
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLignin monomer production integrated into the γ-valerolactone sugar platform†
Jeremy S.
Luterbacher‡
 ab,
Ali
Azarpira
b,
Ali H.
Motagamwala
ab,
Fachuang
Lu
bc,
John
Ralph
bc and
James A.
Dumesic
*ab
ab,
Ali
Azarpira
b,
Ali H.
Motagamwala
ab,
Fachuang
Lu
bc,
John
Ralph
bc and
James A.
Dumesic
*ab
aDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA. E-mail: dumesic@engr.wisc.edu; Fax: +1-608-262-0832
bGreat Lakes Bioenergy Research Center, Wisconsin Energy Institute, 1552 University Ave, Madison, WI 53726, USA
cDepartment of Biochemistry, University of Wisconsin-Madison, Madison, WI 53706, USA
First published on 3rd August 2015
Abstract
We demonstrate an experimental approach for upgrading lignin that has been isolated from corn stover via biomass fractionation using γ-valerolactone (GVL) as a solvent. This GVL-based approach can be used in parallel with lignin upgrading to produce soluble carbohydrates at high yields (≥70%) from biomass without the use of enzymes, ionic liquids, or concentrated acids. The lignin was isolated after an initial hydrolysis step in which corn stover was treated in a high-solids batch reactor at 393 K for 30 min in a solvent mixture consisting of 80 wt% GVL and 20 wt% water. Lignin was isolated by precipitation in water and characterized by 2D HSQC NMR, showing that the extracted lignin was similar to native lignin, which can be attributed to the low acid level and the low extraction temperatures that are achievable using GVL as a solvent. This lignin was upgraded using a two-stage hydrogenolysis process over a Ru/C catalyst. The isolated lignin was first dissolved to form a mixture of 10% lignin, 80% THF, 8.5% H3PO4 and 1.5% H2O, and treated at 423 K under hydrogen. The THF was removed by evaporation and replaced with heptane, forming a biphasic mixture. This mixture was then treated at 523 K in the presence of Ru/C and H2. The resulting heptane phase contained soluble lignin-derived monomers corresponding to 38% of the carbon in the original lignin. By adding 5% methanol during the second catalytic step, we produced additional monomers containing methyl esters and increased carbon yields to 48%. This increase in yield can be attributed to stabilization of carboxylic acid intermediates by esterification. The yield reported here is comparable to yields obtained with native lignin and is much higher than yields obtained with lignin isolated by other processes. These results suggest that GVL-based biomass fractionation could facilitate the integrated conversion of all three biomass fractions.
Broader contextLignocellulosic biomass represents an attractive source of renewable carbon and could be used as a feedstock to substitute fossil-based fuels and chemicals. In addition, lignocellulosic biomass comprises non-edible plants that can grow on marginal lands and thus compete minimally with food production. In the context of increasing environmental issues related to climate change and because of increasing petroleum costs due to diminishing fossil reserves, it is urgent to find sustainable carbon-based substitutes to petroleum. However, growing, harvesting and processing biomass is often costly. Because of the expense of the raw material, the yield for conversion of biomass to useful products must be maximized. Lignocellulosic biomass typically contains about 50–70% polysaccharides (cellulose and hemicellulose) and 15–30% lignin by weight. Lignin is an irregular polymer composed of phenolic subunits. Given the relative uniformity of the carbohydrate polymers compared to lignin and the well-established chemical and biological upgrading pathways developed for sugar production and processing, much research and development has been focused on the carbohydrate platform. Lignin has proven to be more challenging to upgrade. However, because lignin is the most energy-dense fraction of biomass and contains valuable aromatic functionalities, its valorization is an essential step in the development of a successful biorefinery. In this work, we report a lignin valorization pathway from corn lignin for the production of heptane-soluble monomers that can readily be integrated with the production and upgrading of carbohydrates. |
Lignocellulosic biomass is the primary source of fixed renewable carbon on the planet. As such, it is increasingly proposed to be a sustainable alternative to fossil carbon resources. To enable the economic and sustainable production of renewable fuels and chemicals, processes that convert biomass to useful products at high yields must be developed. Thermochemical approaches such as gasification can achieve this goal, notably enabling the recovery of about 70% of the energy content of biomass as synthetic natural gas.1,2 However, due to the energetic and economic requirements of converting these gaseous compounds back to liquids, there is a need to develop targeted biomass upgrading processes that can achieve similar yields and directly produce liquid fuels and/or sustainable chemical alternatives to petrochemicals. In this context, extensive work has focused on the conversion of the polysaccharide fraction of biomass. Depolymerization processes using enzymes,3,4 acids,5,6 solvents,7 or ionic liquids,8 have all achieved high sugar yields (>70%) from biomass.9 However, polysaccharides only represent 60–70% of biomass weight and an even lower fraction of the available energy. Upgrading the remaining biomass fractions, the largest of which is lignin, will be an important prerequisite for developing economical and sustainable processes.
One of the most promising approaches for lignin conversion has been the use of oxidation or hydrogenolysis to produce lignin monomers and other products. Wet oxidation under hydrothermal conditions has shown that simple compounds such as acetic acid could be produced from lignin.10,11 More recently, Stahl and co-workers showed that targeted oxidation of enzymatically extracted lignin could dramatically increase the yield of monomers obtained during depolymerization by formic acid.12 In the late sixties, Pepper et al. treated solvent-extracted spruce in dioxane in the presence of Rh/C and hydrogen, and obtained a 34% yield of monomeric compounds, including 11% guaiacylpropane (propylguaiacol, 2-methoxy-4-propylphenol) and 20% guaiacylpropanol [4-(3-hydroxypropyl)-2-methoxy-phenol].13 More recently, Yan et al. treated birch cell wall isolates (following extractive removal by benzene-extraction) using water–dioxane as a solvent, with added phosphoric acid, in the presence of noble metal catalysts and hydrogen.14 They obtained monomer yields as high as 46% from the “C9 units” of lignin, the phenylpropanoid units comprising the polymer. This yield included 6% guaiacylpropane, 21% syringylpropane, and 15% syringylpropanol (2,6-dimethoxy-4-(3-hydroxypropyl)phenol). These relatively high yields of simple monomeric compounds obtained from untreated biomass are in contrast with those yields obtained from lignin that has been extracted from biomass; for example, treatment of kraft lignin in ethanol/water mixtures and a Pt/Al2O3 catalyst led to ∼17% monomers, with only ∼6% yield of guaiacyl derivatives.15 Recent work by Ferrini and Rinaldi further demonstrated the importance of using native lignin in conversion processes.16 By treating wood pellets in 2-propanol in the presence of RANEY® nickel, the authors obtained non-pyrolytic lignin bio-oil by catalytic transfer hydrogenolysis, while preserving a holocellulose pulp for further hydrolysis. This lignin oil could be completely converted to a colorless volatile fraction by further treatment in 2-propanol with RANEY® nickel. The same two-stage treatment with organosolv lignin yielded a turbid solution that still contained a significant fraction of insoluble polymer. In similar work, sawdust was treated in methanol with Ru/C and H2 to produce “lignin oil” containing a 51% carbon yield of phenolic monomers.17 Following this reaction, a solid pulp containing 78% of the original carbohydrates was recovered mixed with the catalyst. This pulp could be used to produce polyols from the remaining polysaccharides by further hydrogenation. Hydrogenolysis is therefore seen as a promising approach for upgrading lignin, but yields decrease significantly as the lignin stream is increasingly degraded during its isolation from the rest of biomass during processing. This loss of yield is thought to be linked to the propensity of lignin ether bonds to cleave under the conditions required to hydrolyze biomass (a necessary upgrading step to obtain carbohydrates), followed by recondensation to form stable C–C bonds, severely curtailing further upgrading possibilities.18 Lignin-upgrading work has, however, rarely been coupled with structural characterization of the isolated lignin stream. Therefore, explanations for differences in yields obtained from various lignin streams are often qualitative.
We present a method for upgrading lignin isolated from corn stover during an initial batch hydrolysis step that is the first stage of a process for producing concentrated aqueous streams of carbohydrates derived from the hemicelluloses and cellulose.7 Using hydrogenolysis coupled with esterification, we are able to obtain carbon yields of identifiable lignin monomers of up to 48%. In parallel, we have characterized our isolated lignin stream using 2D NMR, and we show that our extracted lignin is remarkably similar to native lignin. Enriched in this lignin is the monocot-specific p-coumarate lignin-associated appendage, a simple ester that attests to the mildness of this process. We propose that our ability to obtain yields that exceed those usually obtained from processed and isolated lignins is due to our biomass treatment process that retains much of the lignin in its native state and these two particular features of our lignin stream.
In our previous work, we used mixtures of γ-valerolactone (GVL) (a biomass-derived solvent) water and H2SO4, at low concentrations, to depolymerize the carbohydrate fraction of biomass.7 GVL promotes biomass deconstruction to sugars by increasing the rate of acid-catalyzed reactions in a manner that rivals treatments with ionic liquids.8 Accelerated polysaccharide hydrolysis was found to be due to the effect of GVL on proton solvation, and to its selective lowering of the apparent activation barrier of polysaccharide hydrolysis compared to that of the competing sugar dehydration reactions to furans that typically plague acid hydrolysis methods.19,20 After optimization of conditions to maximize carbohydrate yield and concentration, we found that the best implementation of our process involved an initial stage to partially depolymerize the hemicellulosic carbohydrate fraction and partially solubilize the lignin fraction, followed by treating the remaining solids in a flow-through reaction setup to depolymerize the remaining polysaccharides (including cellulose). This two-step process led to an overall carbohydrate yield of about 70% for both the C5 and C6 sugars. After biomass fractionation, GVL could be removed and recycled using liquid CO2, which forms a CO2-expanded GVL phase that is no longer miscible with water.7,21 The resulting aqueous sugar solution is concentrated to contain 127 g L−1 carbohydrates, which is appropriate for subsequent purification and fermentation.7,22
In this study, we substituted the small 50 mL glass reactor used in the initial C5 and lignin deconstruction step with a 2-L stirred stainless steel vessel fitted with a custom high-solids mixing system. The latter system led to more effective mixing, and allowed us to reach the optimal extraction condition in 30 instead of 60 min. After 30 min at 423 K, 72.3% of the C5 sugars and 9.4% of the C6 sugars were recovered as soluble sugars (Table 1). At the same time, water-insoluble solids corresponding to 75% of the original lignin were solubilized in the 80/20 GVL/water solvent system (Table 1). To recover these solids for further upgrading, the separated solution was diluted five times with water, filtered, and washed. These solids can also be recovered during CO2 separation, when water-insoluble solids corresponding to about 74% of the original lignin were recovered. As we had shown previously, in addition to the C5 sugars and solubilized lignin yields shown in Table 1, the remaining solids can be treated in a flow-through reactor to reach C6 yields of about 70%.7 Therefore, by treating the extracted lignin recovered here, we are investigating the potential for integrating lignin conversion with a carbohydrate-upgrading platform. This approach differs from recent methods that generally involve the use of a supported metal catalyst in the presence of untreated biomass.14 The disadvantage of these other methods is that they often require the difficult separation of the heterogeneous catalyst and leftover solids, and usually lead to some loss of the C5 sugar fraction that is present during the lignin upgrading reactions. Our method, however, retains the approach of extracting value from the lignin by retaining the lignin in its near-native form, rather than having to deal with condensed lignins after polysaccharide hydrolysis steps.
| Composition [wt%] | ||
|---|---|---|
| Glucan | Xylan | Klason lignin |
| 30.7 | 18.5 | 14.3 |
| Yields [wt%] | ||
|---|---|---|
| C6 sugars | C5 sugars | Extracted lignin |
| 9.4 | 72.3 | 75.4 |
An appealing feature of our GVL-based hydrolysis process is that, even when using a batch process with a 30 min residence time, the resulting lignin fractions are clean and native-like, i.e., relatively carbohydrate free, and with little evidence of lignin degradation other than some β-ether cleavage and concomitant molecular weight reduction. Here we validate this contention by examining the structure and composition of the lignins by NMR. The corresponding 2D HSQC (heteronuclear single-quantum coherence) spectra are particularly detailed and structurally revealing, as is now well demonstrated.23–25
Fig. 1 shows detailed comparisons of the corn GVL lignin (Fig. 1B) along with reference data from the so-called ‘enzyme lignin’ (EL) isolated by laborious conventional methods,26 and the original corn stover whole-cell-wall material. Assignments are made by comparison with quality spectra of isolated lignins and/or whole-cell-wall plant materials.23–25 As revealed in Fig. 1, acidolysis in GVL produces a strikingly clean fraction that contains the various wall aromatics and particularly native-like lignin. In particular, the most sensitive β-ether units A remain prevalent, attesting to the mild nature of the conditions. We suspect that this retention of linkages is partly due to the low temperature of treatment (423 K) that is achievable using GVL, as opposed alternate aqueous or solvent systems. As is usual for corn, phenylcoumaran B and resinol C units usually present in dicot lignins are evident only near the noise level (not shown); monocots in general and corn in particular have been shown previously to be anomalous in this regard.27 Particularly strikingly, the p-coumarate groups that acylate lignin sidechains in corn lignin remain. Esters are normally considered to be sensitive to hydrolysis (although less so to acidic conditions), but these linkages are retained under the mild conditions here and in fact are enriched in these GVL fractions. The novel (non-resinol) β–β-coupling product C′, produced during lignification from sinapyl p-coumarate conjugates, that was recently identified in corn lignins,27 is also readily seen here. Finally, the tricin units T that we recently discovered starting many of the lignin chains in monocots lignins,27,28 are found in this corn GVL lignin; the resolution of the T3 contours even appears to allow distinction of the smaller amount of tricin that has been cleaved and is therefore free from the tricin that remains etherified (in the lignin).
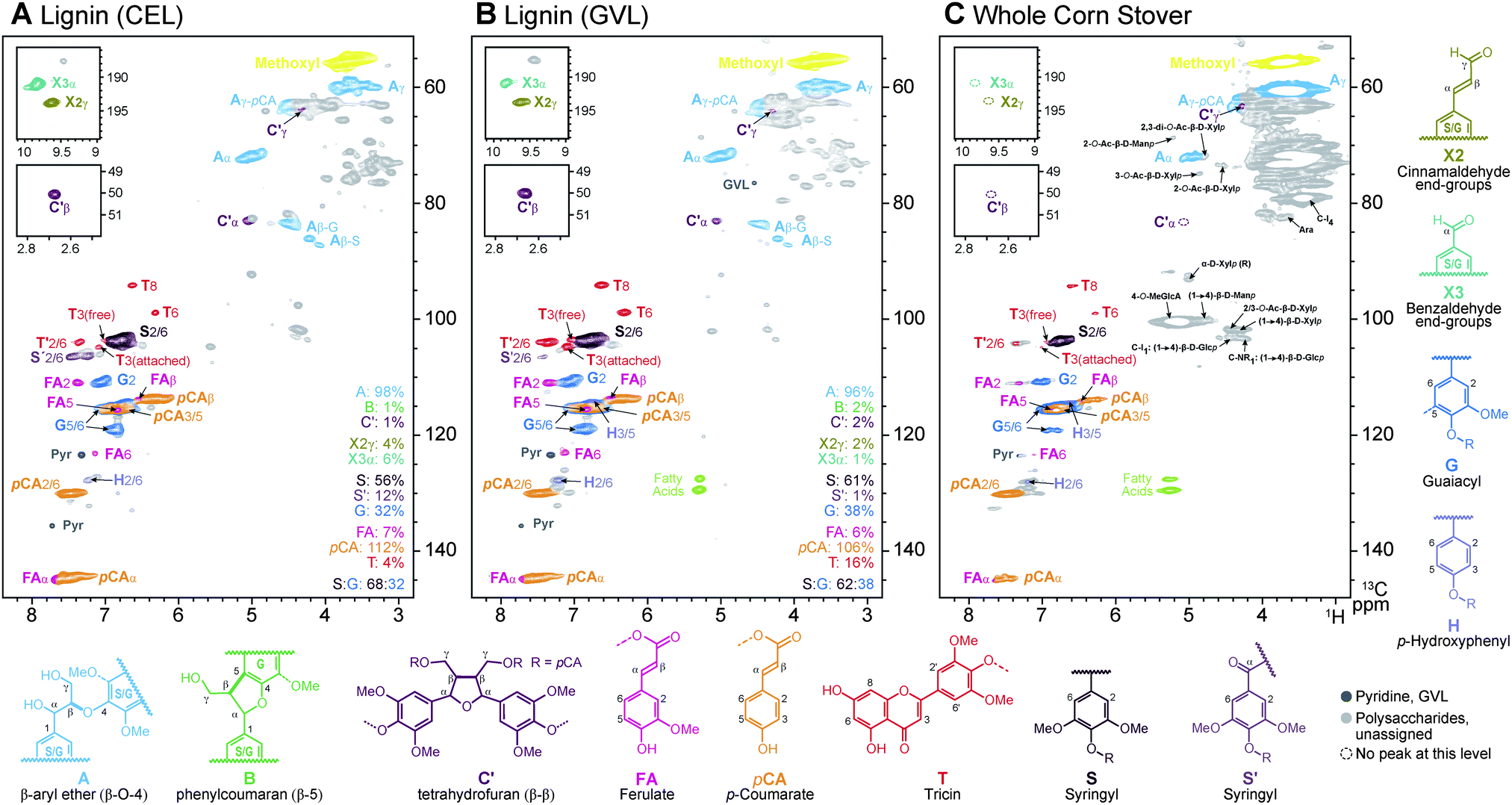 | ||
Fig. 1 2D-HSQC-NMR spectra of corn samples in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMSO-d6:pyridine-d5. (A) Isolated cellulase-digested enzymatic lignin (CEL); (B) GVL-extracted lignin (GVL) reported here, illustrating how clean it is even relative to the CEL in (A); (C) whole cell wall. Lignin and other aromatic correlations are colored and labeled to match the structures below. :1 DMSO-d6:pyridine-d5. (A) Isolated cellulase-digested enzymatic lignin (CEL); (B) GVL-extracted lignin (GVL) reported here, illustrating how clean it is even relative to the CEL in (A); (C) whole cell wall. Lignin and other aromatic correlations are colored and labeled to match the structures below. | ||
All of the features of the lignin revealed by the NMR data suggest that these GVL-derived lignin fractions should be amenable to the same types of conversion reactions as native lignins (in unfractionated biomass material) or the various isolated lignins that essentially preserve the native structure. The advantage with respect to attempting lignin upgrading on whole biomass lies in the simpler conditions necessary to convert the readily solvent-soluble and pure lignin fractions here and, in the case of heterogeneous catalysts, facilitating catalyst recovery. It is for these reasons that the lignins produced via these GVL processes are so appealing for upgrading and, because further hydrolysis produces clean and concentrated sugar streams, the process is promising for biomass fractionation and conversion to sugars.
The extracted and dried lignin was dissolved in tetrahydrofuran (THF), a solvent less subject to hydrogenation than GVL. Like GVL, THF can be produced from biomass and has similar properties. In past work, we have shown that THF can completely solubilize biomass and lead to solubilized C6 and C5 sugar yields close to 70%, similar to GVL.29 Phosphoric acid was added to the mixture because of the reported benefits of the presence of Brønsted acids in addition to metal catalysts for the deoxygenation of syringyl and guaiacyl derivatives.30 During the first stage, the solubilized lignin was treated at 423 K for 4 h in the presence of various noble metal catalysts supported on carbon. The use of phosphoric acid and a carbon-supported metal catalyst would likely not be ideal in an industrial process. Phosphoric acid is an expensive and unsustainable acid, and carbon supports cannot undergo calcination to remove carbonaceous deposits that may occur on the metal nanoparticles. However, the objective of this hydrogenolysis treatment is to assess the deconstruction potential of the lignin rather than to develop a viable industrial process. After initial treatment, the product mixture was notably lighter in color than the feed and contained a number of small lignin-derived compounds. The carbon yield of these small molecules after this first stage was 23% when Ru/C was used (Fig. 2A). The main products included: monomers derived from lignin guaiacyl and syringyl units along with phenylpropanoic acid (dihydrocinnamic acid), cyclohexane propanoic acid (cyclohexylpropanoic acid), and cyclohexene propanoic acid all derived from p-coumarate (following deoxygenation and ring hydrogenation); ferulate analogs were also found to a lesser extent. We also observed several bicyclic products (shown in yellow and orange in Fig. 2A) that we hypothesize to be cyclization products derived from phenylpropanoic acid or its p-coumarate-derived precursors. The presence of small amounts of fatty acids in the recovered compound mixtures indicated that our water-insoluble solids likely contained a small amount of plant waxes; plant materials were used as-received and were not pre-extracted to remove non-cell-wall extractives. Finally, we observed the formation of several methyl-esters (cyclohexanepropionic acid methyl ester and fatty acid methyl esters or FAMEs). An explanation for this behavior is that the methanol liberated during the hydrodeoxygenation of syringyl and guaiacyl units reacted with the carboxylic acid groups on the fatty acids and cyclohexane propionic acids or via transesterification of the original esters (see below).
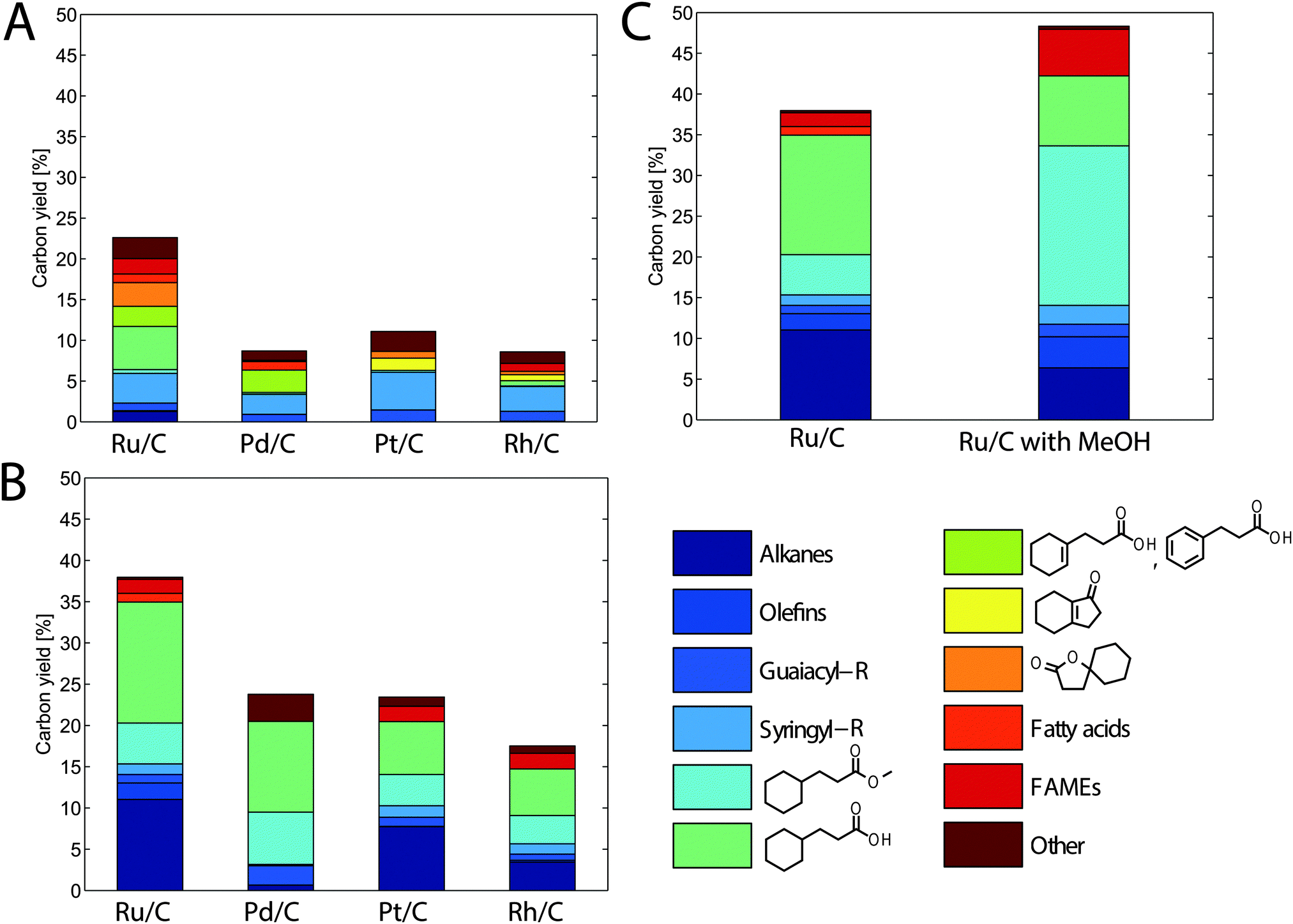 | ||
| Fig. 2 Yields obtained of identified lignin-derived monomers treated in 90% solvent, 8.5% H3PO4 and 1.5% H2O. (A) Effect of catalyst during the first treatment stage in THF (423 K, 4 h). (B) Effect of catalyst during the subsequent second treatment stage in heptane (523 K, 4 h). (C) Effect of adding 5% MeOH during the second stage. Guaiacyl and syringyl-R designate guaiacyl and syringyl monomers, respectively, with R = methyl, ethyl or propyl. FAMEs stands for fatty acid methyl esters. | ||
The other metal catalysts studied (Pt, Pd and Rh) performed significantly worse than Ru/C, with yields that were at most half of those obtained with Ru (Fig. 2A). In addition to single metals, we explored the use of bimetallic alloys, such as RhRe, which has proven to be effective at performing hydrogenolysis of ethers, even without added acids.31 We also studied a RuRe catalyst that was shown to remain active in the presence of sulfuric acid. Such a catalyst could allow us to use H2SO4, which is the acid commonly used for our biomass depolymerization work, instead of H3PO4.32 However, both bimetallic approaches led to lower yields than those obtained with the Ru/C and H3PO4 system (Fig. S1, ESI†).
The low yields achieved during the first stage (<25%) indicated that significant quantities of lignin-derived high molecular weight compounds likely remained in the solution. To further depolymerize these compounds by hydrogenolysis, we explored a higher reaction temperature (523 K) that has proven effective for further lignin breakdown.13,33 At such temperatures, the solvent THF has a high vapor pressure and will also undergo hydrogenolysis; therefore, THF was removed by evaporation, and the remaining solution (containing phosphoric acid, water and lignin derivatives) was contacted with heptane to form a biphasic mixture. Heptane was chosen because of its stability under high-temperature hydrogenation conditions. Each solution was subsequently treated with the same catalyst as in the first stage at 523 K for 4 h, to determine the final attainable yield of lignin monomers recovered in the heptane phase. The trend observed across different metal catalysts during the first stage remained similar to that after the second stage. Ru/C led to the largest carbon yield of 38% (Fig. 2B). The largest contributors to the yield were guaiacyl- and syringyl-derived alkanes – consisting mainly of propyl, ethyl or methyl cyclohexane – and cyclohexane propionic acid or its corresponding methyl ester. Most of the yield increase came in these two categories. Therefore, as demonstrated by the yield increase, the high-temperature hydrodeoxygenation stage serves not only to deoxygenate and fully hydrogenate lignin monomers, but it also leads to further break down solubilized lignin into smaller molecules via hydrogenolysis. We also observed a small increase in the carbon yield of compounds derived from fatty acids (mostly as FAMEs and fully hydrogenated linear alkanes), but their contributions to the total yield (3%) was small.
Similar to the results from the first stage, the other supported metal catalysts that were studied resulted in significantly lower yields than those for Ru/C (Fig. 2B). The use of Pt/C and Rh/C resulted in lower yields for all compounds produced. However, Pd/C was almost as effective at producing p-coumaric acid (and ferulic acid) derivatives (i.e., cyclohexane propionic acid and its methyl ester) but significantly less effective at producing syringyl and guaiacyl derivatives. This result differs from those reported for the processing of unprocessed wood, where palladium was an effective catalyst.14 These differences are likely due to the effects of using extracted versus native lignins or using grass versus wood lignins, highlighting the difficulty of working with this complex polymer mixture. We also used the second high temperature stage with the bimetallic catalyst systems that were mentioned earlier. However, as with the low temperature stage, we observed a significantly lower yield compared to the Ru/C–H3PO4 strategy (Fig. S1, ESI†).
As noted above, we observed the formation of several methyl esters, including cyclohexane propionic acid methyl ester and FAMEs. This formation likely occurs after the liberation of methanol during the hydrogenolysis of syringyl and guaiacyl monomers. Past work has suggested that the formation of these esters may be beneficial for the stabilization of carboxylic acid intermediates and can potentially increase yields.34,35 However, in our case, the formation of methyl esters is likely limited by methanol formation. For this reason, we tried to favor the formation of methyl esters by adding methanol in excess before the second high-temperature stage, with the first stage remaining the same. When doing this addition of methanol for Ru/C, which produced the best yields, we observed that the carbon yield increased to 48% (Fig. 2C). As expected, this increase in yield came almost exclusively from the increased formation of methyl esters, mainly methyl propionate, but also some FAMEs. These yields are comparable to the yields obtained from unprocessed biomass,14,16,17 but our yields have the advantage of using lignin that has been already separated from biomass. Therefore, this process does not require the mixing of a heterogeneous catalyst and a heterogeneous substrate, or the separation of two heterogeneous species. As discussed in the introduction, yields that can be obtained from unprocessed biomass are much higher than those typically obtained from biomass that has been isolated, due to the lignin degradation that often accompanies its extraction. To illustrate this, we have compared our yields (without using methanol) to those obtained using other sources of lignin, including native lignin (i.e., whole corn stover), lignin produced using liquid ammonia pretreatment, and acid insoluble lignin (Table 2). The yield obtained from native lignin (42%) was very similar to that obtained with GVL-extracted lignin (38%). In contrast, the monomer yields obtained using lignin extracted with acid or ammonia were about 2–3 times lower after the second stage. Due to the difficulty of processing high-solids biomass, native lignin was treated at a lower concentration than GVL-extracted lignin (0.9 versus 10 wt%), which could explain the slightly higher yields, given that higher concentrations tend to produce lower yields by promoting condensation and polymerization in biomass conversion reactions.9 This concentration difference, along with the presence of the other biomass fractions, could also explain the different product distribution obtained from native lignin, with a significant increase in the production of aromatic hydrocarbons and a decrease of cyclohexane propionic acid and its derivatives.
:1 ammonia:biomass ratio at 393 K for 30 min) and acid lignin stands for acid extracted lignin (acid-insoluble lignin after treatment with 72% H2SO4 for 60 min at 298 K and 4% H2SO4 for 60 min at 393 K)
| Carbon yield [%] | ||||
|---|---|---|---|---|
| Type of monomer | Native lignin | GVL-lignin | EA-lignin | Acid-lignin |
| 1st stage | ||||
| Propyl-R | — | 2.3 | 0.7 | 0.3 |
| Syringyl-R | — | 3.6 | 0.7 | 0.3 |
| Cyclohexane propionic acid | — | 5.3 | 0 | 1.6 |
| Cyclohexane propionic acid methyl ester | — | 0.5 | 0.2 | 0.01 |
| Other | — | 11.5 | 1.9 | 2.1 |
| Total | — | 22.6 | 3.5 | 4.3 |
| 2nd stage | ||||
| Alkanes | 11.7 | 11.0 | 1.9 | 10.2 |
| Aromatic hydrocarbons | 23.0 | 0.6 | 0.6 | 0.8 |
| Cyclohexane propionic acid methyl ester | 1.6 | 5.0 | 1.5 | 0.5 |
| Cyclohexane propionic acid | 0 | 14.7 | 2.9 | 0.9 |
| Fatty acids methyl esters | 2.7 | 2.0 | 1.3 | 0.7 |
| Other | 3.1 | 4.5 | 2.1 | 4.1 |
| Total | 42.1 | 37.7 | 10.3 | 17.2 |
An interesting feature of the process converting GVL-extracted lignin is that all monomers are recovered in the heptane phase, indicating that these compounds could readily be used as fuel additives due to their hydrocarbon solubility. The cycloalkanes produced typically have 9 carbons, which is very similar to the typical size of alkanes found in fuels. Similarly, methyl-esters have also been extensively used as fuels in the context of biodiesel. The methyl esters produced here are produced from C9 carboxylic acids whereas typical biodiesel mixtures are produced from C12 to C22 fatty acids with C18 often being the major product.36 However, smaller carboxylates (notably pentanoate esters formed from C5 carboxylic acids) have been extensively studied by Shell as a fuel source and deemed fully compatible with gasoline and diesel blending during engine tests.37 Therefore, the alkanes and methyl esters produced here are likely to be compatible as fuel additives. The remaining cyclohexane propionic acid could be more problematic despite its solubility in heptane. However, given the propensity for transesterification processes within biofuel production, it would likely be straightforward to fully convert any remaining carboxylates to methyl esters by adjusting reaction conditions.
Conclusions
The GVL-based hydrolysis process for depolymerization of corn stover allows the separation of an aromatics-rich stream derived from the lignin and cell wall hydroxycinnamates in which these components are essentially unmodified from their original state in the plant cell wall, as revealed by NMR. The solubility of this aromatic fraction allows for more facile hydrodeoxygenation and continuous processing using a supported catalyst, unlike when such processes are applied to whole biomass material. Good yields (up to 48% carbon yields) of heptane-soluble volatile products result from treatment of our GVL-extracted lignin over Ru/C under a hydrogen atmosphere. Although these results are encouraging, future work should focus on developing a process that uses a more sustainable source of acid than phosphoric acid and a catalyst that can easily be regenerated in the event that carbonaceous deposits occur over the active sites. Nevertheless, a significant advantage of this process is that it derives value from a lignin stream that has been easily isolated from whole biomass and its polysaccharides (hemicelluloses and cellulose). In our previous work we showed that cellulose and hemicelluloses can be separately hydrolyzed using the GVL-based process to produce high yields of soluble sugars (≥70%). The results of the present study suggest the possibility of developing an integrated process for upgrading all three biomass fractions.Acknowledgements
This work was funded by the U.S. Department of Energy's Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494). We thank Leonardo Sousa and Bruce Dale from Michigan State University (Lansing, MI) for providing us with ethanol soluble lignin from liquid ammonia pretreatment.References
- A. Duret, C. Friedli and F. Maréchal, J. Cleaner Prod., 2005, 13, 1434–1446 CrossRef PubMed.
- J. S. Luterbacher, M. Froling, F. Vogel, F. Marechal and J. W. Tester, Environ. Sci. Technol., 2009, 43, 1578–1583 CrossRef CAS.
- J. S. Luterbacher, Q. Chew, Y. Li, J. W. Tester and L. P. Walker, Energy Environ. Sci., 2012, 5, 6990 CAS.
- C. E. Wyman, B. E. Dale, R. T. Elander, M. Holtzapple, M. R. Ladisch, Y. Y. Lee, C. Mitchinson and J. N. Saddler, Biotechnol. Prog., 2009, 25, 333–339 CrossRef CAS PubMed.
- R. Rinaldi and F. Schuth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- M. Käldström, N. Meine, C. Farès, R. Rinaldi and F. Schüth, Green Chem., 2014, 16, 2454–2462 RSC.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS PubMed.
- J. B. Binder and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4516–4521 CrossRef CAS PubMed.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- F. Jin, Z. Zhou, A. Kishita and H. Enomoto, J. Mater. Sci., 2006, 41, 1495–1500 CrossRef CAS.
- H. Suzuki, J. Cao, F. Jin, A. Kishita, H. Enomoto and T. Moriya, J. Mater. Sci., 2006, 41, 1591–1597 CrossRef CAS.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- J. M. Pepper and Y. W. Lee, Can. J. Chem., 1969, 47, 723–727 CrossRef CAS.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. T. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS.
- J. Zakzeski, A. L. Jongerius, P. C. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef CAS PubMed.
- P. Ferrini and R. Rinaldi, Angew. Chem., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS.
- J. Li and G. Gellerstedt, Ind. Crops Prod., 2008, 27, 175–181 CrossRef CAS PubMed.
- M. A. Mellmer, C. Sener, J. M. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Angew. Chem., 2014, 53, 11872–11875 CrossRef CAS PubMed.
- M. A. Mellmer, D. Martin Alonso, J. S. Luterbacher, J. M. R. Gallo and J. A. Dumesic, Green Chem., 2014, 16, 4659–4662 RSC.
- J. S. Luterbacher, D. M. Alonso, J. M. Rand, Y. M. Questell-Santiago, J. H. Yeap, B. F. Pfleger and J. A. Dumesic, ChemSusChem, 2015, 8, 1317–1322 CrossRef CAS PubMed.
- J. Han, J. S. Luterbacher, D. M. Alonso, J. A. Dumesic and C. T. Maravelias, Bioresour. Technol., 2015, 182, 258–266 CrossRef CAS PubMed.
- J. Ralph and L. L. Landucci, NMR of Lignins, in Lignin and Lignans; Advances in Chemistry, ed. C. Heitner, D. R. Dimmel and J. A. Schmidt, CRC Press (Taylor & Francis Group), Boca Raton, FL, 2010, ch. 5, pp. 137–234 DOI:10.1201/EBK1574444865.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protoc., 2012, 7, 1579–1589 CrossRef CAS.
- H. Kim and J. Ralph, Org. Biomol. Chem., 2010, 8, 576–591 CAS.
- A. Wagner, Y. Tobimatsu, L. Phillips, H. Flint, B. Geddes, F. Lu and J. Ralph, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6218–6223 CrossRef CAS PubMed.
- W. Lan, F. Lu, M. Regner, Y. Zhu, J. Rencoret, S. A. Ralph, U. I. Zakai, K. Morreel, W. Boerjan and J. Ralph, Plant Physiol., 2015, 167, 1284–1295 CrossRef CAS PubMed.
- J. C. del Rio, J. Rencoret, P. Prinsen, Á. T. Martínez, J. Ralph and A. Gutiérrez, J. Agric. Food Chem., 2012, 60, 5922–5935 CrossRef CAS PubMed.
- J. A. Dumesic, D. Martin Alonso and J. S. Luterbacher, Biomass pretreatment for co-production of high-concentration C5- and C6-carbohydrates and their derivatives, US Pat., application no. 14/136,564, 2013 Search PubMed.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Chem. Commun., 2010, 46, 412–414 RSC.
- M. Chia, Y. J. Pagan-Torres, D. Hibbitts, Q. Tan, H. N. Pham, A. K. Datye, M. Neurock, R. J. Davis and J. A. Dumesic, J. Am. Chem. Soc., 2011, 133, 12675–12689 CrossRef CAS.
- D. J. Braden, C. A. Henao, J. Heltzel, C. C. Maravelias and J. A. Dumesic, Green Chem., 2011, 13, 1755–1765 RSC.
- V. M. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. Li and J. A. Lercher, Chem. – Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed.
- T. Voitl and P. R. von Rohr, ChemSusChem, 2008, 1, 763–769 CrossRef CAS PubMed.
- T. Voitl and P. R. von Rohr, Ind. Eng. Chem. Res., 2010, 49, 520–525 CrossRef CAS.
- S. K. Hoekman, A. Broch, C. Robbins, E. Ceniceros and M. Natarajan, Renewable Sustainable Energy Rev., 2012, 16, 143–169 CrossRef CAS PubMed.
- J. P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., 2010, 49, 4479–4483 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee01322d |
| ‡ Present address: Laboratory of Sustainable and Catalytic Processing, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. |
| This journal is © The Royal Society of Chemistry 2015 |