
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConnected nanoparticle catalysts possessing a porous, hollow capsule structure as carbon-free electrocatalysts for oxygen reduction in polymer electrolyte fuel cells†
Takanori
Tamaki
ab,
Hidenori
Kuroki
ab,
Shun
Ogura
a,
Teruaki
Fuchigami
c,
Yoshitaka
Kitamoto
bc and
Takeo
Yamaguchi
*ab
aChemical Resources Laboratory, Tokyo Institute of Technology, R1-17, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: yamag@res.titech.ac.jp
bKanagawa Academy of Science and Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
cDepartment of Innovative and Engineered Materials, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yoko-hama 226-8502, Japan
First published on 13th October 2015
Abstract
We employ connected nanoparticle catalysts with a porous, hollow capsule structure as carbon-free electrocatalysts for the cathode in polymer electrolyte fuel cells (PEFCs) or proton exchange membrane fuel cells (PEMFCs). The catalysts consist of fused ordered alloy platinum–iron (Pt–Fe) nanoparticles. This unique beaded network structure enables surprisingly high activity for the oxygen reduction reaction, 9 times that of the state-of-the-art commercial catalyst. Because the connected nanoparticle catalysts are formed without sacrificing the high surface area of the nanoparticles and can conduct electrons, the catalysts show good performance in an actual PEMFC without a carbon support. Moreover, the elimination of carbon intrinsically solves the problem of carbon corrosion. Thus, the connected nanoparticle catalysts with a unique structure are a significant advancement over conventional electrode catalysts and will lead to an ultimate solution for PEMFC cathodes.
Broader contextConnected nanoparticle catalysts with much higher activity than that of conventional nanoparticle catalysts will open up a new research area in catalysts for energy and environmental science. We employed the connected nanoparticle catalysts for an oxygen reduction reaction in PEMFCs, which are one of the key devices for next-generation hydrogen energy systems because of their high energy conversion efficiency, even at low temperatures. Fuel cell vehicles based on PEMFCs have been widely developed by many car manufacturers because of their high cruising range and low refueling time.1–3 However, the amount of Pt and the durability of the cathode catalyst are still important problems to be solved in PEMFCs; the amount of Pt used in fuel cell vehicles is approximately ten times higher than that of Pt group metals for exhaust gas catalysts used in the conventional cars. The connected nanoparticle catalysts exhibit sufficiently high activity that minimises the amount of Pt to the level of that of conventional cars. Another important issue, carbon corrosion during the start–stop of PEMFCs, is addressed by the elimination of carbon; the new catalyst is stable under the conditions of the start–stop durability test, which are known to drastically degrade conventional catalysts. Thus, the connected nanoparticle catalysts achieve both high activity and durability. |
To make fuel cell vehicles more affordable and popular, improvement of durability and reduction of costs are necessary,1–5 especially by improving the activity, durability and mass transport of the cathode.4,5 Because of its low activity, a high platinum loading (∼50 g Pt per vehicle5) is required in conventional fuel cell vehicles. Conversely, conventional cars also use Pt group metals (PGMs) to purify exhaust gases. The average quantity of PGMs used in a conventional car is estimated to be ∼4 g.6,7 Thus, a reduction in the quantity of Pt used in PEMFCs to one tenth or a ten-fold increase in the activity of Pt could address the difficulties associated with Pt use in PEMFCs. Degradation of Pt-based catalysts and carbon supports in PEMFCs is another issue. Pt-based nanoparticles themselves degrade during PEMFC operation. Recently reported nanostructured catalysts supported on a high surface area carbon succeeded in achieving high activity and high stability as the catalysts themselves.8–10 The other degradation mode of the cathode in PEMFCs is carbon corrosion that mainly occurs during start–stop operation.2,11–13 Carbon corrosion occurs when hydrogen and oxygen co-exist in the anode, which makes the cathode potential as high as 1.5 V due to a so-called carbon corrosion.11,12 Present fuel cell vehicles implement specific procedures on the system level to eliminate the situation where a high voltage is applied to the cathode.2 If the problem of carbon corrosion is solved on the material level, the control system can be simplified and the robustness of the system can be increased. A possible way to intrinsically solve the problem of carbon corrosion is to employ an unsupported catalyst. Carbon-free nanostructured catalysts have been recently attracting attention as promising cathode catalysts.14–19 In addition to the tolerance to the conditions that accelerate carbon corrosion, carbon-free catalysts enable the formation of thinner electrodes than a conventional PEMFC electrode, a large portion of which is occupied by the carbon support, and a thinner electrode is advantageous for mass transport of oxygen.18,19
Here, we employ connected Pt–Fe nanoparticle catalysts with a porous, hollow capsule structure as a cathode catalyst in PEMFCs for the first time. Because the fused Pt–Fe network structure is formed without sacrificing the high surface area of the nanoparticles and is expected to conduct electrons through the capsules without any support, the catalysts can act as carbon-free electrocatalysts. Considering that the bulk or extended surfaces exhibit much higher activities than conventional nanosized catalysts,15,20–23 the connected catalysts have the potential to exhibit high activity. In addition, the connected Pt–Fe catalysts intrinsically address the carbon corrosion problem and enable the formation of a thin electrode. Thus, the connected Pt–Fe catalysts could solve all problems regarding the PEMFC cathode, namely, activity, durability and mass transport, as schematically shown in the ESI,† Fig. S1.
The connected Pt–Fe catalysts were synthesized via supercritical treatment of Pt–Fe nanoparticles formed on surface-modified silica particles, followed by dissolution of the silica particles in sodium hydroxide.24,25 The details of the experimental procedures are described in the Experimental section in the ESI.†Fig. 1(a) shows transmission electron microscopy (TEM) images of the connected Pt–Fe catalysts. Thermally fused Pt–Fe nanoparticles formed a Pt–Fe network structure with a shell thickness and pore size of ∼10 nm. The X-ray diffraction (XRD) pattern of the catalyst (Fig. S2, ESI†) revealed the formation of a chemically ordered L10 structure and the absence of other impurities such as iron oxides. Catalysts with L10 or L12 structures have recently been reported to exhibit both higher activity and durability compared with disordered alloys.26–32 The crystallite size of the connected Pt–Fe catalysts calculated using the Scherrer equation was approximately 6–7 nm. The Pt/Fe molar ratio of the connected Pt–Fe catalysts as determined using inductively coupled plasma-atomic emission spectroscopy (ICP–AES) was 0.94 ± 0.04, which is close to the feed ratio. Furthermore, the Fe and Pt atoms were evenly distributed in the as-synthesized samples (Fig. 1(b)) using energy dispersive X-ray (EDX) line-scan analysis. After electrochemical pre-treatment (Fig. 1(c)), the Fe atoms near the surface dissolved and the Pt content near the surface was enriched.
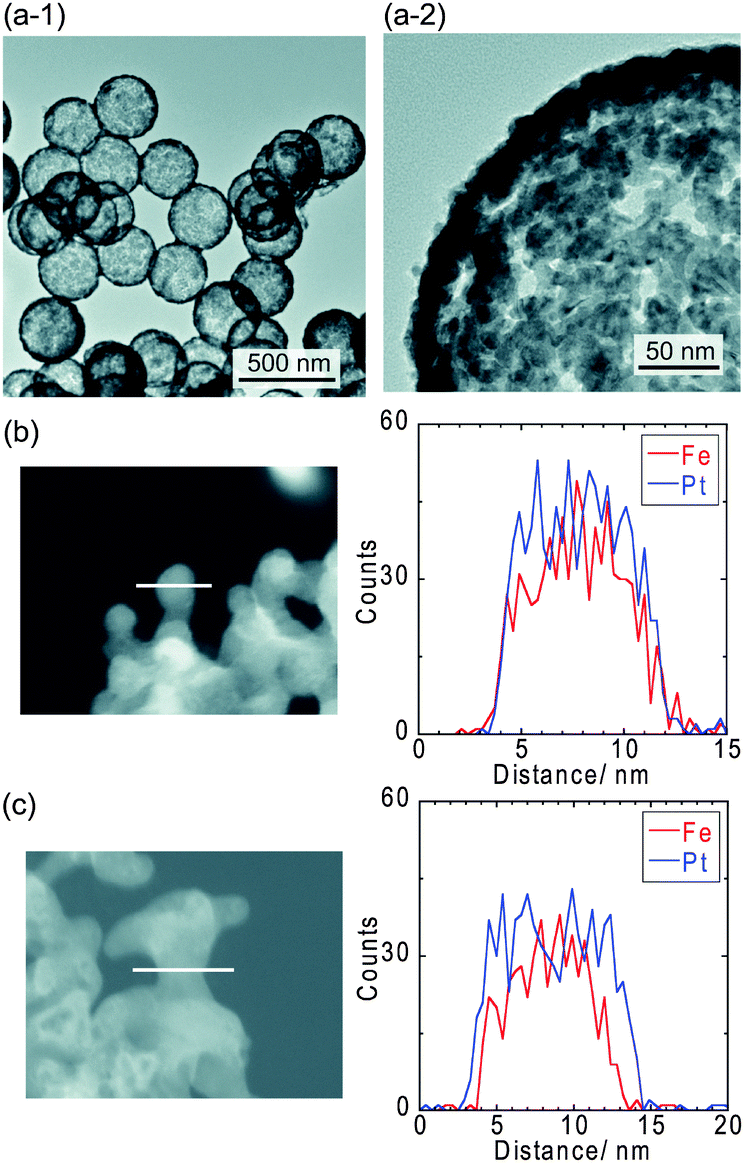 | ||
| Fig. 1 (a) TEM images of the connected Pt–Fe catalysts with a porous hollow capsule structure. EDX line-scans of the connected Pt–Fe catalysts (b) before and (c) after electrochemical pre-treatment. | ||
The oxygen reduction reaction activity of the connected Pt–Fe catalysts was evaluated in an acidic solution. Fig. 2 shows cyclic voltammograms (CVs) (a) and polarization curves (b) of the connected Pt–Fe catalysts after electrochemical pre-treatment and a state-of-the-art commercial Pt/C catalyst supplied by Tanaka Kikinzoku Kogyo (TEC10E50E with 45.8 wt% Pt). The electrochemical surface areas (ECSAs) and mass activities of the catalysts at 0.9 V were calculated from the curves measured in 0.1 M HClO4 (Fig. 2(c)). Then, surface-area-specific activities of the catalysts (hereafter simply denoted as specific activities), which reflect their intrinsic catalytic performance, were calculated by dividing the mass activity by the ECSAs (Fig. 2(d)). For comparison, the specific activity of Pt–Fe nanoparticles with an ordered L10 structure supported on carbon black (Pt–Fe/C) is also shown. The specific activity of the commercial catalyst was 0.27 ± 0.04 mA cmPt−2, which was within the range of reported values for commercial Pt/C catalysts.15,20 The connected Pt–Fe catalysts surprisingly exhibited a much higher specific activity of 2.3 ± 0.2 mA cmPt−2. The total improvement factor or the ratio of the specific activity of the connected Pt–Fe catalysts to that of the commercial Pt/C was ∼9. The specific activity of the connected Pt–Fe catalysts is even higher than that of Pt–Fe/C, implying that the alloying with Fe cannot solely explain the reason for the improvement, although the alloying does increase the activity due to changes in the Pt–Pt distance and electronic structure. Another possible reason for the improvement is the change in the structure and surface composition of the catalyst by the formation of the beaded network structure. The connected structure may resemble, to some extent, that of the bulk or extended surface.
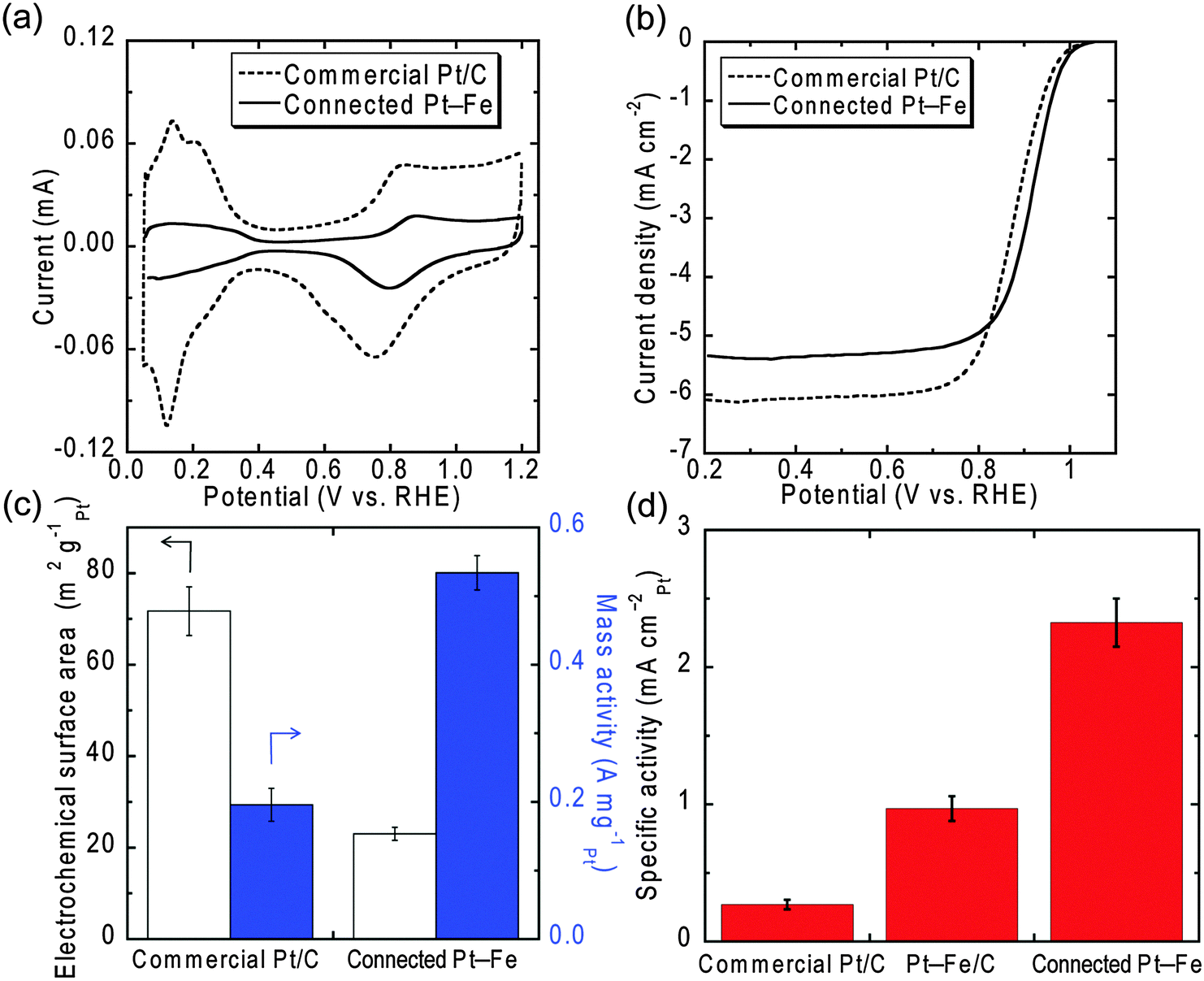 | ||
| Fig. 2 (a) CVs in N2-saturated 0.1 M HClO4 and (b) polarization curves on a rotating disk electrode in O2-saturated 0.1 M HClO4 of the connected Pt–Fe catalysts and a commercial Pt/C. (c) Electrochemical surface areas (ECSAs) for Pt and mass activities and (d) specific activities of the catalysts measured in 0.1 M HClO4. The specific activity of Pt–Fe/C is also shown in (d). The specific activities were calculated by dividing the mass activities by the corresponding ECSA. | ||
MEA was then fabricated using the connected Pt–Fe catalysts as the cathode catalyst. Cross-sectional SEM images of the MEA are shown in Fig. 3(a) and (b). When the Pt loading in the cathode was 0.3 mg cm−2, which is the standard quantity used in conventional electrodes, the thickness of the electrode was 1–1.5 μm, as shown in the left-side electrode in Fig. 3(a) and (b). This value was nearly one-fifth that of a conventional electrode (5–10 μm), a typical example of which is the right-side electrode in Fig. 3(b): the anode composed of commercial Pt/C with a Pt loading of 0.2 mg cm−2, resulting in a thickness of 5–6 μm. This decrease in thickness can be ascribed to the exclusion of the carbon support. Next, the cell performance was evaluated. A current–voltage curve was successfully obtained (Fig. 3(c)), which suggests that the connected Pt–Fe catalysts can conduct electrons through the cathode catalyst layer without a carbon support. The connected Pt–Fe catalysts formed a beaded network structure without sacrificing their high surface area, and thus showed good performance even in the MEA. The ECSA in the MEA calculated from the CV data (Fig. S3 and S4, ESI†) was 14 m2 g−1. The catalyst utilization ratio calculated by dividing the ECSA in the MEA by that measured in acidic solution (Fig. 2(c)) was around 60%. Considering that the catalyst utilization ratios of conventional catalysts were 80–90%,5 triple phase boundaries were well formed in the MEA containing the connected Pt–Fe catalysts. However, due to a slightly larger crystallite size of 6–7 nm, the ECSA of the connected Pt–Fe catalysts was lower than that of the commercial Pt/C, and thus, the net surface area of Pt in the MEA was lower when the Pt loading was set to be the same. The lower surface area of the connected Pt–Fe catalysts can be improved by the decrease in the crystallite size. Turning now to the open circuit voltage (OCV) of the MEA, the value was around 0.91 V, which is slightly lower than that of a conventional MEA, around 1.0 V. However, some literature on Pt-based alloys also reported lower OCV, around 0.95 V.33,34 The hydrogen crossover current density measured under OCV was 1.5 mA cm−2, which is similar to the reported values of conventional standard MEAs,35 and thus not the reason for the low OCV. Then, the durability of the connected Pt–Fe catalysts in the MEA, particularly the effect of eliminating the carbon support, was evaluated using a start–stop durability testing protocol recommended by the Fuel Cell Commercialization Conference of Japan (FCCJ), which mainly accelerates carbon corrosion.12,13 The cell performance of the MEA containing the connected Pt–Fe catalysts did not change before and after the durability test, as shown in Fig. 3(c). The changes in the ECSAs are shown in Fig. 3(d). The ECSA of the conventional commercial Pt/C decreased to less than 50% after 4000 cycles.35 The rapid decrease in the ECSA of conventional Pt/C catalysts was reported to be due to the carbon corrosion.12,13 In contrast, the ECSA of the connected Pt–Fe catalysts remained unchanged, even after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. The retention of the ECSA suggests that the structure of the connected Pt–Fe catalysts was also retained during the durability cycles, which is supported by the TEM images of the catalysts obtained after 10000 cycles (Fig. S5, ESI†). Considering that the ten-year-life of fuel cell vehicles corresponds to 60000 cycles of the durability test,12 the retention of the ECSA of the connected Pt–Fe catalysts after 10000 cycles is promising and thus the catalysts are worth further investigation. EDX-line scans of the connected Pt–Fe catalysts after the start–stop durability test (Fig. 3(e)) revealed that the dissolution of Fe occurred to some extent. Another durability testing protocol, a load-cycle durability test performed in 0.1 M HClO4 at 60 °C (Fig. S6, ESI†), also revealed the decrease in the performance of the catalysts. These results suggest that the dissolution of Fe should be addressed in the connected Pt–Fe catalysts. Previous studies about nanoparticle catalysts supported on carbon show that the increase in the degree of ordered structure29 and the synergistic effects of Cu and the ordered structure32 can suppress the dissolution of Fe and improve the durability of nanoparticle catalysts. These improvements can be applied to the connected nanoparticle catalysts, which will be a topic of future studies. The connected nanoparticle catalysts with high surface areas showed much higher activity and durability than conventional nanoparticle catalysts supported on carbon, and thus, a new research area in catalysts will unfold.
000 cycles. The retention of the ECSA suggests that the structure of the connected Pt–Fe catalysts was also retained during the durability cycles, which is supported by the TEM images of the catalysts obtained after 10000 cycles (Fig. S5, ESI†). Considering that the ten-year-life of fuel cell vehicles corresponds to 60000 cycles of the durability test,12 the retention of the ECSA of the connected Pt–Fe catalysts after 10000 cycles is promising and thus the catalysts are worth further investigation. EDX-line scans of the connected Pt–Fe catalysts after the start–stop durability test (Fig. 3(e)) revealed that the dissolution of Fe occurred to some extent. Another durability testing protocol, a load-cycle durability test performed in 0.1 M HClO4 at 60 °C (Fig. S6, ESI†), also revealed the decrease in the performance of the catalysts. These results suggest that the dissolution of Fe should be addressed in the connected Pt–Fe catalysts. Previous studies about nanoparticle catalysts supported on carbon show that the increase in the degree of ordered structure29 and the synergistic effects of Cu and the ordered structure32 can suppress the dissolution of Fe and improve the durability of nanoparticle catalysts. These improvements can be applied to the connected nanoparticle catalysts, which will be a topic of future studies. The connected nanoparticle catalysts with high surface areas showed much higher activity and durability than conventional nanoparticle catalysts supported on carbon, and thus, a new research area in catalysts will unfold.
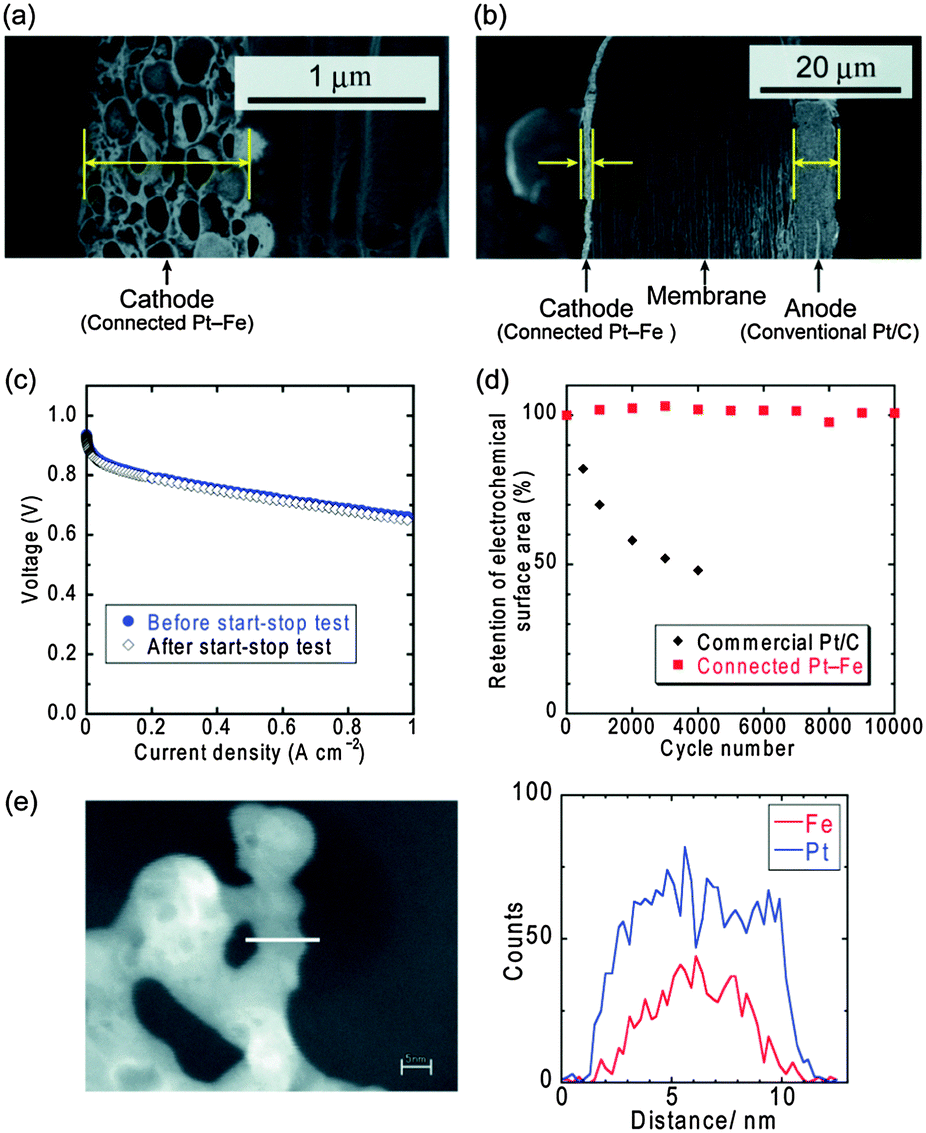 | ||
| Fig. 3 (a) Close-up and (b) entire cross-sectional SEM images of an MEA with a cathode composed of the connected Pt–Fe catalysts. These images were obtained after the start–stop durability test. (c) Current–voltage curves for the MEA at 80 °C before and after the start–stop durability test. The relative humidities of the anode (H2) and the cathode (O2) were 90% and 70%, respectively. (d) Change in the ECSAs of the connected Pt–Fe catalysts and commercial Pt/C as a function of the number of cycles using the MEA at 80 °C. The commercial Pt/C data are cited from a previous report.35 (e) EDX line-scans of the connected Pt–Fe catalysts after 10000 cycles of the start–stop durability test. | ||
Conclusions
Unique connected Pt–Fe nanoparticle catalysts with a beaded network structure were shown to be promising as a cathode catalyst in PEMFCs. The connected Pt–Fe catalysts showed surprisingly high surface-area-specific activity in solution, 9 times that of the state-of-the-art commercial catalyst, because of the formation of a network structure and good performance in an actual PEMFC, namely in an MEA. The carbon-free connected catalysts enabled the formation of a thin cathode with a thickness of 1–1.5 μm, nearly one-fifth that of a conventional electrode. The elimination of the carbon support in the catalyst layer resulted in high durability during start–stop durability tests, which are known to drastically degrade conventional catalysts. Further investigations should be performed to elucidate the reasons for the high activity. Considering that the network structure of the fused metal catalysts can be formed on templates with various shapes, we can now freely design the optimal cathode structure for the mass transport of oxygen and protons and for water management.Acknowledgements
This work was supported by the Kanagawa Academy of Science and Technology. The authors thank Dr Hiroshi Tabata and Ms Kaori Toyama for the cross-sectional SEM images, Material Analysis Suzukake-dai Center, Technical Department, Tokyo Institute of Technology, for the ICP-AES measurements, the Foundation for Promotion of Materials Science and Technology (MST) of Japan for the STEM-EDX line-scan measurements, and the Fuel Cell Cutting-Edge Center Technology Research Association (FC-Cubic) for the evaluation method of the ECSA in the MEA (Fig. S4(a), ESI†).References
- F. T. Wagner, B. Lakshmanan and M. F. Mathias, J. Phys. Chem. Lett., 2010, 1, 2204–2219 CrossRef CAS.
- U. Eberle, B. Mueller and R. von Helmolt, Energy Environ. Sci., 2012, 5, 8780–8798 Search PubMed.
- S. Sekine, K. Kojima, SAE Tech. Paper Series, 2011, 2011-2028-0061.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- H. A. Gasteiger, D. R. Baker, R. N. Carter, W. Gu, Y. Liu, F. T. Wagner and P. T. Yu, in Hydrogen and Fuel Cells: Fundamentals, Technologies and Applications, ed. D. Stolten, Wiley-VCH, Weinheim, 2010, ch. 1, pp. 3–16 Search PubMed.
- Platinum 2013, Johnson Matthey PLC, Royston, Hertfordshire, UK, 2013 Search PubMed.
- Sales of New Vehicles, International Organization of Motor Vehicle Manufacturers (OICA), http://www.oica.net/wp-content/uploads//total-sales-2014.pdf, accessed September 2015.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- C. Baldizzone, S. Mezzavilla, H. W. P. Carvalho, J. C. Meier, A. K. Schuppert, M. Heggen, C. Galeano, J.-D. Grunwaldt, F. Schueth and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 14250–14254 CrossRef CAS PubMed.
- B. Han, C. E. Carlton, A. Kongkanand, R. S. Kukreja, B. R. Theobald, L. Gan, R. O’Malley, P. Strasser, F. T. Wagner and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 258–266 CAS.
- C. A. Reiser, L. Bregoli, T. W. Patterson, J. S. Yi, J. D. L. Yang, M. L. Perry and T. D. Jarvi, Electrochem. Solid-State Lett., 2005, 8, A273–A276 CrossRef CAS PubMed.
- A. Ohma, K. Shinohara, A. Iiyama, T. Yoshida and A. Daimaru, ECS Trans., 2011, 41, 775–784 CAS.
- Y. Hashimasa, T. Shimizu, Y. Matsuda, D. Imamura and M. Akai, ECS Trans., 2012, 50, 723–732 CrossRef PubMed.
- Z. W. Chen, M. Waje, W. Z. Li and Y. S. Yan, Angew. Chem., Int. Ed., 2007, 46, 4060–4063 CrossRef CAS PubMed.
- D. F. van der Vliet, C. Wang, D. Tripkovic, D. Strmcnik, X. F. Zhang, M. K. Debe, R. T. Atanasoski, N. M. Markovic and V. R. Stamenkovic, Nat. Mater., 2012, 11, 1051–1058 CAS.
- J. Kibsgaard, Y. Gorlin, Z. B. Chen and T. F. Jaramillo, J. Am. Chem. Soc., 2012, 134, 7758–7765 CrossRef CAS PubMed.
- W. Liu, P. Rodriguez, L. Borchardt, A. Foelske, J. P. Yuan, A. K. Herrmann, D. Geiger, Z. K. Zheng, S. Kaskel, N. Gaponik, R. Kotz, T. J. Schmidt and A. Eychmuller, Angew. Chem., Int. Ed., 2013, 52, 9849–9852 CrossRef CAS PubMed.
- O. H. Kim, Y. H. Cho, S. H. Kang, H. Y. Park, M. Kim, J. W. Lim, D. Y. Chung, M. J. Lee, H. Choe and Y. E. Sung, Nat. Commun., 2013, 4 DOI:10.1038/ncomms3473.
- E. Antolini and J. Perez, J. Mater. Sci., 2011, 46, 4435–4457 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS PubMed.
- M. Nesselberger, M. Roefzaad, R. F. Hamou, P. U. Biedermann, F. F. Schweinberger, S. Kunz, K. Schloegl, G. K. H. Wiberg, S. Ashton, U. Heiz, K. J. J. Mayrhofer and M. Arenz, Nat. Mater., 2013, 12, 919–924 CrossRef CAS PubMed.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. J. Mayrhofer and M. Arenz, J. Am. Chem. Soc., 2011, 133, 17428–17433 CrossRef CAS PubMed.
- F. J. Perez-Alonso, D. N. McCarthy, A. Nierhoff, P. Hernandez-Fernandez, C. Strebel, I. E. L. Stephens, J. H. Nielsen and I. Chorkendorff, Angew. Chem., Int. Ed., 2012, 51, 4641–4643 CrossRef CAS PubMed.
- T. Fuchigami, R. Kawamura, Y. Kitamoto, M. Nakagawa and Y. Namiki, Langmuir, 2011, 27, 2923–2928 CrossRef CAS PubMed.
- T. Fuchigami, R. Kawamura, Y. Kitamoto, M. Nakagawa and Y. Namiki, Biomaterials, 2012, 33, 1682–1687 CrossRef CAS PubMed.
- J. Kim, Y. Lee and S. H. Sun, J. Am. Chem. Soc., 2010, 132, 4996–4997 CrossRef CAS PubMed.
- X. Li, L. An, X. Y. Wang, F. Li, R. Q. Zou and D. G. Xia, J. Mater. Chem., 2012, 22, 6047–6052 RSC.
- D. L. Wang, H. L. L. Xin, R. Hovden, H. S. Wang, Y. C. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- Q. Li, L. Wu, G. Wu, D. Su, H. Lv, S. Zhang, W. Zhu, A. Casimir, H. Zhu, A. Mendoza-Garcia and S. Sun, Nano Lett., 2015, 15, 2468–2473 CrossRef CAS PubMed.
- B. Arumugam, B. A. Kakade, T. Tamaki, M. Arao, H. Imai and T. Yamaguchi, RSC Adv., 2014, 4, 27510–27517 RSC.
- T. Tamaki, A. Minagawa, B. Arumugam, B. A. Kakade and T. Yamaguchi, J. Power Sources, 2014, 271, 346–353 CrossRef CAS PubMed.
- B. Arumugam, T. Tamaki and T. Yamaguchi, ACS Appl. Mater. Interfaces, 2015, 7, 16311–16321 CAS.
- R. Srivastava, P. Mani, N. Hahn and P. Strasser, Angew. Chem., Int. Ed., 2007, 46, 8988–8991 CrossRef PubMed.
- P. Mani, R. Srivastava and P. Strasser, J. Power Sources, 2011, 196, 666–673 CrossRef CAS PubMed.
- Cell Evaluation and Analysis Protocol Guideline (Japanese), New Energy and Industrial Technology Development Organization, 2012, p. 17 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental section, illustration of the present study (Fig. S1), XRD pattern of the connected Pt–Fe catalysts (Fig. S2), CVs of the connected Pt–Fe catalysts in the MEA before and after the start–stop durability test (Fig. S3), methods to calculate the ECSA in the MEA (Fig. S4) TEM images of the connected Pt–Fe catalysts after the start–stop durability test (Fig. S5), and CVs of the catalysts during the load-cycle durability test in acidic solution (Fig. S6). See DOI: 10.1039/c5ee02450a |
| This journal is © The Royal Society of Chemistry 2015 |