DOI:
10.1039/C5EE01361E
(Review Article)
Energy Environ. Sci., 2015,
8, 2551-2573
Progress and perspectives in exploiting photosynthetic biomolecules for solar energy harnessing
Received
1st May 2015
, Accepted 8th June 2015
First published on 8th June 2015
Abstract
Photosynthetic proteins are emerging as a new class of photovoltaic materials as their nature-designed architecture and internal circuitry are so sophisticated that they carry out the initial light-driven steps of photosynthesis with ≈100% quantum efficiency. Research on bioinspired solar cells has increased in recent years as they promise better efficiency than the conventional p–n junction solar cells that have limited conversion efficiency (34%). Since it is a mammoth task to perfectly mimic the intricate proteins evolved in nature, the idea of interfacing the natural proteins with engineered materials seems to be propitious for developing biohybrid solar cells. Herein, we summarize various approaches in immobilizing the photosynthetic biomolecules in photovoltaic devices and the progress in the photocurrent generation achieved. This review highlights the multidisciplinary nature of photosynthetic biohybrid devices and their future prospects in light of some of the research challenges and discrepancies witnessed by this field. The fascinating aspect of this research area is that it guides the biologists to explore the possibilities of improving protein stability and robustness suitable for solar cells and inspires the solar cell researchers to explore the physics behind the working mechanisms of biohybrid solar cells which can generate novel architectures in future solar energy conversion devices.

Sai Kishore Ravi
| Sai Kishore Ravi received his BE in Materials Science and Engineering from Anna University (CEG) in 2014 and is currently pursuing his PhD under the guidance of Prof. Tan Swee Ching at the Department of Materials Science and Engineering, National University of Singapore. Sai is interested broadly in studying bio-hybrid and nanostructured materials for energy conversion and storage. |

Swee Ching Tan
| Dr Tan Swee Ching received his Bachelor in Physics from the National University of Singapore. He then worked in Hewlett Packard Singapore and Ireland as a Laser Process and Equipment engineer to develop new technology for silicon micromachining. He was honoured the Award for Outstanding Achievement for his contributions to the company. In October 2006, he gained PhD admission to the University of Cambridge Electrical Engineering Department with Scholarships from Cambridge Commonwealth Trust and Wingate Foundations. His PhD work was to use photosynthetic proteins as light absorbing materials for solar cells under the supervision of Professor Sir Mark Welland. After getting his PhD in 2010, Dr Tan then moved to the Department of Materials Science and Engineering at MIT to become a postdoctoral associate working on nanoelectronics. He is currently an Assistant Professor at the Department of Materials Science and Engineering at the National University of Singapore. |
Broader context
Photosynthetic proteins present in plants and microbes naturally possess an excellent ability to harvest solar energy by means of the highly efficient light driven steps of photosynthesis. Exploiting the photovoltaic abilities of these proteins for developing biohybrid solar cells is a new area of research and is a promising approach for solar energy conversion, devoid of any economic and environmental constraints posed by other emerging technologies. This review briefly summarizes the basic structure and function of photosynthetic subsystems with a comprehensive presentation on the progress of the different approaches in integrating photosynthetic biomolecules in material environments. In light of this, this review also highlights the application prospects of this approach and the stability issues faced by the biohybrid devices.
|
1. Introduction
The diminishing reserves of fossil fuels and the environmental concerns in extracting the carbonaceous fuels from the earth's crust necessitate our independence from the non-renewable energy resources.1–3 A promising alternative to these fuels is to make use of carbon-free and profusely available solar energy.1–3 Solar energy is harvested by different approaches namely direct solar energy – electricity conversion, solar energy – chemical energy conversion and solar energy – thermal energy conversion.4 The first approach includes different types of solar cells while the second includes photoelectrochemical water splitting5 for hydrogen production6 and photoelectrochemical/photocatalytic reduction7 of CO2 to liquid fuels. These two approaches have witnessed considerable improvements due to bioinspiration.8,9 A major development in the domain of solar cells came with Michael Gratzel's work on Dye Sensitized Solar Cells (DSSCs).10–13 As opposed to the conventional solid state photovoltaic devices where the semiconductor performs both the tasks of light absorption and charge carrier transport, the functions in the Gratzel cell are decentralized and the light absorption is performed by an organic sensitizer (dye) held in a mesoporous and nanocrystalline scaffold.10–13 This design of a DSSC is an analogue of natural photosynthesis, where the function of chlorophyll is adopted by the synthetic dye and a cyclic electron flow as in photosynthesis has been facilitated by a redox mediator.8,9 The domain of solar-fuel generation also has similar bioinspirations, where the water-oxidation catalysts used in fuel cells are analogues of the oxygen evolving complex present in photosystem II of higher plants.8
Scientists of all times have been enthralled by materials engineered by nature. The level of sophistication and miniaturization found in nature has far surpassed that in the manmade.14 It is not a hyperbole to regard nature as an implicit school of materials science as nature has tactically experimented over ages and arrived at the best solutions, employing the principles of physics, chemistry and engineering.14–16 It is high time that engineers resort to bioinspiration in improving their devices, as the biological systems and mechanisms, in the process of continuing evolution, have been engineered by nature to be highly efficient, making them worthy models for design and engineering.16 Some microbes and plants found in nature have a superior system and mechanism for light harvesting and energy conversion. Their quantum efficiencies17 are higher than that of the manmade solar cells.18–20 Photosynthesis is an exemplary model for solar cell research as it is the prime mover powering the biological world, the mechanism behind the energy storage in fossil fuels and the sustainer of the earth's oxygenated atmosphere.21 The architecture and the internal circuitry of the photosynthetic systems are very sophisticated that the initial light driven steps have ≈100% quantum efficiency.18,22,23 Bioinspiration of photosynthesis in solar cells has instigated novel research perspectives which are, in close pace, moving towards devising a high efficiency solar cell. Artificial photosynthesis is one novel approach that tries to emulate the natural photosynthetic systems by employing intricate biomolecular complexes to execute the light harvesting and charge separation.24 Extensive research is being done to make synthetic complexes that match the sophistication and functions of the natural photosynthetic biocomplexes.
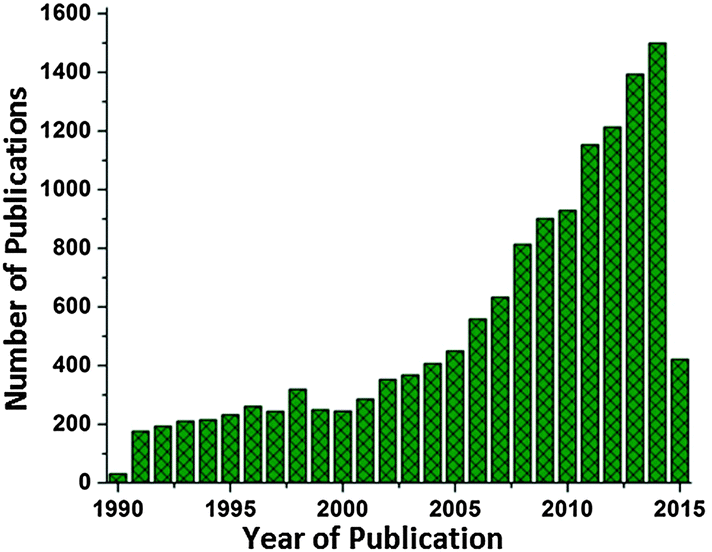
Though there is good progress in the supramolecular research emulating natural complexes, the mimics are still a far cry from the molecular circuitry of the photosynthetic protein complexes.25,26 This limitation thus gives rise to a new idea of making hybrid devices involving the biomolecular complexes effectively interfaced with manmade materials.27 This research perspective has gathered much interest in the recent years and there is good progress in the number of research studies (Fig. 1) utilizing the photovoltaic abilities of natural photosynthetic systems for various device applications like solar cells, photodetectors, biosensors and solar fuel cells.27 In this review, we shall discuss the recent developments and approaches in the in vitro employment of photosynthetic biocomplexes with a focus on solar cell applications.
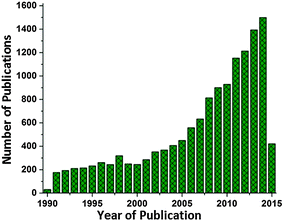 |
| | Fig. 1 Number of publications on photosynthetic biocomplexes utilized for solar energy harvesting. The plot includes the studies on photosynthetic apparatus from photosynthetic bacteria (RCs/RC–LH1/other subsystems/whole cell), higher plants (photosystems) and photosynthetic algae (subsystems/whole cell) for photovoltaic and solar fuel applications. The results are obtained from the Web of Science database (as on 21 Apr 2015). | |
2. Nature's photosynthetic apparatus
The profound strategy equipped by the biological systems to harvest solar energy is worthwhile to be appreciated for designing a biohybrid device. Organisms capable of deriving some of their cellular energy from light are termed as photosynthetic organisms.28 There are two types of them in general. One of them is the oxygenic phototroph that produces molecular oxygen, resulting from the oxidation of water; the other type is called the anoxygenic phototroph that does not involve the production of oxygen and instead of oxidation of water, a different electron donor is oxidized.28–30 Though there are some differences in their operation, the general principles of photon absorption and energy transduction are the same for both.30 Photosynthesis takes place by a systematic sequence of operations performed by various subsystems like photosynthetic pigments, light harvesting (LH) antenna systems, reaction centers (RCs) etc.29 The characteristics of these photosynthetic subsystems often differ with species and may even vary within the same organism; an exhaustive description on their classification, structure and function may be found in the literature.28,31,32
2.1 Role of photosynthetic pigments
In general, at the first phase of photosynthesis, photon/light absorption takes place aided by photosynthetic pigments namely chlorophyll and a few other accessory pigments like carotenoids.28 It is the architecture or the arrangement of these pigments that greatly supports the feasibility of photosynthesis rather than just the chemistry of the pigments.28 Considering the intensity of solar light and the dimensions of the pigment molecules, it has been estimated that a single chlorophyll molecule can only absorb ten photons per second, thus signifying the need for a well-designed arrangement of pigment molecules apposite for an efficient photon absorption process.28 It is an interesting fact that the photosynthetic pigments perform different roles at different sites of the photosynthetic apparatus. Some pigments are involved in a photophysical action (light absorption) while some carry out photochemical reactions. It was found that only less than 1% of the pigments in the photosynthetic apparatus are photochemically active and the rest of the pigments are involved only in the light absorption.28,33 The photon absorption creates an electronic excited state in an antenna pigment molecule, which is migrated from one molecule to the other and finally trapped within a site called the RC by various mechanisms.28
2.2 Role of light harvesting antenna complexes
The arrangement of pigments can often be referred to as an antenna system, as the arrangement enables collection and concentration of light suitable to facilitate an energy conversion.28 The photophysical properties of the pigments are fine-tuned by nature by an elegantly engineered biopolymer material called protein that offers the biological systems the required the degree of specificity, efficiency and control to accomplish a biological function.33 Thus the antenna systems exist as an assemblage of pigment and protein where the photosynthetic pigments are bound to the proteins in highly specific associations to ensue in an effective light absorption.28,33 The antenna complex concentrates the collected light energy to the RC where the photochemical reaction takes place.28,33 The structure of the antenna complex is not unique, because different photosynthetic organisms, as a part of evolution, adapt their light gathering systems in different ways to suit their environments.29 The mechanism of energy transfer to RCs varies with the type of antenna system (the type depends on the relative arrangement of pigments with the lipid membrane) present in the photosynthetic apparatus of an organism.28 In principle, the function of an antenna system, regardless of the species, is to efficiently absorb the light energy and efficiently deliver it to the RC.
2.3 Role of reaction center (RC) complexes
The RC is a complex organization of pigment molecules and redox active cofactors held in a precise three dimensional configuration by a protein scaffold.34 The RC is embedded in a photosynthetic membrane as a multi-subunit protein complex containing chlorophyll and other cofactors, with the extremely hydrophobic peptides threading the membrane back and forth.28 The general process that occurs in an RC is presented in Fig. 2. The pigments in the RC are chemically identical to those in the antenna complex but the environment in the RC renders these pigments fit for photochemical functions.28 In general, the photochemistry is affected by a special dimer of pigments (P) in the RC that acts as the primary electron donor for the subsequent electron transport process and it is into this dimer the antenna complex concentrates the light energy, making it electronically excited.28 As the excited electronic state is a highly reducing agent, it rapidly loses an electron to an acceptor molecule (A) and generates an ion pair state P+A−.28 In this primary process of photosynthesis, the electronic excitation energy is transformed into a chemical redox energy which is highly prone to be lost as heat, as the physical proximity of the highly oxidizing species P+ with the highly reducing species A− may easily deem a backflow of electrons to P+ from A−.28 But such a recombination is tactfully avoided by nature through a series of extremely expeditious secondary reactions that spatially separate the positive and negative charges.28,29
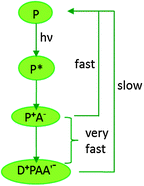 |
| | Fig. 2 General electron transfer scheme in photosynthetic reaction centers. Light excitation promotes a pigment (P) to an excited state (P*), where it loses an electron to an acceptor molecule (A) to form an ion-pair state P+A−; secondary reactions separate the charges, by transfer of an electron from an electron donor (D) and from the initial acceptor A to a secondary acceptor (A′). This spatial separation prevents the recombination reaction. [Adapted from ref. 28, with permission from John Wiley and Sons.] | |
In anoxygenic phototrophs, RCs exist as structurally separated operational units whereas in oxygenic phototrophs, RCs exist as an integral part of larger complexes called photosystems which are capable of oxidizing water.29,33 There are two different photosystems namely PS I and PS II; both of which are found only in oxygenic phototrophs like cyanobacteria, algae and plants, where they carry out the oxygenic photosynthesis by a coordinated sequence of actions.35 There are two kinds of RC complexes namely RC type I and RC type II, based on the identity of the electron acceptor present. The type I uses the FeS cluster as a terminal electron acceptor, and the examples of which are the RCs found in green sulphur bacteria and heliobacteria. The purple and green filamentous bacteria have the type II RC where the electron acceptor is pheophytin and quinone.29,36 Despite the structural differences, the key function of both RCs is the charge separation process which is attractive for the solar energy research. A comprehensive account on the structure and function of these photosynthetic subsystems is available in the literature.37,38
3. Photosynthesis for solar cells
3.1 Phases inspired
Photosynthesis in organisms involve a series of processes led by different components described above; but only the initial steps involving the light absorption and charge separation are essentially the most useful phases for solar cell applications. The primary stages of photosynthesis involve a sequence of actions starting with photon absorption which is an extremely swift process taking place in a few femtoseconds followed by fast photochemical reactions (a few ns to ps) and electron transport processes (a few μs).28,31 For biohybrid solar cells, the biocomplexes (RCs or photosystems) are intended to perform only these initial phases of photosynthesis in a manmade material environment. The initial steps are then followed by much slower biochemical (few ms) and physiological and ecological reactions involving synthesis and transport of stable products spanning a few seconds, which are not of direct use for solar cells though of some use in biofuels.28,31
3.2 What component of photosynthetic apparatus is useful for biohybrid devices?
The types of photosynthetic biocomplexes that can be used for solar energy harnessing have increased in the recent years thanks to the advancements in biochemistry and genetic engineering which have made it possible to extract different functional units of photosynthetic apparatus from different species and to improve their functions by genetic modifications. The RCs are some of the most widely studied photosynthetic components for employment in solar cells. While some studies only use the core RCs for the purpose, there are also a few studies using the RCs with the surrounding light harvesting complexes, in view of obtaining an improved performance.22,27,39–45 Photosystems I and II are also studied for photoelectrochemical applications. Some biochemical separations from photosystems are also being used, an example of which is the membrane fragments of PS II called PSII particles (BBY- or KM-).38 Biocomplexes of a higher structural level like chloroplasts46 and chromatophores47,48 are also used in biohybrid devices. Apart from utilizing the function of a protein biocomplex in a device, it has also been a well-known approach to use the photosynthetic pigments like chlorophyll and its derivatives in devices like DSSCs.49–52
4. Reaction centers – structure and function
4.1 The concept of reaction centers
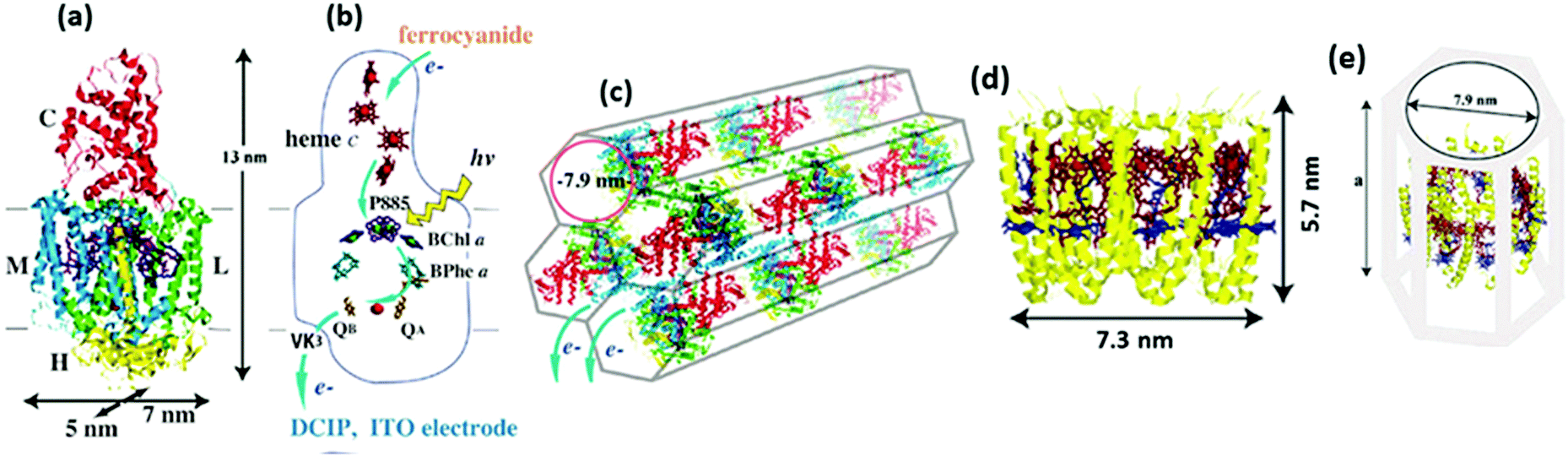
The RCs in anoxygenic phototrophs like purple bacteria have a simple and well-understood structure (Fig. 3) compared to the photosystems of cyanobacteria and higher plants.53 Though the structures of photosystems are more complex than RCs, there are some structural similarities between the RCs and the photosystems that facilitate a better understanding of the highly intricate photosystems.53 It has been found that the PS II shares some similarities to the RC in purple bacteria (i.e. RC type II) while the PS I resembles some structural aspects of the RC in green sulphur bacteria (i.e. RC type I).36,54,55 In a strict sense, the RC has to be defined as a minimal unit capable of photochemical charge separation between the primary electron donor and the primary electron acceptor which is then followed by stabilization of the separated charges.38 The photosystems are sometimes referred to as reaction centers, but the minimal unit responsible for the photochemistry is often not easily isolatable.29,38,54 Photosystems are in a way different from RCs as they are not the minimal units performing the photochemical reactions; being a complex assemblage of several constituents like pigments, antennae and proteins present in addition to the core system primarily carrying out the photochemistry.38 Several attempts have been made to biochemically separate the smallest structural units from the photosystems to make them as simple as RCs but they pose a few functional limitations.38 Since the RCs of purple bacteria have been extensively studied and are devoid of some of the structural complexities found in the photosystems, understanding their in vitro behaviour becomes more promising for various device applications. These RCs are also known to be more robust than those found in the photosystems of algae and higher plants.34 Though in a true sense, the photosystems are not to be classified as RCs, they have nevertheless been researched for use in bio-hybrid devices over years and are discussed in a few recent reviews.56,57 Thus, this review would mainly discuss the different research perspectives developed over years in employing the bacterial RCs in biohybrid devices and their progress in the photoelectric performance, highlighting some of the present problems and the future prospects.
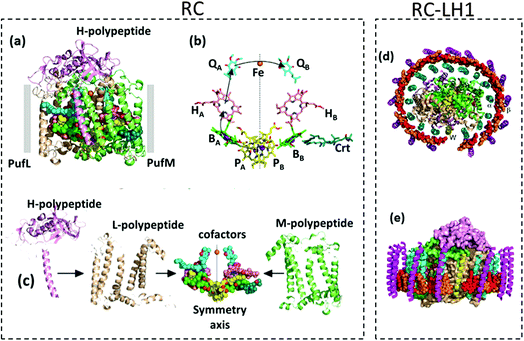 |
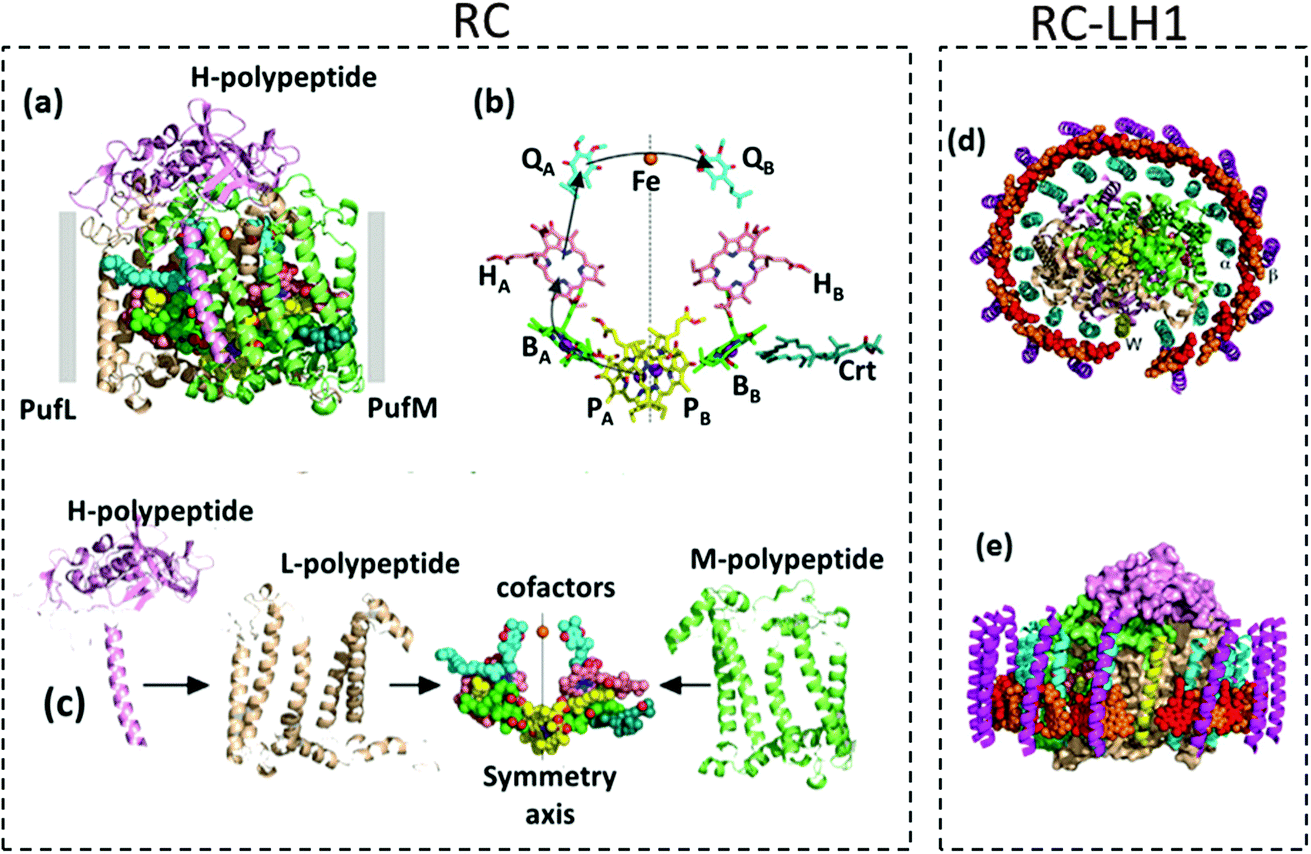
| | Fig. 3 (a) Structure of RC – the ten cofactors are held in place by a scaffold consisting of PufL and PufM, (b) the BChl, BPhe and ubiquinone cofactors form two membrane-spanning branches. Mg atoms of BChl and non-heme Fe are shown as spheres; the arrows show the route of electron transfer. (c) Individual structure of each RC subsystem. (d) and (e) Structures of RC–LH1 – the central RC is surrounded by an LH1 antenna pigment–protein comprising an inner ring of 16 α-polypeptides (cyan ribbons) and an outer ring of 16 β-polypeptides (magenta ribbons), each of which has a single membrane-spanning α-helix; sandwiched between these concentric LH1 protein cylinders is a ring of 32 BChls (shown as spheres alternating red/orange). [Reproduced from ref. 34 (with permission from Elsevier Limited), ref. 44 (with permission from John Wiley and Sons) and ref. 59.] | |
4.2 Purple bacterial reaction centers
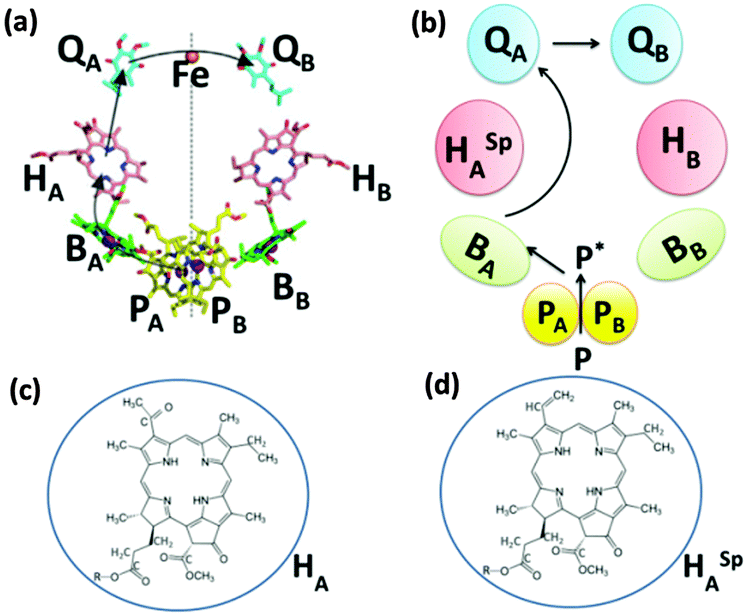
A comprehensive elucidation of the structural model and characterization of bacterial RCs deciphering their functions and mechanisms is available in several research articles.17,30,34,53,54,58,59 One of the widely used purple bacterial RCs for photovoltaic applications is extracted from Rhodobacter sphaeroides. The RC complex of this bacteria contains three polypeptides namely H, L and M that encase ten cofactors which are namely four Bacteriochlorophyll (BChl), two Bacteriopheophytin (BPhe), two ubiquinone molecules, a photoprotective carotenoid and a non-heme iron atom.34 The structure of the RC and the arrangement of its subunits and cofactors are often described with respect to the photosynthetic membrane of which the RC is inherently a part of and from which it has been isolated. The structure of the membrane is available in the literature28 and is not discussed here. The two polypeptides L and M, also referred to as PufL and PufM, are arranged around an axis of 2-fold rotational pseudo symmetry that runs perpendicular to the plane of the photosynthetic bilayer membrane28,59 and forms a scaffold that holds the cofactors in a precise configuration.34,59 The BChl, BPhe and the ubiquinone molecules are arranged at the interface of the L and M polypeptides in two membrane spanning branches named A and B that are related by a twofold pseudo symmetry.34,59 The cofactors located in the A and B branches are denoted by a subscript A and B respectively. Near the periplasmic side of the membrane, two of the BChl molecules form a dimer called ‘special pair’. These two closely spaced and excitonically coupled molecules are called PA and PB, as they are located in the A and B branch, respectively, which are shown by the yellow carbons in Fig. 3.34,59,60 Near the special pair there exist two monomeric bacteriochlorophyll molecules named BA and BB which are often called accessory bacteriochlorophylls, depicted by the green carbons in Fig. 3.34,59 These are then followed by the two BPhes (HA and HB) and the two quinones (QA and QB). The single carotenoid is embedded in the M-polypeptide, adjacent to BB.34,59 The non-heme iron is located right on the symmetry axis between the two quinones.34,59 The structure of the RC and the location of its cofactors are shown in Fig. 3(a) and (b) respectively. Although it is possible to isolate the RC as a discrete fully-functional photochemical unit, the so-called “RC–LH1 core complex” having an LH1 antenna pigment protein encircling the RC (Fig. 3(d) and (e)), which is found to naturally exist in all characterized photosynthetic purple bacteria, is promising to enhance the light absorption and hence the photoelectric performance in devices.34,44 In Rhodobacter sphaeroides the LH1 antenna consists of a large number of polypeptides that are basically of two types named α and β, each of which has a single transmembrane helix together with BChl and carotenoid pigments, which performs the function of light absorption.34 In nature, the LH1 protein forms a hollow cylinder that only partially surrounds the RC due to the presence of a PufX polypeptide,44,61 which can be removed by genetic engineering to obtain a more thermally stable RC–LH1 that has an enlarged LH1 antenna completely surrounding the central RC core.44 When light is incident on the RC complex, the photochemical process begins by the formation of a singlet excited state of the BChl special pair.59,60 In the case of RC–LH1, the light is first absorbed by the BChls and the carotenoids of the LH1 protein and the excitation energy is passed to the BChl special pair (P) that acts as a trap for the electronic energy in the RC core, thereby forming an excited singlet state P*. In RCs without LH1, the P* is directly formed by the light absorption of BChl molecules present in the special pair.59,60 The P* formed is a strong reducing agent and acts as the primary electron donor that transfers an electron to the adjacent BA molecule forming the P+BA− radical within 3 ps. The electron is then passed to HA forming P+HA− in about 1 ps and further transferred to QA forming P+QA− in approximately 200 ps.34,60 This is followed by a rather slow charge separation process in which the electron is transferred to the secondary quinone QB in a span of several microseconds aided by a few other electron or proton carriers.60 The formation of such a charge separated state P+QB− is the key principle utilized by the photosynthetic biohybrid solar cells for the photocurrent generation. A way of achieving this is to reduce the photooxidized special pair P+ by an electrode with the electrons being delivered to the counter electrode by an electrolyte containing an electron mediator, thereby generating a photocurrent. Different approaches have been attempted in the past few decades in devising an efficient method by which the separated charges are utilized for photocurrent generation in these photoelectrochemical cells.
5. Ways to interface RCs to electrodes
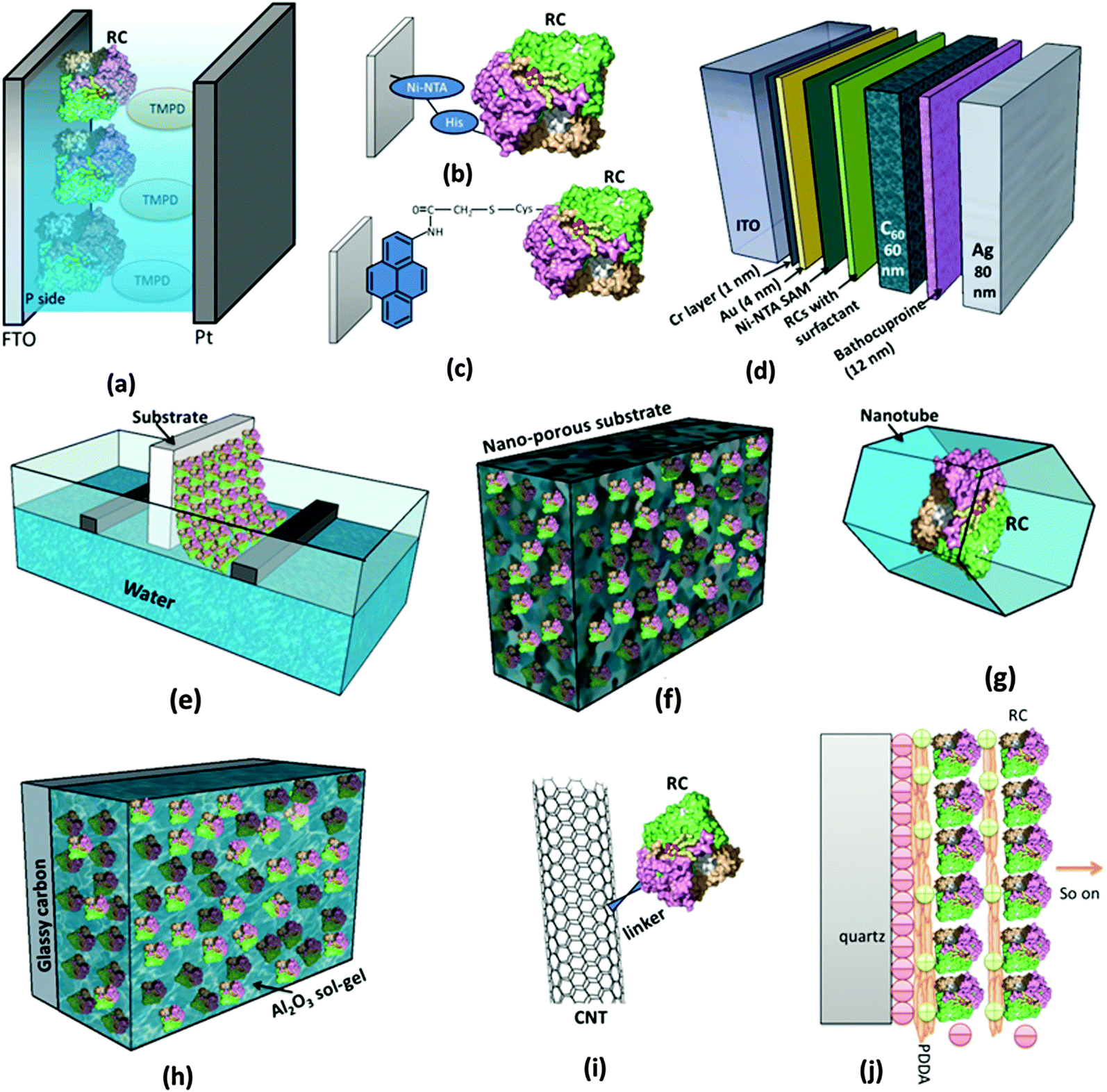
As in Fig. 4, for RCs to be employed in vitro in a photoelectrochemical device, effective immobilization on electrode is critical as it is important to retain the natural function of the biomolecules and efficiently transfer the photoinduced electrons to the electrode.62 Besides this specific use as photoelectrodes in solar cells, immobilizing biomolecules on a substrate in general has a basic benefit in characterizing them, as microstructural imaging and photochemical studies become convenient as proteins are intact on a substrate.62,63 A few recent reviews have elicited the various aspects of the protein immobilization on electrodes.64–66 The Langmuir–Blodgett (LB) method was widely being used to coat electrodes with RC layers and attempts were made to control the orientation of the RCs on the electrodes.67–71 The LB method involves consecutive crossing of an air–water interface by which a compact monomolecular layer of amphiphilic molecules is coated on the substrate with a well-defined molecular arrangement and orientation.64 Artificial lipid bilayers called liposomes were also used to immobilize the proteins on the electrode as the liposome film on the electrode can provide a nearly native environment for the proteins.64 One commonly used method for interfacing the RCs with the electrode is to use chemical functional groups or cross linkers that facilitate covalent binding of RCs with the electrode; nevertheless, it is also possible to attach RCs to the bare electrode surface by a simple physical adsorption.66 Both the chemical and physical binding methods suffer some disadvantages of structural and functional degradation.66 These drawbacks of physical and chemical binding are overcome by precoating the electrode with a self assembled monolayer (SAM), where an ultrathin ordered film is prepared based on the spontaneous molecular assembly using bifunctional reagents thereby providing an easy way to control the orientation and conformation of protein on the electrode surface.64,66 Attaching a genetically engineered poly-histidine (His) tag to RCs has been found to be of great use to control the orientation of RCs on the electrode surface.72 This is generally achieved by coating the electrode with a commercially available Ni2+ resin called Ni-NTA (Ni-nitrilotriacetic acid) which has a very high affinity for His-tagged proteins.26,39,62,73 There are also a few studies on immobilization of RCs in the polymer gel and sol–gel matrix. The use of nanomaterials as electrodes has also been increasingly studied for immobilization of RCs.64
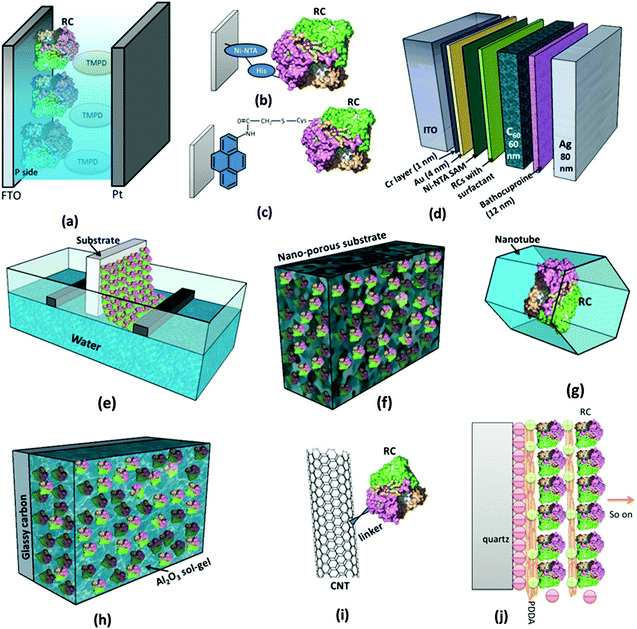 |
| | Fig. 4 Different ways of employing RCs in a photoelectrochemical set up. (a) RCs added to the electrolyte.44,45 (b) Use of the genetically engineered His-tag to immobilize RCs to the Ni-NTA modified electrode.72 (c) Attachment of RCs to the electrode by chemical linkers.77 (d) RCs stabilized in a solid state device.26 (e) Mono/multilayers of RCs coated on the electrode by the Langmuir–Blodgett (LB) method. (f) Immobilization of RCs on the nanostructured electrode.90 (g) Entrapping RCs inside nanopores and nanotubes.104,106 (h) RCs entrapped in sol–gel medium.86 (i) Binding RCs to nanotubes similar to other proteins as in ref. 101. (j) Electrostatically bound multilayers of RCs immobilized on the electrode.79,85 [RC image in figures (a)–(j) reproduced from ref. 34, with permission from Elsevier Ltd.] | |
6. Perspectives in incorporation of RCs into biohybrid solar cells and their performances
6.1 Bare electrodes
One of the earliest attempts of incorporating RCs in solar cells was by Janzen and Seibert in 1980 when they constructed a photoelectrochemical cell with RCs coated on the SnO2 coated glass electrode.74 The cell had a two electrode configuration with a photoactive RC coated working electrode and a Pt or a SnO2 counter electrode in a 0.1 M Na2SO4 electrolyte containing suitable buffer solutions.74 RCs were immobilized using a rather simple method of dipping the electrode in a concentrated suspension of RCs, thereby the RCs are physically adsorbed to the electrode when dried.74,75 When using platinized platinum as a working electrode, the charge separation resulting from the photochemical reaction in RCs could not be utilized, as there was supposedly a rapid back reaction from the electrode to the oxidized RC.74,75 To overcome this, an SnO2 semiconductor electrode was then used to obtain a better electrical coupling with the RC's photoinduced charge separation, a photocurrent of 0.3 μA cm−2 was thereby achieved on illumination of the cell with light of wavelengths greater than 600 nm.74 The use of an antimony doped SnO2 electrode as a working electrode and the addition of secondary quinone to RCs yielded a better photocurrent of 0.46 μA cm−2. The addition of secondary quinone was found to change the kinetics of electron transfer and stabilize the primary charge separation in a longer time duration thus facilitating enough time for the tunnelling of electrons through the SnO2/RC interface.75 Although no attempt was made to orient the RCs in these pioneering studies, it has been predicted that a better photoresponse could be possible with controlled orientation.74
6.2 Use of chemical linkers
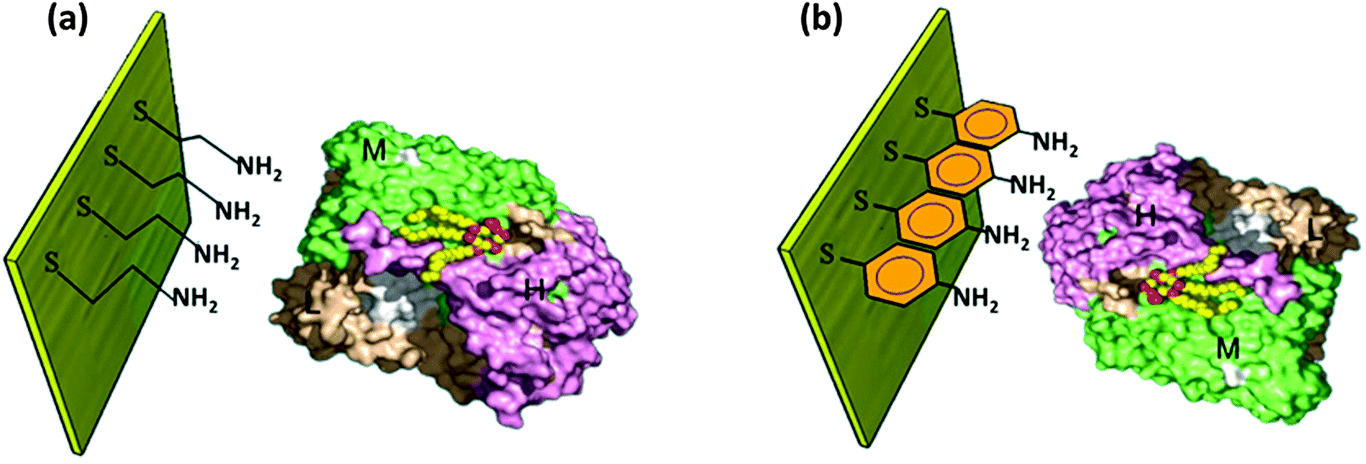
A number of perspectives then engendered to control RC orientation, one being the widely used method of making SAMs. Oriented immobilization of RC monolayers on the platinum electrode76 and the pyrolytic graphite electrode77 modified by organic functional groups has been studied. With the use of bifunctional agents with condensed aromatic groups and cysteine thiol groups, the photocurrent was found to increase greatly as the RCs were oriented by these linkers.77 Different bifunctional agents have been used to establish site specific binding of RCs with electrodes.78,79 When aminothiophenol (ATP) was used, the bonding site was non-heme iron and the electron transfer was observed between the non-heme iron and the primary quinone whereas when mercaptoethylamine (MEA) was used the electron transfer was between the primary donor (P) and the bacteriopheophytin (Bphe) resulting in a greater photocurrent than that of ATP.78 The schematic of the RCs immobilized on the Au electrode with the two kinds of bifunctional agents is shown in Fig. 5. The RC-MEA modified photoelectrode exhibited a higher photocurrent (40 nA cm−2) than the RC-ATP (30 nA cm−2) which underscores the importance of employing an appropriate chemical linker to improve the binding of RCs with the electrode and to desirably orient them for improved electron transfer efficiency. The importance of selecting the chemical linker has also been realized in effectively adsorbing RCs on the electrode.39 The terminating group in the chemical linker used for the SAM has been found to affect adsorption of RC complexes on the electrode.39 The complexes exhibited a higher stability and were well adsorbed to a gold electrode with amino group terminated SAMs while they were partially stable with carboxyl ended chemical linkers and were found to be greatly denatured when SAMs with terminal methyl groups were used.39 Thus the choice of chemical linker has a greater role in affecting the orientation of the RC complexes on the electrode. The selection of an electrolyte and its constituents are also vital for the photoelectric performance but by and large the addition of appropriate electron transfer mediators has been found to significantly enhance the electron transfer efficiency and hence the photocurrent. The use of ubiquinone-10 as a diffusionally mobile electron transfer mediator in the solution was also found to improve the electron transfer between the RC and the electrode thereby increasing the photocurrent.76,77 Cytochrome c, ferrocene80 and methyl viologen39 have also been used for the purpose. It is a general approach to improve the electron transfer by two electron transfer mediators (a donor and an acceptor). Ubiquinone and cytochrome c are often added to the electrolyte to serve as an electron acceptor and a donor respectively.20,81–83
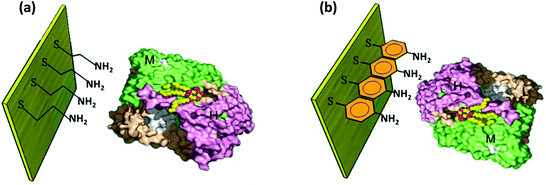 |
| | Fig. 5 RC bound to the electrode by a chemical linker. (a) Mercaptoethylamine (MEA) and (b) aminothiophenol (ATP). [Adapted from ref. 78, with permission from Elsevier Ltd, RC image in figures reproduced from ref. 34, with permission from Elsevier Ltd.] | |
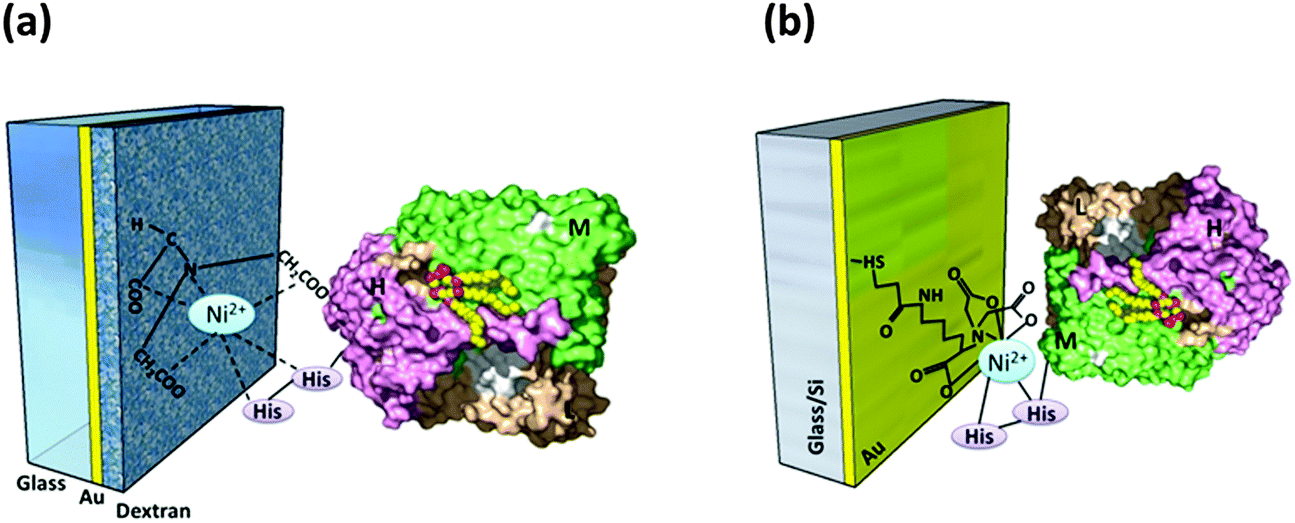
6.3 Genetic modifications
Genetically engineered poly-histidine tags are often attached to different subunits of RCs to vary the orientation. Two differently tagged RCs were studied for their orientation on the Ni-NTA modified dextran coated gold electrode: one of which had a hexameric histide tag (His6) added to the H subunit of the RC (HHisRC) and the other had the His6 tag attached to the M subunit of the RC (MHisRC). Upon irradiation of light the electrode with HHisRC (Fig. 6(a)) exhibited a much stronger photoinduced displacement current72 than that with MHisRC evincing the fact the H subunit being relatively hydrophilic (compared to M and L) more strongly interacts with the hydrophilic dextran matrix than M and thus making unidirectional orientation of HHisRC possible as opposed to MHisRC.72 Better orientation and electron transfer properties were obtained mainly when HHisRCs were bound to that SAM coated gold electrode with specific linkers that make the SAM surface moderately hydrophilic.84 Nevertheless, the MHisRC configuration (Fig. 6(b)) where the primary donor of the RC faces the substrate has also been found to retain the photochemical and electron transfer activity yielding a significant photocurrent on illumination.26,62 Trammel et al. obtained a sustained cathodic photocurrent of 30 nA cm−2 for the MHisRC configuration which was attributed to the photoinduced reduction of the primary donor followed by electron transfer through the L branch and finally the transfer to ubiquinone 10 that acted as an electron acceptor.62 A number of reports following this had a detailed explanation on the photoelectric performances of different orientations which is discussed in section 6.5 of this review.
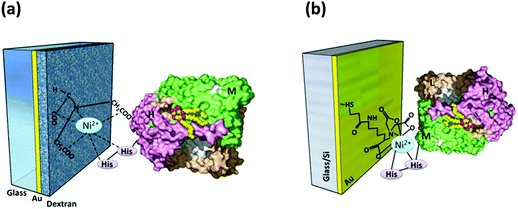 |
| | Fig. 6 (a) HHisRC on the Ni-NTA modified electrode. [Adapted with permission from ref. 72.†] (b) MHisRC on the Ni-NTA modified electrode. [Adapted from ref. 62, with permission from Elsevier Ltd.] (RC image in the figures reproduced from ref. 34, with permission from Elsevier Ltd.) | |
6.4 Electrostatic adsorption
6.4.1 Effect of RC multilayers.
A superior photoelectric performance was obtained when multiple layers of RCs were coated to the electrode.79 Since by principle, polyelectrolytes adsorb to an oppositely charged surface, reaction centers which are generally negatively charged can be adsorbed to the electrode by making the electrode surface positively charged.85 Alternatively, the first layer of RCs can be chemically linked to an electrode and the other RC layers could be assembled layer by layer by electrostatic interactions. The stacking of multiple RC layers was prepared by controlling the pH so that the negatively charged RCs electrostatically adsorbed to a positively charged polyelectrolyte namely poly(diallyldimethylammoniumchloride) (PDDA) and the RC multilayer was bound to the Au electrode by a bifunctional agent 2-mercaptoacetic acid (MAA).79 The photoelectrode with 24 layers of RCs yielded a photocurrent of 77 nA cm−2, while a monolayered RC electrode yielded just 8.5 nA cm−2.79 Electrostatic binding of RC multilayers to a substrate is possible without the use of chemical linkers by employing an electrode which by itself has the required surface charge. A clean quartz electrode which is negatively charged in nature can bind to the negatively charged RC layer by using positively charged PDDA polyion glue in between.85
6.4.2 RCs embedded in sol–gel.
Employing sol–gel modified electrodes has also been a useful approach for a better photoelectric performance. The RCs were immobilized on a glassy carbon electrode by using a positively charged hydrophilic Al2O3 sol–gel matrix that electrostatically binds the negatively charged RCs.86 Under optimum experimental conditions (neutral pH, H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al = 200:1), high photocurrents of the order of a few micro amperes were obtained.86
:Al = 200:1), high photocurrents of the order of a few micro amperes were obtained.86
6.5 Effect of different orientations of RCs
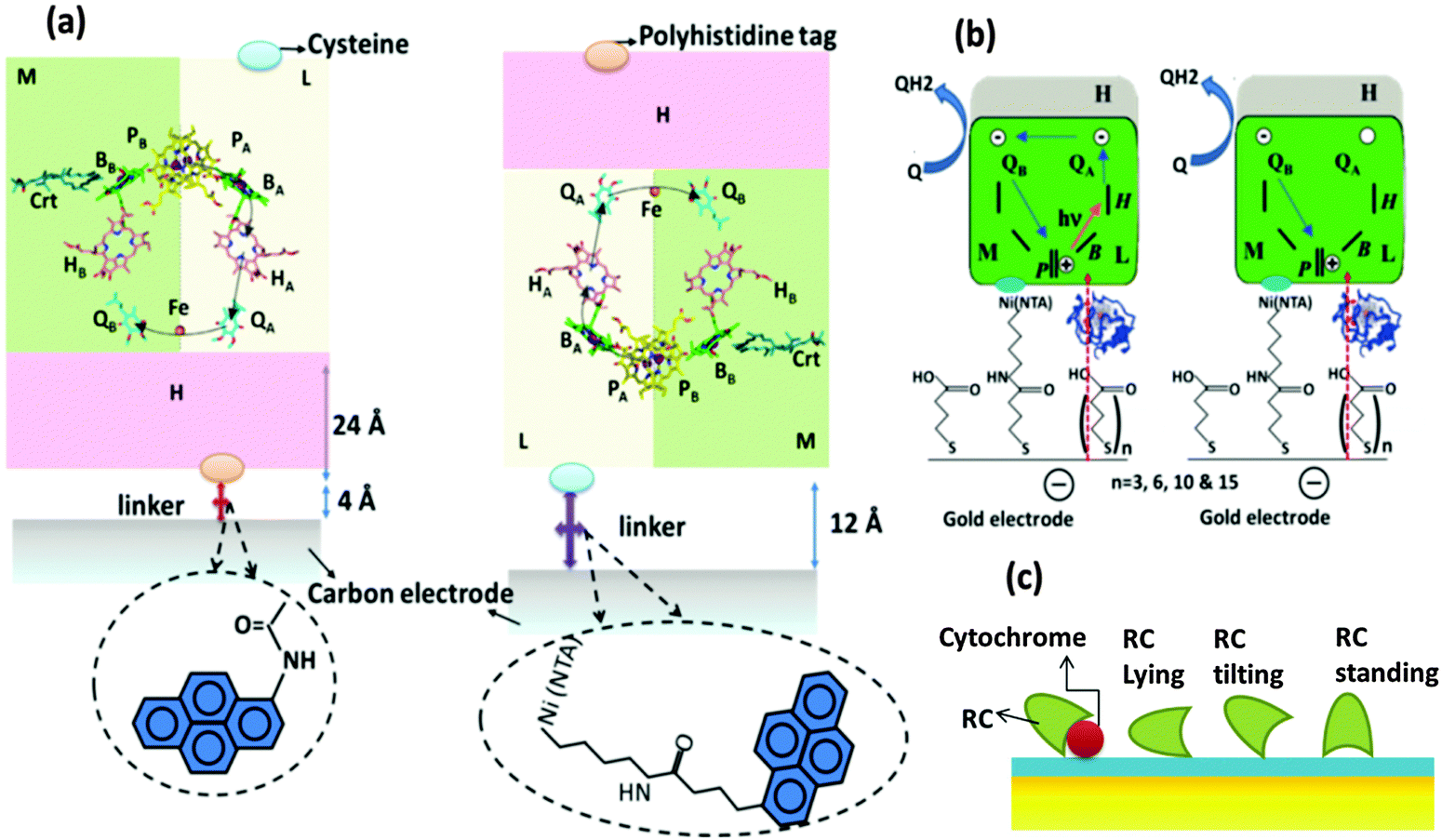
The need for oriented RC layers on the electrode for an improved performance has been found necessary even in the earliest attempts of RC immobilization in solar cells but the photoelectric performances of different orientations were not extensively studied until recently. Two opposite RC orientations, one with a primary donor (P-side) facing the electrode and the other with the acceptor (H subunit side) facing the electrode, have been elaborately studied.81 The two orientations of the proteins on a carbon coated gold grid electrode are shown in Fig. 7(a). In the first orientation, the RC is bound to the electrode by means of a bifunctional linker with one end having an NTA group charged with Ni2+ suitable to bind with the His tag of the RC and the other end having a pyrene group to attach to the carbon electrode and in the second type, N-(1-pyrene)iodoacetamide was used as a linker that binds to the RC H-subunit through a single cysteine group.81 Ubiquione-10 and cytochrome c (reduced by Na2S2O4) were used as diffusible electron transfer mediators. Two profound conclusions were drawn from the photoelectric studies of these two orientations: (1) the photosynthetic RCs act as a photorectifiers making the photocurrent always flow in one direction i.e. from the primary donor to the primary acceptor. This applies well to both the orientations in Fig. 7(a) where photocurrent is anodic if the H subunit side faces the electrode and cathodic if the P side faces the electrode. (2) The orientation of RCs with the P side bound to the electrode exhibits a higher photocurrent and reaches the photochemical steady state approximately an order of magnitude faster than that orientation with the H-subunit bound to the electrode.81 Reasonable explanations were put forth for the decreased photocurrent observed in the case of a H subunit facing the electrode. Difference in surface coverage of RCs could not be a reason as equally high surface coverage of RCs was ensured for both the orientations.81 Though the length of the bifunctional linker used to bind H to the electrode is shorter in length (4 Å) than that used to bind the P-side to the electrode (12 Å), the electron transfer efficiency in the former orientation is lesser.81 However, X-ray crystallographic studies revealed that the actual distance between the final electron acceptor and the electrode bound to the H subunit is 28 Å considering both the thickness of the H subunit (24 Å) and the linker length (4 Å).81 It has been estimated that a variation of 20 Å in the distance between the electron donor and the electron acceptor in a protein would change the electron transfer rate by 1012 fold.87 Thus, the mediocre performance of the orientation with the H subunit bound to the electrode is attributed to the higher electron tunnelling distance between the electron acceptors and the electrode, owing to the presence of the relatively thick H subunit.81 A similar account on the importance of the protein orientation to minimize the distance of the electron transfer pathway is elucidated by Kondo et al.39 with the study of photoelectrodes modified by RC–LH1 isolated from Rhodopseudomonas palustris, where the orientation of the RC complex with the H-chain facing the electrode was found to be more favourable than the opposite orientation.39
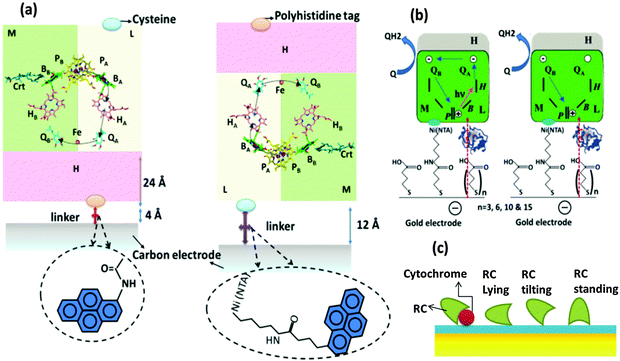 |
| | Fig. 7 (a) Two possible ways of RC binding and ET pathways between RCs and the electrode. P-primary electron donor (special pair), B-monomeric bacteriochlorophyll, H-bacteriopheophytin, Qa and Qb-primary and secondary electron acceptors (quinones). [Adapted from ref. 31 and 81, with permission from Elsevier Ltd.] (b) Photoinduced- and dark-electron transfers in RC-Cyt-SAM-gold electrode. [Adapted with permission from ref. 82, Copyright © 2007, American Chemical Society.] (c) Different orientations of RC with cytochrome c on NTA SAM modified electrode. [Adapted with permission from ref. 18, Copyright © 2006, American Chemical Society.] | |
The effect of the dependence of electron transfer kinetics on the distance between the electron acceptor and the electrode has been systematically studied using a series of MHisRCs modified Ni-NTA SAM coated gold electrodes having SAMs of different thicknesses with the number (n) of methylene units in the linker molecule (n = 3, 6, 10 and 15) being the measure of SAM thickness (Fig. 7(b)).82 In the photoelectric measurements, two electron transfer mediators were added namely ubiquinone (Q2) that acts an electron acceptor and cytochrome c that acts as an electron donor and also serves as a conductive wire in coupling the working electrode with the RC's special pair.82 The photoelectric studies proved a significant dependence on linker lengths and thus the SAM thicknesses. The photocurrent was found to be independent of the distance (linker length) when the RCs are at shorter distances from the electrode and it decreases to a great extent with the distance from the electrode. A maximum steady state photocurrent of 167 nA cm−2 was observed when 7-carboxyheptyl disulphide acid having 6 methylene units was used as a linker. The linkers of lengths 3 and 10 methylene units yielded photocurrents of 161 and 158 nA cm−2 which are still close to the maximum photocurrent obtained, but there was a drastic decrease in the photocurrent to about 25 nA cm−2 when the linker length was 15 methylene units. The study highlights the importance of the protein's proximity to the electrode to achieve a sound RC-electrode junction for high photoelectric efficiency.82 These observations are also in congruence with the photoelectrochemical cell reported by Kondo et al.39 where the photoelectric performance of RC–LH1 immobilized on the gold electrode coated with SAMs of different linker lengths of alkanethiols NH2(CH2)nSH (n = 2, 6, 8, 11) was studied.39 The photocurrent was found to be maximum for the linker length n = 6 and decreasing with increasing linker length.39 The photocurrent decrease was due to the increase in the distance between the electrode and the RC–LH1 with the maximum current at a separation distance of 1 nm corresponding to the linker length n = 6 and lower currents for n = 8 and 11 that resulted in higher separation distances of 1.4 and 2.1 nm respectively.39 It is interesting to note that the maximum photocurrent was not observed at the lowest linker length (n = 2) as the adsorption of the RC complexes to the electrode was poor. It was found that the adsorption of the complexes increased with increasing linker length which being a conflicting condition for improving the electron transfer efficiency, a trade off was essential and was achieved at a linker length of 6 methylene units. The effectiveness of cytochrome c in improving the electrical coupling of RCs and the electrode has been studied. An Ni-NTA SAM coated gold electrode was used to immobilize the RCs with His tagged M subunits.18 A time dependent improvement in photocurrent was observed upon addition of cytochrome c to the electrolyte and the photocurrent increased 20 to 40 times higher than the initial value after a few minutes of incubation which was observed to occur with both the oxidized and the reduced forms of cytochrome c.18 Cytochrome c was assumed as sitting on the SAM surface and the RC was assumed to have a single point of contact with the SAM. In the absence of cytochrome, three possible RC orientations were explained (Fig. 7(c)). In the first case, the RC was assumed to lie on the SAM surface. In the second case, the RC was assumed to be oriented to SAMs in the same way if cytochrome was present whereas in the third case, the RC was assumed to stand on the surface of SAMs with its primary donor facing the SAM surface.18 With the assumption that the open area around the RC was surrounded by water in all the three cases, it was found that the electron transfer could be possible in the absence of cytochrome only when the RC is close to the standing position.18 The addition of cytochrome c was found to offer a shorter electron tunnelling path and a more effective electron transfer, thus improving the photoelectric performance acting as a conductive wire connecting the RC's special pair P and the electrode.18 Further to this study, Mahmoudzadeh et al.83 reported the study of photoelectric performances of different configurations of RC modified photoelectrodes with varying RC-electrode distances, in light of their electron tunnelling probabilities and mechanisms.83,88
6.6 Nanocrystalline and nanoporous electrodes
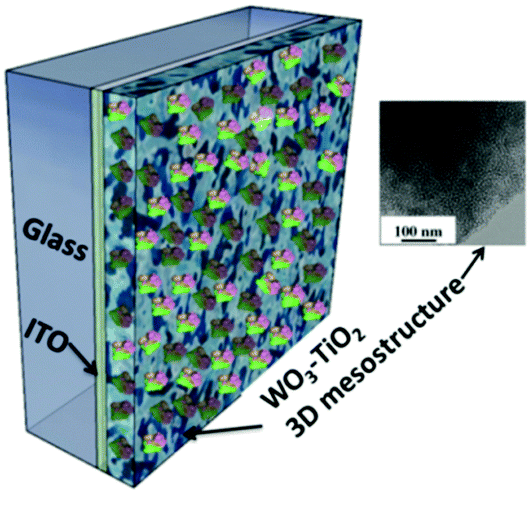
Immobilization of RCs on nanocrystalline electrodes has been found to improve the photoelectric performance of the devices. RCs have been immobilized in a nanoporous nanocrystalline TiO2 film coated on ITO-glass by anodic electrodeposition.89 The larger surface area in the porous matrix facilitates higher adsorption of RCs leading to an increased photochemical activity even without the use of any chemical linkers. A photocurrent of 8 μA cm−2 was achieved with a biophotovoltaic cell with the RC modified nanoporous TiO2 working electrode and Pt counter electrode with an electrolyte containing sodium dithionite.89 The photoelectric performance of TiO2 doped amorphous WO3 and WO3–TiO2 nanoclusters was found to be superior to that of individual TiO2 and WO3. RCs were immobilized on a tailored three-dimensional (3D) wormlike mesoporous WO3–TiO2 electrode (Fig. 8) that had a number of features favourable for an enhanced photoelectric performance notably the well matched energy levels of WO3–TiO2 with RCs.90 The immobilized RC had retained the natural function and activity due to the mesoporous structure that had open pores of size ≈7 nm matching the dimension of the RC, an ideal hydrophilic surface and suitable surface charge.64 The performance stability of the RC immobilized mesoporous electrode was reasonably good that the photocurrent decreased only 15% after a continuous illumination of 1 hour, which is promising for constructing bioelectronic devices though inadequate for solar cells.64
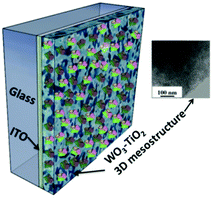 |
| | Fig. 8 RCs immobilized to the WO3–TiO2 3D mesostructure; inset – TEM image of the mesoporous structure of WO3–TiO2. [Adapted with permission from ref. 90, Copyright © 2005, American Chemical Society.] | |
An equally good photoelectric performance was exhibited by a thick film (4 μm) of the nanocrystalline porous TiO2 photoelectrode embedded with RCs. The porous electrode was prepared by annealing a paste of TiO2 nanocrystalline powders with polyethylene glycol and a few stabilizing additives layered over an ITO glass at 550 °C.91 The RCs were adsorbed to the porous electrode by incubating the electrode in the RC solution and the absorption was found to be complete within 24 hours of incubation. The photocurrent from the RC modified electrode was significantly higher than that from the unmodified, which was due to the additional injection of electrons from RCs to the conduction band of TiO2.91
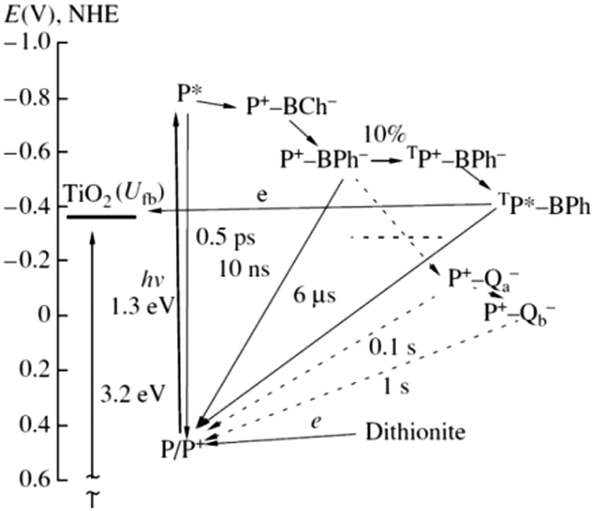
The immobilization of RCs in the porous electrode offers a wider range of surface energy states and hence electron transfer pathways which is regarded as an added advantage besides the high protein loading capacity.91 The various electron transfer pathways possible is schematically shown in Fig. 9. The fact that the halftime of P+ and BPh− reverse recombination is about 10 ns and the potential electron donors being located at a greater distance from the RC surface makes it necessary for an efficient electron transferred from the excited state P* of the RC to TiO2 to occur within a few nanoseconds which is difficult to be realized as there are not many ways to increase the life time of the charge separated state in the absence of a natural electron donor like cytochrome c.91 Achieving a direct electron transfer from RCs to the electrode is hardly possible that a mediator based transport is often required in these photoelectrochemical cells.83,92 An alternative to using an electron mediator could be a long lived P dimer triplet93,94 which is formed as a result of the ion radical pair P+ BPh− transiting from the dimer in the singlet state to the triplet state when the quinone acceptors are chemically reduced and unable to participate in electron transfer events.91 This triplet state of the pair TP+–BPh− has a formation probability of 10% at room temperature and recombines to form the triplet excitation state TP* that has a higher lifespan of 6 μs.31,91 As the energy level of the TP* is very close to the energy bands of TiO2, a low rate electron transfer is possible to occur from TP* to TiO2 spanning a few microseconds.91
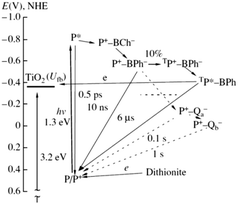 |
| | Fig. 9 Possible pathways of electron transfer in the TiO2–RC photovoltaic cell. Dashed lines denote the electron transfer events that did not take place in the RC due to a dark reduction of the quinone acceptors by dithionite. The potential is shown relative to the standard hydrogen electrode; Ufb is the conduction band potential of the semiconductor. [Reproduced from ref. 91, with permission from Springer.‡] | |
6.7 Effect of pigment substitution
Highly enhanced photocurrent was further achieved using a pigment exchanged RC adsorbed onto the nanostructured WO3–TiO2 matrix.64,95 A pigment replaced RC mutant containing spinach pheophytin in place of bacteriopheophytin was used to alter the energetics and kinetics of the electron transfer process. Such RCs (RC-Phe) entrapped in the nanoporous electrode exhibited a significant delay in the excitation transfer and the relatively slower charge separation was attributed to the higher energy level of P+Phe− than that of P+Bchl−.64,95 Native RCs and pheophytin replaced RCs (RCPhe) were also studied for their photoelectric performance in SAM coated gold electrodes.96 A thin film of 2-mecraptoacetic acid (MAA) and polydimethylsiallylammonium chloride (PDDA) was coated on the gold electrode to form the SAM.96 Native RCs (termed as wild type RCWT in Fig. 10(a)) and the RC mutant (RCPhe in Fig. 10(b)) were assembled on separate SAMs forming two different photoelectrodes namely RCWT-PDDA-MAA-Au and RCPhe-PDDA-MAA-Au.96 It was observed that the short circuit photocurrent obtained for the RCPhe-PDDA-MAA-Au (∼45 nA cm−2) was about 15% greater than that for RCWT-PDDA-MAA-Au (∼30 nA cm−2).96 Since both the photoelectrodes had almost the same protein loading, the differences in photoelectric performances were mainly attributed to the relatively increased electron injection for the SAMs with pheophytin replaced RCs.96 The electron transfer process for the mutant RC is found to be different from native RCs and it is shown in Fig. 10. The enhanced photoelectric performances are principally affected by the higher population and longer lifetime of P* or P+BChl− in the mutant RC as opposed to wild type RCs.96 The wider energy gap between P+QA− and P/P+ was also found to contribute to the superior photoelectric performance of mutant RCs.96
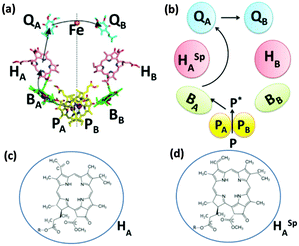 |
| | Fig. 10 (a) Electron transfer in native RC. [Reproduced from ref. 34, with permission from Elsevier Ltd.] (b) Electron transfer in the mutant RCphe. (c) Molecular structure of bacteriopheophytin in native RC. (d) Molecular structure of spinach pheophytin (HspA) in the mutant RCphe. [Adapted from ref. 95.] | |
6.8 RCs bound to nanotubes
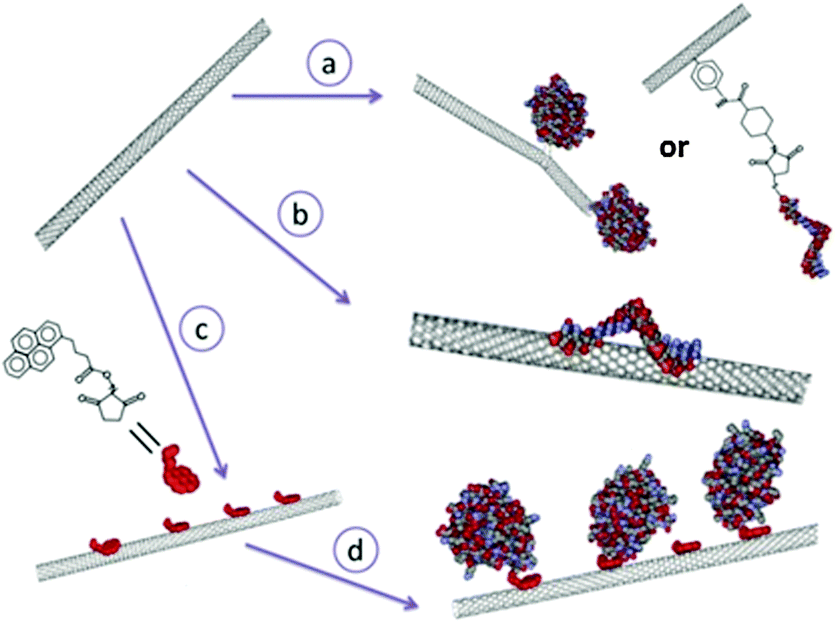
Nanotubes offer a number of attractive advantages for protein immobilization. Firstly, they provide larger inner volumes related to the dimensions of the tube which can be occupied by desired chemical or biomolecular species; the inner and outer surfaces have distinct properties enabling differential modification suitable for chemical or biofunctionalization; the open ends of the tubes render the inner surfaces accessible favouring easier addition of biomolecules.97 Though the most commonly used nanotubes are of carbon, there are also other materials like silica,98 boron nitride99etc. that are used for protein immobilization. Carbon nanotubes are promising materials for bioelectrochemistry principally due to the possibility of bringing them close to the redox potentials of proteins.100 The review by Wenrong Yang et al.101 discusses the various methods of modification of carbon nanotubes with biomolecules grouped under three major heads namely the covalent attachment, non-covalent attachment and hybrid approach (Fig. 11). In the covalent approach, the biomolecule is chemically attached to the nanotube by means of a bifunctional spacer group or by direct reaction with a preferred site of the biomolecule or by chemical binding to the carboxylic acid functionalized nanotube.101 In the non-covalent approach, the hydrophobic nature of the CNT surface is exploited to physically adsorb suitable complementary biomolecules.101 These hydrophobic interactions can take place both in the internal and external surfaces of CNTs but the biomolecules are non-specifically bound to the CNT by this method.101 In the hybrid approach, a small anchor molecule is first non-covalently adsorbed to the CNT and the biomolecules are then chemically linked (covalently bound) to the anchor molecules.101 Functionalization of various protein molecules to CNTs101–103 has been demonstrated but the attempts of encapsulation of biomolecules in CNTs were less fruitful as they could produce only a meagre quantity and it was difficult to obtain a uniform distribution.20 The immobilization of a biomolecule in the small internal cavity of a nanotube is rather difficult and limits the size of the biomolecule.101
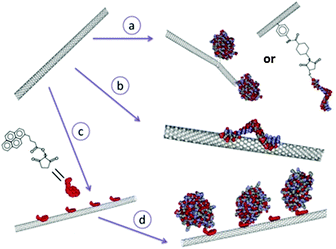 |
| | Fig. 11 The three main approaches for modifying carbon nanotubes with biomolecules: the covalent approach (route a), non-covalent approach (route b) and hybrid approach where a small molecule ‘anchor’ is first non-covalently absorbed to the carbon nanotube (route c), followed by a chemical reaction between the anchor and the biomolecules of interest (route d). [Adapted with permission from ref. 101.] | |
RCs have nevertheless been encapsulated in CNTs as these nano-organized materials in CNTs promise high functional density, increased stability, one dimensional mass and electron transfer.20 Photoelectric performance of reaction centers immobilized on HOPG has been compared with RCs encapsulated in a parallelly aligned, densely packed multiwalled CNTs open at one end and electrically contacted at the other end.20 Based on the electron-transfer properties, the open ends of CNTs tend to resemble the edge planes of HOPG, while the walls take after the properties of the basal planes of HOPG.100 The CNT electrode was fabricated by a template assisted chemical vapour deposition growth technique. The CNTs in the array were separated by Al2O3 spacer that would electrically isolate the CNTs and prevent the binding of RCs to the outer surface of CNTs and the other side of the CNT array-oxide film was covered with a sub-micrometer layer of gold.20 RCs were immobilized to the HOPG electrodes using a bifunctional linker that has a pyrene group at one end for the attachment to the electrode and a Ni(NTA) group at the other end for binding the RCs that are genetically engineered with a polyhistide tag at M subunit (MHisRCs).20 For CNT electrodes, RCs were allowed to penetrate into the CNTs by diffusion and capillary motion is effected by the incubation of the RC solution with CNTs at 4 °C for 1–2 h.20 The bifunctional linker was used for CNT electrodes as well, where the pyrene end of the linker non covalently binds the electrode and the Ni(NTA) end binds the His tagged M subunit of the RC.20 It was found that the protein loading in CNT electrodes with a linker was several folds greater than that of the HOPG electrodes and the native CNT electrodes.20 The photoelectric studies of CNT and HOPG photoelectrodes revealed that the photocurrent is cathodic indicating that the reaction centers are ordered with the P side facing the electrode. When RCs are bound to HOPG without any linkers, a small photocurrent (30 nA cm−2) was obtained and in the presence of the linker, the photocurrent increased to about 314 nA cm−2. The RCs bound to CNTs via a linker yielded a higher photocurrent of 1414 nA cm−2. It is interesting to note that the electron transfer is faster with CNT photoelectrodes than that of HOPG, the RCs being bound to both the carbon surface in the same way, which was attributed to the lower internal resistance and higher unidirectional conductance of CNTs along the length of the nanotube as opposed to that of planar graphite.20
Novel materials with hexagonal honeycomb structured pores are attractive for loading protein complexes of any size as it is possible to control the diameter of the tubular pores by chosen process routes.104,105 RCs104 and light harvesting complexes106 from a thermophilic purple photosynthetic bacterium, thermochromatium tepidum, were successfully adsorbed to a folded-sheet silica mesoporous material (FSM) and the binding of RCs to FSM of different pore sizes was studied. Interestingly, it was found that the protein complex was capable of retaining the photosynthetic function inside the material only when the pore size matched the size of the complex.104,106 The interior surface of the pores being hydrophobic, the pores are expected to provide the protein complex an environment similar to the hydrophobic membrane.104,106 As shown in Fig. 12(a) and (b), the RC complex has four polypeptide subunits, L, M, H, and C binding various cofactors including the P885, which is a special pair of bacteriochlorophyll a (BChl a) that donates an electron to the bacteriopheophytin a (BPhe a) within a few picoseconds followed by electron transfer to the electron acceptor menaquinone QA in less than 1 ns and further gets rereduced by an electron from one of the four hemes c in the C subunit, a detailed description of its structure and mechanism may be found elsewhere.107–110 FSMs of different pore diameters namely 2.7 nm, 7.9 nm and 9 nm were used to study the photochemical capability of the proteins in the porous matrix. The RC may be understood as a cylindrical unit with 5 × 7 nm cross section along the membrane surface and a 13 nm height normal to the membrane.104 A maximum absorption of the RC was evident in FSM with 7.9 pore diameter that fits the RC well as the pore diameter closely matches the cross sectional dimensions of the reaction center complex (Fig. 12(c)).104 While it was possible to adsorb 0.29 g of RCs per gram of silica in FSM with 7.9 nm diameter, the maximum protein adsorbtion possible in the other two FSMs was much lower; in FSM with 2.7 nm pores, the adsorption amount of RC was 0.02 g g−1 of silica and for that with 9 nm pores, it was 0.1 g g−1 of silica.104 An intact structure and photochemical activity was found to be possible only with the FSM of fitting pore size which was also witnessed when light harvesting complexes LH2 were loaded to various FSMs.106 As shown in Fig. 12(d–f), the light harvesting complex adsorbed well with the FSM with 7.9 nm pores as the dimensions of the LH2 complex fit to the pore size.106 A high adsorption capacity of 1.1 g of LH2 per gram of silica was possible with FSM of 7.9 nm pores.106 This is higher than that obtained for the RC as the size of the LH2 is more close to the pore diameter than that of the RC, which is suggestive of an enhanced photoelectric peformance possible if RCs with LH2 can be immobilized to these porous materials owing to a higher adsorption capacity.
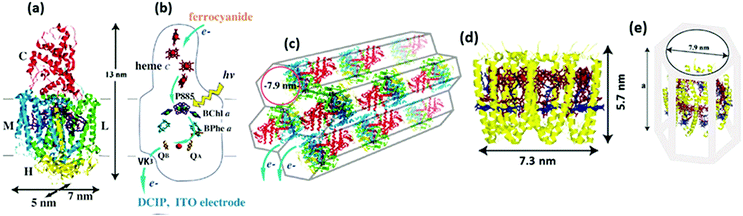 |
| | Fig. 12 (a) Structure of the RC complex-(L, M, H, and C are the RC's subunit polypeptides), (b) electron transfer cofactors and the pathway inside the RC, (c) schematic view of the RC inside silica nanopores in FSM7.9. (d) Molecular structure of the LH2 complex, (e) schematic view of the LH2-FSM7.9 conjugate. The value for ‘a’ in (e) was estimated to be 9.2–11.4 nm upon the binding of 1.11 mg of LH2 per mg of FSM7.9 based on the results in the specific study. [Reproduced with permission from ref. 104 (Copyright © 2010, American Chemical Society) and ref. 106 (Copyright © 2006, American Chemical Society).] | |
6.9 RCs in solid state devices
It was for the first time that the RCs were used in an electrolyte-less energy device when Rupa Das et al. reported the photoelectric performance of an RC immobilized solid state set up (Fig. 4(d)).26 Surfactant like peptides111–114 have been used to stabilize the RC complexes and a subnanometer thick layer of amorphous organic semiconductor has been deposited in between the RC and the metal electrode to serve as a solid state antenna enhancing the light absorption.26 Rupa Das et al. constructed an RC based solid state electronic device with a conductive ITO coated glass electrode, coated further by a nanolayer of the gold anode (with a Cr adhesion layer in between Au and ITO) to which MHisRCs are oriented by an Ni-NTA SAM on the gold surface.26 The RC layer is further coated by a preferentially electron transporting fullerene C60 followed by a layer of bathocuproine (BCP) and finally by a layer of silver that acts as a cathode. This solid state device has exhibited the highest photocurrent of 0.12 mA cm−2 under an excitation intensity of 10 W cm−2.26
6.10 Photosynthetic proteins in solution
6.10.1 Use of two mediators.
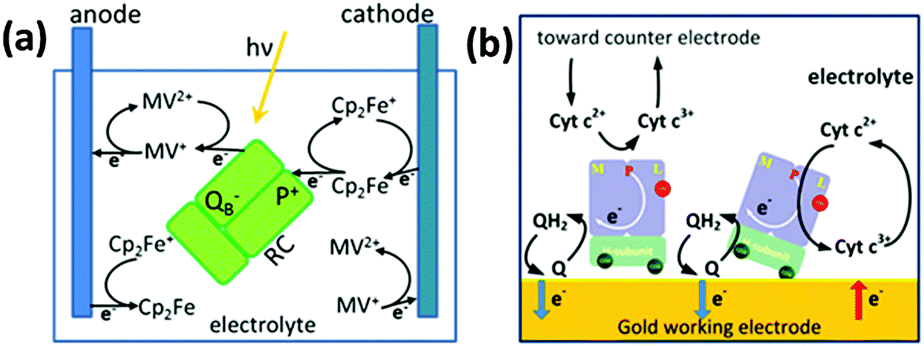
Takshi et al.80 studied the photoelectric performance of a photoelectrochemical cell with the RCs dissolved in the electrolyte (Fig. 13(a)). Since it was found that a significant fraction of electron transfer occurs through electron transfer mediators even when the RCs are directly attached to the electrode, this study attempted to improve the performance by making the charge transfer fully diffusion controlled and without the use of any direct electron transfer as the RCs are not bound to the electrode.80,92 Two electron transfer mediators namely ferrocene and methyl viologen that were added to the electrolyte prevent the charge recombination by effecting faster redox reactions and transfer the charges to the electrodes.80 The reactions involve oxidation of Cp2Fe to Cp2Fe+ by an electron transfer from ferrocene to P and reduction of MV2+ to MV+ by the electron transfer from QB to methyl viologen.40,80 The products formed MV+ and Cp2Fe+ interact to form MV2+ and Cp2Fe, the rate of which increased as the concentration of the MV+ and Cp2Fe+ produced by the redox reaction increased with time and a steady state was reached when the recombination rate reached a limit called generation rate.80 In this study, the photoelectrochemical cell works in such a way that the two electrodes used have different kinetic rates for the two mediators, thus oxidation would be favourable on one electrode and the reduction on the other.80 A cathodic photocurrent was obtained when the reduction rate of Cp2Fe+ on an electrode surface exceeds the recombination reaction rate and the oxidation rate of MV+.80 A steady state photocurrent was said to be possible if the oxidation rate of MV+ at the anode is higher than the recombination rate and the reduction rate of Cp2Fe+.80,115 A photocurrent of approximately 400 nA cm−2 was obtained upon illumination of the photoelectrochemical setup employing the HOPG cathode, Pt anode, electrolyte having 15 μM of the RC, 0.75 mM of Cp2Fe and 0.75 mM of MV2+, with a light of intensity 2.8 mW cm−2.80 The photocurrent was found to increase linearly with light intensity and nonlinearly with RC concentration in the electrolyte.80 It was suggested that a better photocurrent is still possible with a higher RC concentration, the maximum concentration used in the work being 15 μM.80 Recently, a photo electrochemical cell with RCs added to electrolyte containing ubiquinone-10 and cytochrome c has been reported where the RCs showed an increased propensity to bind to the gold electrode due to a cysteine tag in the H subunit (Fig. 13(b)).116 It was found that about 78% of the RCs bind preferentially to the gold electrode while the rest bind to the counter electrode or float freely in the electrolyte negligibly affecting the photocurrent generation. Though the RCs were in direct contact with the gold electrode, the photocurrent was close to zero in the absence of the two electron transfer mediators, suggesting the least possibility of a direct electron transfer and the importance of the mediators in achieving the electron transfer.116
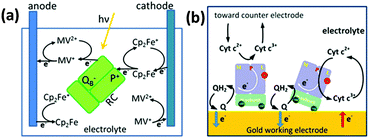 |
| | Fig. 13 (a) Reaction centers added to the electrolyte with two electron transfer mediators (ferrocene and methyl viologen). The thick arrows represent reactions with high reaction rate constants, while the thin arrows represent reactions that reduce cell efficiency. [Adapted from ref. 80.] (b) Reaction centers added to electrolyte with two electron transfer mediators (ubiquinone-10 and cytochrome c). [Reproduced with permission from ref. 116, Copyright © 2012, American Chemical Society.] | |
6.10.2 Use of a single mediator.
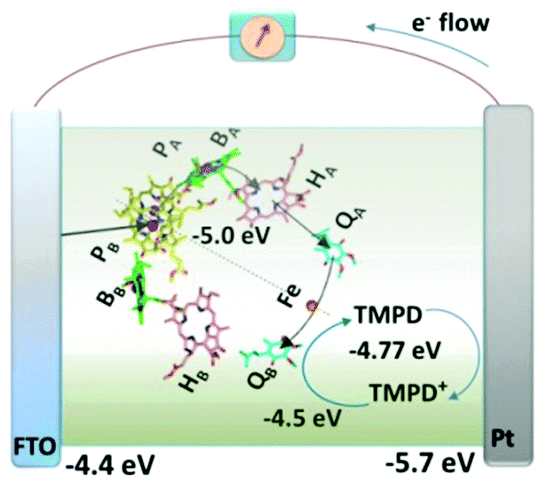
Unlike the observation in the above discussed work, the RCs added to the electrolyte were still found to adhere to the electrode without the use of any chemical linkers or tags. Employing a single redox mediator can achieve this direct electrical contact between the RCs and the electrode which may be understood as an electrolyte that by itself also a good electron transfer mediator.44 The photoelectrochemical cell employed in the study involved RC–LH1 complexes from Rhodobacter Sphaeroides and the fabrication procedure was simple where a mixture of the protein and the mediator N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) was injected into a 10 μL cavity formed between a fluorine-doped tin oxide (FTO) front electrode and a Pt rear electrode joined together by a sealing foil (Fig. 14).44,45 The FTO electrode acted most preferentially as the working electrode as the RCs being hydrophilic in nature tend to adhere more to the hydrophilic FTO electrode than to the relatively hydrophobic Pt electrode.44 Thus a majority of RC–LH1 complexes were believed to bind to the FTO electrode with any of the two possible terminals of the complex and the electrons were shuttled from the other terminal of the complex to the Pt counter electrode.44
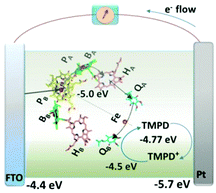 |
| | Fig. 14 Mechanism proposed for operation of the RC and RC–LH1 cells with TMPD as the single redox mediator. Current-supporting RCs (as shown) or RC–LH1 complexes (not shown) are oriented with the P-side close to the FTO-glass electrode. Arrows indicate the route of electron transfer through the RC (blue), through the TMPD/TMPD+ pool to the Pt electrode (orange) and into the P-side of the RC from the FTO-glass electrode (green). Electron transfer within the RC from the P bacteriochlorophyll dimer to the QB quinone occurs via a monomeric bacteriochlorophyll (BA), bacteriopheophytin (HA), and quinone (QA). [Adapted from ref. 44, with permission from John Wiley and Sons.] | |
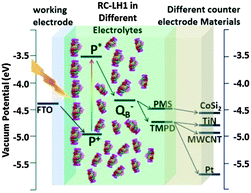 |
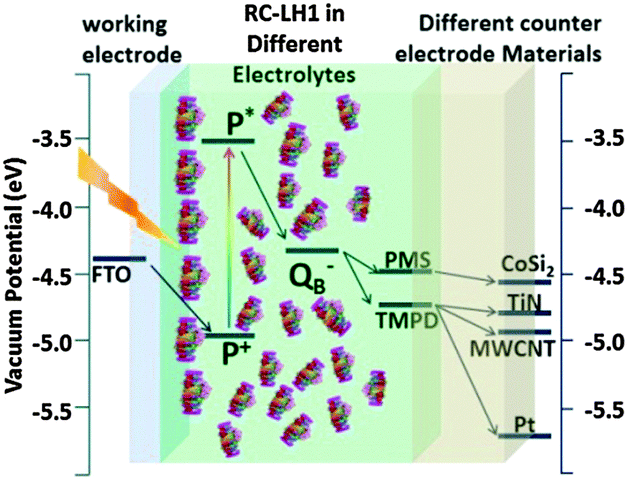
| | Fig. 15 Schematic showing the vacuum potentials of the key components of RC–LH1 based photoelectrochemical cell. Photoexcitation of P to P* changes its redox potential and triggers electron transfer to the QB quinone. With TMPD or PMS (for CoSi2 electrode) as mediator a flow of direct current to the back electrode is observed. [Adapted from ref. 27, with permission from John Wiley and Sons.] | |
7. Where are we heading to?
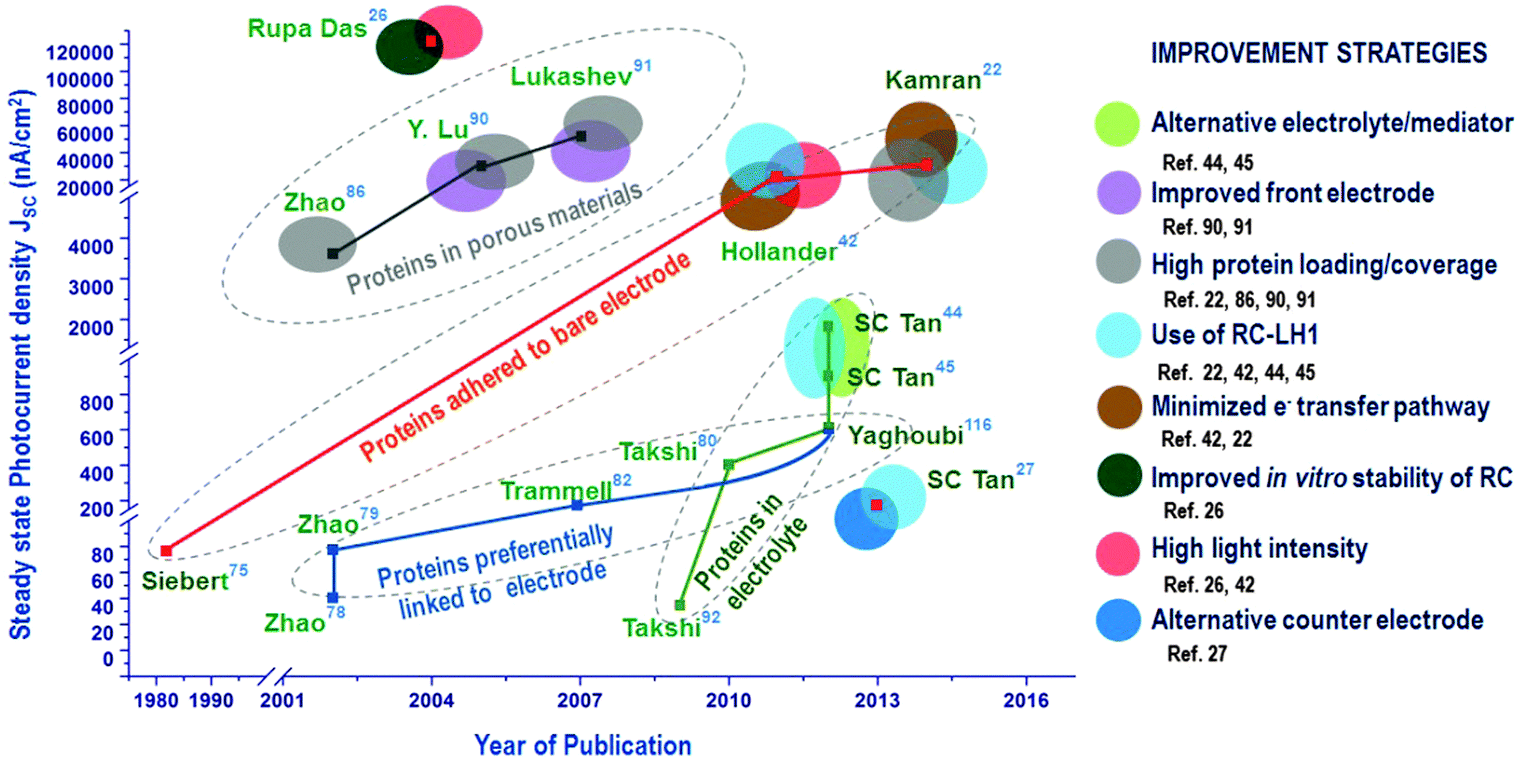
While enormous efforts have been directed to control the orientation of RCs by chemically modifying the electrodes, in recent years, an improved photoelectric performance has been proved possible even without any linkers and by more novel immobilization techniques with no effort to orient the RCs on the electrode.44,117 Adding the RCs to the electrolyte is one such an approach that just uses a bare electrode where the photocurrent generation is aided by electron transfer mediators. Hollander et al.42 showed that a high photocurrent density of 3980 nA cm−242 was possible when the bare electrode was dipped in the solution containing RCs and electron transfer mediators, an approach similar to that used in the pioneering studies74,75 where simple dipping of the electrode in RC suspension yielded around 300 nA cm−2.74 The only difference in these recent studies is the use of the electron transfer mediators that increases the efficiency of electron transfer and hence the photocurrent. RCs with a light harvesting complex are also being increasingly studied for their photovoltaic applications. The maximum photocurrent density reported so far for biohybrid solar cells with RC–LH1 is 45 μA cm−2 obtained for RC–LH1 from Rhodopseudomonas acidophila.22 However, the highest photocurrent reported for RC so far is 120 μA cm−2 which is obtained from an RC based solid state device.26 The progress in the photoelectric performance of the RC-based biohybrid solar cells achieved over decades is worthwhile to be understood is very encouraging. The progress of steady state photocurrent densities achieved in the four main approaches used in RC-solar cells is presented in the road map (Fig. 16). The various improvement strategies adopted so far by researchers are also presented alongside the photocurrents as this may be handy to compare and devise future techniques to improve the performance of these solar cells (Fig. 16). One classic approach is the adsorption of proteins to an unfunctionalized electrode which after a great number of years have gathered interest as they have been proved to be one of the effective methods when used with a higher protein loading (i.e. the amount of protein) and a minimal protein–electrode distance. The pioneering studies of Janzen and Seibert74,75 directed the research more towards protein orientation on electrodes which was then utilized by a majority of researchers by attaching the proteins to electrodes functionalized by chemical linker molecules and by engineering special tags in the protein to ensure uniform orientation. The progress achieved by this approach of ‘preferential linking to electrode’ has been less appreciable though it spanned more than a decade. Over this period there were also a few attempts to improve photocurrent by increasing the protein content in the solar cell which was promising and yielded more attractive results with the idea of using nanoporous materials tailor-made to match the size and energy levels of the proteins. A recently developed approach is the employment of RCs in the electrolyte which has shown great progress in a short time span especially due to the use of alternative electrolytes. It is also evident from the road map that using RC–LH1 has also been a very useful strategy to obtain a better photoelectric performance. It is also interesting to note that two different approaches may coexist as in ref. 116 where the RCs are added to the electrolyte while some extent of oriented attachment to the electrode was also attempted by using mutant RCs with an external Cys-group that tends to preferentially attach to the electrode.116 Unlike other strategies, using very high light intensity may not be very useful as solar cells ultimately are to use the sunlight, which is discussed in the next section of this review. As also notable in the roadmap, some aspects like the counter electrode material and the protein stability in the solar cell have rarely been explored and are worthy to be our future research avenues in this field.
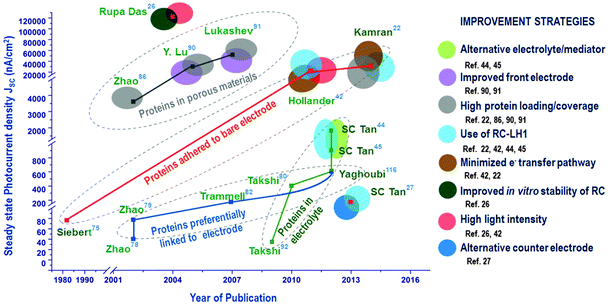 |
| | Fig. 16 Progress in photocurrent generation in photosynthetic protein based photovoltaic devices. | |
7.1 Shortcomings in reporting the performance of biohybrid solar cells
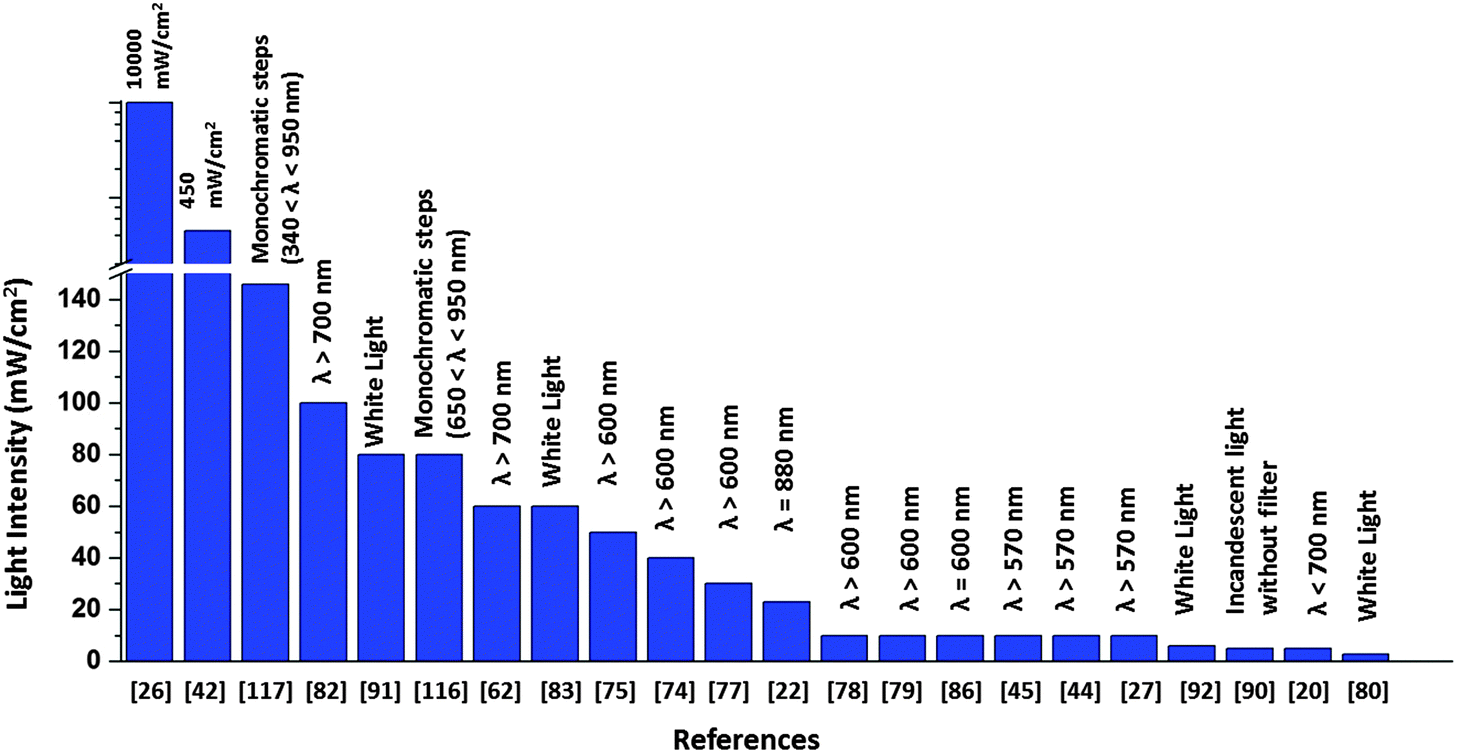
There is a major disparity in the light intensity used for illuminating the solar cell, which greatly influences the value of photocurrent generated. The light intensity used in the literature ranges from 0.1 mW cm−2 to as high as 105 times greater intensities (Fig. 17). As these research studies are oriented towards devising an efficient biohybrid solar cell, it would be realistic to use intensities comparable to the intensity of natural sunlight. A relatively very high value of 120 μA cm−2 was obtained when the RC implanted photovoltaic device was illuminated with a laser light of intensity 10 W cm−226 which is far greater than that of sunlight, a value comparable to light intensity from 100 suns as mentioned by Kamran et al.22 As suggested by Henry Snaith,118 the light source used in the characterization of any solar cell must closely match the terrestrial solar spectrum, which is possible by using a xenon lamp with appropriate light filters118 Although the laser light26 used in the work makes it hard to realize the device as an RC based solar cell, several issues related to the stability and robustness of the complexes have been addressed in the device construction using protective coatings and surfactants which can be adopted by new protein-based solar devices to improve their performance.26 The light intensity used for the obtained photocurrent is in many cases not reported39–41,119 Another issue is that the active area of the device is not mentioned in some reports which makes it difficult to compare the photocurrent of their devices. This is because a much bigger area will contribute to a higher current output. Reporting the photocurrent density or the active area of illumination is important as a mere high value of photocurrent with the active area unspecified can be ambiguous. Open-circuit voltage output is often not reported in most studies which makes it impossible to calculate the overall conversion efficiency of the devices. The solar cell efficiency being dependent on both photocurrent density and open circuit voltage, the focus is mainly on improving the photocurrent density while the latter is often ignored. The need for a standardized measurement is thus highly felt among the biohybrid solar cells, which is now crucial to be addressed in order to ensure much constructive and meaningful research in future.
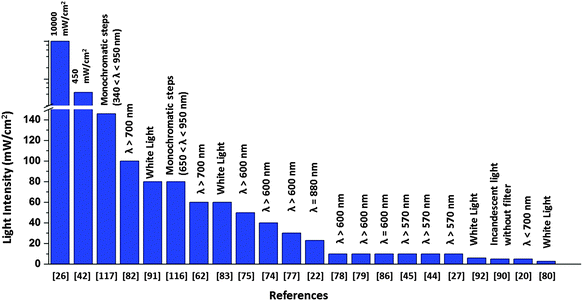 |
| | Fig. 17 Intensity and wavelength range adopted in the literature for solar cell illumination. | |
7.2 RCs in solar cells – fish out of water!
Enormous efforts have been made to improve the useful life period of these cells are way lesser than the conventional solar cells. This is evident with most of the reported studies on photosynthetic protein-based solar cells. Though high photocurrents are achieved with these cells, they are short-lived and they can hardly produce any photocurrent after a week which is indicative of some kind of degradation with time.80 As the major photovoltaic components in these solar cells are biomolecular complexes, it is vital to understand their vulnerabilities in a foreign environment. The biomolecular complexes often lack a protective environment in the solar cells which deteriorates their functionalities, ensuing in a short-lived, solar cell. The in vitro stability of the RCs needs to be improved to devise a more useful and realistic solar cell. As RCs are isolated from their native environment, they are prone to conformational changes as the stabilizing effect offered by the membrane lipids is lost. Lipids play a vital role in affecting the biophysical and electron transfer properties and promote structural stability and flexibility, binding the light harvesting cofactors and filling the intra protein cavities.34 The stability of these biomolecules is mostly affected by two main stress factors – light and temperature, which cause their denaturation.120 Denaturation of proteins involves a loss of structural integrity that occurs due to the separation of secondary structural subunits and unfolding of domains outside the membrane.121
7.2.1 Temperature induced and light induced denaturation of RCs.
The structural integrity of an RC can be predicted from the absorption spectrum. As RCs denature, the characteristic absorbance bands of the RC cofactors are seen displaced and shifted, that is suggestive of unfolding of RCs, where some of the cofactors are no more bound to it.121,122 The stability of RCs is also studied by discerning their conformation changes through specialized techniques like Surface Enhanced Resonance Raman Scattering Spectroscopy, Fluorescence Resonance Energy Transfer etc.123–125 The RCs in their native heterogeneous environment of lipids have a greater resistance to thermal denaturation and are stable up to 70 °C.121 When isolated, the possible structural changes associated with the thermal effects can be understood by studying the energetics and kinetics of the denaturation process.121,125 Hughes et al.121 proposed a kinetic model for the thermal denaturation of the Rba. Sphaeroides RCs that demonstrates the likelihood of an intermediate state with respect to heating times.121 When RCs were held at a high temperature for a very short period and cooled down to room temperature, the spectral properties lost at the high temperature state were regained, while this did not happen so with longer holding periods at a high temperature.121 Since it is known that a complete reversibility from the denatured state is impossible, there must be some intermediate state from which complete reversibility to the native state was possible.121 As shown in Fig. 18, at low temperatures, RCs are in the native state (N) which upon heating follow a kinetic pathway to the denatured state (D) involving an off-pathway intermediate state (I).121 The intermediate state is interpreted as a misfolded RC with a distorted structure but with a significant fraction of cofactors still bound to RCs in such a way that the transition from I to N is reversible and this reversible transition is coupled to an irreversible transition to the denatured state where the cofactors are unbound from the RCs and the polypeptides are separated leading to the unfolding of the protein.121
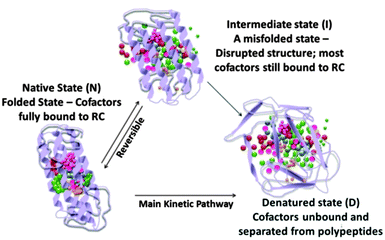 |
| | Fig. 18 A schematic of thermal denaturation of RCs as described in ref. 121. The spheres of various colours represent the RC cofactors and the ribbons represent the polypeptides. (The scheme is illustrative and does not represent the true structure of the RC.) | |
Upon continuous illumination with intense light, a light induced denaturation occurs by either or all of the three mechanisms – 1. singlet oxygen sensitization, 2. reduction of quinone (QA) to quinol (QAH2) and 3. localized heating.120 The temperature induced denaturation has also been found to be linked with light stress as an intense illumination often creates thermal effects.120 It has been found that, upon continuous illumination, the light induced changes in the redox states of the RC have a greater effect on its thermal stability and in turn its photochemical activity compared to the effect of the light intensity and the duration of illumination.120 Detailed mechanisms of the light induced and temperature induced damage in RCs have been discussed in the literature.120,121
7.2.2 How to improve stability?.
The stability of the protein-based solar cells has been found to last only for a few hours upon continuous illumination. Upon continuous illumination, a 15% reduction in photocurrent was observed in a protein-based photoelectrochemical cell90 after 1 hour, while a 60% reduction was observed after 10 hours of continuous illumination in another.42 When stored under dark conditions at low temperatures, the solar cells have exhibited extended lifetime as the denaturation is prevented by minimizing both the stress factors.42 The stability has also been found to increase by removing oxygen from the environment which has been realized by implanting RCs under anaerobic conditions and by perfectly sealing the device.22,116 One major approach to improve the stability of RCs in the solar cell is to achieve an RC-compatible environment which can be best achieved by mimicking the native membrane of RCs. In contrast to the isolated RCs, the photosynthetic membranes used as the photovoltaic component in the solar cell exhibited more resistance to stress factors and the functionality of RCs was maintained for three days when continuously illuminated.126 The LH1 antenna complex surrounding the RC has been found to increase its robustness and improve the resistance to aerobic condition and intense lights126 The use of non-conventional surfactants and various lipid–surfactant systems in preserving the RC's functionality in vitro has been discussed in a few reviews.127,128 While one way to improve RCs’ stability is to make the material environment more conducive to RCs, another approach is to make the RCs more robust and resistant to harsh environments and stress factors. An example of the latter is to increase the carotenoid content in the complex as the carotenoids are known to play photoprotective roles and improve the robustness of RCs.126 Recently, a copolymer–lipid environment has been found to be capable of improving the in vitro stability of RCs.129 This area deserves more research focus to devise better material systems that can offer a more protective and conducive environment for RCs, so that efficient solar cells with higher lifetimes can be made.
7.3 Prospects of photosynthetic biohybrid devices
An attractive aspect of these biohybrid devices is that they employ a low-cost biodegradable photovoltaic material that is abundantly available and can easily be extracted from the photosynthetic organisms with no harm to the environment. At present these devices are under laboratory research and quite far from commercialization mainly due to the inferior photoelectric performance and the stability issues. The photoelectric performance has a great scope for improvement by developing new device architectures employing new electrode and electrolyte materials envisaging the photosynthetic energy transfer and electron transfer pathways possible in the design. Apart from the direct photovoltaic applications, photosynthetic proteins also find use in solar fuel generation and biosensing applications.
7.3.1 New device architectures.
Developing solid state devices with different lipid–surfactant environments for proteins is one promising research direction as the use of surfactants has already been proven to increase the usable life of the device to several weeks.26 Many viable combinations of electrode materials and redox mediators that can be employed in biohybrid devices can be identified by considering the energy level match for ensuring the cyclic electron flow and the stability of the biomolecule in the environment. A number of metal silicides can be the potential counter electrode materials in a set up as in Fig. 15. TiSi2 (ϕ ≈ −4.6 eV)130 that has a work function and resistivity131 similar to CoSi2 can be used with the PMS mediator while Ti5Si4 (ϕ ≈ −5.6 eV)130 can be used with the TMPD mediator as the work function is similar to that of Pt. Considering the high optical transparency to visible light and the high carrier mobility, graphene can be a good front electrode material to immobilize photosynthetic proteins.132 Device architectures with a better photoelectric performance would be possible by controlling the mode and the rate of electronic energy transfer (EET) possible in the molecular circuitry of the device. In a photosynthetic apparatus, the excitation energy absorbed by a pigment molecule is transferred to another molecule separated by distances upto several tens of angstroms by a process of resonance energy transfer.133 Depending on the electronic coupling between the pigment molecules and the coupling of the photosynthetic protein with its environment, the energy transfer mode could be either an incoherent EET (Förster energy transfer) or a coherent EET or a relaxation dominant mode.134 This principle can be used in designing a biohybrid solar cell by engineering the molecular circuitry by linking different natural and synthetic molecules for improved power conversion efficiency. The Förster resonance energy transfer (FRET) has been utilized in different hybrid systems of biomolecules linked with semiconductor quantum dots (QDs). New biohybrid systems with FRET coupled molecules can be designed by following two main conditions: 1. an optimum proximity between the light harvesting biomolecule (the excited state donor) and the ground state acceptor molecule, close enough to enable the short range interaction and far enough to prevent the overlapping of molecular orbitals thus avoiding the quenching of excitation states,135,136 2. a sizable spectral overlap between the donor emission profile and the acceptor absorption profile.135 A high FRET efficiency is possible by a high donor to acceptor ratio, which makes QD a potential acceptor material as the large surface area of QD enables a higher coverage of biomolecules over it.135
Different QD–LH137 and QD–polypeptide135 hybrid systems have been designed employing the principles of FRET, which can be potential designs for biohybrid solar cells. This concept has also been utilized in DSSCs to enable stronger light absorption in a wide range of wavelengths, by coupling an energy relay dye to the sensitizing dye via FRET in the electrolyte, where the energy relay dye often has a strong absorption at lower wavelengths while the sensitizing dye is redshifted.138 Photosynthetic biomolecules are also promising to be alternative sensitizers139 in DSSCs, so as to replace the costly and non-eco-friendly Ru based dyes with natural dyes which also offer a wider panchromatic absorption range.140,141 Due to the presence of natural pigment molecules, these DSSCs face similar stability issues which need research focus.140 Employing hybrid plasmonic nanostructures with photosynthetic proteins is also a potential way in designing devices with a high photoelectric performance. Hybrid devices with different light harvesting complexes coupled to semicontinuous metal nanostructures have shown an improved light absorption due to the plasmon-induced increase in fluorescence of the complexes.142,143 While the stability issues are avoided by ideally having the photosynthetic proteins in their native environment, there is a new research approach where the device accommodates the entire photosynthetic bacteria and derives electric energy from its photosynthetic activity.144 These devices can be good models for understanding the photosynthetic activity in man-made environments and can aid in developing better materials and device architectures for biohybrid solar cells. Though the photoelectric performance of the biohybrid solar cells are not on par with the conventional solar cells, with a research focus on the stability issues, they can be scaled up in the near future for application in low power electronics like sensors and wireless devices.
7.3.2 Solar fuel generation.
Photosynthetic proteins are integrated in energy harvesting devices both for generating photocurrents and for generating chemical fuels.145 Photosystems found in plants and cyanobacteria are mainly used for the latter purpose. PS II is an efficient water oxidizing complex that efficiently performs a light driven water oxidation.146,147 During photosynthesis, the electrons generated by the PSII assisted water oxidation are utilized by PSI to produce a stable reductant NADPH which is a biological equivalent of H2.147,148 As NADPH is not a useful source of stored bond energy, biohybrid solar fuel cells generally make use of a catalyst to enable H2 production from PS I.147,149 Hydrogenase is a natural enzyme employed by photosynthetic organisms to catalyse the H2 evolution.147 A number of solar fuel cell systems have been developed over the past decade, ranging from fully biological systems with enzyme catalysts to biohybrid and biomimetic systems.147,149 Linking the proteins to catalytic nanoparticles via molecular wires has been a common approach to facilitate a direct electron transfer.147,150 Plant's photosystems are extensively researched for developing hybrid artificial photosynthetic systems for photocatalytic hydrogen evolution and CO2 reduction.151 While it is challenging in artificial photosynthesis to perfectly mimic the protein biomolecules, there is also a new approach where the biomimetic materials are employed in conjunction with the natural proteins in the energy harvesting devices. Certain ruthenium based catalysts have been found to be the reasonable mimics of the oxygen evolving complex of PSII with comparable efficiencies.25 PS II together with such synthetic photocatalysts has been integrated in a water-splitting device for solar-chemical energy conversion which could serve as a model for developing new bioartificial photosystems for autonomous solar water splitting.146
7.3.3 Photosynthetic biosensing.
Apart from the mainstream research on developing solar cells, the photovoltaic abilities of the photosynthetic proteins also find application in herbicide detectors85,152–155 and phototransistors.156 There is an increasing concern to detect the hazardous chemicals like pesticides, insecticides, herbicides etc. that contaminate water resources and enter the food chain.151 The photosynthetic proteins are highly sensitive to these chemical substances as they often target and inhibit photosynthesis or the energy transport enzymes.153 The presence of inhibitor molecules generally affects the process of photosynthesis by either blocking the electron flow or by reducing the oxygen concentration or by increasing the fluorescence, which are often realized as detectable signals in a PS II biosensor.153 Mathematic models are often required for PSII biosensors to compute the concentration of herbicides from the signals obtained.153 Photoelectrochemical cells based on purple bacterial RCs can be used to detect specific herbicide species and their concentration can directly be obtained from the degree of photocurrent attenuation without the need of complex mathematical models.153 The concept of improving stability by employing whole bacterial cell has also been utilized in biosensors as it is critical for these sensors to preserve the proteins’ stability and specificity to specific chemical species.154
8. Conclusion and outlook
In summary, the photovoltaic capabilities of the RCs have been highlighted in relevance to their structure and function in the nature's photosynthetic apparatus. The various strategies adopted so far by the researchers to utilize RCs in photovoltaic devices have been discussed. The progress in the photocurrent generation is presented discerning the key factors improved in various approaches over the years. With an interdisciplinary approach, we still have a large room to explore newer and better ways of exploiting natural photosynthetic biomolecules for solar energy harnessing. Researching on alternative electrode–electrolyte systems and new device architectures would offer a great scope for more efficient and low cost solar cells. The unresolved stability concerns of the photosynthetic protein-based solar cells have been underscored emphasizing the need for future research. As reflected from the nature of this field and its recent progress, a research focus on materials engineering in conjunction with biomolecular and genetic engineering can undoubtedly take the photosynthetic solar energy harvesting to newer dimensions and improve the commercialization prospects.
However, one striking backlog in this field is the pronounced dissonance in measuring and reporting the photoelectric performance, which needs our attention. It is high time that the researchers in this field adhere to performance measurement standards that would make the future studies more pragmatic and foster fruitful research.
Acknowledgements
SC Tan acknowledges the financial support from MOE AcRF 1 (R-284-000-134-112).
Notes and references
-
Solar Cell Materials: Developing Technologies, ed. G. J. Conibeer and A. Willoughby, John Wiley & Sons, 2014 Search PubMed.
-
Advanced Energy Materials, ed. A. Tiwari and S. Valyukh, John Wiley & Sons, 2014 Search PubMed.
- E. W. McFarland, Energy Environ. Sci., 2014, 7, 846–854 CAS.
- K. Kalyanasundaram and M. Graetzel, Curr. Opin. Biotechnol., 2010, 21(3), 298–310 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- M. A. Modestino and S. Haussener, Annu. Rev. Chem. Biomol. Eng., 2015, 6, 1.1–1.2 Search PubMed.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- I. McConnell, G. Li and G. W. Brudvig, Chem. Biol., 2010, 17(5), 434–447 CrossRef CAS PubMed.
- G. F. Moore and G. W. Brudvig, Annu. Rev. Condens. Matter Phys., 2010, 2(1), 303–327 CrossRef.
- F. Bella, C. Gerbaldi, C. Barolob and M. Gratzel, Chem. Soc. Rev., 2015, 44, 3431–3473 RSC.
- J. H. Yum, E. Baranoff, S. Wenger, M. K. Nazeeruddin and M. Gratzel, Energy Environ. Sci., 2011, 4, 842–857 CAS.
- K. G. Reddy, T. G. Deepak, G. S. Anjusree, S. Thomas, S. Vadukumpully, K. R. V. Subramanian, S. V. Nairb and A. S. Nair, Phys. Chem. Chem. Phys., 2014, 16, 6838–6858 RSC.
- C. P. Lee, R. Y. Y. Lin, L. Y. Lin, C. T. Li, T. C. Chu, S. S. Sun, J. T. Lin and K. C. Ho, RSC Adv., 2015, 5, 23810–23825 RSC.
- B. Bensaude-Vincent, H. Arribart, Y. Bouligand and C. Sanchez, New J. Chem., 2002, 26(1), 1–5 RSC.
- C. Sanchez, H. Arribart and M. M. G. Guille, Nat. Mater., 2005, 4(4), 277–288 CrossRef CAS PubMed.
-
Y. Bar-Cohen, Biomimetics: nature-based innovation, CRC press, 2011 Search PubMed.
- R. J. Cogdell, T. H. P. Brotosudarmo, A. T. Gardiner, P. M. Sanchez and L. Cronin, Biofuels, 2010, 1(6), 861–876 CrossRef CAS.
- N. Lebedev, S. A. Trammell, A. Spano, E. Lukashev, I. Griva and J. Schnur, J. Am. Chem. Soc., 2006, 128(37), 12044–12045 CrossRef CAS PubMed.
-
N. Lebedev, A. Spano, S. Trammell, I. Griva, S. Tsoi and J. M. Schnur, In Photonic Devices + Applications, International Society for Optics and Photonics, 2007, p. 665614 Search PubMed.
- N. Lebedev, S. A. Trammell, S. Tsoi, A. Spano, J. H. Kim, J. Xu, M. E. Twigg and J. M. Schnur, Langmuir, 2008, 24(16), 8871–8876 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42(12), 1890–1898 CrossRef CAS PubMed.
- M. Kamran, J. D. Delgado, V. Friebe, T. J. Aartsma and R. N. Frese, Biomacromolecules, 2014, 15(8), 2833–2838 CrossRef CAS PubMed.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332(6031), 805–809 CrossRef CAS PubMed.
- P. D. Frischmann, K. Mahata and F. Wurthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- A. A. Boghossian, M. H. Ham, J. H. Choi and M. S. Strano, Energy Environ. Sci., 2011, 4(10), 3834–3843 CAS.
- R. Das, P. J. Kiley, M. Segal, J. Norville, A. A. Yu, L. Wang, S. A. Trammell, L. E. Reddick, R. Kumar, F. Stellacci, N. Lebedev, J. Schnur, B. D. Bruce, S. Zhang and M. Baldo, Nano Lett., 2004, 4(6), 1079–1083 CrossRef CAS.
- S. C. Tan, F. Yan, L. I. Crouch, J. Robertson, M. R. Jones and M. E. Welland, Adv. Funct. Mater., 2013, 23(44), 5556–5563 CrossRef CAS.
-
R. E. Blankenship, Molecular mechanisms of photosynthesis, John Wiley and Sons, 2014 Search PubMed.
- R. E. Blankenship, Plant Physiol., 2010, 154(2), 434–438 CrossRef CAS PubMed.
- X. Hu, T. Ritz, A. Damjanovic, F. Autenrieth and K. Schulten, Q. Rev. Biophys., 2002, 35(01), 1–62 CrossRef CAS PubMed.
-
B. Ke, Photosynthesis photobiochemistry and photobiophysics, Springer, 2001, vol. 10 Search PubMed.
- M. F. Hohmann-Marriott and R. E. Blankenship, Annu. Rev. Plant Biol., 2011, 62, 515–548 CrossRef CAS PubMed.
- G. Renger, Curr. Sci., 2010, 98(10), 1305–1319 CAS.
- M. R. Jones, Prog. Lipid Res., 2007, 46(1), 56–87 CrossRef CAS PubMed.
- K. Nguyen and B. D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2014, 1837(9), 1553–1566 CrossRef CAS PubMed.
- N. Nelson and W. Junge, Annu. Rev. Biochem., 2015, 84, 26.1–26.25 CrossRef PubMed.
-
Photosynthetic protein complexes: a structural approach, ed. P. Fromme, John Wiley & Sons, 2008 Search PubMed.
-
Oxygenic photosynthesis: the light reactions, ed. D. R. Ort, C. F. Yocum and I. F. Heichel, Springer, 1996, vol. 4 Search PubMed.
- M. Kondo, Y. Nakamura, K. Fujii, M. Nagata, Y. Suemori, T. Dewa, K. Iida, A. T. Gardiner, R. J. Cogdell and M. Nango, Biomacromolecules, 2007, 8(8), 2457–2463 CrossRef CAS PubMed.
- Y. Suemori, M. Nagata, Y. Nakamura, K. Nakagawa, A. Okuda, J. I. Inagaki, K. Shinohara, M. Ogawa, K. Iida, T. Dewa, K. Yamashita, A. Gardiner, R. J. Cogdell and M. Nango, Photosynth. Res., 2006, 90(1), 17–21 CrossRef CAS PubMed.
- Y. Suemori, K. Fujii, M. Ogawa, Y. Nakamura, K. Shinohara, K. Nakagawa, K. Shinohara, K. Nakagawa, M. Nagata, K. Iida, T. Dewa, K. Yamashita and M. Nango, Colloids Surf., B, 2007, 56(1), 182–187 CrossRef CAS PubMed.
- M. J. den Hollander, J. G. Magis, P. Fuchsenberger, T. J. Aartsma, M. R. Jones and R. N. Frese, Langmuir, 2011, 27(16), 10282–10294 CrossRef CAS PubMed.
- M. Kondo, K. Iida, T. Dewa, H. Tanaka, T. Ogawa, S. Nagashima, K. Shimada, H. Hashimoto, A. T. Gardiner, R. J. Cogdell and M. Nango, Biomacromolecules, 2012, 13(2), 432–438 CrossRef CAS PubMed.
- S. C. Tan, L. I. Crouch, M. R. Jones and M. Welland, Angew. Chem., Int. Ed., 2012, 51(27), 6667–6671 CrossRef CAS PubMed.
- S. C. Tan, L. I. Crouch, S. Mahajan, M. R. Jones and M. E. Welland, ACS Nano, 2012, 6(10), 9103–9109 CrossRef CAS PubMed.
- Y. Amao, A. Tadokoro, M. Nakamura, N. Shuto and A. Kuroki, Res. Chem. Intermed., 2014, 40(9), 3257–3265 CrossRef CAS.
- J. W. Harrold Jr, K. Woronowicz, J. L. Lamptey, J. Awong, J. Baird, A. Moshar, M. Vittadello, P. G. Falkowski and R. A. Niederman, J. Phys. Chem. B, 2013, 117(38), 11249–11259 CrossRef PubMed.
- K. Woronowicz, S. Ahmed, A. A. Biradar, A. V. Biradar, D. P. Birnie, T. Asefa and R. A. Niederman, Photochem. Photobiol., 2012, 88, 1467–1472 CrossRef CAS PubMed.
- S. Sakai, T. Noji, M. Kondo, T. Mizuno, T. Dewa, T. Ochiai, H. Yamakawa, S. Itoh, H. Hashimoto and M. Nango, Langmuir, 2013, 29(17), 5104–5109 CrossRef CAS PubMed.
- X. F. Wang, H. Tamiaki, L. Wang, N. Tamai, O. Kitao, H. Zhou and S. I. Sasaki, Langmuir, 2010, 26(9), 6320–6327 CrossRef CAS PubMed.
- H. Furukawa, N. Inoue, T. Watanabe and K. Kuroda, Langmuir, 2005, 21(9), 3992–3997 CrossRef CAS PubMed.
- L. K. Tsui, J. Huang, M. Sabat and G. Zangari, ACS Sustainable Chem. Eng., 2014, 2(9), 2097–2101 CrossRef CAS.
- M. M. Leonova, T. Y. Fufina, L. G. Vasilieva and V. A. Shuvalov, Biochemistry, 2011, 76(13), 1465–1483 CAS.
- W. Hillier and G. T. Babcock, Plant Physiol., 2001, 125(1), 33–37 CrossRef CAS PubMed.
-
The Biophysics of Photosynthesis, ed. J. Golbeck and A. Est, Springer, 2014 Search PubMed.
- R. Tel-Vered and I. Willner, ChemElectroChem, 2014, 1(11), 1778–1797 CrossRef CAS.
- F. Wang, X. Liu and I. Willner, Adv. Mater., 2013, 25(3), 349–377 CrossRef CAS PubMed.
-
H. J. M. de Groot, Modern Magnetic Resonance, Springer, Netherlands, 2006, pp. 327–333 Search PubMed.
- M. R. Jones, Biochem. Soc. Trans., 2009, 37(2), 400 CrossRef CAS PubMed.
- W. Zinth and J. Wachtveitl, ChemPhysChem, 2005, 6(5), 871–880 CrossRef CAS PubMed.
- K. Holden-Dye, L. I. Crouch and M. R. Jones, Biochim. Biophys. Acta, Bioenerg., 2008, 1777(7), 613–630 CrossRef CAS PubMed.
- S. A. Trammell, L. Wang, J. M. Zullo, R. Shashidhar and N. Lebedev, Biosens. Bioelectron., 2004, 19(12), 1649–1655 CrossRef CAS PubMed.
- H. J. Butt, T. Muller and H. Gross, J. Struct. Biol., 1993, 110(2), 127–132 CrossRef CAS.
- Y. Lu, J. Xu, B. Liu and J. Kong, Biosens. Bioelectron., 2007, 22(7), 1173–1185 CrossRef CAS PubMed.
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8(6), 1775–1789 CrossRef CAS PubMed.
- L. Nagy, M. Magyar, T. Szabó, K. Hajdu, L. Giotta, M. Dorogi and F. Milano, Curr. Protein Pept. Sci., 2014, 15(4), 363 CrossRef CAS PubMed.
- Y. Yasuda, Y. Hirata, H. Sugino, M. Kumei, M. Hara, J. Miyake and M. Fujihira, Thin Solid Films, 1992, 210, 733–735 CrossRef.
- Y. Yasuda, H. Sugino, H. Toyotama, Y. Hirata, M. Hara and J. Miyake, Bioelectrochem. Bioenerg., 1994, 34(2), 135–139 CrossRef CAS.
- Y. Yasuda, H. Toyotama, M. Hara and J. Miyake, Thin Solid Films, 1998, 327, 800–803 CrossRef.
- T. Ueno, J. Miyake, T. Fujii, M. Shirai, T. Arai, Y. Yasuda and M. Hara, Supramol. Sci., 1998, 5(5–6), 783–786 CrossRef CAS.
- M. Hara, Y. Asada and J. Miyake, Mater. Sci. Eng., C, 1997, 4(4), 321–325 CrossRef.
-
C. Nakamura, M. Hasegawa, Y. Yasuda and J. Miyake, In Twenty-First Symposium on Biotechnology for Fuels and Chemicals, Humana Press, 2000, pp. 401–408 Search PubMed.
- J. O. Goldsmith and S. G. Boxer, Biochim. Biophys. Acta, Bioenerg., 1996, 1276(3), 171–175 CrossRef.
- A. F. Janzen and M. Seibert, Nature, 1980, 286, 584 CrossRef CAS.
- M. Seibert, A. F. Janzen and M. Kendall-Tobias, Photochem. Photobiol., 1982, 35(2), 193–200 CrossRef CAS.
- A. A. Solov'ev, E. Y. Katz, V. A. Shuvalov and Y. E. Erokhin, Bioelectrochem. Bioenerg., 1991, 26(1), 29–41 CrossRef.
- E. Katz, J. Electroanal. Chem., 1994, 365(1), 157–164 CrossRef CAS.
- J. Zhao, Y. Zou, B. Liu, C. Xu and J. Kong, Biosens. Bioelectron., 2002, 17(8), 711–718 CrossRef CAS PubMed.
- J. Zhao, B. Liu, Y. Zou, C. Xu and J. Kong, Electrochim. Acta, 2002, 47(12), 2013–2017 CrossRef CAS.
- A. Takshi, J. D. Madden, A. Mahmoudzadeh, R. Saer and J. T. Beatty, Energies, 2010, 3(11), 1721–1727 CrossRef CAS.
- S. A. Trammell, A. Spano, R. Price and N. Lebedev, Biosens. Bioelectron., 2006, 21(7), 1023–1028 CrossRef CAS PubMed.
- S. A. Trammell, I. Griva, A. Spano, S. Tsoi, L. M. Tender, J. Schnur and N. Lebedev, J. Phys. Chem. C, 2007, 111(45), 17122–17130 CAS.
- A. Mahmoudzadeh, R. Saer, D. Jun, S. M. Mirvakili, A. Takshi, B. Iranpour, E. Ouellet, E. T. Lagally, J. D. W. Madden and J. T. Beatty, Smart Mater. Struct., 2011, 20(9), 094019 CrossRef.
- T. Mikayama, T. Miyashita, K. Iida, Y. Suemori and M. Nango, Mol. Cryst. Liq. Cryst., 2006, 445(1), 291–581 CAS.
- M. Giustini, M. Autullo, M. Mennuni, G. Palazzo and A. Mallardi, Sens. Actuators, B, 2012, 163(1), 69–75 CrossRef CAS.
- J. Zhao, N. Ma, B. Liu, Y. Zhou, C. Xu and J. Kong, J. Photochem. Photobiol., A, 2002, 152(1), 53–60 CrossRef CAS.
- C. C. Moser, J. M. Keske, K. Warncke, R. S. Farid and P. L. Dutton, Nature, 1992, 355(6363), 796–802 CrossRef CAS PubMed.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402(6757), 47–52 CrossRef CAS PubMed.
- Y. Lu, Y. Liu, J. Xu, C. Xu, B. Liu and J. Kong, Sensors, 2005, 5(4), 258–265 CrossRef CAS.
- Y. Lu, M. Yuan, Y. Liu, B. Tu, C. Xu, B. Liu, D. Zhao and J. Kong, Langmuir, 2005, 21(9), 4071–4076 CrossRef CAS PubMed.
- E. P. Lukashev, V. A. Nadtochenko, E. P. Permenova, O. M. Sarkisov and A. B. Rubin, Dokl. Biochem. Biophys., 2007, 415(1), 211–216 CrossRef CAS PubMed . MAIK Nauka/Interperiodica.
- A. Takshi, J. D. W. Madden and J. T. Beatty, Electrochim. Acta, 2009, 54(14), 3806–3811 CrossRef CAS.
- R. J. Cogdell, T. G. Monger and W. W. Parson, Biochim. Biophys. Acta, Bioenerg., 1975, 408(3), 189–199 CrossRef CAS.
- T. G. Monger, R. J. Cogdell and W. W. Parson, Biochim. Biophys. Acta, Bioenerg., 1976, 449(1), 136–153 CrossRef CAS.
- Y. Lu, J. Xu, Y. Liu, B. Liu, C. Xu, D. Zhao and J. Kong, Chem. Commun., 2006, 785–787 RSC.
- J. Xu, Y. Lu, B. Liu, C. Xu and J. Kong, J. Solid State Electrochem., 2007, 11(12), 1689–1695 CrossRef CAS.
- C. R. Martin and P. Kohli, Nat. Rev. Drug Discovery, 2003, 2(1), 29–37 CrossRef CAS PubMed.
- M. Kang, L. Trofin, M. O. Mota and C. R. Martin, Anal. Chem., 2005, 77(19), 6243–6249 CrossRef CAS PubMed.
- C. Zhi, Y. Bando, C. Tang and D. Golberg, J. Am. Chem. Soc., 2005, 127(49), 17144–17145 CrossRef CAS PubMed.
- J. J. Gooding, R. Wibowo, J. Liu, W. Yang, D. Losic, S. Orbons, F. J. Mearns, J. G. Shapter and D. B. Hibbert, J. Am. Chem. Soc., 2003, 125(30), 9006–9007 CrossRef CAS PubMed.
- W. Yang, P. Thordarson, J. J. Gooding, S. P. Ringer and F. Braet, Nanotechnology, 2007, 18(41), 412001 CrossRef.
- G. Gruner, Anal. Bioanal. Chem., 2006, 384(2), 322–335 CrossRef CAS PubMed.
- M. Alvaro, P. Atienzar, P. de la Cruz, J. L. Delgado, V. Troiani, H. Garcia, F. Langa, A. Palkar and L. Echegoyen, J. Am. Chem. Soc., 2006, 128(20), 6626–6635 CrossRef CAS PubMed.
- I. Oda, M. Iwaki, D. Fujita, Y. Tsutsui, S. Ishizaka, M. Dewa, M. Nango, T. Kajino, Y. Fukushima and S. Itoh, Langmuir, 2010, 26(16), 13399–13406 CrossRef CAS PubMed.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121(41), 9611–9614 CrossRef CAS.
- I. Oda, K. Hirata, S. Watanabe, Y. Shibata, T. Kajino, Y. Fukushima, S. Iwai and S. Itoh, J. Phys. Chem. B, 2006, 110(3), 1114–1120 CrossRef CAS PubMed.
- I. Fathir, T. Mori, T. Nogi, M. Kobayashi, K. Miki and T. Nozawa, Eur. J. Biochem., 2001, 268(9), 2652–2657 CrossRef CAS PubMed.
- D. Garcia, P. Parot and A. Verméglio, Biochim. Biophys. Acta, Bioenerg., 1987, 894(3), 379–385 CrossRef CAS.
- T. Nozawa, T. Fukada, M. Hatano and M. T. Madigan, Biochim. Biophys. Acta, Bioenerg., 1986, 852(2), 191–197 CrossRef CAS.
- T. Nozawa, J. T. Trost, T. Fukada, M. Hatano, J. D. McManus and R. E. Blankenship, Biochim. Biophys. Acta, Bioenerg., 1987, 894(3), 468–476 CrossRef CAS.
- C. Valery, F. Artzner and M. Paternostre, Soft Matter, 2011, 7, 9583–9594 RSC.
- H. Tsutsumi and H. Mihara, Amino Acids, Pept., Proteins, 2013, 38, 122–150 Search PubMed.
- I. W. Hamley, Angew. Chem., Int. Ed., 2014, 53, 6866–6881 CrossRef CAS PubMed.
- G. Colherinhas and E. Fileti, J. Phys. Chem. C, 2014, 118, 9598–9603 CAS.
- B. A. Gregg and M. C. Hanna, J. Appl. Phys., 2003, 93(6), 3605–3614 CrossRef CAS.
- H. Yaghoubi, Z. Li, D. Jun, R. Saer, J. E. Slota, M. Beerbom, R. Schlaf, J. D. Madden, J. T. Beatty and A. Takshi, J. Phys. Chem. C, 2012, 116(47), 24868–24877 CAS.
- S. M. Mirvakili, J. E. Slota, A. R. Usgaocar, A. Mahmoudzadeh, D. Jun, M. N. Mirvakili, J. T. Beatty and J. D. W. Madden, Adv. Funct. Mater., 2014, 24, 4789–4794 CrossRef CAS.
- H. J. Snaith, Nat. Photonics, 2012, 6(6), 337–340 CrossRef CAS.
- M. Ogawa, K. Shinohara, Y. Nakamura, Y. Suemori, M. Nagata, K. Iida, A. T. Gardiner, R. J. Cogdell and M. Nango, Chem. Lett., 2004, 33(6), 772–773 CrossRef CAS.
- Z. Tokaji, J. Tandori and P. Maroti, Photochem. Photobiol., 2002, 75(6), 605–612 CrossRef CAS PubMed.
- A. V. Hughes, P. Rees, P. Heathcote and M. R. Jones, Biophys. J., 2006, 90, 4155–4166 CrossRef CAS PubMed.
- N. Harris and P. Booth, Biochim. Biophys. Acta, 2012, 1818, 1055–1066 CrossRef CAS PubMed.
- R. Picorel, R. E. Holt, R. Heald, T. M. Cotton and M. Seibert, J. Am. Chem. Soc., 1991, 113, 2839–2843 CrossRef CAS.
- R. Picorel, M. Bakhtiari, T. Lu, T. M. Cotton and M. Seibert, Photochem. Photobiol., 1992, 56(2), 263–270 CrossRef CAS.
- C. A. S. A. Minetti and D. P. Remeta, Arch. Biochem. Biophys., 2006, 453, 32–53 CrossRef CAS PubMed.
- G. J. Magis, M. J. den Hollander, W. G. Onderwaater, J. D. Olsen, C. N. Hunter, T. J. Aartsma and R. N. Frese, Biochim. Biophys. Acta, 2010, 1798, 637–645 CrossRef CAS PubMed.
- J. L. Popot, Annu. Rev. Biochem., 2010, 79, 737–775 CrossRef CAS PubMed.
- C. Hein, E. Henrich, E. Orbán, V. Dötsch and F. Bernhard, Eng. Life Sci., 2014, 14(4), 365–379 CrossRef CAS.
- D. J. K. Swainsbury, S. Scheidelaar, R. van Grondelle, J. A. Killian and M. R. Jones, Angew. Chem., Int. Ed., 2014, 53, 11803–11807 CrossRef CAS PubMed.
-
L. J. Chen and W. W. Wu, in Silicon and Silicide Nanowires: Applications, Fabrication, and Properties, ed. Y. Huang and K. N. Tu, Pan Stanford Publishing, 2013, ch. 4, pp. 121–186 Search PubMed.
- T. Yu, S. C. Tan, Z. X. Shen, L. W. Chen, J. Lin and A. See, Appl. Phys. Lett., 2002, 80(13), 2266–2268 CrossRef CAS.
- D. Gunther, G. LeBlanc, D. Prasai, J. R. Zhang, D. E. Cliffel, K. I. Bolotin and G. K. Jennings, Langmuir, 2013, 29(13), 4177–4180 CrossRef CAS PubMed.
- D. Beljonne, C. Curutchet, G. D. Scholes and R. J. Silbey, J. Phys. Chem. B, 2009, 113(19), 6583–6599 CrossRef CAS PubMed.
- A. Chenu and G. D. Scholes, Annu. Rev. Phys. Chem., 2014, 66, 69–96 CrossRef PubMed.
- I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11(1), 17–45 RSC.
- R. van Grondelle and V. I. Novoderezhkin, Nature, 2010, 463(7281), 614–615 CrossRef CAS PubMed.
- F. J. Schmitt, E. G. Maksimov, P. Hätti, J. Weißenborn, V. Jeyasangar, A. P. Razjivin, V. Z. Paschenko, T. Friedrich and G. Renger, Biochim. Biophys. Acta, Bioenerg., 2012, 1817(8), 1461–1470 CrossRef CAS PubMed.
- B. E. Hardin1, E. T. Hoke, P. B. Armstrong, J. H. Yum, P. Comte, T. Torres, J. M. J. Frechet, M. K. Nazeeruddin, M. Gratzel and M. D. McGehee1, Nat. Photonics, 2009, 3(7), 406–411 CrossRef.
- D. Yu, M. Wang, G. Zhu, B. Ge, S. Liu and F. Huang, Sci. Rep., 2015, 5, 9375 CrossRef CAS PubMed.
- N. A. Ludin, A. A. A. Mahmoudb, A. B. Mohamad, A. A. H. Kadhum, K. Sopian and N. S. A. Karim, Renewable Sustainable Energy Rev., 2014, 31, 386–396 CrossRef CAS.
- H. Hug, M. Bader, P. Mair and T. Glatzel, Appl. Energy, 2014, 115, 216–225 CrossRef CAS.
- N. Czechowski, P. Nyga, M. K. Schmidt, T. H. P. Brotosudarmo, H. Scheer, D. Piatkowski and S. Mackowski, Plasmonics, 2012, 7(1), 115–121 CrossRef CAS PubMed.
- S. R. Beyer, S. Ullrich, S. Kudera, A. T. Gardiner, R. J. Cogdell and J. Kohler, Nano Lett., 2011, 11(11), 4897–4901 CrossRef CAS PubMed.
- H. Lee and S. Choi, Lab Chip, 2015, 15, 391–398 RSC.
- O. Yehezkeli, R. Tel-Vered, J. Wasserman, A. Trifonov, D. Michaeli, R. Nechushtai and I. Willner, Nat. Commun., 2012, 3, 742 CrossRef PubMed.
- W. Wang, J. Chen, C. Li and W. Tian, Nat. Commun., 2014, 5, 4647 CAS.
- C. E. Lubner, R. Grimme, D. A. Bryant and J. H. Golbeck, Biochemistry, 2010, 49(3), 404–414 CrossRef CAS PubMed.
- S. C. Silver, J. Niklas, P. Du, O. G. Poluektov, D. M. Tiede and L. M. Utschig, J. Am. Chem. Soc., 2013, 135(36), 13246–13249 CrossRef CAS PubMed.
- S. Fukuzumi, Curr. Opin. Chem. Biol., 2015, 25, 18–26 CrossRef CAS PubMed.
- C. E. Lubner, P. Knorzer, P. J. N. Silva, K. A. Vincent, T. Happe, D. A. Bryant and J. H. Golbeck, Biochemistry, 2010, 49(48), 10264–10266 CrossRef CAS PubMed.
- F. Wen and C. Li, Acc. Chem. Res., 2013, 46(11), 2355–2364 CrossRef CAS PubMed.
- J. Masojídek, P. Souček, J. Máchová, J. Frolík, K. Klem and J. Malý, Ecotoxicol. Environ. Saf., 2011, 74(1), 117–122 CrossRef PubMed.
- D. J. K. Swainsbury, V. M. Friebe, R. N. Frese and M. R. Jones, Biosens. Bioelectron., 2014, 58, 172–178 CrossRef CAS PubMed.
- M. T. Giardi, V. Scognamiglio, G. Rea, G. Rodio, A. Antonacci, M. Lambreva, G. Pezzotti and U. Johanningmeier, Biosens. Bioelectron., 2009, 25(2), 294–300 CrossRef CAS PubMed.
- D. G. Varsamis, E. Touloupakis, P. Morlacchi, D. F. Ghanotakis, M. T. Giardi and D. C. Cullen, Talanta, 2008, 77(1), 42–47 CrossRef CAS PubMed.
- L. Frolov, Y. Rosenwaks, S. Richter, C. Carmeli and I. Carmeli, J. Phys. Chem. C, 2008, 112(35), 13426–13430 CAS.
Footnotes |
| † C. Nakamura, M. Hasegawa, Y. Yasuda and J. Miyake, Applied Biochemistry and Biotechnology, Self-assembling photosynthetic reaction centers on electrodes for current generation, Springer, 2000, vol. 84–86, pp. 401–408, Fig. 1, Copyright © 2000, Humana Press Inc. With kind permission from Springer Science and Business Media. |
| ‡ E. P. Lukashev, V. A. Nadtochenko, E. P. Permenova, O. M. Sarkisov, A. B. Rubin, Doklady Biochemistry and Biophysics, Electron phototransfer between photosynthetic reaction centers of the bacteria Rhodobacter sphaeroides and semiconductor mesoporous TiO2 films, Springer, 2007, vol. 415, pp. 211–216, Fig. 4, Copyright © 2007, Pleiades Publishing, Ltd. With kind permission from Springer Science and Business Media. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence