDDS requires a narrower pH range than EDTA for the extraction and solvent/chelator regeneration processes. The complete process flow chart is shown in Fig. 14.
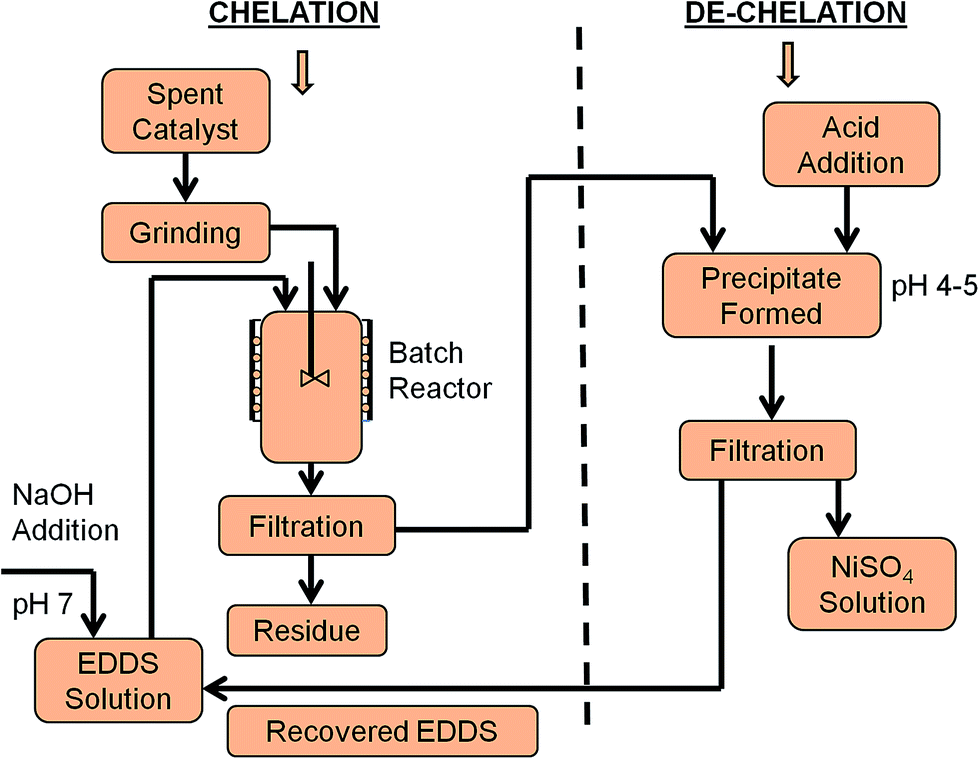 | ||
| Fig. 14 Process flow chart for solid–liquid extraction and regeneration of chelating agent. | ||
Goel et al.8 studied the exchange behavior of Ni from primary reforming waste catalyst used in the fertilizer industry towards the chelating agents EDTA and DTPA. It was concluded that the extraction of the metal depends on the complexing affinity of the chelating agent for the metal and on the affinity between the solid and metal. EDTA has six binding sites (4 acid and 2 amines sites) that make coordination bonds with the metal, while DTPA has eight sites (3 amine and 5 acid sites), which show stronger binding capacity and higher extraction efficiency than EDTA, but this cannot be recovered easily due to the complicated process to break the coordination bonds.
The extraction efficiency of recovered EDTA was investigated and a significant extraction of Co and Mo was observed for up to five cycles using recovered EDTA under optimum reaction conditions;9 however, a nearly 20% loss in extraction efficiency was observed for the fifth cycle of recovered EDTA compared with the fresh EDTA. The loss in extraction efficiency can be related to a loss in the number of metal-binding sites due to the repetitive precipitation of EDTA9 and the added number of impurities during each cycle.13,15
Therefore, it can be concluded that metal extraction from spent catalysts using chelating agents is a good idea on the economical and ecological levels. Due to its recoverable nature, this process is more economical than any other process. Table 4 provides an economic evaluation of the chelation process in comparison with other methods employed in the literature. It can be seen from Table 4 that the material cost of chelation technology is less than that of other methods, although it may also be seen that the cost factor of $0.62 for alkaline leaching and $0.61 for chelation technology are nearly equal to each other. However, the fact cannot be denied that the alkaline reagent cannot be reused for the next alkaline leaching cycles, while the chelating agent can be reused at least four times for chelation experiments without a significant loss in extraction efficiency. Thus, by comparing the cost factor for 4 cycles using these two methods, the cost for the alkaline method will be $2.44 but is only $0.74 for chelation using recovered EDTA. Thus, the possibility of recycling combined with good extraction efficiency reduces the process cost significantly and makes the process more attractive than other available methods.
| Technology | Reaction condition | Extraction yield | Chemicals needed | Amount of chemicals and prices | Material cost for the process | Ref. |
|---|---|---|---|---|---|---|
| Acid leaching | 50% H2SO4, S/L = 1/20, 85 °C, 2.5 h | 94% nickel | Sulphuric acid | 10 g → catalyst 200 ml solution, 100 ml H2SO4 ($4.25 for 500 ml H2SO4, Fisher Scientific) = (4.25/500) × 100 = $0.85 | $0.85 for 10 g catalyst processing | Abdel-Aal & Rashad190 |
| Acid leaching | 67% H2SO4, S/L = 1/14, 80 °C, 2.33 h | 85% nickel | Sulphuric acid | 10 g catalyst → 140 ml solution, 93.8 ml H2SO4 ($4.25 for 500 ml H2SO4, FS) = (255/500) × 93.8 = $0.79 | $0.79 for 10 g catalyst processing | Idris et al.,191 |
| Bioleaching | 30 °C, 175 rpm shaking, 21 days treatment | 83% Ni, 90% V, 30% Mo | 9K medium – (NH4)2SO4,KCl, H2HPO4, MgSO4·7H2O, Ca(NO3)2, agar gel, FeSO4, catalyst digestion – HCl, bacterial cultures | 10 g catalyst, 70 ml solution of 9K medium + 30 ml FeSO4 solution to make 100 ml solution. 0.21 g of (NH4)2SO4 × 0.092 $ g−1 = $0.019, 0.01 g of KCl × 0.074 $ g−1 = $0.00073, 0.05 g of H2HPO4 × 0.131 $ g−1 = $0.0065, 0.05 g of MgSO4·7H2O × 0.0078 $ g−1 = $0.00033, 0.001 g of Ca(NO3)2 × 0.142 $ g−1 = $0.00098, 0.5 g agarose gel × 0.9128 $ g−1 = $0.456, 4.91 g FeSO4 × 0.011 $ g−1 = $0.054, 50% HCl = 50 ml HCl × 0.0067 $ ml−1 = $0.34 | $0.87 + price of bacterial culture | Belochini et al.,192 |
| Bioleaching | 35 °C, 180 rpm shaking, 240 h treatment, particle size – 106 μm | 85–95% Ni, V | 9K medium, bacterial culture, 10% HCl for dilution, HgCl2 | 9K medium price same as above + HCl and HgCl2, price according to requirement | $0.87 + price of bacterial culture | Kim et al.,193 |
| Alkaline leaching followed by acid washing | Time – 1 h, temperature – 30 °C, and pulp density – 10% | 97% Ni, 99% V, 84% Mo | (NH4)2CO3·H2SO4 | 10 g → 250 ml solution, 40 g L−1 (NH4)2CO3 ≥ 10 g in 250 ml solution, 10 g (NH4)2CO3 × 0.0505 $ g−1 = $0.5055, 1 M H2SO4 = 13.6 ml in 250 ml solution, 13.6 ml H2SO4 × 0.0085 $ g−1 = $0.1156 | $0.621 | Mishra et al.,32 |
| Chelation technology | 0.4 M EDTA, S/L = 1:15, 4 h, 130 °C |
>90% Ni, 88% Mo, 84% Co | EDTA, NaOH/H2SO4 (98% conc. = 18 mol L−1) for adjusting pH | 10 g → 150 ml solution, 0.4 M EDTA ≥22.3 g/150 ml solution, 22.3 g EDTA × 0.0276 $ g−1 = $0.616, 0.1 M NaOH ≥ 0.02 g in 5 ml solution, 0.02 g × 0.00733 $ g−1 = $0.000147, 0.2 M H2SO4 = 0.108 ml in 10 ml solution, 0.108 ml × 0.0085 $ ml−1 = $0.00091 | $0.617 (for one run using fresh EDTA), 0.74$ to conduct 4 runs using recovered EDTA | Chauhan et al., 2012 (ref. 15), 2013 (ref. 9) |
This process is also ecofriendly due to the easy recovery of the chelating agent used for the process, and the fact that no corrosive environment is used and no hazardous byproducts are liberated during the process. The high efficiency of metal extraction and moderate thermodynamic stabilities of the metal complexes make this technology more favorable than any other technology for metal recovery. Chelation technology (solid–liquid extraction) takes place in two steps while pyrometallurgical processes (which have a high temperature requirement) can be performed in just one step; nevertheless, chelation technology offers an advantage over pyrometallurgical processes in terms of a lower energy consumption. Another crucial advantage of chelation technology is related to the final product formation. It is always easier to prepare salts, oxides or complexes using a liquid-phase process which can be reused in catalyst preparation, whereas a high temperature process would yield the metals in the metallic state.
However, many things still remain to be found out in this particular research area, and not much work has been reported in the literature on the extraction of metals from spent catalysts. The search for new biodegradable chelating agents may be helpful in the development of new ecotechnology processes. The generation of new adsorbent beds and resins by coating with chelating agents will provide a direction to develop a continuous process for metal extraction on an industrial scale. The recovery of the chelating agent is another important research area that still needs more investigation. The literature still does not state complete recovery and efficient recycling of chelating agents; therefore, new methods need to be found to give efficient recovery during the process.
5.2. Chelating agents used for metal extraction from soil
Soil contamination due to exposure to heavy metals has become a serious concern that affects crop yields and soil productivity and leads to bioaccumulation of metals in the ecosystem. Heavy metal sorption at the soil surface can be explained on the basis of the surface complexation concept. The active functional groups present on the soil surface (S) bind with metal (M) through covalent interactions, which can be explained by eqn (14).194| S–OH + Mn+ ⇌ S–OMn+−1 + H+ | (14) |
It can be seen from eqn (14) that metal sorption to the soil matrix is a pH-dependent reaction. Surface adsorption sites become saturated and sometimes the possibility of surface polymerization increases with increasing metal concentrations in the soil matrix, which may result in metal precipitation. Various soil remediation techniques are being adopted these days in order to remove metal pollutants from soil or transform these metals into their non-toxic forms. Davis and Singh195 compared chlorination, acid leaching and chelation technology using EDTA and DTPA to extract Zn from an artificially contaminated soil column. The highest removal efficiency was obtained using the chelation process at an optimized flow rate. Kocialkowski et al.196 evaluated the performance of different chelating agents for the extraction of the heavy metals Cu, Zn, Pb and Mn from arable soil samples and concluded that EDTA was the best chelating agent for the extraction of all the metals. Lim et al.197 investigated the ability of EDTA to remove the divalent metals Cd, Pb and Ni from soil. They also developed a cost-effective closed-loop utilization of EDTA by recovering and reusing it for metal extraction. Hong et al.118 investigated the extraction, recovery, and biostability of DTPA as a remediation agent for soils contaminated with metals. It was observed that DTPA was capable of extracting Pb from the contaminated soils and could be recovered by the use of cationic and anionic precipitants under alkaline pH conditions. Palma et al.198 investigated the effectiveness of the extractant EDTA, its structural isomer EDDS, rhamnolipids and citric acid for heavy metal removal from a contaminated harbour sediment by varying the concentration of chelating agent and the overall washing time. EDTA and EDDS showed a comparable metal extraction efficiency (∼70%) whereas a lower removal efficiency was achieved using rhamnolipids and citric acid. Tandy et al.104 compared the traditional chelating agent EDTA with the biodegradable chelating agents [S,S]-EDDS, IDSA, MGDA and NTA for the extraction of heavy metals from contaminated soil. The authors optimized the reaction time to 24 h and a higher concentration of chelating agent was considered favourable for higher metal extraction efficiency. A lower pH dependence and smaller difference in extraction efficiency was also observed for various chelating agents at higher molar concentrations of complexing agents. Extraction with EDDS at pH = 7 showed the optimum extraction efficiency for Cu, Zn, and Pb, and loss of Ca and Fe from the soil. The work was extended to compare the efficiency of batch and column extraction of Cu, Zn, and Pb from polluted soils using the biodegradable chelating agent EDDS.199Table 5 gives a list of chelating agents in the literature that have been used for metal extraction from soil.
| Type of soil | Chelating agent | Heavy metals | Ref. |
|---|---|---|---|
| Superfund site soil | EDTA | Pb | Ellis et al.200 |
| Soil from battery recycling facility | EDTA, NTA | Pb | Elliot and Brown12 |
| Quartz-rich sediment and soil samples | NTA | Pb, Ni, Zn | Howard and Shu201 |
| Wetland soil | Acetic acid, EDTA, DTPA | Zn, Fe, Mn, Ca, Cd, Mg, K, Al, Na, Cu, Pb | Sistani et al.202 |
| Industrially contaminated soil, agricultural soil | EDTA, EDDS, IDSA, MGDA, NTA | Zn, Cu, Pb, Cd, Ni | Tandy et al.104 |
| Soil from 25–50 cm below ground surface near a bay | EDDS | Cu, Zn, Pb | Yip et al.203 |
Yip et al.204 employed EDDS for the extraction of Cu, Zn and Pb from artificially- and field-contaminated soil samples and developed an empirical model using the initial metal distribution to estimate the extraction efficiency at equilibrium. It was also observed that higher extraction rates can be achieved in multi-metal-contaminated soil samples than mono-metallic-contaminated soils due to the presence of a large proportion of carbonated and exchangeable fraction of heavy metals. Batch kinetic and equilibrium experiments were conducted to study the influence of the EDDS to metal molar ratios (MRs), solution pH and soil to solution ratio on metal extraction from contaminated soil under EDDS deficiency (i.e. MR < 1).205
Nowadays, phytoextraction is an appealing technology for metal extraction from soils and is also known as ‘green remediation’, which involves desorption of metals from the soil matrix and metal mobilization to the rhizosphere for uptake by plant roots. It can be categorized into phytoextraction (plant harvesting to accumulate metals in shoots), phytomining (use of plant biomass), phytovolatilization (use of microbes to transform soil elements into volatile forms), and phytostabilization (conversion of heavy metals into less toxic and biodegradable forms). However, due to the limited number of plant species with a high capacity to accumulate metals and to produce a large amount of biomass, chelation technology has been incorporated into phytoextraction to improve the uptake of metals by high biomass plants.206,207 Blaylock et al.208 demonstrated the application of Brassica juncea (Indian mustard) as a hyper-accumulator with synthetic chelating agents to extract Pb from soil. It was observed that the combined effect of EDTA and acetic acid led to a nearly two times higher accumulation of Pb in the mustard shoots compared to the extraction efficiency of EDTA alone. Vassil et al.209 also reported similar results for the extraction of Pb using Indian mustard along with EDTA and concluded that the coordination of Pb transport by EDTA enhances the mobility of insoluble Pb2+ ions within the plants, whereas high concentrations of Pb–EDTA may cause reductions in the transpiration rate and shoot water content due to the presence of free protonated EDTA (H-EDTA) in the hydroponic solution. Luo et al.210 investigated chemically-enhanced phytoextraction using EDTA, EDDS and citric acid for the intake of Cu, Pb, Zn and Cd by corn (Zea mays L.) and bean (Phaseolus vulgaris L.) plants. The results indicated that EDTA is a more efficient chelating agent than EDDS in the extraction of Pb and Cd, whereas EDDS is more effective in the extraction of Cu and Zn. Meers et al.211 also compared the performance of the biodegradable chelating agents EDDS, NTA and citric acid with EDTA in enhancing shoot accumulation of Cd, Cu, Cr, Ni, Pb and Zn in Helianthus annuus. Similar results were obtained to justify the applicability of EDDS for phytoextraction of heavy metals. Evangelou et al.212 investigated the influence of EDDS and EDTA on the uptake of Cd and Cu from soil with tobacco, Nicotiana tabacum. The effects of EDTA and citric acid on the phytoextraction of heavy metals (Cd, Cu, Pb and Zn) from industrially-contaminated soil using the hyper-accumulator Sedum alfredii Hance were investigated by Sun et al.213
Thus, valuable studies conducted by many researchers have endorsed the use of chelating agents in phytoextraction to augment the metal extraction process. However, some areas are still under investigation for the successful application of chelating agent-induced phytoextraction. An effective method for prevention of leaching into groundwater, the need for more field studies to give a better outlook on phytoextraction possibilities, and justified reasons for the different observations made by various researchers are the most important factors to investigate before phytoextraction can be considered as a promising solution.
6. Future research aspects – statistical and computational approaches for chelation
Chelation experiments are performed in the laboratory and need lots of effort and accuracy to optimize the metal extraction process. Therefore, in the present computational era, it is prudent to find new technologies to minimize experimental effort and to achieve better accuracy in observations. Design of experiments, computational fluid dynamics, and molecular and computational simulations can be considered a better approach for designing and optimizing metal extraction processes. Mohd Salim et al.214 employed a face-centered cube design (FCD) in response surface methodology (RSM) to evaluate the relationship between the extraction parameters and the metal chelating activity of Centella asiatica (CA) and Erythroxylum cuneatum (EC). The experiments were designed for 30 experimental runs, including six replications at centre point, and concluded that response surface methodology reduced the extraction time, temperature and stirring speed. This methodology also helped to subsequently improve the chelating activity of the plants in comparison to the conventional method. Chauhan et al.112 explained that the ‘one variable at a time (OVAT)’ approach does not consider the interaction effect of process variables and cannot be an efficient approach for multivariate analysis. Therefore, a Box–Behnken design (BBD) was coupled with RSM to optimize the process parameters using statistical analysis and to provide an enhanced quality of information. Response surface plots demonstrated the optimum reaction conditions (molar ratio of chelating agent to Ni, S/L and reaction time; MR = 3.6, S/L = 1/22.3, reaction temperature of 85.2 °C, reaction time of 8.2 h and pH = 7.3) for Ni extraction from spent catalysts using various aminopolycarboxylate chelating agents. It was also suggested that optimization studies using computational methods may prove useful on the industrial scale to minimize manual effort and to achieve better efficiency with low processing costs.Pinto et al.148 performed computer simulations in order to assess the selectivity of various chelating agents for transition metals in medium with an excess of Ca and Mg ions and the ability of compounds to complex Ca and Mg ions at high concentrations. The results demonstrated the pH ranges for various chelating agents at which they can complex more than 80% of the respective transition metals (Fe, Mn, Cu, Pb, Cd, and Zn) in an excess of Ca and Mg ions. The metal speciation calculations and computer simulations at different pH ranges may provide an idea about the most favorable chelating agents in different industrial processes. It was also suggested that NTA, EDDS, MGDA, and PDA can be used in the paper and pulp industries due to its favorable pH range.148 Experimental studies215 also suggest a similar observation at a specific pH range in the pulp and paper industries. It was also concluded that still, no biodegradable chelating agent has superior efficiency to EDTA for Pb removal from soil, which is in accord with the experimental studies.103,105,114,115,120
Density functional theory (DFT) methods have also been employed for the static and dynamic structure calculations of various chelating agents to investigate the coordination properties of chelating agents with metal ions.216–219 Chen et al.83 calculated the free energy changes for the hydration and complexation processes and demonstrated that the metal complexation process is a substitution reaction of the coordinated water molecules by the chelating agents. Degradation pathways were also deduced for EDTA and EDDS by calculating the dissociation energies of the various C–N bonds of the ligands. Sillanpaa et al.220 performed geometry optimizations, continuum-solvation and mixed cluster–continuum modelling, and Car–Parrinello molecular dynamics simulations to study the complexation of amino polycarboxylic acids with different divalent metal ions. The work presented a good estimation of the metal ion complexation constant with different ligands. Paola et al.221 investigated the Jahn–Teller distortion and effect of spin states on different palladium clusters and concluded that ligand and kinetic effects play a significant role in driving the formation of ligand-stabilized clusters. Coskuner et al.222 demonstrated the stable nature of the crystalline structure of octahedral Al–EDTA in aqueous solution.
Thus, computational approaches may reduce manual effort and provide an enhanced quality of information in different research aspects. However, more studies are still needed in the area of computational simulation to make it more convenient to researchers. Computational fluid dynamics may also be helpful in order to study the hydrodynamics associated with the metal extraction process and to provide an approximation of the various process parameters in order to perform pilot plant studies.
7. Summary and outlook
Chelation technology for the extraction of heavy metals from contaminated sites (soil, water, industrial waste) is drawing great attention at present in the development new promising green methods, which should be efficient on the ecological and economical levels. Various R&D studies are currently underway to identify process parameters which may affect the formation and stability of metal–ligand complexes. Higher extraction efficiency, reusability of chelating agents and diverse applicability make it a convincing technology; however, a number of challenges still need to be addressed. The synthesis of new biodegradable mobilizing agents and the identification of their degradation pathways, by means of molecular simulations or biological methods, are open research fields with immense opportunities for further development in the area of chelation technology. Different possible methods to recover and recycle the chelating agents should also be explored to bring about an efficient ‘closed loop’ chelation process. The successful application of chelation technology for metal extraction from contaminated sites is a very elegant example of a ligand substitution mechanism, though the industrial application of the process is still restricted by the lack of adequate knowledge about competing reactions that may affect the metal–ligand complexation. More experimental studies will be helpful to provide a better understanding of the ligand substitution mechanism and to explore the diverse applicability of chelation technology. It is conceived that modern computational tools may result in an ongoing renaissance of research activities in chelation technology to carve-out new territory for metal–ligand complexation. Thus, future attempts should focus on the sustainability, economics and environmental impact of the process to meet the growing industrial demand.References
- G. H. Brundtland, For World Commission on Environment and Development, Our Common Future, Oxford University Press, Oxford, UK, 1987 Search PubMed.
- International Energy Outlook 2013, U.S. Energy Information Administration, U.S. Department of Energy, Washington DC, 2013, DOE/EIA-0484(2013) Search PubMed.
- J.-M. Lee, Fluid Phase Equilib., 2012, 319, 30–36 CrossRef CAS PubMed.
- M. Regel-Rosocka and F. J. Alguacil, Rev. Metal., 2013, 49, 292–316 CrossRef CAS.
- A. Stark, Energy Environ. Sci., 2011, 4, 19–32 CAS.
- M. A. Barakat, Arabian J. Chem., 2011, 4, 361–377 CrossRef CAS PubMed.
- F. J. Deive, A. Rodriguez, A. Varela, C. Rodrigues, M. C. Leitao, J. A. M. P. Houbraken, A. B. Pereiro, M. A. Longo, M. A. Sanroman, R. A. Samson, L. P. N. Rebelo and C. S. Pereira, Green Chem., 2011, 13, 687–696 RSC.
- S. Goel and A. Gautam, Hydrometallurgy, 2010, 101, 120–125 CrossRef CAS PubMed.
- G. Chauhan, K. K. Pant and K. D. P. Nigam, Ind. Eng. Chem. Res., 2013, 52, 16724–16736 CrossRef CAS.
- Hazardous waste listings 2008, http://www.epa.gov/osw/hazard/wastetypes/pdfs/listing-ref.pdf, (accessed February 2014) Search PubMed.
- J. Paz-Ferreiro, H. Lu, S. Fu, A. Méndez and G. Gascó, Solid Earth, 2014, 5, 65–75 CrossRef PubMed.
- H. A. Elliott and G. A. Brown, Water, Air, Soil Pollut., 1989, 45, 361–369 CrossRef CAS.
- S. Goel, K. K. Pant and K. D. P. Nigam, J. Hazard. Mater., 2009, 171, 253–261 CrossRef CAS PubMed.
- K. R. Vuyyuru, K. K. Pant, V. V. Krishnan and K. D. P. Nigam, Ind. Eng. Chem. Res., 2010, 49, 2014–2024 CrossRef CAS.
- G. Chauhan, K. K. Pant and K. D. P. Nigam, Ind. Eng. Chem. Res., 2012, 51, 10354–10363 CrossRef CAS.
- W. C. Leung, M. F. Wong, H. Chua, W. Lo and C. K. Leung, Water Sci. Technol., 2000, 41, 233–240 CAS.
- T. A. Kurniawan, G. Y. S. Chan, W. H. Lo and S. Babel, Sci. Total Environ., 2006, 366, 409–426 CrossRef CAS PubMed.
- T. A. Kurniawan, G. Y. S. Chan, W. H. Lo and S. Babel, Chem. Eng. J., 2006, 118, 83–98 CrossRef CAS PubMed.
- A. J. Pedersen, Biomass Bioenergy, 2003, 25, 447–458 CrossRef CAS.
- L. R. Skubal, N. K. Meshkov, T. Rajh and M. Thurnauer, J. Photochem. Photobiol., A, 2002, 148, 393–397 CrossRef CAS.
- R. K. Srivastav, S. K. Gupta, K. D. P. Nigam and P. Vasudevan, Int. J. Environ. Stud., 1993, 45, 43–50 CrossRef CAS.
- K. D. P. Nigam, R. K. Srivastav, S. K. Gupta and P. Vasudevan, Environmental Modeling & Assessment, 1998, 4, 249–248 Search PubMed.
- R. L. Chaney, M. Malik, Y. M. Li, S. L. Brown, E. P. Brewer, J. S. Angle and A. J. M. Baker, Curr. Opin. Biotechnol., 1997, 8, 279–284 CrossRef CAS.
- P. Ziarati and S. Alaedini, J. Environ. Anal. Toxicol., 2014, 4, 208–211 Search PubMed.
- M. C. Steele and J. Pichtel, J. Environ. Eng., 1998, 124, 639–645 CrossRef CAS.
- W. Jiang, T. Tao and Z. Liao, Open J. Soil Sci., 2011, 1, 70–76 CAS.
- M. H. Shariat, N. Setoodeh and R. A. Dehghan, Miner. Eng., 2001, 14, 815–820 CrossRef CAS.
- D. D. Sun, J. H. Tay, H. K. Cheong, D. L. K. Leung and G. Qian, J. Hazard. Mater., 2001, B87, 213–223 CrossRef.
- B. B. Kar, P. Datta and V. N. Misra, Hydrometallurgy, 2004, 72, 87–92 CrossRef CAS.
- B. B. Kar, B. V. R. Murthy and V. N. Misra, Int. J. Miner. Process., 2005, 76, 143–147 CrossRef CAS PubMed.
- W. Mulak, A. Szymczycha, A. Lesniewicz and W. Zyrnicki, Physicochem. Probl. Miner. Process., 2006, 40, 69–76 CAS.
- D. Mishra, G. R. Chaudhury, D. J. Kim and J. G. Ahn, Hydrometallurgy, 2010, 101, 35–40 CrossRef CAS PubMed.
- L. Zeng and C. Y. Cheng, Hydrometallurgy, 2010, 101, 141–147 CrossRef CAS PubMed.
- K. H. Park, B. R. Reddy, D. Mohapatra and C. W. Nam, Int. J. Miner. Process., 2006, 80, 261–265 CrossRef CAS PubMed.
- D. Santhiya and Y. P. Ting, J. Biotechnol., 2005, 116, 171–184 CrossRef CAS PubMed.
- D. J. Kim, D. Mishra, J. G. Ahn, G. R. Chaudhury and D. E. Ralph, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2009, 44, 1585–1591 CrossRef CAS PubMed.
- A. Bharadwaj and Y.-P. Ting, Bioresour. Technol., 2013, 130, 673–680 CrossRef CAS PubMed.
- F. Gerayeli, F. Ghojavand, S. M. Mousavi, S. Yaghmaei and F. Amiri, Sep. Purif. Technol., 2013, 118, 151–161 CrossRef CAS PubMed.
- F. Amiri, S. M. Mousavi, S. Yaghmaei and M. Barati, Biochem. Eng. J., 2012, 67, 208–217 CrossRef CAS PubMed.
- F. Amiri, S. Yaghmaei, S. M. Mousavi and S. Sheibani, Hydrometallurgy, 2011, 109, 65–71 CrossRef CAS PubMed.
- F. Beolchini, V. Fonti, F. Ferella and F. Vegliò, J. Hazard. Mater., 2010, 178, 529–534 CrossRef CAS PubMed.
- R. Banda, T. H. Nguyen, S. H. Sohn and M. S. Lee, Hydrometallurgy, 2013, 133, 161–167 CrossRef CAS PubMed.
- K. Mazurek, Hydrometallurgy, 2013, 134–135, 26–31 CrossRef CAS PubMed.
- P. K. Parhi, K. H. Park and G. Senanayake, J. Ind. Eng. Chem., 2013, 19, 589–594 CrossRef CAS PubMed.
- H. Al-Sheeha, M. Marafi, V. Raghavan and M. S. Rana, Ind. Eng. Chem. Res., 2013, 52, 12794–12801 CrossRef CAS.
- S. P. Barik, K.-H. Park, P. K. Parhi and J. T. Park, Hydrometallurgy, 2012, 111–112, 46–51 CrossRef CAS PubMed.
- S. Huang, Z. Zhao, X. Chen and F. Li, Int. J. Refract. Met. Hard Mater., 2014, 46, 109–116 CrossRef CAS PubMed.
- A. Katsiapi, P. E. Tsakiridis, P. Oustadakis and S. Agatzini-Leonardou, Miner. Eng., 2010, 23, 643–651 CrossRef CAS PubMed.
- K. Mazurek, K. Białowicz and M. Trypuć, Hydrometallurgy, 2010, 103, 19–24 CrossRef CAS PubMed.
- Y. Chen, Q. Feng, Y. Shao, G. Zhang, L. Ou and Y. Lu, Miner. Eng., 2006, 19, 94–97 CrossRef CAS PubMed.
- A. K. Upadhyay, J.-C. Lee, E.-Y. Kim, M.-S. Kim, B.-S. Kim and V. Kumar, J. Chem. Technol. Biotechnol., 2013, 88, 1991–1999 CAS.
- M. W. Ojeda, E. Perino and M. d. C. Ruiz, Miner. Eng., 2009, 22, 409–411 CrossRef CAS PubMed.
- Y. Shen, W. Li, J. Wu, S. Li, H. Luo, S. Dai and W. Wu, Dalton Trans., 2014, 10023–10032 RSC.
- A. Stojanovic, D. Kogelnig, L. Fischer, S. Hann, M. Galanski, M. Groessl, R. Krachler and B. K. Keppler, Aust. J. Chem., 2010, 63, 511–524 CrossRef CAS.
- L. Fischer, T. Falta, G. Koellensperger, A. Stojanovic, D. Kogelnig, M. Galanski, R. Krachler, B. K. Keppler and S. Hann, Water Res., 2011, 45, 4601–4614 CrossRef CAS PubMed.
- A. E. Visser, R. P. Swatloski, M. W. Reichert, R. Mayton, S. Sheff, A. Wierzbicki, J. H. Davis and R. D. Rogers, Environ. Sci. Technol., 2002, 36, 2523–2529 CrossRef CAS.
- G. Z. Kyzas and M. Kostoglou, Materials, 2014, 7, 333–364 CrossRef CAS PubMed.
- A. L. Iskander, E. M. Khald and A. S. Sheta, Annals of Agricultural Sciences, 2011, 56, 43–48 CrossRef PubMed.
- O. E. Abdel Salam, N. A. Reiad and M. M. ElShafei, J. Adv. Res., 2011, 2, 297–303 CrossRef PubMed.
- A. Dubey, A. Mishra and S. Singhal, Int. J. Environ. Sci. Technol., 2014, 11, 1043–1050 CrossRef CAS PubMed.
- Y. Zhang, R. Zheng, J. Zhao, F. Ma, Y. Zhang and Q. Meng, BioMed Res. Int., 2014, 2014, 1–8 Search PubMed.
- G. Annadurai, R. S. Juang and D. J. Lee, Water Sci. Technol., 2003, 47, 185–190 CAS.
- P. d'Abzac, F. Bordas, E. Joussein, E. D. van Hullebusch, P. N. L. Lens and G. Guibaud, Environ. Sci. Pollut. Res., 2013, 20, 4509–4519 CrossRef PubMed.
- P. d'Abzac, F. Bordas, E. Joussein, E. Van Hullebusch, P. N. L. Lens and G. Guibaud, Environ. Sci. Technol., 2010, 44, 412–418 CrossRef PubMed.
- R. Mikutta, A. Baumgärtner, A. Schippers, L. Haumaier and G. Guggenberger, Environ. Sci. Technol., 2012, 46, 3866–387 CrossRef CAS PubMed.
- S. Ahmady-Asbchin, Y. Andres, C. Gerente and P. Le Cloirec, Bioresour. Technol., 2008, 99, 6150–6155 CrossRef CAS PubMed.
- V. K. Gupta and A. Rastogi, Colloids Surf., B, 2008, 64, 170–178 CrossRef CAS PubMed.
- C. A. Rios, C. D. Williams and C. L. Roberts, J. Hazard. Mater., 2008, 156, 23–35 CrossRef CAS PubMed.
- E. I. Basaldella, P. G. Vazquez, F. Iucolano and D. Caputo, J. Colloid Interface Sci., 2007, 313, 574–578 CrossRef CAS PubMed.
- P. Tang, C. K. Lee, K. S. Low and Z. Zainal, Environ. Technol., 2003, 24, 1243–1251 CrossRef CAS PubMed.
- E. Repo, J. K. Warchol, T. A. Kurniawan and M. E. T. Sillanpaa, Chem. Eng. J., 2010, 161, 73–82 CrossRef CAS PubMed.
- L. Wang, L. Yang, Y. Li, Y. Zhang, X. Ma and Z. Ye, Chem. Eng. J., 2010, 163, 364–372 CrossRef CAS PubMed.
- K. Dzulkefly, M. J. Haron, W. H. Lim and C. C. Woon, J. Oleo Sci., 2002, 51, 749–751 CrossRef CAS.
- D. Kolodynska, Desalination, 2010, 263, 159–169 CrossRef CAS PubMed.
- J. Lv, K. Y. Wang and T. S. Chung, J. Membr. Sci., 2008, 310, 557–566 CrossRef CAS PubMed.
- M. G. Khedr, Desalination, 2008, 222, 135–145 CrossRef CAS PubMed.
- R. Klaassen, P. Feron and A. Jansen, Desalination, 2008, 224, 81–87 CrossRef CAS PubMed.
- T. Mohammadi, A. Razmi and M. Sadrzadeh, Desalination, 2004, 167, 379–385 CrossRef CAS PubMed.
- T. Mohammadi, A. Mohebb, M. Sadrzadeh and A. Razmi, Sep. Purif. Technol., 2005, 41, 73–82 CrossRef CAS PubMed.
- L. Wang, N. Wang, L. Zhu, H. Yu and H. Tang, J. Hazard. Mater., 2008, 152, 93–99 CrossRef CAS PubMed.
- E. Gkika, A. Troupis, A. Hiskia and E. Papaconstantinou, Appl. Catal., B, 2006, 62, 28–34 CrossRef CAS PubMed.
- V. S. Bryantsev, M. S. Diallo and W. A. Goddard III, J. Phys. Chem. B, 2008, 112, 9709–9719 CrossRef CAS PubMed.
- L. Chen, T. Liu and C. Ma, J. Phys. Chem. A, 2010, 114, 443–454 CrossRef CAS PubMed.
- D. Kołodyńska, in Expanding Issues in Desalination, ed. R. Y. Ning, 2011, ISBN: 978-953-307-624-9 Search PubMed.
- Transparency Market Research, 2012, Global Industry Analysis, Size, Share, Growth, Trends and Forecast, 2013–2019, http://www.transparencymarketresearch.com, accessed January 2014 Search PubMed.
- J. L. Means, T. Kucak and D. A. Crerar, Environ. Pollut., Ser. B, 1980, 1, 45–60 CrossRef CAS.
- J. Pastor, A. M. Aparicio, A. Gutierrez-Maroto and A. J. Hernandez, Sci. Total Environ., 2007, 378, 114–118 CrossRef CAS PubMed.
- Chemical Insight and Forecasting, Chemical Economics Handbook, IHS Chemical, 2012, http://www.ihs.com/products/chemical/planning/ceh/chelating-agents.aspx?pu=1%26rd=chemihs, accessed December 2013 Search PubMed.
- Global Industry Analyst, Chelating Agent: A Global Strategic Business Report, PRWEB, California, 2012, http://www.prweb.com/releases/chelating_agents_market/EDTA_market/prweb9485451.htm, accessed January 2014 Search PubMed.
- W. G. Jackson, J. A. McKeon and S. Cortez, Inorg. Chem., 2004, 43, 6249–6254 CrossRef CAS PubMed.
- K. Bowman-James, Acc. Chem. Res., 2005, 38, 671–678 CrossRef CAS PubMed.
- G. T. Morgan and H. D. K. Drew, J. Chem. Soc., Trans., 1920, 117, 1456–1465 RSC.
- A. M. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC.
- J. Reedijk, Platinum Met. Rev., 2008, 52, 2–11 CrossRef CAS.
- P. Almarin, Y. A. Zolotov and V. A. Bodnya, Pure Appl. Chem., 1971, 25, 667–680 Search PubMed.
- H. Hyvönen, Ph.D. Thesis, University of Helsinki, 2008.
- G. Schwarzenbach, Helv. Chim. Acta, 1952, 35, 2344–2359 CrossRef CAS.
- A. W. Adamson, J. Am. Chem. Soc., 1954, 76, 1578–1579 CrossRef CAS.
- R. H. Myres, Inorg. Chem., 1978, 17, 953–958 Search PubMed.
- C. S. Chung, J. Chem. Educ., 1984, 61, 1062–1064 CrossRef CAS.
- P. Prapaipong and E. L. Shock, Geochim. Cosmochim. Acta, 2001, 65, 3931–3953 CrossRef CAS.
- V. Vallet, U. Wahlgren and I. Grenthe, J. Am. Chem. Soc., 2003, 125, 14941–14950 CrossRef CAS PubMed.
- Y. A. Zolotov, Extraction of Chelate Compounds, Ann Arbor-Humphrey Science Publications, Ann Arbor, London, 1970 Search PubMed.
- S. Tandy, K. Bossart, R. Mueller, J. Ritschel, L. Hauser, R. Schulin and B. Nowack, Environ. Sci. Technol., 2004, 38, 937–944 CrossRef CAS.
- Z. A. Begum, I. M. M. Rahman, H. Sawai, S. Mizutani, T. Maki and H. Hasegawa, Water, Air, Soil Pollut., 2013, 224, 1381–1402 CrossRef.
- T. T. Lim, J. H. Tay and J. Y. Wang, J. Environ. Eng. Div., 2004, 130, 59–66 CrossRef CAS.
- K. Fischer and H. P. Bipp, Water, Air, Soil Pollut., 2002, 138, 271–288 CrossRef CAS.
- U. Chatreewongsin, Dissertation, Faculty of the Virginia Polytechnic Institute and State, 2000.
- D. Fangueiro, A. Bermond, E. Santos, H. Carapuca and A. Duarte, Anal. Chim. Acta, 2002, 459, 245–256 CrossRef CAS.
- Z. Zou, R. Qiu, W. Zhang, H. Dong, Z. Zhao, T. Zhang, X. Wei and X. Cai, Environ. Pollut., 2009, 157, 229–236 CrossRef CAS PubMed.
- Z. A. Begum, I. M. M. Rahman, H. Sawai, Y. Tate, T. Maki and H. Hasegawa, J. Chem. Eng. Data, 2012, 57, 2723–2732 CrossRef CAS.
- G. Chauhan, K. K. Pant and K. D. P. Nigam, Green Process Synth., 2013, 2, 259–271 CAS.
- C. Kim, Y. Lee and S. K. Ong, Chemosphere, 2003, 51, 845–853 CrossRef CAS.
- N. Manouchehri, S. Besancon and A. Bermond, Anal. Chim. Acta, 2006, 559, 105–112 CrossRef CAS PubMed.
- Z. A. Begum, I. M. M. Rahman, Y. Tate, Y. Egawa, T. Maki and H. Hasegawa, J. Solution Chem., 2012, 41, 1713–1728 CrossRef CAS.
- Z. A. Begum, I. M. M. Rahman, Y. Tate, H. Sawai, T. Maki and H. Hasegawa, Chemosphere, 2012, 87, 1161–1170 CrossRef CAS PubMed.
- M. A. M. Kedziorek, A. Dupuy and A. C. M. Bourg, Environ. Sci. Technol., 1998, 32, 1609–1614 CrossRef CAS.
- P. K. Hong, X. Cai and Z. Cha, Environ. Pollut., 2008, 153, 14–21 CrossRef CAS PubMed.
- G. In, Y.-S. Kim and J.-M. Choi, Bull. Korean Chem. Soc., 2008, 29, 969–973 CrossRef CAS.
- P. C. Vandevivere, H. Saveyn, W. Verstraete, T. C. J. Eijtel and D. Schowanek, Environ. Sci. Technol., 2001, 35, 1765–1770 CrossRef CAS.
- J.-C. Chao, A. Hong, R. W. Okey and R. W. Peters, Proceedings of the 1998 Conference on Hazardous Waste Research, 1998, pp. 142–160 Search PubMed.
- A. Martell and R. Smith, Critical Stability Constants, Plenum Press, New York City, NY, USA, 1974 Search PubMed.
- T. P. Knepper and H. Weil, Vom Wasser, 2001, 97, 193–232 CAS.
- J. Byegard, G. Skarnemark and M. Skålberg, J. Radioanal. Nucl. Chem., 1999, 241, 281–290 CrossRef CAS.
- BASF, Trilon® B Marken, 2006, http://www.performancechemicals.basf.com/evwcmsin/internet/en_GB/function/conversions:/publish/content/EV/EV5/products/ca/doc/trilon_b.pdf, accessed February 2014.
- G. Andegregg, Pure Appl. Chem., 1982, 54, 2693–2758 Search PubMed.
- G. Anderegg, F. Arnauld-Neu, R. Delgado, J. Felcman and K. Popov, Pure Appl. Chem., 2005, 77, 1445–1495 CrossRef CAS.
- Z. Jusys, R. Pauliukaite and A. Vaskelis, Phys. Chem. Chem. Phys., 1999, 1, 313–318 RSC.
- H. A. Azab and A. Hassan, Bull. Soc. Chim. Fr., 1989, 1989, 599–602 Search PubMed.
- H. Hyvonen, M. Orama, H. Saarinen and R. Aksela, Green Chem., 2003, 5, 410–414 RSC.
- E. Rasanen, R. Pajarre, A. van Heiningen, R. Aksela, P. Stenius and P. Koukkari, Proceedings of Iberoamerican Congress on Pulp and Paper Research, Brazil, 2002 Search PubMed.
- M. Orama, H. Hyvonen, H. Saarinen and R. Aksela, J. Chem. Soc., Dalton Trans., 2002, 24, 4644–4648 RSC.
- S. Tandy, A. Ammann, R. Schulin and B. Nowack, Environ. Pollut., 2006, 142, 191–199 CrossRef CAS PubMed.
- A. E. Martell, R. M. Smith and R. J. Motekaitis, NIST Critically Selected Stability Constants of Metal Complexes V6.0, NIST, Gaithersburg, USA, 2001 Search PubMed.
- K. Pirkanniemi, Doctoral Dissertation, Department of Environmental Sciences, University of Kuopio, 2007 Search PubMed.
- C. Oviedo and J. Rodriguez, Quim. Nova, 2003, 26, 901–905 CrossRef CAS PubMed.
- J. C. Friedly, D. B. Kent and J. A. Davis, Environ. Sci. Technol., 2002, 36, 355–363 CrossRef CAS.
- M. Sillanpaa, Rev. Environ. Contam. Toxicol., 1997, 132, 85–111 Search PubMed.
- S. Schullerer and H. J. Brauch, Vom Wasser, 1989, 72, 21–29 CAS.
- A. C. Alder, H. Siegrist, W. Gujer and W. Giger, Water Res., 1990, 24, 733–742 CrossRef CAS.
- EAC, European Aminocarboxylates Committee, 2002, pp. C-014.
- ECB, European Chemical Bureau, European Union Risk Assessment Report, 2004, vol. 51, pp. 160 Search PubMed.
- WHO, World Health Organization, 1st addendum to ed. 3, 1, recommendations, Geneva, Switzerland, 2006, pp. 515 Search PubMed.
- M. Bucheli-Witschel and T. Egli, FEMS Microbiol. Rev., 2001, 25, 69–106 CrossRef CAS PubMed.
- B. Nörtemann, in ACS Symposium Series, ed. B. Nowack and J. M. VanBriesen, Washington DC, 2003, pp. 150–170 Search PubMed.
- Test No. 301: Ready Biodegradability, in OECD Guidelines for Testing of Chemicals, Section 3, OECD Publishing, 1992, DOI:10.1787/9789264070349-en.
- V. Sykora, P. Pitter, I. Bitternova and T. Lederer, Water Res., 2001, 35, 2010–2016 CrossRef CAS.
- S. S. Pinto, I. F. F. Neto and H. M. V. M. Soares, Environ. Sci. Pollut. Res., 2014, 21, 11893–11906 CrossRef PubMed.
- H. A. Painter, K. Miura, Y. Tosima, S. Nakamura, T. Sigeoka, A. Tanoue, M. Takatsuki and M. Kitano, OECD Guidelines for testing of chemicals, Paris, France, 1993, p. 1 Search PubMed.
- W. Guhl and J. Steber, Chemosphere, 2006, 63, 9–16 CrossRef CAS PubMed.
- P. Gerike and W. K. Fischer, Ecotoxicol. Environ. Saf., 1979, 3, 159–173 CrossRef CAS.
- EAC, European Aminocarboxylates Committee, 2003, pp. B-013.
- M. Borowiec, M. Huculak, K. Hoffmann and J. Hoffmann, Pol. J. Chem. Technol., 2009, 11, 1–3 CrossRef PubMed.
- A. B. Lanham, M. Carvalheira, A. M. Rodrigues, V. V. Cardoso, M. J. Benoliel, M. T. Barros, M. J. Morgado, H. M. V. M. Soares, P. C. Lemos and M. A. M. Reis, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2011, 46, 553–559 CrossRef CAS PubMed.
- M. Aoki and Y. Hara, US Patent US6465676, 2002.
- J. G. Martins, I. F. F. Neto, I. S. S. Pinto, E. V. Soares, M. T. Barros and H. M. V. M. Soares, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2014, 49, 1–11 CrossRef PubMed.
- M. Sillanpaa and K. Pirkanniemi, Environ. Technol., 2001, 22, 791–801 CrossRef CAS PubMed.
- S. Korhonen, S. Metsarinne and T. Tuhkanen, Ozone: Sci. Eng., 2000, 22, 279–286 CrossRef.
- J. Ramo, M. Sillanpaa, M. Orama, V. Vickackaite and L. Niinisto, J. Pulp Pap. Sci., 2000, 26, 125–131 CAS.
- B. Nortemann, Appl. Environ. Microbiol., 1992, 58, 671–676 CAS.
- R. A. Thomas, K. Lawlor, M. Bailey and L. E. Macaskie, Appl. Environ. Microbiol., 1998, 64, 1319–1322 CAS.
- J. J. Lauff, D. B. Steele, L. A. Coogan and J. M. Breitfeller, Appl. Environ. Microbiol., 1990, 56, 3346–3353 CAS.
- M. Witschel, S. Nagel and T. Egli, J. Bacteriol., 1997, 179, 6937–6943 CAS.
- T. Kluner, D. C. Hempel and B. Nortemann, Appl. Microbiol. Biotechnol., 1998, 49, 194–201 CrossRef CAS.
- T. Uetz, R. Schneider, M. Snozzi and T. Egli, J. Bacteriol., 1992, 174, 1179–1188 CAS.
- J. Bohuslavek, J. W. Payne, Y. Liu, H. J. Bolton and L. Xun, Appl. Environ. Microbiol., 2001, 67, 688–695 CrossRef CAS PubMed.
- J. W. Payne, H. J. Bolton, J. A. Campbell and L. Xun, J. Bacteriol., 1998, 180, 3823–3827 CAS.
- T. Uetz and T. Egli, Biodegradation, 1993, 3, 423–434 CrossRef CAS.
- Y. Liu, T. M. Louie, J. Payne, J. Bohuslavek, H. Bolton Jr and L. Xun, Appl. Environ. Microbiol., 2001, 67, 696–701 CrossRef CAS PubMed.
- H. Bolton, D. C. Girvin, A. E. Playmale, S. D. Harvey and D. J. Workman, Environ. Sci. Technol., 1996, 30, 931–938 CrossRef CAS.
- A. J. Francis, C. J. Dodge and J. B. Gilow, Nature, 1992, 356, 140–142 CrossRef CAS.
- B. Nörtemann, Appl. Microbiol. Biotechnol., 1999, 51, 751–759 CrossRef.
- Comprehensive market search report, 2012, http://www.strategyr.com/Chelating_Agents_Market_Report.asp, accessed January 2014 Search PubMed.
- S. H. Hutner, L. Provasoli, A. Schatz and C. P. Haskins, Proc. Am. Philos. Soc., 1950, 94, 152–170 CAS.
- L. Jacobson, Plant Physiol., 1951, 26, 411–413 CrossRef CAS PubMed.
- S. M. Altmann and E. M. Crook, Nature, 1953, 171, 76–77 CrossRef CAS.
- SCOPUS, http://www.scopus.com, accessed March 2014; SCOPUS, http://www.scopus.com, accessed 2014.
- R. P. Silvy, Appl. Catal., A, 2004, 261, 247–252 CrossRef CAS PubMed.
- M. Marafi and A. Stanislaus, Resour., Conserv. Recycl., 2008, 52, 859–873 CrossRef PubMed.
- R. Crnojevich, E. I. Wiewiorowski, L. R. Tintin and A. B. Case, Applied Technology, 1990, 42, 42–45 CAS.
- W. Shen, T. Li and J. Chen, Procedia Environ. Sci., 2012, 16, 253–256 CrossRef CAS PubMed.
- P. P. Sun and M. S. Lee, Hydrometallurgy, 2011, 110, 91–98 CrossRef CAS PubMed.
- M. K. Sinha, S. K. Sahu, P. Meshram and B. D. Pandey, Hydrometallurgy, 2014, 147–148, 103–111 CrossRef CAS PubMed.
- D. Mishra, J. G. Ahn, D. J. Kim, G. R. Chaudhury and D. E. Ralph, J. Hazard. Mater., 2009, 167, 1231–1236 CrossRef CAS PubMed.
- I. Asghari, S. M. Mousavi, F. Amiri and S. Tavassoli, J. Ind. Eng. Chem., 2013, 19, 1069–1081 CrossRef CAS PubMed.
- I. Gaballah, E. Allain and M. Djona, Light Met., 1994, 29, 1153–1161 Search PubMed.
- M. A. Rabah, I. F. Hewaidy and F. E. Farghaly, Powder Metall., 1997, 40, 283–288 CrossRef CAS PubMed.
- J. M. Juneja, S. Singh and D. K. Bose, Hydrometallurgy, 1996, 41, 201–209 CrossRef CAS.
- I. Gaballah and M. Djona, Metall. Mater. Trans. B, 1995, 26, 41–50 CrossRef.
- E. A. Abdel-Aal and M. M. Rashad, Hydrometallurgy, 2004, 74, 189–194 CrossRef CAS PubMed.
- J. Idris, M. Musa, C.-Y. Yin and K. H. K. Hamid, J. Ind. Eng. Chem., 2010, 16, 251–255 CrossRef CAS PubMed.
- F. Belochini, V. Fontia, F. Ferella and F. Vegliò, J. Hazard. Mater., 2010, 178, 529–534 CrossRef PubMed.
- D. J. Kim, D. Pradhan, J. G. Ahn and S. W. Lee, Hydrometallurgy, 2010, 103, 136–143 CrossRef CAS PubMed.
- A. P. Davis, Rev. Int. Contam. Ambiental, 2000, 16, 169–174 CAS.
- A. P. Davis and I. Singh, J. Environ. Eng., 1995, 121, 174–185 CrossRef CAS.
- W. Z. Kociałkowski, J. B. Diatta and W. Grzebisz, Pol. J. Environ. Stud., 1999, 8, 149–154 Search PubMed.
- T.-T. Lim, P.-C. Chui and K.-H. Goh, Chemosphere, 2005, 58, 1031–1040 CrossRef CAS PubMed.
- L. D. Palma, O. Gonzini and R. Mecozzi, Chemoecology, 2011, 27, 97–106 Search PubMed.
- L. Hauser, S. Tandy, R. Schulin and B. Nowack, Environ. Sci. Technol., 2005, 39, 6819–6824 CrossRef CAS.
- W. D. Ellis, T. C. Fogg, and A. N. Tafuri, Proceedings of the Twelfth Annual Research Symposium, USEPA Hazardous Waste Engineering Research Laboratory, Cincinnati, OH, 1986 Search PubMed.
- J. L. Howard and J. Shu, Environ. Pollut., 1996, 91, 89–96 CrossRef CAS.
- K. R. Sistani, D. A. Mays, R. W. Taylor and C. Buford, Commun. Soil Sci. Plant Anal., 1995, 26, 2167–2180 CrossRef CAS.
- T. C. M. Yip, D. C. W. Tsang, K. T. W. Ng and I. M. C. Lo, Environ. Sci. Technol., 2009, 43, 831–836 CrossRef CAS.
- T. C. M. Yip, D. C. W. Tsang, K. T. W. Ng and I. M. C. Lo, Chemosphere, 2009, 74, 301–307 CrossRef PubMed.
- D. Y. S. Yan, T. C. M. Yip, M. M. T. Yui, D. C. W. Tsang and I. M. C. Lo, J. Hazard. Mater., 2010, 178, 890–894 CrossRef CAS PubMed.
- Z. Li and L. M. Shuman, Soil Sci., 1996, 161, 226–241 CrossRef CAS PubMed.
- J. W. Huang and S. D. Cunningham, New Phytol., 1996, 134, 75–84 CrossRef CAS PubMed.
- M. J. Blaylock, D. E. Salt, S. Dushenkov, O. Zakharova, C. Gussman, Y. Kapulnik, B. D. Ensley and I. Raskin, Environ. Sci. Technol., 1997, 31, 860–865 CrossRef.
- A. D. Vassil, Y. Kapulnik, I. Raskin and D. E. Salt, Plant Physiol., 1998, 117, 447–453 CrossRef CAS PubMed.
- C. Luo, Z. Shen and X. Li, Chemosphere, 2005, 59, 1–11 CrossRef CAS PubMed.
- E. Meers, A. Ruttens, M. J. Hopgood, D. Samson and F. M. G. Tack, Chemosphere, 2005, 58, 1011–1022 CrossRef CAS PubMed.
- M. W. H. Evangelou, U. Bauer, M. Ebel and A. Schaeffer, Chemosphere, 2007, 68, 345–353 CrossRef CAS PubMed.
- Y.-b. Sun, Q.-x. Zhou, J. An, W.-t. Liu and R. Liu, Geoderma, 2009, 150, 106–112 CrossRef CAS PubMed.
- R. J. Mohd Salim, M. I. Adenan, A. Amid, M. H. Jauri and A. S. Sued, Biotechnol. Res. Int., 2013, 2013, 1–5 CrossRef PubMed.
- S. S. Pinto, O. S. Ascenso, M. T. Barros and H. M. V. M. Soares, Int. J. Environ. Sci. Technol., 2014 DOI:10.1007/s13762-013-0480-0.
- M. Buhl, Inorg. Chem., 2005, 44, 6277–6283 CrossRef PubMed.
- T. Marino, M. Toscano, N. Russo and A. Grand, J. Phys. Chem. B, 2006, 110, 24666–24673 CrossRef CAS PubMed.
- I. Georgieva and N. J. Trendafilova, J. Phys. Chem. A, 2007, 111, 13075–13087 CrossRef CAS PubMed.
- Y. Huang, A. Zhong, C. Rong, X. Xiao and S. Liu, J. Phys. Chem. A, 2008, 112, 305–311 CrossRef CAS PubMed.
- A. J. Silanpaa, R. Aksela and K. Laasonen, Phys. Chem. Chem. Phys., 2003, 5, 3382–3393 RSC.
- N. Paola, S. Marek and A. Reinhart, Phys. Chem. Chem. Phys., 2003, 5, 3372–3381 RSC.
- O. Coskuner and E. A. A. Jarvis, J. Phys. Chem. A, 2008, 112, 2628–2633 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |