
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-performance silicon-based multicomponent battery anodes produced via synergistic coupling of multifunctional coating layers†
Jung-In
Lee
a,
Younghoon
Ko
a,
Myoungsoo
Shin
a,
Hyun-Kon
Song
a,
Nam-Soon
Choi
a,
Min Gyu
Kim
*b and
Soojin
Park
*a
aDepartment of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), UNIST-gil 50, Ulsan 689-798, Republic of Korea. E-mail: mgkim@postech.ac.kr; Fax: +82-52-217-2909; Tel: +82-52-217-2515
bBeamline Research Division, Pohang Accelerator Laboratory, POSTECH, Pohang 790-784, South Korea. E-mail: spark@unist.ac.kr
First published on 28th May 2015
Abstract
Nanostructured Si-based materials are key building blocks for next-generation energy storage devices. To meet the requirements of practical energy storage devices, Si-based materials should exhibit high-power, low volume change, and high tap density. So far, there have been no reliable materials reported satisfying all of these requirements. Here, we report a novel Si-based multicomponent design, in which the Si core is covered with multifunctional shell layers. The synergistic coupling of Si with the multifunctional shell provides vital clues for satisfying all Si anode requirements for practical batteries. The Si-based multicomponent anode delivers a high capacity of ∼1000 mA h g−1, a highly stable cycling retention (∼65% after 1000 cycles at 1 C), an excellent rate capability (∼800 mA h g−1 at 10 C), and a remarkably suppressed volume expansion (12% after 100 cycles). Our synthetic process is simple, low-cost, and safe, facilitating new methods for developing electrode materials for practical energy storage.
Broader contextLithium-ion batteries (LIBs) are now emerging as some of the powerful energy storage devices for large-size electric devices including electric vehicles and energy storage systems. In order to meet high power and gravimetric energy density which are required by large-size electric devices, Si has attracted much attention because of the highest theoretical specific capacity (>3580 mA h g−1), environmentally friendliness, and a low cost. However, Si-based anodes show severe capacity fading during cycling due to the huge volume change (>300%) during the lithiation/delithiation process. Nanostructuring (e.g., nanoparticles, nanowires, nanotubes, etc.) of Si electrodes can overcome pulverization. However, nanomaterials are difficult to be used for practical applications in industry due to some drawbacks, such as a high synthetic cost, a low volumetric energy density, a serious side reaction, and safety issues. Here, we report a novel Si-based multicomponent design, in which the Si core is covered with multifunctional shell layers. The synergistic coupling of Si with the multifunctional shell provides vital clues for satisfying all Si anode requirements for practical batteries. The Si-based multicomponent anode delivers a high capacity of ∼1000 mA h g−1 and a highly stable cycling retention (∼65% after 1000 cycles at 1 C), an excellent rate capability (∼800 mA h g−1 at 10 C). Our synthetic process is simple, low-cost, and safe, facilitating new methods for developing electrode materials for practical energy storage. |
Introduction
Rechargeable batteries are the most viable method for powering portable consumer electronics, electric vehicles, and large-scale stationary energy storage.1–3 Compared to traditional lead-acid batteries and nickel-based batteries, lithium-ion batteries (LIBs) are the most popular rechargeable batteries due to their relatively high energy density, long cycling life, lightweight design, and good environmental compatibility.4,5 However, conventional LIB technology is reaching the limit of its gravimetric/volumetric energy density and power capabilities, reducing its capacity to power newly emerging electronic devices and or act as large-scale batteries.6Among the promising candidates for anode materials, silicon (Si) has several advantages including a high theoretical capacity (3579 mA h g−1 for Li15Si4 at room temperature), a low reaction potential against the cathode material, environmental compatibility, low toxicity, and low cost.7–10 However, for practical implementation of Si anodes in LIBs, several critical issues, including high-power, low-heat generation, accommodation of a large volume change, and high tap density, should be considered. One major problem with Si anodes is the huge volume change (>300%) during the lithiation/delithiation process, leading to the pulverization of Si particles (well above the particle size of 150 nm), which consequently results in the fast capacity loss of the electrode.9,11–13 The cracking and pulverization problem of Si anodes can be reduced or avoided by reducing the particle size to the nanometre range and by suitably designing Si-based materials.14–18
Another critical factor in Si anode implementation requires controlling the formation of solid-electrolyte-interface (SEI) layers on the Si surface caused by the decomposition of liquid electrolytes. Typically, electrolyte decomposition occurs due to the low potential of the Si anode (<0.5 V versus Li/Li+), resulting in electronic insulating SEI layer formation on the Si electrode surface during the lithiation process.19 The continuous growth of SEI layers during repeated cycling leads to a degradation in the electrochemical performance of Si anodes through (i) continuous consumption of electrolytes and lithium ions, (ii) loss of electrical contact between the current collector and the Si electrode, (iii) an increase in the lithium diffusion path, (iv) degradation of the structural integrity caused by mechanical stress, and (v) a large amount of heat generation.9,15,20–22 For these reasons, the formation of a stable SEI layer is critical for implementing fast charging/discharging and stable cycling with minimal heat generation. Several strategies have been developed to control stable SEI formation using additional protective additives, double-walled Si nanotubes, and Si-carbon yolk–shell structures, leading to a significant improvement in the cycling life and rate capabilities with low areal mass loading (0.01–1 mg cm−2).23–26 Although Si particle nanostructuring significantly improves the high power and long-term cycling stability, it gives rise to new fundamental challenges, including a serious side reaction caused by high surface area, low tap density, and large heat generation. Moreover, large-scale and low-cost synthetic strategies for nanostructured Si-based materials with desirable electrochemical performances remain an important challenge. Several nanostructured and novel structured Si-based materials have shown high lithium storage capacity, stable cycling stability, and excellent rate capability at an acceptable mass loading level.15,26–28 However, for practical LIB applications, Si-based materials should meet various conditions such as high-power, low-heat generation, low volume expansion, high tap density, and low-cost processing. So far, there have been no reliable Si-based materials reported that satisfy all of these requirements, and thus, new design protocols are highly desirable.
Herein, we design a novel core–shell structured Si-based multicomponent, in which the core is active Si and the multifunctional outer shells are composed of lithium silicate (Li2SiO3 and Li2Si2O5) and lithium titanate (Li4Ti5O12). Synergistic coupling of the multifunctional shell layers has several advantages: (1) lithium silicate with good mechanical properties prevents fracture of Si particles; (2) lithium titanate provides an effective path for Li-ion diffusion; (3) both lithium silicate and lithium titanate promote the formation of a stable SEI layer on the Si electrode surface; (4) multifunctional coating layers significantly reduce the exothermic heat and accommodate a large volume change during the battery operation; and (5) low tap density of Si particles is partially solved by the lithium titanate covering the Si surface. The newly developed Si-based multicomponent anodes exhibit a reversible specific capacity of 1000 mA h g−1 based on the total electrode weight, a highly stable cycling retention (∼65% after 1000 cycles at a discharge/charge rate of 1 C at an areal density of 1.5 mA h cm−2), an excellent rate capability (a specific capacity of 89% at 10 C compared to 0.2 C) and a remarkably suppressed volume expansion (12% after 100 cycles at 0.5 C rate).
Experimental
Synthesis of Si-based multicomponents
The Si nanoparticles (Alfa Aesar, an average diameter of 100 nm) were treated with hydrogen peroxide (H2O2, Samchun chemical) and dried in a convection oven at 70 °C. In a typical process, 0.5 g of as-treated Si particles and 0.82 g (8 mmol) of lithium acetate dihydrate (CH3COOLi·2H2O, Sigma Aldrich) were dispersed in 10 mL of ethanol under stirring. After 1 h, 1.4 g (5 mmol) of titanium(IV) isopropoxide (Sigma Aldrich) was added to the above solution with stirring for 2 h and dried in a convection oven at 70 °C. To control the thickness of the coating layer, the amounts of lithium acetate dihydrate and titanium(IV) isopropoxide were adjusted with a fixed molar ratio (Li/Ti = 8/5). The Si-multi-65 sample was prepared with 2.5 mmol of titanium(IV) isopropoxide and 4 mmol of lithium acetate dehydrate, while the Si-multi-80 particles were synthesized with 1.25 mmol of titanium(IV) isopropoxide and 2 mmol of lithium acetate dehydrate. The mixture was then dried and ground. Finally, the powder was thermally treated at 610 °C for 12 h in an argon atmosphere to obtain the Si-based multicomponents.Physical characterization
The microstructure of the as-synthesized Si-based multicomponents was characterized using a high-power X-ray diffractometer (XRD) on a Rigaku D/MAX at 2500 V using Ni-filtered Cu-Kα radiation. The TEM images were taken using a high-resolution transmission electron microscope (HRTEM, JEM-2100F) operating at an accelerating voltage of 200 kV with a normal transmission electron microscope (JEM-2100) operating at 150 kV. Raman spectra were obtained from micro-Raman (Witec, Alpha 300R) with a He–Ne laser operating at λ = 532 nm. The nitrogen adsorption and desorption isotherms were measured using an automatic physisorption analyser (Micromeritics, ASAP 2020) at 77 K in the relative pressure range of P/P0 from 0.05 to 0.3 to determine the BET surface area. The X-ray photoelectron spectroscopy (XPS) analysis was performed using a K-alpha XPS (Thermo Fisher). To analyse the thermal properties of electrodes with electrolytes, the DSC profiles were obtained from METTLER TOLEDO DSC 1 at a heating rate of 5 °C from 30 to 400 °C.In situ XAFS measurements
In situ Ti K-edge X-ray absorption spectra, X-ray absorption near-edge structure (XANES), and extended X-ray absorption fine structure (EXAFS) were collected on the BL10C beam line (WEXAFS) at the Pohang light source (PLS-II) with top-up mode operation under a ring current of 300 mA at 3.0 GeV. The Si(111) crystal pair was used for Ti K-edge XAFS measurements (absorption edge energy of 4966 eV) for better energy resolution. During the discharging and charging processes, real-time Ti K-edge X-ray absorption spectroscopic data were recorded for an LTO-coated Si anode assembled in a home-made Swagelok-type in situ electrochemical cell with polyimide film windows in the transmittance mode using Ar/N2 gas-filled ionization chambers (IC-SPEC, FMB Oxford) for the incident and transmitted X-ray photons. Energy calibration was simultaneously performed for each measurement with the reference Ti metal foil placed in front of the third ion chamber. The data reductions of the experimental spectra to normalized XANES and Fourier-transformed radial distribution functions (RDFs) were performed through the standard XAFS procedure.Electrochemical test
The electrochemical performance test was performed using a galvanostatic cycler with a coin-type half-cell (2016R type). The cells were composed of the as-obtained samples (as a working electrode) and lithium metal (as a counter electrode). The working electrodes of about 1.5 mg cm−2 were prepared by mixing 60 wt% active material, 20 wt% super-P carbon black, and a 20 wt% poly(acrylic acid)/sodium carboxymethyl cellulose (PAA/CMC, wt/wt = 50/50) binder. High mass loading (3 mg cm−2) working electrodes were also prepared by mixing 80 wt% active material, 10 wt% super-P carbon black, and a 10 wt% PAA/CMC binder. The electrolyte was composed of 1.3 M LiPF6 with ethylene carbonate/diethyl carbonate (EC/DEC, 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 vol%) and a 10 wt% fluorinated ethylene carbonate (FEC) additive. The coin cells were galvanostatically charged and discharged under different current densities between 0.01 and 1.2 V using a WBCS3000K8 (WonATech Co., Ltd). The cyclic voltammetry results of the cells were obtained using a potentiostat/galvanostat (VSP classic, Bio-Logic) at a scan rate of 0.1 mV s−1 from 0.01–2.5 V. In situ electrochemical impedance of the coin cells was analysed by two channels of the multichannel potentiostat (Bio-Logic/VSP-300) in the frequency range of 10−3 to 106 Hz. Impedance data were fitted with a proposed equivalent circuit using software (Princeton Applied Research, ZSimpWin).
:70 vol%) and a 10 wt% fluorinated ethylene carbonate (FEC) additive. The coin cells were galvanostatically charged and discharged under different current densities between 0.01 and 1.2 V using a WBCS3000K8 (WonATech Co., Ltd). The cyclic voltammetry results of the cells were obtained using a potentiostat/galvanostat (VSP classic, Bio-Logic) at a scan rate of 0.1 mV s−1 from 0.01–2.5 V. In situ electrochemical impedance of the coin cells was analysed by two channels of the multichannel potentiostat (Bio-Logic/VSP-300) in the frequency range of 10−3 to 106 Hz. Impedance data were fitted with a proposed equivalent circuit using software (Princeton Applied Research, ZSimpWin).
Results and discussion
Synthesis of Si-based multicomponents with multifunctional coating layers
Core-shell-structured Si-based multicomponents were synthesized using a simple sol–gel process, in which double-shell coating layers consisting of lithium silicate (Li2SiO3 and Li2Si2O5, denoted LS) and lithium titanate (Li4Ti5O12, denoted LTO) are uniformly coated on the Si surface, as described in Fig. 1a. Commercial Si nanoparticles (Si NPs) are treated with hydrogen peroxide to activate the Si surface with a hydroxyl group, and the activated Si can be well mixed with both Li and Ti precursor solutions. The as-treated Si NPs were dispersed in the Li precursor solution, and a controlled amount of Ti precursor was subsequently added to react directly with the Li precursor on the Si surface. Finally, the Si NPs coated with Li/Ti precursors were thermally treated at 610 °C for 12 h under an argon environment to develop the multifunctional coating layers of Li2SiO3, Li2Si2O5, and Li4Ti5O12. When we used an excess Li precursor, the Li precursors reacted with the native SiO2 layer on the Si surface to produce lithium silicate and reacted with the Ti precursor to form spinel Li4Ti5O12.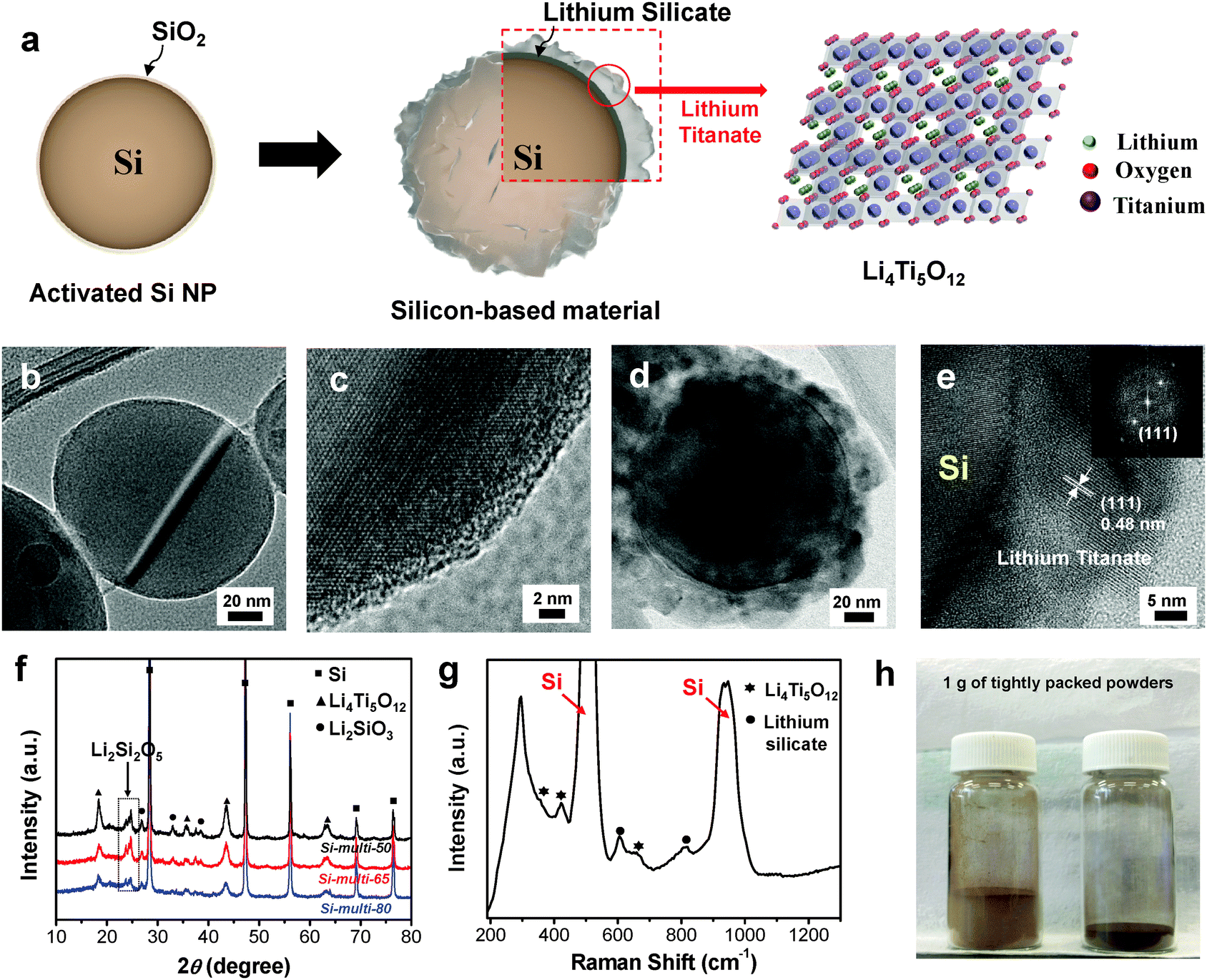 | ||
| Fig. 1 (a) Schematic illustration showing the synthetic process of the Si-based multi-components. (b) A TEM image showing Si nanoparticles with the native SiO2 layer and (c) its magnification showing the crystalline Si core and a 2 nm-thick SiO2 layer on the Si surface. (d) A TEM image of Si-multi-50 with 20–30 nm-thick coating layers and (e) its magnification showing the crystalline lithium titanate phase (Inset: the FFT pattern). (f) The XRD patterns of Si-multi-50 (black), Si-multi-65 (red), and Si-multi-80 (blue) particles. (g) Raman spectrum of Si-multi-50 showing the existence of the lithium silicate and lithium titanate phases. (h) Photographs of pristine Si NPs (left) and Si-multi-50 (right) containing 1 g of tightly packed powder. | ||
We used transmission electron microscopy (TEM) measurements to investigate the morphology of Si NPs with native oxides and the spatial locations of LTO from the core–shell-structured Si particles. The Si NPs have diameters of 50–150 nm (Fig. 1b) and have a SiO2 native oxide layer with a thickness of ∼2 nm on the Si surface (Fig. 1c). When the molar ratio of Si:Li:Ti was 17:8:5 (denoted Si-multi-50), the TEM image of the resulting products shows that 20–30 nm-thick layers are uniformly coated on the Si surface (Fig. 1d). From the magnified TEM image, we can see that the 0.48 nm spacing is consistent with the (111) plane of spinel Li4Ti5O12, as shown in Fig. 1e and in the selected area electron diffraction (SAED) pattern in the inset. Moreover, elemental mapping analyses for Si, O, and Ti in the Si-multi-50 products (Fig. 1d) show that each element is uniformly distributed on the Si surface, indicating that the shell layers are uniformly coated on the Si particles (ESI,† Fig. S1a–d). The spatial locations of LS and LTO can be estimated by elemental line mapping of TEM (ESI,† Fig. S1e and f). The Ti and O ions are uniformly distributed to the outer shell. However, the amount of Ti ions quickly decreased at a distance of ∼25 nm from the outermost shell, while the amount of O ions remained unchanged, indicating that new LS layers (with thicknesses of <5 nm) exist on the Si surface where the Ti ions disappeared (ESI,† Fig. S1e and f). Meanwhile, the LS and LTO shell thickness can be easily controlled by tuning the amount of Li and Ti precursors during the synthesis. Through inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis of the resulting products, the contents of Si and Ti were determined (ESI,† Fig. S2).
The microstructures of the LS and LTO coating layers were characterized by X-ray diffraction (XRD) measurements (Fig. 1f). The XRD pattern of the Si-multi-50 sample shows that LS exists as Li2SiO3 and Li2Si2O5, while LTO can be assigned as a spinel Li4Ti5O12 structure.29–31 As the Si content was increased, the XRD intensity of the coating layers (LS and LTO) gradually decreased, and vice versa. To investigate the microstructure of the coating layers, Raman spectroscopy was employed (Fig. 1g). The Raman spectrum of Si-multi-50 shows that peaks of LS related to Si–O–Si vibration appear at 600 cm−1 and 835 cm−1, corresponding to Li2Si2O5 and Li2SiO3, respectively.32 Furthermore, the Raman scattering peaks appearing at ∼400 cm−1 are attributed to Li4Ti5O12 [ref. 33]. This result indicates that the shell layers on the Si surface are composed of two different LS compounds (Li2Si2O5 and Li2SiO3) and one LTO compound (Li4Ti5O12).
Because the multi-phase shell layers are uniformly coated on the Si nanoparticles, the resulting Si-based multicomponents show a significantly increased tap density, which is one of the important parameters for practical rechargeable batteries.34 The Si NPs have a tap density of 0.14 g cm−3, while the Si-based multicomponents (Si-multi-50) have a tap density of 0.58 g cm−3, corresponding to an increase of 300% (Fig. 1h). Furthermore, nitrogen adsorption/desorption analysis was performed to measure the surface area of the pristine Si and Si-based multicomponents (ESI,† Fig. S3). The Si-based multicomponents showed a typical adsorption–desorption hysteresis loop corresponding to a mesoporous structure due to the coating layers,35,36 while the Si NPs exhibit a non-porous structure. The Brunauer–Emmett–Teller (BET) analysis indicated that the surface areas of the Si NP, Si-multi-50, Si-multi-65 (molar ratio of Si:Li:Ti = 17:4:2.5), and Si-multi-80 (molar ratio of Si:Li:Ti = 17:2:1.25) were 21 m2 g−1, 32 m2 g−1, 30 m2 g−1, and 26 m2 g−1, respectively. LTO coating material in the Si-multicomponent particles is nanocrystalline as confirmed by TEM and XRD measurements. Void spaces between the crystalline LTO are contributed to increasing surface area of the Si-multicomponent. That is why the surface area of Si-multi-50 is increased compared to pristine Si. Meanwhile, when the LTO contents became larger, the surface area of Si-multicomponents was also increased. In addition, X-ray photoelectron spectroscopy (XPS) is a powerful technique for confirming the chemical state of metal ions in the species formed on the Si surface. The XPS spectra were obtained from the pristine Si nanoparticles and the Si-based multicomponents (ESI,† Fig. S4). The binding energy of Si 2p in the pristine Si (Si 100%) is located at around 99 eV and 103 eV for Si0+ and Si4+, respectively, indicating that a native SiO2 layer was formed on the Si surface.37 On the other hand, the Si 2p XPS spectra of the Si-based multicomponents only showed the peak at 103 eV, implying complete coverage of the coating layers on the Si surface. Furthermore, the valences of O2− and Ti4+ determined from the LS and LTO shells on the Si surface are clearly determined from the XPS analyses38 (ESI,† Fig. S4b and c).
Electrochemical performance as an anode material for LIBs
To investigate the electrochemical properties of pristine Si and Si-based multicomponents, cyclic voltammetry (CV) measurements were performed with a half-cell from 2.5 V to 0.01 V (versus Li/Li+) at a 0.1 mV s−1 scan rate after the first cycle (Fig. 2a and b). As shown in Fig. 2a, a cathodic peak appeared at 0.20 V, which corresponds to a series of Li–Si alloy formation. Two anodic peaks were clearly observed at about 0.53 and 0.39 V, respectively. These peaks are related to the de-alloying of the Li–Si alloy.39 The cathodic and anodic peaks of the Si-multi-50 profile (Fig. 2a and b, red line) were broadened and shifted because Si-multi-50 includes a larger amount of Li2Si2O5 than the other multicomponents. The reaction with Li2Si2O5 and Li+ ions is confirmed from the cathodic peak at around 0.27 V and from anodic peak at around 0.34–0.39 V.40 In particular, the other cathodic and anodic peaks are observed at 1.54 V and 1.58 V, respectively, corresponding to the spinel Li4Ti5O12 reaction with Li+ (Fig. 2b). The current density is two orders of magnitude lower than redox peaks of Si particles due to a significant capacity difference between Si and Li4Ti5O12. This result confirms the existence of electrochemically active Li4Ti5O12.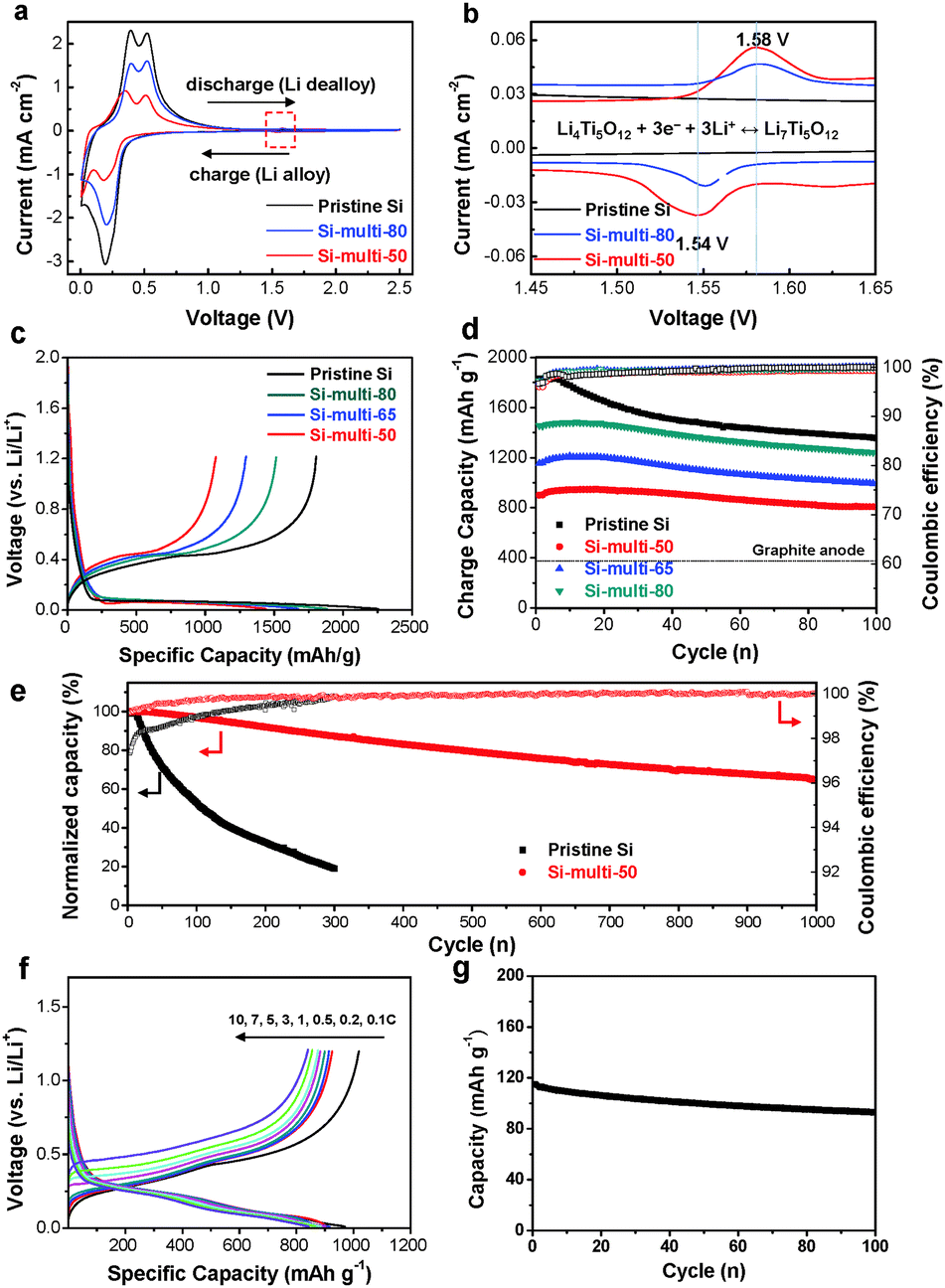 | ||
| Fig. 2 (a) Cyclic voltammogram (CV) of pristine Si, Si-multi-50, and Si-multi-80 electrodes obtained from 2.5 V to 0.01 V (versus Li/Li+) at a 0.1 mV s−1 scan rate. (b) The CV data taken from the red square box shown in (a). (c) The first cycle voltage profiles of the pristine Si and Si-multicomponent electrodes. (d) Cycle retentions of four different Si-based electrodes obtained at 0.1 C. (e) Long-term cycling properties of the pristine Si and Si-multi-50 electrodes obtained at a 1 C discharge–charge rate. (f) Rate capabilities of the Si-multi-50 electrode obtained at various charge rates with a fixed discharge rate of 0.2 C. (g) Cycle retention of the Si-multi-50 (anode)-LiCoO2 (cathode) full-cell at a charging–discharging rate of 1 C. | ||
When the spinel Li4Ti5O12 on the Si surface was lithiated to 0.01 V, it is converted to rock-salt Li7Ti5O12 at around 1.5 V and to the Li8.5Ti5O12 phase at 0.05 V.41 Subsequent cycling of the Si-based multicomponent anode between 0.01 V and 1.2 V leads to a formation of lithium Li7Ti5O12 coating layers that can significantly enhance the electronic conductivity of the Si materials. Spinel Li4Ti5O12 is an insulator with an electron conductivity as low as 10−13–10−9 S cm−1, while Li7Ti5O12 shows a high electronic conductivity of 10−2 S cm−1 [ref. 42]. Additionally, the Li7Ti5O12 phase provides a three-dimensional network of channels for facile Li-ion diffusion.
Based on the positive effect of multifunctional Li7Ti5O12 coating layers on the Si anode, the cycling performance of the Si-based multicomponent electrodes was evaluated using charge/discharge cycling from 0.01 V to 1.2 V at room temperature (Fig. 2c–g). All reported capacities are based on the total mass of Si and the coating materials. The typical mass loading of the electrodes was ∼1.5 mg cm−2. As shown in Fig. 2c, the discharge (delithiation) capacity of the Si-multi-50 electrode is 1032 mA h g−1 for the first cycle at a rate of 0.05 C (1 C is the charge or discharge in 1 h) with an initial coulombic efficiency of 78.5%, while the pristine Si electrode exhibits a discharge capacity of 2077 mA g−1 (an initial coulombic efficiency of 83%). Because the Si-multi-50 electrode contains 50% Si, its specific capacity is low, but it is still more than two times higher than conventional graphite anodes (372 mA h g−1). By controlling the thickness of the coating layers, the specific capacity of Si-based multicomponent electrodes can be tuned as shown in Fig. 2c. The charge capacity retention of Si-based electrodes shows different results by controlling the Si contents within the Si-based multicomponents at a rate of 0.1 C (Fig. 2d). The cycling performance of the four different Si-based electrodes, which include pristine Si, Si-multi-50, Si-multi-65, and Si-multi-80, was compared using the same cycling condition. After 100 cycles, the capacity retention of the pristine Si electrode was 74.2%, while that of the Si-multi-50, Si-multi-65, and Si-multi-80 electrodes was 89.7%, 86.1%, and 85.3%, respectively, implying that the multifunctional coating layers play a key role in highly stable cycling performance.
In particular, the Si-multi-50 electrode exhibited a highly stable cycling property at a 1 C discharge/charge rate (Fig. 2e). Under this condition, the first cycle charge capacities of pristine Si and Si-multi-50 are 1200 mA h g−1 and 650 mA h g−1, respectively. To more clearly compare the capacity retention, the charge capacity of each electrode was normalized. The pristine Si electrode showed a capacity retention of 20% after 300 cycles. In contrast, the capacity retention of the Si-multi-50 electrodes at the 200th, 500th, 800th, and 1000th cycle was 92%, 80%, 70%, and 65%, respectively. Because the charge capacity only degrades ∼3.5% per 100 cycles over 1000 cycles, the Si-multi-50 electrode demonstrates an outstanding long-term cycling stability. Interestingly, the Si-multi-50 electrodes showed a high coulombic efficiency per cycle (>99.5%) after 50 cycles, which is one of the critical factors in practical rechargeable batteries. Moreover, when the Si-multi-50 electrode with a high active mass loading (3 mg cm−2) was cycled at a 0.1 C rate in the range of 0.01–1.2 V, it still showed good cycling stability (a capacity retention of 90% after 50 cycles) compared to the pristine Si electrode (a capacity retention of 44%) (ESI,† Fig. S5). These results suggest that the functional coating layers in the Si-based multi-components may act as a helpful buffer for large volume change and provide an effective pathway for Li-ion diffusion during repeated cycles. In particular, the Si-multicomponent electrode with a high mass loading (1.5–3.0 mg cm−2) showed significantly improved electrochemical properties, compared to previous reports (ESI,† Table S1).
In addition, to use Si-based electrodes in high-power LIBs, the charge and discharge rate should be investigated. The rate capabilities of the Si-multi-50 electrode were evaluated with charge rates in the range of 0.1–10 C with a fixed discharge rate of 0.2 C (Fig. 2f). The Si-multi-50 electrode exhibited an excellent rate capability (∼800 mA h g−1 at a high rate of 10 C with a specific capacity of 89% compared to the 0.2 C rate). Moreover, when the discharge rate capability was tested at rates of 0.1–10 C at a fixed charge rate of 0.2 C, a high specific capacity of 50% compared to that of the 0.2 C rate was exhibited even at the 10 C rate (ESI,† Fig. S6). For control experiments, when the discharge rate capabilities of the pristine Si nanoparticle and carbon-coated Si nanoparticle electrodes were tested at a fixed charge rate of 0.2 C, the specific capacities significantly declined at a rate of 3 C (ESI,† Fig. S6). These results imply that the Si-multi-50 electrode has multifunctional layers which can provide an effective pathway for lithium ions and enhance the electrical conductivity of Si.
The electrochemical performances of the Si-based multicomponent electrodes were tested in a half-cell. In order to claim its relevance as a promising anode for practical LIB applications, we tested a full cell (an energy density of 3.0 mA h cm−2) where the Si-multi-50 anode was coupled with a LiCoO2 cathode. The electrochemical performance of the Si-multi-50/LiCoO2 system was tested at charge/discharge = 1.0 C/1.0 C at a voltage range of 2.5–4.2 V. The charge/discharge voltage profiles of the full-cell are shown in the ESI,† Fig. S7. The first charge/discharge profile of the Si/LiCoO2 cell shows a coulombic efficiency of 86% in the pre-cycle at a C/10 rate (ESI,† Fig. S7). Subsequent long-term cycling (up to 100th cycle at 1 C charging–1 C discharging rate) in a full-cell showed good a capacity retention of 82% (Fig. 2g). It suggests that the Si-multi-50 electrode is a promising candidate as an anode material for practical LIB applications.
The role of the surface LTO layer in the enhanced kinetic electrochemical performance of the surface-modified Si anode material can be directly determined with atomic-selective in situ Ti K-edge XAFS characterization during the first lithiation. In the first discharge, the initial spinel Li4Ti5O12-like XANES spectral feature effectively shifts towards a lower energy region in the voltage plateau at 1.5 V and thereafter shows constantly lithiated-Li7Ti5O12 phase characteristics even in the 5th and 50th cycles (Fig. 3a). This result demonstrates that the Ti4+ state in the coated Li4Ti5O12 layer is initially reduced to be a mixed valent Ti3.4+ state at 1.5 V. During consecutive 5th and 50th cycles in the voltage window of 0.01–1.2 V, the partially-reduced Ti3.4+ state remains constant with a fully lithiated spinel Li7Ti5O12 phase on the surface of the core Si phase. This result can also be supported with local structural variations around the Ti ion during the electrochemical reaction (Fig. 3b).
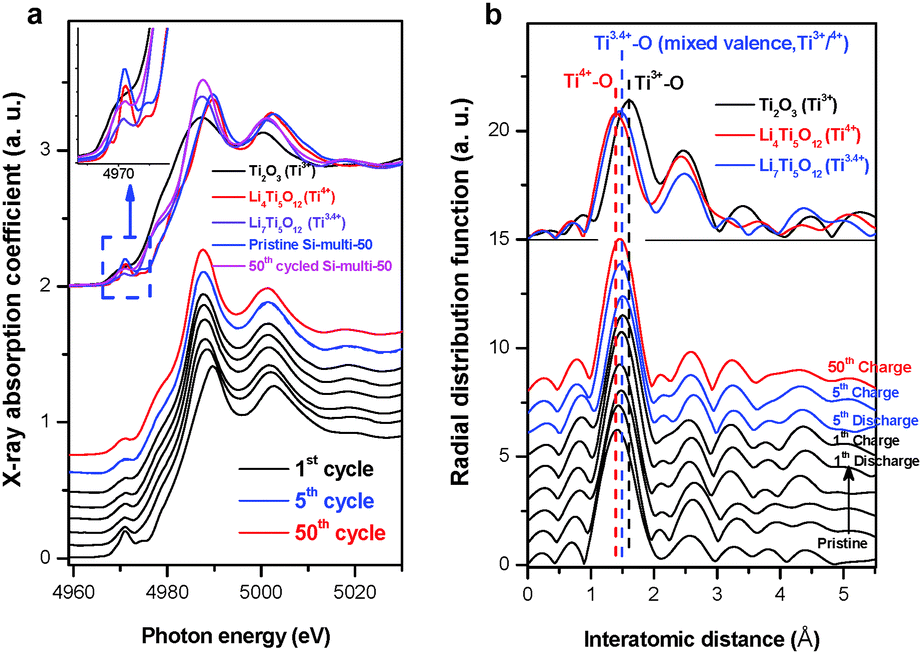 | ||
| Fig. 3 Structural variation of the surface LTO in Si-based multicomponent electrodes. (a) Normalized Ti K-edge XANES spectra during the first discharge in the voltage region of 2.5 V and 0.01 V as well as the 5th and 50th cycles in the voltage region of 0.01 and 1.2 V. (b) Corresponding radial distribution functions of k3-weighted EXAFS spectra. | ||
The Ti4+–O bond length of the initial Li4Ti5O12-phase increases to the longer Ti3.4+–O bond length during the first discharge, and the radial distribution functions (RDFs) hereafter remain constant at an intermediate RDF feature between Ti3+ and Ti4+ through the following electrochemical reactions. As a result, the lithiated Li7Ti5O12 phase on the surface is no longer an electrochemically active site on the Si anode in the considered voltage region. These spectroscopic results for the surface LTO layer can prove the formation of stable SEI layers on the intrinsically brittle Si material, which prevents the core Si anode from being pulverized and provides an effective Li-ion diffusion pathway. Furthermore, the enhancement of electrical conductivity of the lithiated Li7Ti5O12 surface layer due to the mixed valence state of Ti3+/Ti4+ can promote the rate capability of the LTO-coated Si anode material through fast Li-ion conduction in the interface region compared to the pristine Si anode.43
Lithium-ion diffusivity of Si-based electrodes
In an effort to provide a more comprehensive understanding of the effect of multifunctional coating layers (LS and LTO) on the excellent electrochemical properties of Si-based multicomponents, in situ galvanostatic electrochemical impedance spectroscopy (GS-EIS) was performed on the electrodes after the 1st and 60th cycles at a rate of 1 C because the lithiation/delithiation kinetics of the electrodes are highly dependent on the resistances associated with the mass transfer and charge transfer process.The in situ GS-EIS results are displayed in Fig. 4a–c and are fitted to an equivalent circuit (Fig. 4e). Lithium-ions (Li+ ions) migrate through the electrolyte into the electrode surface (solution resistance, RS), enter into the SEI layer (RSEI), and are alloyed from the SEI layer to the surface of the electrode materials (charge transfer resistance, RCT), and finally diffuse to the inter-space of the electrode materials (Warburg impedance, ZW).44 Although the starting point of RCT of the Si-multi-50 electrode was higher than the pristine Si (Fig. 4a) and carbon-coated Si electrodes (Fig. 4b) due to the porous surface of the Si-multi-50 electrode and the low electronic conductivity of lithium titanate, the RCT value of the Si-multi-50 electrode continuously decreased during lithiation after the 1st cycle (Fig. 4c).
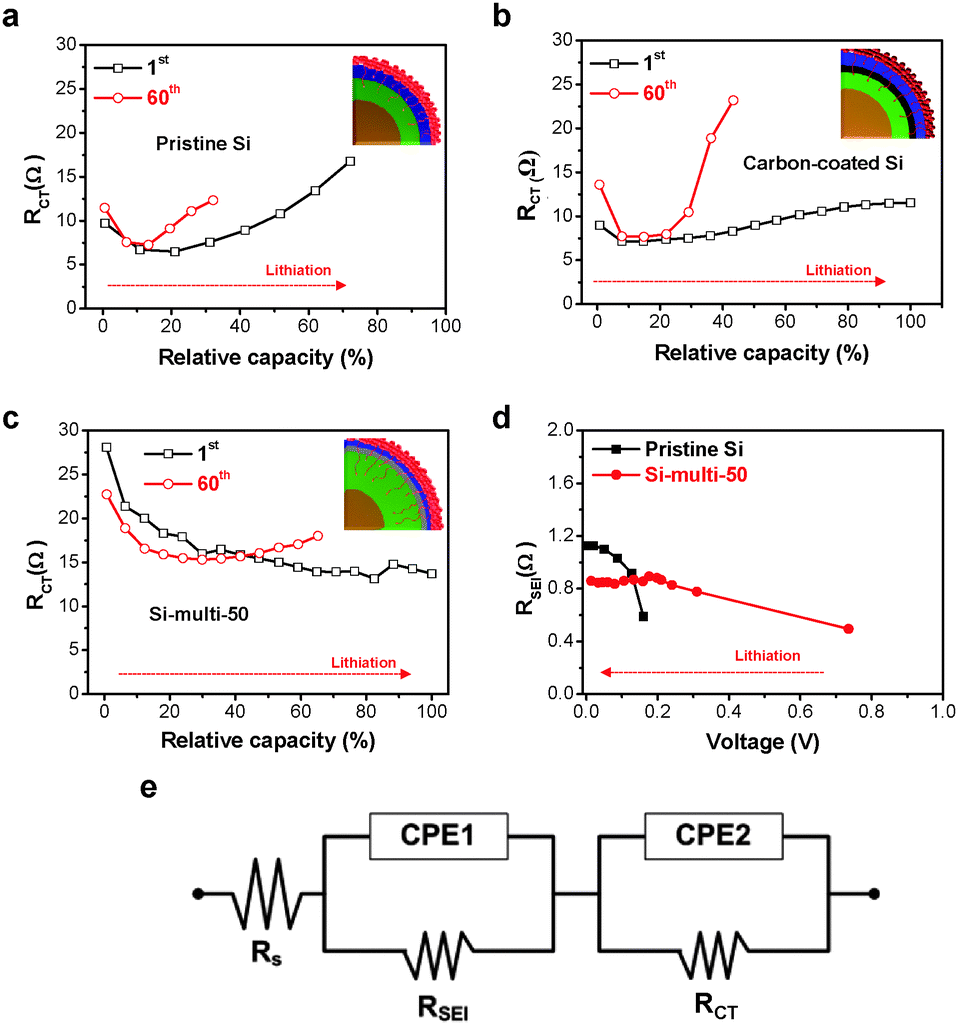 | ||
| Fig. 4 Structural in situ galvanostatic electrochemical impedance results. (a) The pristine Si, (b) carbon-coated Si, and (c) Si-multi-50 electrodes obtained during the lithiation process at a 1 C rate in the 1st and 60th cycle. The polarization resistance (RCT) change of each electrode was monitored as a function of relative capacity. (d) The RSEI value measured in the pristine Si and Si-multi-50 electrodes during the lithiation process. (e) An equivalent circuit used for electrochemical impedance analysis. | ||
This result indicates that the initial access of Li+ ions on the Si-multi-50 electrode surface is not easy, but immediately after the first access, the Li+ ions quickly reach the Si active layers to form lithium alloy materials. Moreover, at a rate of 1 C, the Si-multi-50 electrode delivered fully available capacity up to 100%. After the 60th cycle, the RCT starting point of the Si-multi-50 electrode was reduced due to the stabilization of the LTO layer on the Si particle surface,45 but the capacity of the Si-multi-50 electrode was still maintained at more than 70%, compared to the capacity in the first cycle. In contrast, the pristine Si and the carbon-coated Si electrodes showed capacity retention of 30–40% compared to the 1st cycle, with a drastic increase in RCT during the lithiation process.
In addition, the RSEI values of both the Si-multi-50 and pristine Si electrodes were monitored during the lithiation process after the 60th cycle (Fig. 4d). The RSEI value of the Si-multi-50 electrode was maintained at 0.8 Ω, while the pristine Si nanoparticle electrode was significantly increased up to 1.2 Ω due to unstable SEI layer formation. These results demonstrate that the multi-functional coating layers on the Si-multi-50 surface directly affect the electrical conductivity of the whole Si-based electrode. Because the Li+ ion diffusivity of the electrode material is also an important factor affecting the cycling retention and charge–discharge rate capability, we used a galvanostatic intermittent titration technique (GITT) for the Si-multi-50 (Fig. 5b) and the pristine Si electrodes (Fig. 5a) after the 5th cycle. Various values for the Li+ ion diffusivity in amorphous Si (ranging from 10−14 to 10−9 cm2 s−1) were measured through GITT, CV, and EIS analyses.46–50 In this work, the calculated Li+ ion diffusivity of the Si-multi-50 electrode after the 5th cycle is 8.36 × 10−11 cm2 s−1, which is several times higher than that of the pristine Si electrode (1.56 × 10−11 cm2 s−1) at around 0.5 V (Fig. 5c). This result indicates that Li+ ions transport much more quickly in the Si-multi-50 electrode than the pristine Si electrode because the Li7Ti5O12 layer (discussed in Fig. 3) provides an effective pathway for lithium ions which minimizes the Li+ ion transport resistance. Additionally, the total internal resistance was determined by GITT. The internal resistance of the electrodes involves the combined influence of three parameters: interface electron transfer, electronic conduction, and Li+ ion transport.51 In the lithiation process from 0.6 V to 0.01 V, the internal resistance of the Si-multi-50 and pristine Si electrodes was almost the same and gradually decreased until deep lithiation due to the enhancement of the Si electronic conductivity from Li–Si alloying (Fig. 5d). However, the internal resistance of the pristine Si NP was about five times higher than the Si-multi-50 electrode during the delithiation process from 0.01 to 1.0 V, indicating a remarkable increase in the electron transfer resistance of the pristine Si electrode. The Li7Ti5O12 layer on the Si-multi-50 surface plays an important role in assisting Li+ ion transport and reducing internal resistance during the lithiation/delithiation process, as evidenced by in situ EIS and GITT analyses.
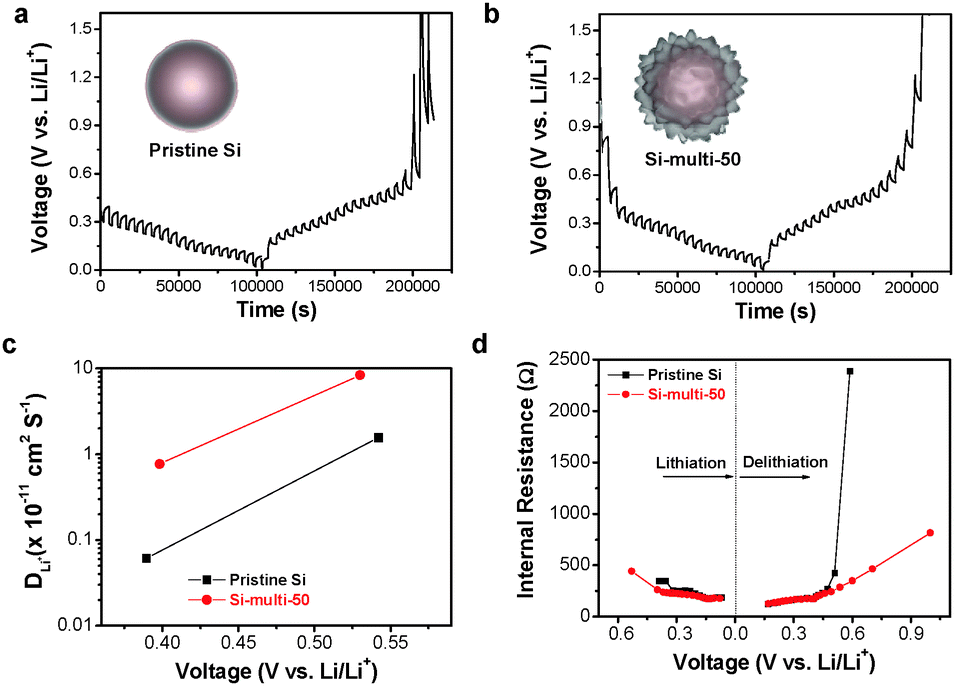 | ||
| Fig. 5 The GITT profiles of the (a) pristine Si and (b) Si-multi-50 electrodes obtained during the lithiation/delithiation process. (c) The diffusion coefficient of Li+ ions calculated from the pristine Si and Si-multi-50 electrodes between 0.55 V and 0.3 V. (d) The variation of the internal cell resistance determined from the pristine Si NPs (black) and the Si-multi-50 (red) electrodes during the lithiation/delithiation process. | ||
Characterization of Si-based electrodes
The formation of a stable SEI layer in Si-based electrodes is another key factor for determining a high electrochemical performance including cycling stability and rate capability. The SEI layer on the Si-based electrodes is formed due to electrolyte decomposition at a low potential range (<1.0 V versus Li/Li+).10,19 The SEI layers formed at the lithiated state on the surface of the Si electrode can be broken at the delithiated state owing to the huge volume expansion of Si. This exposes a fresh Si surface to the electrolyte, and a new SEI is formed on the fresh Si surface. Accordingly, the SEI layer becomes thicker with continuous consumption of electrolytes and Li+ ions during repeated cycling. Thus, preventing direct contact between the Si electrodes and the electrolyte is important for obtaining cycling stability.10,19 The Si-multi-50 electrode with multi-functional coating layers is not directly in contact with the electrolyte owing to the full coverage of lithium silicate and lithium titanate on the Si surface. As mentioned above, lithium silicate is an electron insulator, but Li+ ion conductors can hamper the decomposition of the electrolyte and maintain Li+ ion diffusion.29,40 Moreover, electronic conductivity and Li+ ion diffusion were improved by the Li7Ti5O12 layer and were maintained during cycling between 0.01–1.2 V. As a result, the formation of a stable SEI layer was expected for the Si-multi-50 electrode.The Si-multi-50 electrode showed a uniform SEI layer and a stable Li7Ti5O12 phase after 100 cycles, as shown in the TEM image, and the dimension of the cycled-Si-based particles does not significantly change compared to the original Si-multi-50 electrode (ESI,† Fig. S8c and d). In contrast, the pristine Si electrode formed a thick SEI layer due to the continuous growth of SEI layers during repeated cycles, and the Si nanoparticles were significantly changed as remarkably expanded particles (ESI,† Fig. S8a and b).
To provide a more comprehensive understanding for the SEI layers formed on the Si electrode surface after 100 cycles, we performed X-ray photoelectron spectroscopy (XPS) analysis. Fig. 6 shows the F 1s, Li 1s, and Si 2p XPS spectra, and their deconvolution for the pristine Si electrode as well as for the Si-multi-50 electrode. The F 1s core level peaks assigned to LiF appeared at 685.5 eV for both electrodes because the fluorinated ethylene carbonate (FEC), which is used as a reducible additive, produced an SEI layer primarily composed of LiF (Fig. 6a and d).19,52 A noticeable feature in the F 1s XPS spectra of Fig. 6a and d is that the Si-multi-50 electrode exhibits discernibly increased peak intensity attributed to LiF compared to the pristine Si. Further evidence is given via comparison of the Li 1s XPS spectra of the electrode surface (Fig. 6b and e). The relative fraction of the LiF peak in the Si-multi-50 electrode SEI had a much stronger intensity than that of the pristine Si electrode SEI. The FEC additive undergoes electrochemical decomposition to form Li2CO3, C–F containing polymer species, and LiF by the reaction of the C–F containing polymer species ((CH2–CHF)n) with the Li ions and electrons.53
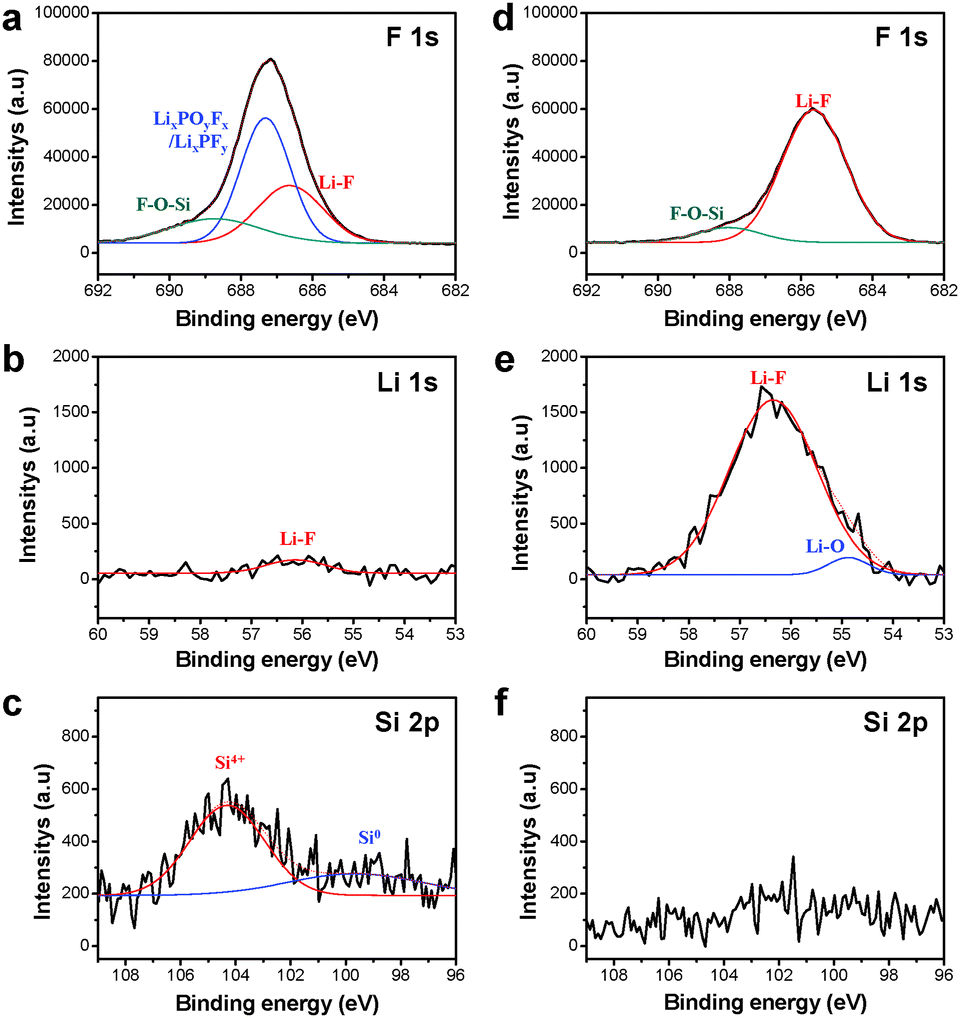 | ||
| Fig. 6 XPS characterization of the SEI layer on the electrode surface after 100 cycles. (a) The F 1s, (b) Li 1s, and (c) Si 2p XPS spectra of the pristine Si NP electrodes. (d) The F 1s, (e) Li 1s, and (f) Si 2p XPS spectra of the Si-multi-50 electrodes. | ||
This result suggests that multifunctional coating layers on the Si electrode help form a stable SEI layer on the Si-multi-50 electrode, avoiding undesirable electrolyte decomposition. The F1s XPS spectrum obtained from the pristine Si electrode after 100 cycles exhibited a relatively strong peak corresponding to LixPOyFz and LixPFy at 687 eV, compared to the Si-multi-50 electrode. This result demonstrates that the multi-functional coating layers in the Si-multi-50 electrode effectively suppress the decomposition of LiPF6 salt. In addition, the peaks corresponding to SiOx (Si4+) and metallic Si (Si0), which were not observed in the Si-multi-50 electrode, appeared at ∼104 and 99.5 eV for the pristine Si electrode, respectively (Fig. 6c and f). The active Si surface may be exposed to the electrolyte solution by cracking of the Si nanoparticles without multifunctional coating layers during cycling, and Si, which is not covered by SEI, may be detectable in the Si 2p XPS spectra. From the XPS results, the Si-multi-50 electrode clearly maintains the FEC-derived SEI layer upon lithiation and delithiation processes and thus exhibits excellent long-term cycling stability.
The Si-multi-50 electrode with multi-functional coating layers exhibited several advantages as a high-performance LIB anode material. First, the lithium titanate (Li7Ti5O12) layer enhanced the electronic conductivity and Li+ ion diffusivity of the Si particles during charge/discharge cycling. Generally, Li4Ti5O12 is a poor electronic conductor (ca. 10−13 S cm−1) because the oxidation state of Ti in Li4Ti5O12 is +4 [ref. 54]. However, the Li7Ti5O12 and Li8.5Ti5O12 layers formed in the lithiation step are good electronic conductors because the Ti oxidation state is changed to +3.4 and +3, respectively.54 Second, lithium silicate, which is electrochemically stable in a wide voltage range, not only provides structural integrity to the Si-multi electrodes but also promotes stable SEI layer formation on the Si electrode surface. The synergistic coupling of both multifunctional coating layers plays a key role in hampering direct contact between Si and the electrolyte. Thus, the continuous formation of an unstable SEI layer on the Si electrode could be restrained during repeated cycling. Third, the Si-based multicomponent structure can overcome handicaps of nanostructured materials including the low tap density, serious side reaction, and huge exothermic heat evolution. Finally, the multicomponent layers on the Si surface prevent the significant volume expansion of the Si-based electrode after cycling (ESI,† Fig. S9). The pristine Si electrode expanded to 100% after 100 cycles at a 0.1 C rate, while the Si-multi-50 exhibited less volume expansion (37%) under the same conditions. This result demonstrates that the multifunctional coating layers act as physical buffer layers which can alleviate the huge Si volume expansion.
Conclusion
In summary, we have developed Si-based multicomponents with multifunctional coating layers (consisting of lithium silicate and lithium titanate) for high-performance lithium-ion battery anodes using a simple sol–gel process. The multifunctional coating layers not only increased electronic conductivity to enhance rate capability but also promoted the formation of a stable SEI layer on the Si electrode surface, enhancing the cycling properties. The resulting Si-based multicomponent showed highly stable long-term cycling properties (a capacity retention of 65% after 1000 cycles at a 1 C discharge–charge rate), a high reversible capacity of 1000 mA h g−1 and an excellent rate capability (a specific capacity of 89% at 10 C compared to 0.2 C). These results demonstrate that Si-based multicomponents could be promising anode materials for practical lithium ion battery applications. We strongly believe that our synthetic route could be extended to the development of other high-performance anode materials for rechargeable batteries.Acknowledgements
This work was supported by the IT R&D program of MOTIE/KEIT (10046309).Notes and references
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed
.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS PubMed
- Y. Yang, S. Jeong, L. Hu, H. Wu, S. W. Lee and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13013–13018 CrossRef CAS PubMed
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966–4985 CrossRef CAS PubMed
- B. Key, R. Bhattacharyya, M. Morcrette, V. Seznéc, J.-M. Tarascon and C. P. Grey, J. Am. Chem. Soc., 2009, 131, 9239–9249 CrossRef CAS PubMed
- V. Chevrier and J. Dahn, J. Electrochem. Soc., 2009, 156, A454–A458 CrossRef CAS PubMed
- U. Kasavajjula, C. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003–1039 CrossRef CAS PubMed
- H. Wu, G. Zheng, N. Liu, T. J. Carney, Y. Yang and Y. Cui, Nano Lett., 2012, 12, 904–909 CrossRef CAS PubMed
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed
- M. T. McDowell, S. W. Lee, C. Wang, W. D. Nix and Y. Cui, Adv. Mater., 2012, 24, 6034–6041 CrossRef CAS PubMed
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS PubMed
- M.-H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS PubMed
- J. Saint, M. Morcrette, D. Larcher, L. Laffont, S. Beattie, J.-P. Pérès, D. Talaga, M. Couzi and J.-M. Tarascon, Adv. Funct. Mater., 2007, 17, 1765–1774 CrossRef CAS PubMed
- M. Obrovac and L. Krause, J. Electrochem. Soc., 2007, 154, A103–A108 CrossRef CAS PubMed
- N.-S. Choi, K. H. Yew, K. Y. Lee, M. Sung, H. Kim and S.-S. Kim, J. Power Sources, 2006, 161, 1254–1259 CrossRef CAS PubMed
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 CAS
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS PubMed
- Y. M. Lee, J. Y. Lee, H.-T. Shim, J. K. Lee and J.-K. Park, J. Electrochem. Soc., 2007, 154, A515–A519 CrossRef CAS PubMed
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS PubMed
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Nat. Chem., 2013, 5, 1042–1048 CrossRef CAS PubMed
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H.-W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed., 2008, 120, 10305–10308 CrossRef PubMed
- J.-I. Lee, K. T. Lee, J. Cho, J. Kim, N.-S. Choi and S. Park, Angew. Chem., Int. Ed., 2012, 51, 2767–2771 CrossRef CAS PubMed
- M. R. Mohammadi and D. J. Fray, J. Sol-Gel Sci. Technol., 2010, 55, 19–35 CrossRef CAS PubMed
- T. Tang, Z. Zhang, J.-B. Meng and D.-L. Luo, Fusion Eng. Des., 2009, 84, 2124–2130 CrossRef CAS PubMed
- P. Li, B. Ferguson and L. Francis, J. Mater. Sci., 1995, 30, 4076–4086 CrossRef CAS
- T. Fuss, A. Moguš-Milanković, C. S. Ray, C. Lesher, R. Youngman and D. E. Day, J. Non-Cryst. Solids, 2006, 352, 4101–4111 CrossRef CAS PubMed
- K. Mukai, Y. Kato and H. Nakano, J. Phys. Chem. C, 2014, 118, 2992–2999 CAS
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 CAS
- J. Yu, Y. Su and B. Cheng, Adv. Funct. Mater., 2007, 17, 1984–1990 CrossRef CAS PubMed
- T. Amatani, K. Nakanishi, K. Hirao and T. Kodaira, Chem. Mater., 2005, 17, 2114–2119 CrossRef CAS
- R. Tan, Y. Azuma and I. Kojima, Surf. Interface Anal., 2006, 38, 784–788 CrossRef CAS PubMed
- M.-S. Song, R.-H. Kim, S.-W. Baek, K.-S. Lee, K. Park and A. Benayad, J. Mater. Chem. A, 2014, 2, 631–636 CAS
- B. Boukamp, G. Lesh and R. Huggins, J. Electrochem. Soc., 1981, 128, 725–729 CrossRef CAS PubMed
- W.-S. Chang, C.-M. Park, J.-H. Kim, Y.-U. Kim, G. Jeong and H.-J. Sohn, Energy Environ. Sci., 2012, 5, 6895–6899 CAS
- Z. Zhong, C. Ouyang, S. Shi and M. Lei, ChemPhysChem, 2008, 9, 2104–2108 CrossRef CAS PubMed
- N. Takami, K. Hoshina and H. Inagaki, J. Electrochem. Soc., 2011, 158, A725–A730 CrossRef CAS PubMed
- H. Kim, M. G. Kim and J. Cho, Adv. Energy Mater., 2012, 2, 1425–1432 CrossRef CAS PubMed
- Y. Ko, Y.-G. Cho and H.-K. Song, RSC Adv., 2014, 4, 16545–16550 RSC
- L. Shen, X. Zhang, E. Uchaker, C. Yuan and G. Cao, Adv. Energy Mater., 2012, 2, 691–698 CrossRef CAS PubMed
- N. Ding, J. Xu, Y. X. Yao, G. Wegner, X. Fang, C. H. Chen and I. Lieberwirth, Solid State Ionics, 2009, 180, 222–225 CrossRef CAS PubMed
- W. Si, I. Mönch, C. Yan, J. Deng, S. Li, G. Lin, L. Han, Y. Mei and O. G. Schmidt, Adv. Mater., 2014, 26, 7973–7978 CrossRef CAS PubMed
- K. Yoshimura, J. Suzuki, K. Sekine and T. Takamura, J. Power Sources, 2007, 174, 653–657 CrossRef CAS PubMed
- J. Xie, N. Imanishi, T. Zhang, A. Hirano, Y. Takeda and O. Yamamoto, Mater. Chem. Phys., 2010, 120, 421–425 CrossRef CAS PubMed
- Y. Son, M. Park, Y. Son, J.-S. Lee, J.-H. Jang, Y. Kim and J. Cho, Nano Lett., 2014, 14, 1005–1010 CrossRef CAS PubMed
- H. Zhang and P. V. Braun, Nano Lett., 2012, 12, 2778–2783 CrossRef CAS PubMed
- M.-J. Chun, H. Park, S. Park and N.-S. Choi, RSC Adv., 2013, 3, 21320–21325 RSC
- H. Nakai, T. Kubota, A. Kita and A. Kawashima, J. Electrochem. Soc., 2011, 158, A798–A801 CrossRef CAS PubMed
- X. Yao, S. Xie, H. Nian and C. Chen, J. Alloys Compd., 2008, 465, 375–379 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee01493j |
| This journal is © The Royal Society of Chemistry 2015 |