
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithium salts for advanced lithium batteries: Li–metal, Li–O2, and Li–S
Reza
Younesi
*ab,
Gabriel M.
Veith
c,
Patrik
Johansson†
de,
Kristina
Edström
be and
Tejs
Vegge
a
aDepartment of Energy Conversion and Storage, Technical University of Denmark, Frederiksborgvej 399, P.O. Box 49, DK-4000 Roskilde, Denmark. E-mail: reyo@dtu.dk
bDepartment of Chemistry-Ångström Laboratory, Uppsala University, Box 538, SE-75121 Uppsala, Sweden. E-mail: reza.younesi@kemi.uu.se
cOak Ridge National Laboratory, Materials Science and Technology Division, Oak Ridge, TN 37831, USA
dDepartment of Applied Physics, Chalmers University of Technology, SE-41296 Göteborg, Sweden
eALISTORE-ERI European Research Institute, 33 rue Saint Leu, 80039 Amiens, France
First published on 1st June 2015
Abstract
Presently lithium hexafluorophosphate (LiPF6) is the dominant Li-salt used in commercial rechargeable lithium-ion batteries (LIBs) based on a graphite anode and a 3–4 V cathode material. While LiPF6 is not the ideal Li-salt for every important electrolyte property, it has a uniquely suitable combination of properties (temperature range, passivation, conductivity, etc.) rendering it the overall best Li-salt for LIBs. However, this may not necessarily be true for other types of Li-based batteries. Indeed, next generation batteries, for example lithium–metal (Li–metal), lithium–oxygen (Li–O2), and lithium–sulfur (Li–S), require a re-evaluation of Li-salts due to the different electrochemical and chemical reactions and conditions within such cells. This review explores the critical role Li-salts play in ensuring in these batteries viability.
Broader contextBatteries are one of the most efficient energy storage systems, which in combination with renewable energy sources like solar, wind, and hydro-power can decrease our society's dependence on fossil fuels. Today's rechargeable batteries are, however, far from being fully competitive for large-scale applications such as electric vehicles and grid storage. In order to meet the increasing energy storage/conversion demands in the best possible way, various battery concepts referred to as beyond Li-ion batteries, such as lithium–metal, lithium–oxygen and lithium–sulfur batteries, have been subject to intense research in recent years. These advanced lithium batteries still suffer from issues often associated with the properties of their electrolytes. Indeed, electrolytes based on aprotic solvents and Li-salts commonly used in commercial Li-ion batteries have failed in advanced lithium batteries. Here, we present an overview of the different requirements that Li-salts need to fulfill to be implemented in any lithium batteries, as well as studies investigating their roles in advanced lithium batteries. |
1. Introduction
The successful integration of solar, wind, and hydro power with energy storage systems like batteries will play a pivotal role in reforming our society from being dependent on fossil fuels to being able to rely more on renewable energy sources.1,2 A major challenge is to design better batteries for large-scale applications such as electric vehicles (EVs) and grid storage, which will require higher energy and power densities, as well as a significant reduction in cost.1,2 In response to this challenge, various “new” lithium battery concepts such as the lithium–metal (Li–metal), lithium–oxygen (Li–O2), and lithium–sulfur (Li–S) batteries are now being intensely studied. These “advanced lithium batteries” are still in their early stages of research and all suffer from several obstacles like poor utilization of Li–metal, high cost, safety concerns, capacity fading, complicated chemistries and reactions, etc. as reviewed in several papers.3–9 A critical parameter for all these battery systems is “the role of the electrolyte”. Coulombic efficiency and dendrite formation of Li–metal as well as the degradation of electrolytes by the intermediate and final reaction products in Li–O2 and Li–S batteries are directly influenced by the electrolyte.2. Li-salts in aprotic electrolytes
Most electrolytes investigated for rechargeable intercalation batteries and advanced cell chemistries are based on a matrix of aprotic organic solvent(s); other electrolyte concepts like ionic liquids (IL), solids/ceramics, polymers, and aqueous based systems are also investigated, but generally have lower ionic conductivities and higher production costs, or lead to batteries with less power density, limiting their utilization.10–17 ILs and solid/ceramic are still hampered by expensive manufacturing and purification process, which turn them inappropriate for large scale applications in commercial lithium batteries. Similar challenges govern rechargeable aqueous lithium batteries using solid state electrolytes and Li–metal anodes, despite recent works showing promising results.18–20 As an alternative, full cell rechargeable aqueous lithium batteries using Li2SO4 and LiNO3 salt and metal oxide electrodes can offer cheap and environmental friendly batteries. However, the narrow potential window of aqueous electrolytes, compared to aprotic electrolytes, implies limited energy densities. On the other hand, the high ionic conductivity of aqueous electrolytes (1–2 orders of magnitudes higher than aprotic electrolytes) results in very high power densities.Aprotic organic solvent based electrolytes for LIBs, allowing for both high energy densities and appreciable power densities, contain at least two components: the solvent matrix and the Li-salt. These components together provide a medium to transfer charge between the electrodes via the Li+ cations and the counter-anions. While several studies have previously reviewed the impact of the solvent chemistry on the performance of lithium batteries,10–16 the role of the Li-salts has comparatively gained less attention.14–16,21–24 A large number of new anions with simple linear or more complex cyclic structures have been synthesized in the last three decades, and the anion chemistry of the salt has been shown to have a large impact on many properties of interest. In general, an electrolyte needs to meet several criteria concurrently to be considered for LIBs, which are comprehensively discussed in ref. 10–16. Below, we specifically address the demands put forth by the “advanced lithium batteries”.
2.1. Li-salts for re-chargeable batteries
Early studies on lithium batteries with metal anodes in the 1970–1980's were limited to Li-salts with a few anions like hexafluoroarsenate (AsF6−), perchlorate (ClO4−), hexafluorophosphate (PF6−), tetrafluoroborate (BF4−), and trifluoromethanesulfonate or triflate (Tf) (CF3SO3−). A schematic with the chemical structures of these and several other anions discussed in this work is presented in Fig. 1.25–28 With the development of LIBs it became clear that LiAsF6 and LiClO4 were inappropriate for commercial cells, due to safety and toxicity concerns. Similarly, the relatively low conductivities of electrolytes based on LiTf made also this salt less popular. Later, with the commercialization of graphite anodes, the LiPF6 and LiBF4 became the most popular Li-salts as they, together with solvents such as ethylene carbonate (EC), could form a solid electrolyte interphase (SEI) on graphite, making rechargeable lithium batteries commercially viable.29–34 Ultimately, LiPF6 became, and still is, the dominant salt in commercial LIBs. This success is not because LiPF6 is considered “the best” salt in all categories. In fact, LiPF6 is not the leading salt in any of the properties important for batteries like: ionic conductivity – relatively good,35 sensitivity to hydrolysis – poor,36 or thermal stability – poor,37,38 but its success is due to the ability to provide the best balance of these and other properties, as well as the formation of a proper SEI on the graphite anode and a protective layer on the aluminum cathode current collector. This is a clear example showing the complexity of the criteria the Li-salt should fulfill with respect to the several different components of a cell. For specific applications, other salts are often competitive, e.g. LiBF4 has shown to improve the performance of LIBs at high (50–80 °C) and low temperatures (−20 °C),39–41 but its overall drawback is moderate ionic conductivity in the resulting electrolytes.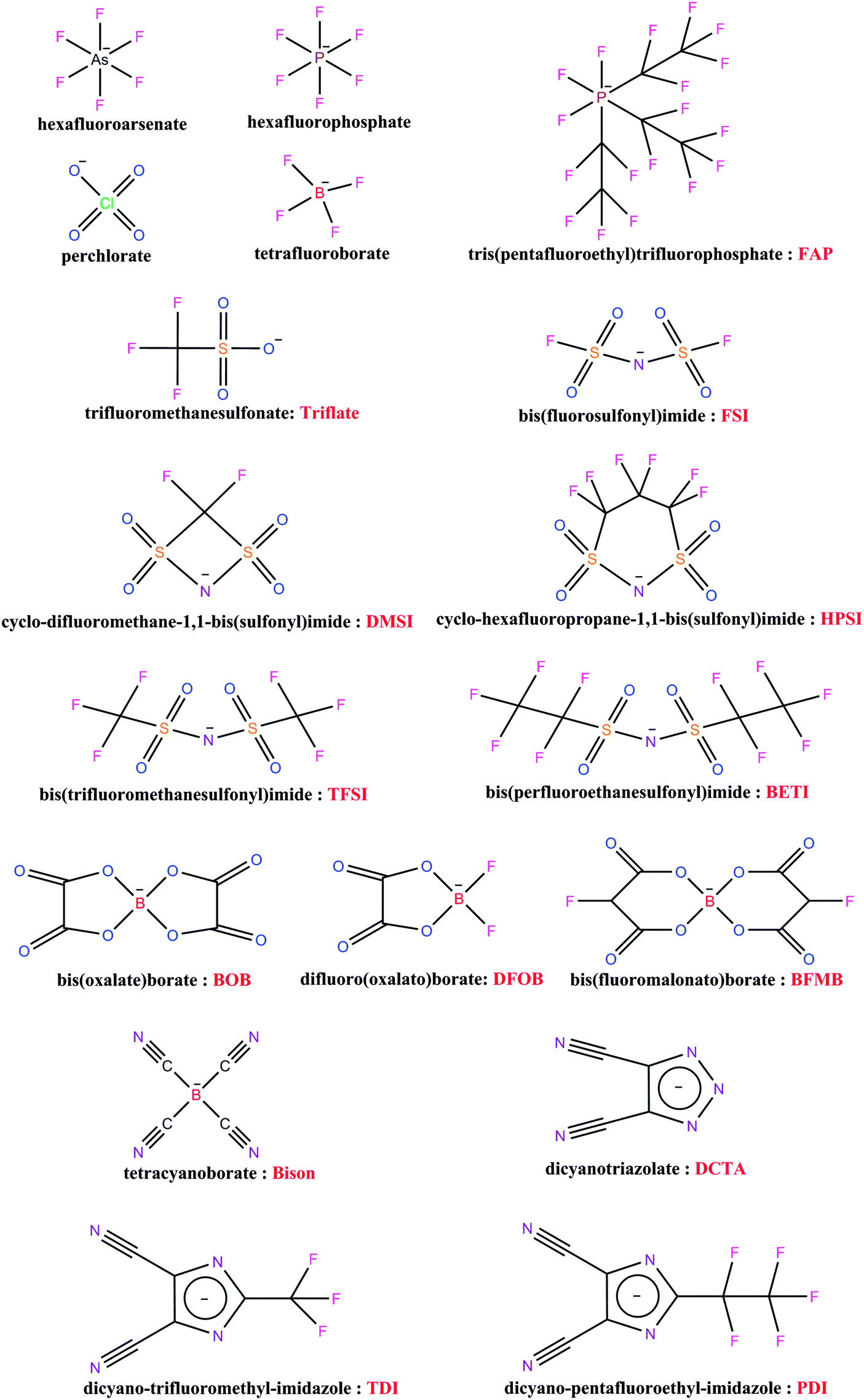 | ||
| Fig. 1 Chemical structures of several of the anions reviewed. | ||
Several salts inspired by LiPF6 and LiBF4 have been synthesized in recent years in an attempt to design salts with improved thermal, ionic or other properties. For example, there was an evolution from anions comprised of ligands around a central atom (e.g. PF6−, ClO4−) to large complex anions e.g. bis(trifluoromethanesulfonyl)imide (TFSI or sometimes TFSA)42 and organic ligand based anions e.g. bis(oxalate)borate (BOB).43 One category of Li-salts comprehensively studied for LIBs contains sulfonyl groups. Triflate is the simplest anion in this family, while imide-based anions with two x-fluorosulfonyl (x = 1–5) groups like bis(fluorosulfonyl)imide (FSI), TFSI, and bis(perfluoroethanesulfonyl)imide (BETI or sometimes PFSI) have recently attracted more attention.44–46 The common issue with these anions is the aluminum corrosion by their electrolytes, but a proper electrolyte solvent or additive can be applied to reduce the corrosion.47 Also, two new Li-salts of this family, lithium-cyclo-difluoromethane-1,1-bis(sulfonyl)imide (LiDMSI) and lithium-cyclo-hexafluoropropane-1,1-bis(sulfonyl)imide (LiHPSI) have been reported to form a stable SEI on a graphite anode and passivate an Al current collector significantly better than LiTFSI.48 Other derivations include compounds combining both a mixture of chemical components for these larger bulk anion components such as tris(pentafluoroethyl)trifluorophosphate (FAP),36 inspired and derived from PF6−, a family of perfluoroalkyltrifluoroborates CnF(2n+1)BF3 (n = 1–4),49 as alternatives to BF4−, and lithium difluoro(oxalato)borate (LiDFOB), with its combination of different ligands of fluorine and oxalate.50
All aforementioned anions (except ClO4− and BOB) contain fluorine (F) atoms in their structure, which increases safety concerns. In this regard, BOB and more recent F-free anions such as tetracyanoborate (Bison)51–54 and dicyanotriazolate (DCTA or sometimes TADC)54–56 are interesting candidates for LIBs. These examples have distinct and unique advantages, but also suffer from issues that have prevented them from replacing LiPF6. The BOB anion is known to take part in forming interphases on both the anode and cathode to improve cell performance, but LiBOB has limited solubility in most aprotic solvents. Bison and DCTA both have high thermal stabilities, but relatively low oxidation potentials and low ionic conductivities of their Li-salt electrolytes. There have been attempts to improve the properties of these salts by adding F-species at the expense of increased safety risks and production costs. Several borate-based anions have been synthesized including bis(fluoromalonato)borate (BFMB) to tune the properties of the BOB anion.57 Similarly, dicyano-trifluoromethyl-imidazole (TDI) and dicyano-pentafluoroethyl-imidazole (PDI) as well as other imidazole or benzimidazole based anions have shown to be more promising compared to DCTA.58,59
Despite the fact that none of the Li-salts mentioned above have been used commercially for LIBs with graphite anodes and 3–4 V cathodes, the “advanced lithium batteries” with different cathode and anode materials may well be better suited. This review therefore makes a first attempt to reveal how the choice of the Li-salt possibly (or not) can meet the different demands that each of these batteries entails. Below, we therefore step through the basic requirements of electrolytes – pointing out how each of the new concepts differs from LIBs and the role of the Li-salt in this respect.
2.2. Solubility
Unlike in protic solvents (e.g. water, ammonia, etc.) in which anion solvation occur by hydrogen bond formation, in aprotic (often just quoted as “non-aqueous”) solvents, the absence of acidic protons implies that the dissolution of a Li-salt primarily is by solvent–Li+ interactions.15,16 Hence, two steps are involved: the dissociation of Li+ from the anion, overcoming the lattice energy of the salts, and the consequential formation of coordination bonds between Li+ and electron lone-pairs of the solvent molecules. Besides the importance of solvent properties like polarity, viscosity, etc., the strength of the Li+–anion interaction is critical. Due to this, many simple Li-salts such as LiCl, LiF, Li2O, etc., are excluded from electrolyte usage since their strong cation–anion interactions result in high lattice energies and thus poor solubilities in relevant aprotic solvents. Therefore, focus has been on using Li-salts of weakly coordinating anions (WCAs),60 made through delocalization of the negative charge over the whole anion via electron-drawing substituents like –F or –CF3, all in order to decrease the Li+–anion interaction.16 Similarly, and to overcome the safety and cost issues, F is now substituted by N in several novel WCAs.58,61 To explore and screen for new promising Li-salts ab initio and DFT calculations have been extensively used to qualitatively address the relative strengths of the Li+–anion interaction using isolated ion-pairs and including the WCA chemistry variations above.62–64 Two recent representative examples using this strategy for distinctly different families of new WCAs aimed at battery usage are the studies by Carboni et al.65 and Scheers et al.66 Given the parameters above, the Li-salt solubility is usually the first consideration when investigating new Li-salts. Moving from LIBs to next generation batteries does not change this notion considerably; the same type of solvents as for LIBs are used and also the preferred salt concentration targets are the same to obtain the maximum conductivity.2.3. Ionic conductivity
Given that next generation batteries with Li–metal anode batteries, including the Li–O2 and Li–S concepts, in general are proposed for high power application like EVs, any selection of a Li-salt should enable to provide a high Li+ transport rate.The Li+ cation conductivity (σLi+) originate from both the total ionic conductivity and the cation transference number (tLi+). Given that the tLi+ in non-aqueous solvents is usually smaller than 0.5, the ionic conductivity plays a critical role in the battery performance.49 In principle, the solvation of a Li-salt in a solvent increases the electrolyte conductivity as a function of salt concentration (by increasing the number of charge carriers). Further increases in Li+ concentration results in lower ionic mobilities (while still increasing the ionic charge densities, this is outweighed by re-combination of ions to neutral contact ion-pairs). The conductivity of any aprotic Li-salt based electrolyte is, however, dependent not only on the salt concentration, but also the anion, the solvent composition, and the temperature. For example, the conductivity of LiClO4 in propylene carbonate (PC) reaches a maximum of 1.5 mS cm−1 at a low salt concentration of 0.08 M, while any further concentration increase results in a lower conductivity.67 In stark contrast, using a solvent mixture of PC and EC, the highest conductivity, 15 mS cm−1, is achieved at a salt concentration of 1 M.67
It is commonly assumed in the LIB literature that a 1 M salt concentration provides the highest conductivity, and thus, the focus has been mainly on the composition of solvent mixture. Table 1 shows the ionic conductivity of a few selected Li-salts dissolved in some typical electrolyte solvents/solvent combinations. It should be noted that the measure of total ionic conductivity is most often used as a screening criterion for electrolytes, without considering the more relevant contribution of the lithium ion part of the conductivity, σLi+.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvent mixtures (a – volume or b – weight) at 20–25 °C.14,15,58,70–72
:1 solvent mixtures (a – volume or b – weight) at 20–25 °C.14,15,58,70–72
| Li-salt anion | Solvent (mixture) | Ionic conductivity [mS cm−1] |
|---|---|---|
| EC: ethylene carbonate; DMC: dimethyl carbonate; DME: 1,2-dimethoxyethane or “monoglyme” or “G1”; DOL: 1,3-dioxolane; TEGDME: tetraethylene glycol dimethyl ether or “tetraglyme” or “G4”. | ||
| ClO4− | EC–DMCa | 10.1 |
| DME–DOLa | 7 | |
| TEGDME–DOLa | 5 | |
| AsF6− | EC–DMCb | 11.1 |
| PF6− | EC–DMCb | 10.8 |
| BF4− | EC–DMCb | 4.9 |
| Triflate | EC–DMCa | 3.1 |
| DME–DOLa | 2 | |
| TFSI | EC–DMCb | 9 |
| DME–DOLa | 11 | |
| TEGDME–DOLa | 7 | |
| BETI | DME–DOLb | 11.1 |
| BOB | DME | 14.9 |
| DCTA | EC–DMCb | 2.7 |
| TDI | EC–DMCb | 6.7 |
| PDI | EC–DMCb | 6.3 |
A novel approach recently proposed is to use highly concentrated or “solvent-in-salt” electrolytes, in which the Li-salt is the dominant component, and surprisingly large tLi+ are achieved.68,69 The higher salt concentration means a higher electrolyte cost, but the approach opens new possibilities to overcome electrolyte degradation, especially interesting for Li–O2 and Li–S batteries, since the solubility and reaction path of intermediate species in these batteries may be tuned by the salt concentration. Also, the cycling efficiency of Li–metal has shown to increase in highly concentrated electrolytes.68,69
2.4. Donor number (DN)
For any type of battery electrolytes the Gutmann73 donor number (DN) is an often used measure for solvents; depicting the electron donating properties and hence the ability to interact with acceptors such as protons, Li+, and other cations. The higher DN, the higher basicity of the solvent, resulting in strong interaction with hard Lewis acids such as Li+. While the DN of Li-salts seldom is mentioned, as most anions have similar and relatively small DN, there is a notable exception of Tf with its rather high DN (Table 2).| Anion | DN |
|---|---|
| PF6− | 2.5 |
| AsF6− | 2.5 |
| TFSI | 5.4 |
| BF4− | 6.0 |
| ClO4− | 8.4 |
| Tf | 16.9 |
Especially in Li–O2 batteries the role of the DN have recently been highlighted,74–76 revealing its influence on solubility and life-time of superoxide (O2−) as well as the discharge reaction pathway and the final capacity of the cells. Altering the Li-salt using anions with low DN (e.g. PF6− or TFSI) may not have any crucial impact based on the DN, but using LiTf thus might.76 In Li–S battery electrolytes the DN is an important parameter in controlling the solubility of polysulfide species,77 even if more studies on this aspect are needed, and also here LiTf is a popular electrolyte salt.
2.5. Electrochemical stability window (ESW) and SEI
From a thermodynamic stability standpoint, a battery should contain an electrolyte with the ESW located beyond the reduction and oxidation potential window of the anode and the cathode, respectively. The difference between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) of the electrolyte defines the theoretical ESW, which is influenced by the chemistry of both solvent and Li-salt.15 These properties can easily be computed using DFT approaches for different choices of anions, often WCAs, focusing on the inherent stability vs. oxidation79,80 – giving a screening of the thermodynamic limit to some appreciable accuracy. However, the commercialization of LIBs in the 1990's was launched for cells with alkyl carbonate electrolytes, which are thermodynamically unstable in contact with graphite anodes. The success of graphite anodes relies on the formation of a passivation layer called the solid electrolyte interphase (SEI), which acts as a barrier for electron transfer between the anode and the electrolyte resulting in kinetic stability of the cell and prevents exfoliation during cycling from solvent intercalation. The SEI forms by decomposition of the electrolyte, and thus, the chemistry of electrolyte solvents and salts affects the composition and properties of the SEI. A similar concept is considered for cathodes with the electrochemical potential below HOMO of electrolyte, where decomposition of the electrolyte can form cathode/electrolyte passivation layer often called the solid permeable interphase (SPI).81 Aluminum is commonly used as the cathode current collector due to its low price and the anodic (oxidative) decomposition of the electrolyte resulting in the formation of a passivation layer on the aluminum, suppressing corrosion in the following cycles.82Fig. 2 shows the influence of different anions on the corrosion potential and maximum corrosion current density of aluminum electrodes in electrolytes containing Li-salt dissolved in propylene carbonate and N,N-diethyl-N-methyl-N-(2-methoxyethyl)-ammonium bis(trifluoromethyl-sulfonyl)azanide (PC and DMMA-TFSA).83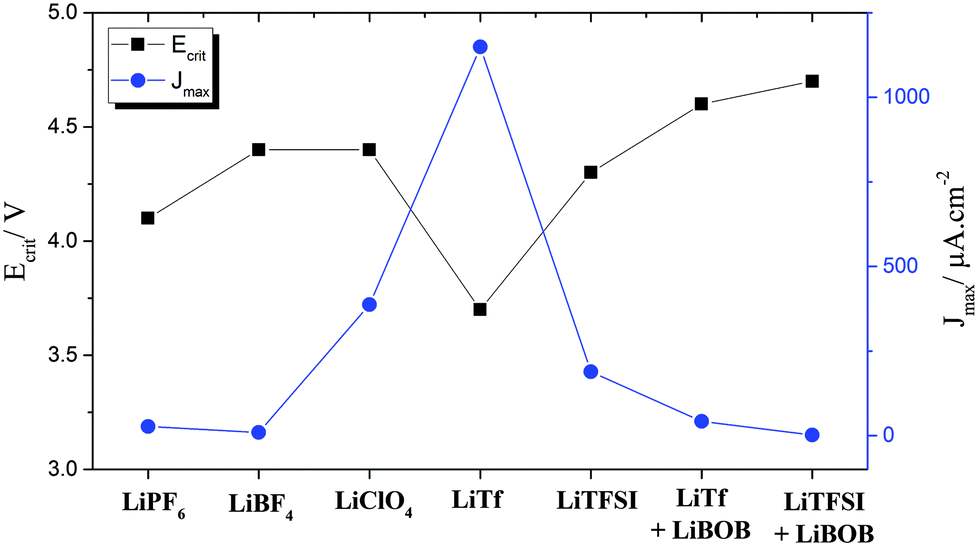 | ||
| Fig. 2 Critical corrosion potentials and maximum corrosion current densities observed on Al electrodes in electrolytes containing different Li-salts. Total concentration of salts was 0.5 mol Li-salt in 1 kg of electrolyte mixture of PC and DMMA-TFSA. LiBOB concentration was 0.15 mol kg−1. Data obtained from ref. 83. | ||
The potential of lithium (−3.04 vs. the standard hydrogen electrode) is higher than the LUMO of aprotic electrolytes, which usually results in the continuous decomposition of electrolyte unless a proper SEI protects the electrolyte – and as all the next generation batteries considered here use Li–metal anodes, this must be considered carefully. In contrast, on the cathode side of Li–O2 and Li–S batteries, the anodic decomposition of electrolytes should be less of an issue as the thermodynamic potentials of cell reactions are below ∼3 V vs. Li+/Li°, which is within the ESW of most aprotic electrolytes. However, overpotentials originating from poor conductivity of reaction products (particularly in Li–O2 cells) can potentially increase the potential of the charging reactions, leading to a higher risk of anodic decomposition.
2.6. Thermal stabilities
It is well known that LiPF6 has poor thermal stability due to the weak P–F bond and the auto-decomposition reaction of PF6− = >PF5 + F−, which can result in formation of toxic gaseous, HF and several other decomposition products, even at moderately high temperatures.84 The strategies to overcome this issue can be divided to 3 categories: (i) replacing LiPF6 by Li-salts with higher thermal stabilities, (ii) using a co-salt to improve the thermal stability of LiPF6-based cells, (iii) adding additives to increase the thermal safety of LiPF6-based cells. The first two approaches have initiated several works to synthesise e.g. sulfone- and boron-based anions with higher thermal stabilities.85Table 3 shows the melting points and initial decomposition temperatures of several Li-salts. However, the thermal stability of the Li-salt is not necessarily an adequate measure to improve the thermal stability of batteries. In fact, the solvent chemistry as well as the electrode composition and morphology together with the Li-salt all influence the thermal stability of the cells. For example, LiBOB, as compared to LiPF6, has been shown to decrease the reactivity of mesocarbon microbeads negative electrodes, while it increases the reactivity with the LiCoO2 positive electrode.86 The graphite electrode has a higher thermal stability in an LiPF6 EC/DMC electrolyte compared to an identical cell with the LiBF4 salt, even though the LiPF6 salt has lower decomposition temperature as compared to LiBF4.29 Such studies have been mainly performed for Li-ion batteries with electrolytes and electrodes close to commercial cells, and Li–metal electrodes have rarely been the subject of these thermal stability investigations.87–90 The introduction of novel anions showing promising results for Li–metal anodes thus requires a new look at the thermal stability, while the challenge to meet several other requirements in addition to the thermal stability of course remains.2.7. Chemical stability in contact with reaction products
Electrolytes should be either (i) thermodynamically stable or (ii) kinetically stabilized in contact with battery components including electrodes, as discussed above. Likewise, electrolytes should be stable in contact with reaction products formed during cycling. This is particularly important for the Li–O2 and Li–S concepts in which new species are formed in each cycle, in contrast to conventional LIBs based on intercalation electrodes.It should be emphasized that the degradation of Li-salts (or solvents) by intermediate/final products in Li–O2 and Li–S batteries are not to be confused with the concept of partial decomposition of Li-salts on the anode and cathode surfaces in LIBs to form the SEI and/or protective layers on the anode and/or the current collectors (see above). In contrast, in Li–O2 batteries the intermediate/final products continuously form in each discharge and consequently even a very small amount of electrolyte degradation make for pre-mature battery failure (which is observed today).
A most noteworthy contamination in batteries is traces of water and the hydrolytic sensitivity most Li-salt anions demand special care to be taken. This basically means extra costs for the extreme purification of electrolyte salts and solvents as well as for assembling cells in water-free environments. More importantly, the hydrolysis of the F-containing anions is often accompanied with HF formation, which is a serious safety issue. HF can also degrade electrolytes and electrode materials, especially at high temperatures or high voltages.
PF6 is well known to be poor in terms of hydrolysis as it can readily degrade to phosphorus pentafluoride (PF5) and lithium fluoride (LiF), and the former can easily undergo hydrolysis to form hydrofluoric acid (HF).95 Hence, commercial LIBs usually contain a mixture of scavengers, at the expense of extra cost, to adsorb produced HF. Under some circumstances HF is actually believed to play key role in formation of a flexible SEI on Li–metal, which can increase cycling efficiency of the electrode (see Section 4).
For Li–O2 and Li–S batteries, the role and influence of any contaminant water in the reaction pathways and the formation of reaction products are under debate.75,96,97 This is notably critical to assess for the Li–O2 concept as these cells are open to the surrounding atmosphere, and thus the Li-salt should remain robust in contact with traces of water originating also from any “pure” oxygen feed.
2.8. Production cost of Li-salt
The final cost of any Li-salt depends on several parameters such as cost and toxicity of reagents, synthesis procedure, waste produced, the salt purification, etc. Also, the scalability of the process(es) and the intellectual property rights will eventually influence the cost. Notably, large efforts have been devoted to develop low-cost and scalable methods to synthesize LiPF6 with a very high purity. In early works, LiF and PF5 were reacted in liquid anhydrous hydrogen fluoride or in an organic solvent such diethyl ether.98,99 However, H2O and HF are two common impurities resulting and requires further purification of LiPF6 by recrystallization in a dry organic media. Later, a method developed by Wiesboeck et al., LiF and PF5 were reacted in the presence of acetonitrile at high pressure and low temperatures (−40 to −80 °C) to form a tetra-acetonitrolo lithium hexafluorophosphate, Li(CH3CN)4PF6, complex, to be dissociated into very high purity LiPF6 and CH3CN.100 Similarly Li(CH3CN)4PF6 can also be produced by a direct reaction between a HPF6 acid solution and LiOH in acetonitrile.101 The general trend is to improve the synthesis methods and production time, by simplifying the procedure while increasing the purity of the produced LiPF6.102–106To our knowledge, there is no report discussing all synthesis procedures for the different Li-salts. Since many of the Li-salts have only been synthesized in a small scale with different purities within academia and with no available information about the expenses of the synthesis process, it is impossible to truly compare the total cost for the large scale production of different salts. The purchase price of commercialized Li-salts may, however, give a reasonable first approximation comparison and in Table 4 we report these data based on three different providers. Note that to obtain a 1 M electrolyte using heavier (higher molecular weight) salt requires more mass of the salt, resulting higher cost and lower gravimetric energy density of cells, and hence also the molecular weight of the Li-salt needs to be accounted for (e.g. to prepare 1 M electrolyte using the LiTFSI salt compared to the LiBF4 salt, almost 3 times more in mass is needed). The large cost differences are likely due to the different purities of the Li-salts; for example Sigma-Aldrich provides “battery grade” Li-salts, usually with a purity above 99.99%, while the salts produced by BASF have lower purities (no comparative information is provided by EMD Millipore).
| Salt | Molecular weight [g mol−1] | Sigma-Aldrich | BASF | EMD Millipore | ||
|---|---|---|---|---|---|---|
| Estimated pricea [USD per kg] | Purity (%) | Priceb [USD per kg] | Purity (%) | Estimated pricea [USD per kg] | ||
| a The prices are estimated according to the available prices in the website of Sigma-Aldrich and EMD Millipore for small quantities of 10–250 g in March–April 2015. b The prices are obtained from BASF for 1 kg of the salts in March–April 2015. | ||||||
| LiPF6 | 151.9 | 8700 | ≥99.99 | 500 | 99.8 | |
| LiBF4 | 93.7 | 28000 |
99.99 | 650 | 97.5 | 5800 |
| LiAsF6 | 195.9 | 15000 |
98 | — | — | |
| LiClO4 | 106.4 | 2700 | 99.99 | 500 | 99.0 | |
| LiBOB | 193.8 | 5200 | — | 650 | 99.0 | |
| LiTf | 156 | 8000 | 99.995 | 650 | 99.5 | |
| LiTFSI | 287.1 | 4100 | 99.95 | 650 | 99.9 | 3800 |
| LiDFOB | 143.8 | 9500 | 99 | — | — | |
The cost of the Li-salt is, however, not yet a limiting factor for neither Li–metal, Li–O2, or Li–S batteries, since these are still in their early development and it is difficult to predict the Li-salt ultimately to be implemented in commercial cells.
3. Applying current knowledge to future battery chemistries
In Section 2, many of the important criteria used to evaluate Li-salts for Li-ion batteries were discussed. These criteria (solubility, conductivity, stability, SEI formation) are likely to also be critical for future Li-based rechargeable batteries. However, with these newer chemistries other factors also become important. For example, in metal–air batteries, the electrolyte needs to be stable in contact with O2 and its reduced species. The next three sections discuss the role of the Li-salt in Li–metal, Li–O2 and Li–S batteries with a particular emphasis on the differences in reaction chemistry of these systems as compared to the LIB's. There is still no complete predictive insight allowing rational pre-selection of a specific Li-salt for specific battery chemistries. With this in mind, the status of the many different Li-salt selections for tests made for each of the three next generation batteries is reviewed below.4. Li-salts for Li–metal batteries
In theory, Li–metal is the best anode material for any lithium-based battery technologies. It has a high volumetric energy density (2046 mA h cm−3) and the highest gravimetric energy density (3862 mA h g−1) of any known material.107 It also has a flat voltage profile and the lowest redox potential (−3.04 V vs. SHE) of any alkali metal. As is commonly known in the LIB field, however, metallic Li electrodes suffer from two fatal flaws. First, metallic Li is prone to form dendrites during cycling, which increases the risk of catastrophic fires or loss of capacity when the dendrites separate from the bulk of the Li.108 Second, Li electrodes continuously form a SEI layer with cycling, causing significant Coulombic efficiency losses, consumption of Li, and increases in cell resistance.108 This effect is often evident in studies using Li-based half-cells such as the data shown in Fig. 3. Here, the same batch of LiNi1/3Mn1/3Co1.3O2 cathode is cycled versus graphite (full cell) or a Li–metal anode (half-cell). For the half-cell there is an apparent loss of capacity at ca. 60 cycles due to the insulating SEI layer being formed. These two problems need to be solved because the use of a Li–metal anode is required for advanced battery chemistries like Li–O2 and Li–S in order to realize dramatic improvements in energy density, vehicle range, and cost requirements envisioned.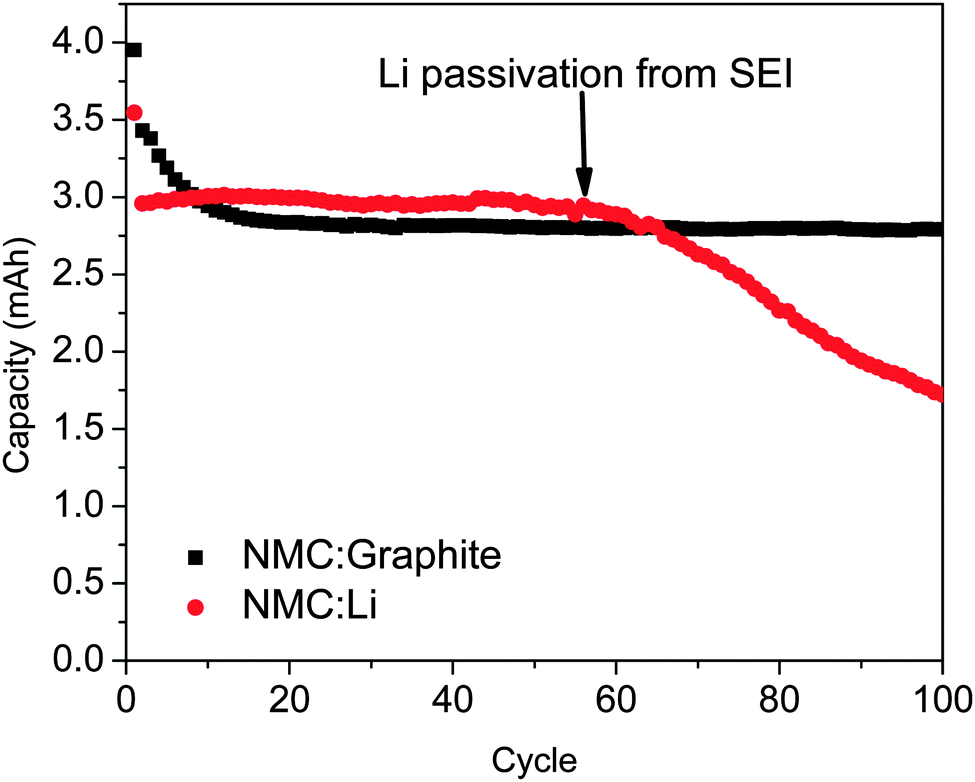 | ||
Fig. 3 Capacity versus cycle number measured for LiNi1/3Mn1/3Co1.3O2 (NMC) versus Li–metal ( ) and graphite (■) using a standard 1.2 M LiPF6 in EC/DMC (3:7 wt%) electrolyte at 20 °C. The capacity fade at ∼60 cycles for the Li anode is due to SEI formation. ) and graphite (■) using a standard 1.2 M LiPF6 in EC/DMC (3:7 wt%) electrolyte at 20 °C. The capacity fade at ∼60 cycles for the Li anode is due to SEI formation. | ||
There are two general methods to prevent these losses for Li–metal anodes. First, one could introduce a solid electrolyte to act as a physical barrier.109 This barrier should be strong enough to prevent dendrites from pushing towards the cathode while simultaneously preventing liquid electrolyte from coming in contact with the metallic Li thus preventing SEI formation. Modelling performed by Monroe and Newman indicated that the lithium dendrite formation can be inhibited when the solid electrolytes shear modulus is twice that of metallic Li, or greater than about 7 GPa at room temperature.110,111 The potential choices of solid electrolytes are limited due to instability in contact with Li–metal and low ionic conductivities,112 however, under the right conditions, lithium can be cycled thousands of times with very little loss of capacity.113,114 Advances in protected Li–anodes will require significant research on the solid–solid interface, formation of thin ceramic sheets, and optimization of electrolyte chemistries. The second method to stabilize Li–metal anodes is through a protective layer formed by a chemical reaction. An SEI layer would fall into this category as well as a flexible polymeric material.115
Attempts to develop electrolyte chemistries to stabilize the Li–metal surface have been performed for a long time. In all cases, the solvent and the Li-salt decompose to form an SEI layer. The anions of the Li-salts are decomposed forming the inorganic component of the SEI layer as has been widely discussed.108 No predictive correlation between the choices of Li-salts and solvents and the best SEI exists, despite the relatively low number of suitable battery Li-salts. In some cases, like LiBOB, the SEI layer is too thick and highly resistive, which lowers the power and rate performance of the cell.116 Other salts such as LiBF4 do not form stable dense SEI layers on Li, however thinner SEI layers built using LiBF4 do result in a low charge transfer resistances enabling operations at low temperatures and high rates.116 In yet other cases, the reduction of the anion is not sufficient to form a suitable SEI layer to prevent the continuous consumption of the electrolyte. The desire is to develop the optimal chemistry that will be compatible with Li–metal as well as current state-of-the-art high voltage cathode materials. There have been hundreds of studies that have focused on understanding the origin of the SEI layer on Li–metal as a function of solvent mixed with a salt. Here, we only intent to highlight key findings on Li–metal–SEI chemistry and point out what we believe are the most promising research directions or particularly interesting results.
For the passivation of Li–metal anodes there are only a few solvent/salt systems which produce a stable SEI that can enable long term cycling.14 A commonality to these solvent/salt systems is that they are not purely based on carbonates/LiPF6, in contrary to electrolytes for commercial LIBs. For example, electrolytes of DOL and LiClO4 or LiAsF6 provide excellent Li passivation resulting in cycling efficiencies greater than 96–98%.117 The lithium deposition is smooth and results in very little dendrite formation. The origin of this stability is the polymerization of DOL, which forms an elastic barrier on the Li surface that “breaths” as the surface undergoes volume changes.108,118 This system suffers when the electrolyte gets consumed during high rate applications and tends to form smaller Li particles.117 These smaller Li particles are then passivated which consumes all available electrolyte within the cell. A second stable system is 2-methyltetrahydrofuran (2-MeTHF) with LiAsF6, which cycles with >96% efficiency. It is believed that the 2-MeTHF provides a good surface because it reacts very slowly with the Li–metal.119 Finally, the LiBETI salt has been reported to cycle Li with around 97% efficiency in a EC–DME solvent mixture.120
The results above have led to a search for other potential electrolyte chemistries, with the gained knowledge that cyclic oxygen containing solvent molecules and an inorganic salt capable of forming LiF appear to be essential for a dense and stable SEI layer. Indeed, nearly all the Li-salts under investigation, with the exception of LiClO4 and LiBOB, are fluorinated. Fluorinated salts tend to be more hygroscopic and thus contain some more water (ppms), which changes the SEI chemistry significantly.
One of the most interesting sets of studies to correlate chemical reactivity of electrolyte components with Li passivation was reported by Odziemkowski and Irish.121,122 These authors measured the magnitude and rate of reaction of mixtures of several salts (LiAsF6, LiClO4, LiBF4, LiPF6 and LiN(CF3SO2)2) with various solvents (THF, 2-MeTHF and PC) in contact with pure Li–metal. They found the LiAsF6 salt to react most rapidly with Li, forming a passivating film.121,122 This explains the suitability of LiAsF6 to form a stable SEI layer. The other salts react slower and take longer to form a passivating film. With these slower reacting salts, LiPF6 and LiBF4 had the largest reactivity, as indicated with the largest corrosion potential maxima. This data shows that reduction of the salt anion may be most critical to the passivation of Li. The influence of the solvent was not as dramatic, although there was clear evidence that solvent plays a role in the stability, especially with time. Subsequent studies by Rahner showed that the reactivity of the salts depended strongly on the solvent properties and salt concentrations.123 Indeed, at low salt concentrations of LiClO4, the anion reduction and PC decomposition is faster than at high concentrations of LiClO4. This correlation between the Li-salt and solvent indicates there is a possibility to optimize the formation chemistry of the SEI layer although this chemistry maybe far from optimal for battery operation.
To understand some of the effects of salts on the SEI formation Nishikawa et al. have performed holographic interferometry measurements to measure the concentration of gradients of Li+ as a function of potential and time near dendrites and a Li surface in PC with LiClO4 and LiPF6.124 Their results show an incubation period, which depends on the salt and the applied potential, which mediates dendrite growth and roughness. For example at low potentials (0.5 mA cm−2), the concentration of Li+ around a Li–dendrite was the same as the concentration of Li around a dendrite free surface until about 25 seconds. At this point, the concentration of Li+ dropped significantly as the dendrite grew. Interestingly, the incubation period around the dendrite in LiPF6 based electrolytes was longer than the period for LiClO4 based electrolytes at a current density of 0.5 mA cm−2. At higher applied currents (>1.0 mA cm−2), the incubation period is reversed such that LiClO4 reacts faster. The authors attribute these changes in incubation periods to SEI formation and electrolyte impurities.124 Within LiPF6 electrolytes, there is a small concentration of water, which reacts with LiPF6 to form small concentrations of HF. This HF reacts with the Li–metal to form a dense LiF surface. LiClO4 does not suffer from the same reactions with water so there are less inorganic salts on the Li–metal surface. With the application of a potential, the less passivated Li surface (LiClO4/PC) will rapidly react to form dendrites, whereas the LiF terminated surface requires more current to break up the LiF surface leading to longer incubation times. At higher rates, there is not enough time for the HF to diffuse and react with the Li surface leading to lower incubation times. Interestingly, the longer incubation times needed to break down the LiF layer would be consistent with the widely reported smoother lithium deposits using LiPF6 salts compared to LiClO4. Electrochemical quartz crystal microbalance measurements (EQCM) support the more stable, or thinner, SEI produced with LiPF6 compared to LiClO4 and other salts such as LiTf consistent with the arguments laid out above.125 Subsequent work by Nishikawa et al. found the SEI layer to affect the morphology and growth rates of the Li dendrites. They reported that LiPF6 dendrites were more hemispherical, while LiClO4 dendrites were more like whiskers. The dendrites grow linearly with square root of time, which indicates an ionic mass transfer rate in solution. These results clearly show the importance of ionic conductivity in the electrolyte, which is a function of Li-salt species.126 Larger anions are less mobile in solution and tend to have higher Li-dissociation constants. Under circumstances where Li dendrites are formed, this higher Li transfer may accelerate dendrite formation.14
Throughout all the studies of Li–metal electrodes, the most common salt used is LiPF6. Regardless, LiPF6 does not successfully passivate metallic Li surfaces or enable extended cycling of Li. Consequently, there has recently been a desire to discover new Li-salts, which may form a stable SEI through the formation of more “flexible” decomposition products. These new salts can be broken down into a few categories. The first are based on borate structures. The simplest borate structure is LiBOB, Fig. 1, which very effectively forms an SEI layer. Unfortunately, the layer is too resistive to be suitable for extended cycling at high rates and the solubility of LiBOB is too low (0.8 M in PC/EC/EMC in 1:1:3 wt%).116 However, the ability of the BOB anion to polymerize into an extended network presents an opportunity to formulate a more flexible SEI layer, provided the right salt chemistry can be obtained. Numerous fluorinated borate based salts have been fabricated including LiBFMB57,127 and LiDFOB.50,116,128 Both of these Li salts have much higher solubility in carbonate solvents than LiBOB and Li transference numbers greater than LiPF6 (tLi+ = 0.47 LiBFMB; 0.33 LiDFOB; 0.24 LiPF6).127 Schedlbauer et al. reported a study of the cyclability of Li in LiPF6 and LiDFOB electrolytes,129 and found that the LiDFOB electrolytes had much higher Coulombic efficiencies compared to LiPF6 upon cycling, 95–97% vs. 75% after 500 cycles. Subsequent work by Schedlbauer et al. revealed that the LiDFOB salt improves cycling efficiencies at a variety of cycling rates (0.1–1.0 mA cm2).130 These results demonstrate a significant improvement in cyclability and promising potential, though still not on par with graphite electrodes. A second class of designer Li-salts are based on imide chemistry.48 Two new salts with a ring structure, LiDMSI and LiHPSI,48 are built from the LiTFSI structure, but have added or removed F atoms to form a strained ring structure. To our knowledge, there are no reports on the compatibilities with Li–metal.
In addition to designing new Li-salts, there have been several reports of the addition of simple Li-salts to Li–metal surfaces and a corresponding increase in cyclability. For example, Li terminated with Li3N was shown to cycle with about 85% Coulombic efficiency compared to unterminated Li (70% efficiency) in the same LiPF6 based electrolyte.131 The Li3N appears to promote the formation of a more complete SEI layer lowering parasitic currents while simultaneously reducing Li transport resistances by almost 50%. Similar to Li3N, terminating the Li surface with Li2CO3 also shows an almost 5-fold decrease in diffusion resistances compared to uncoated electrodes after 7 cycles in LiPF6 electrolytes.132 It was recently demonstrated that the addition of LiNO3 stabilizes a Li–metal anode during operation in a Li–air cell.133,134 These authors reported the LiNO3 reacts with or inhibits the formation of species that are electrochemically active above 3.4 V (vs. Li/Li+). The LiNO3 also enables cycling of the Li–metal in an oxygen rich environment without excessive SEI formation.133 It has also been shown that O2 gas can stabilize SEI on Li–metal anode leading to increase in Coulombic efficiency in a solution of LiClO4 in dimethyl sulfoxide134 or LiTFSI in N,N-dimethylacetamide.135 Finally, it was recently reported that the addition of 0.05 M CsPF6 to a LiPF6/PC electrolyte effectively reduces dendrite formation with cycling.136 Albeit, the efficiency of the cycled Li is not much better than the standard electrolyte (76.5 vs. 76.6%).136 The ability to prevent dendrites is a major accomplishment and advancement for the production of safer Li-ion battery cells.
All of the examples described above involve electrolyte chemistries with salt concentrations around 1 M. Several research groups have begun focusing on electrolytes with salt concentrations significantly higher than standard electrolytes. For example, comparing a 3.27 mol kg−1 LiBETI to a 1.28 mol kg−1 LiBETI solution in PC revealed a significant enhancement in Li–metal cyclability.68 Indeed, the efficiency increased to nearly 80% after 40 cycles at high salt concentrations, while it dropped to 0 at low concentrations. From TEM data, the authors showed a thinner SEI layer with the higher LiBETI concentrations and a thicker SEI layer with lower LiBETI concentrations (20 vs. 25 nm). In addition, the thinner SEI suppresses dendrite formation. The authors believe the solvation of the Li ions is changed sufficiently to modify the SEI formation reaction. Recently, Zhang et al. showed that a high Coulombic efficiency of 98.4% could be achieved for >1000 cycles of lithium plating/stripping at a current density of 4 mA cm−2 when the concentration of LiFSI in DME was increased to 4 M. The average Coulombic efficiency over 500 cycles could be increased to 99.1% at a lower current density of 0.2 mA cm−2. These high Coulombic efficiencies are referred to limited side reactions of DME with Li–metal due to the increased concentration of Li-salt as well as to the formation of a highly compact SEI.137 In another example, Yamada et al. recently reported that a supersaturated 4.2 M LiTFSI acetonitrile solution showed a significant increase in stability and cyclability of a graphite anode compared to a traditional 1 M solution.138 The origin of this stability is likely due to changes in the Li+ ion solvation. This is significant because acetonitrile is typically reduced by Li, but these high salt concentrations seem to prevent/reduce the solvent decomposition. In these reports, the reversibility was not as high as desired, but significant improvements were observed.
5. Li-salts for Li–O2 batteries
The Li–O2 battery, often called the Li–air battery, in its non-aqueous implementation holds the promise of providing a specific energy considerably higher than conventional LIBs (∼2000 vs. ∼600 W h kg−1).5 The non-aqueous Li–O2 battery is, in its most simple design, comprised of a Li–metal anode, an electrolyte, and a porous cathode wherein oxygen gas is reduced and reacts with Li+ cations (Fig. 4).4–7 The oxygen reduction reaction (ORR) and plausible subsequent reactions are described below. Lithium peroxide (Li2O2) is the dominant discharge product.139,140| O2 + Li+ + e− → LiO2 E° = 3.0 V vs. Li+/Li° |
| LiO2 + Li+ + e− → Li2O2 E° = 3.1 V vs. Li+/Li° |
| 2LiO2 → Li2O2 + O2 disproportionation reaction |
| O2 + 2Li+ + 2e− → Li2O2 E° = 2.96 V vs. Li+/Li° |
| O2 + 4Li+ + 4e− → 2 Li2O E° = 2.91 V vs. Li+/Li° |
| Li2O2 + 2Li+ + 2e− → 2 Li2O E° = 2.87 V vs. Li+/Li° |
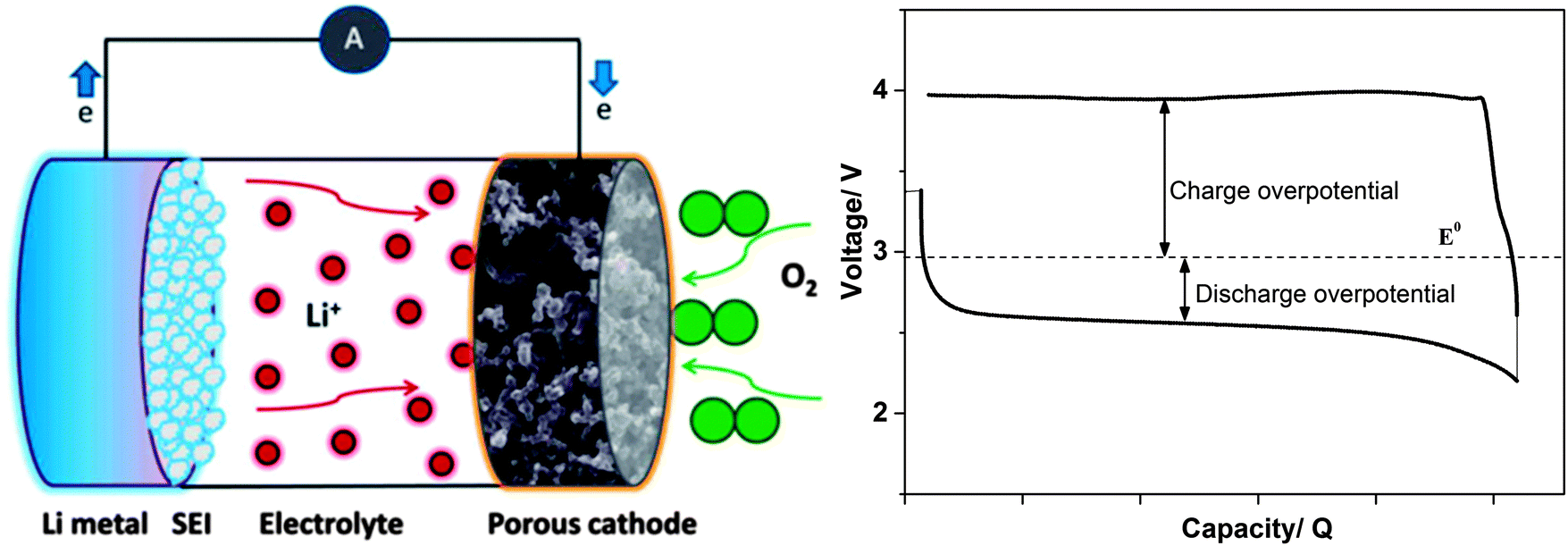 | ||
| Fig. 4 A schematic drawing of a Li–O2 cell (left) and typical galvanostatic discharge–charge curves showing overpotentials (right). | ||
The thermodynamic potential of the reactions is within the ESW of most aprotic electrolytes, and thus, the ORR products are predicted to only be Li–O compounds. Therefore, the passivation to mainly worry about is the Li–metal anode and reactions with water/CO2 from the air. Nevertheless, it is now well accepted that the discharge products contain different compounds formed from the degradation of the electrolyte in contact with intermediate and final reaction products.141–146 Superoxide radical anion (O2−) and lithium superoxide (LiO2) formed during ORR can attack solvents or Li-salts, instead of reacting with Li+ cations to form the final reaction products (see, Fig. 5). The charge reaction or oxygen evolution reaction (OER) is accompanied with overpotentials originating from poor conductivity of Li2O2 or side products such as Li2CO3.147–151 As a result, the anodic (oxidative) stability of electrolyte solvents and salts may become a challenge for electrolytes with limited ESW.
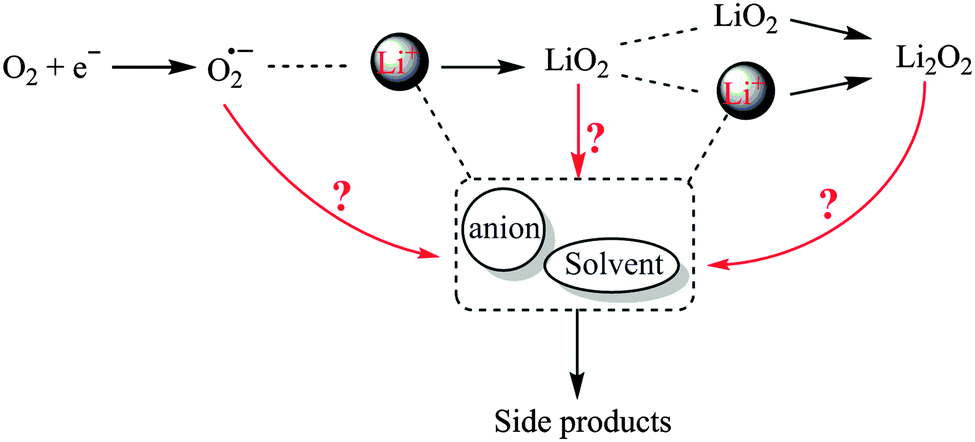 | ||
| Fig. 5 A schematic illustration of possible parasitic reactions of O2− and LiO2 with Li+, solvents, and anions resulting in Li2O2 and/or side products in Li–O2 cells. | ||
Beyond the different reaction processes, the choice of Li-salts impacts the viscosity, the oxygen solubility, and the wettability of the electrolytes.72,152–154Fig. 6 shows discharge curves in cells with different Li-salts dissolved in a DME:DOL solvent mixture, displaying that the discharge capacity increases as: LiBr > LiTriflate > LiTFSI > LiBETI. This was attributed to both the viscosity and the oxygen solubility of the electrolytes being altered by the Li-salts.72 Equally important, a Li-salt should promote the formation of a proper SEI on the Li–metal anode (see Section 4).155
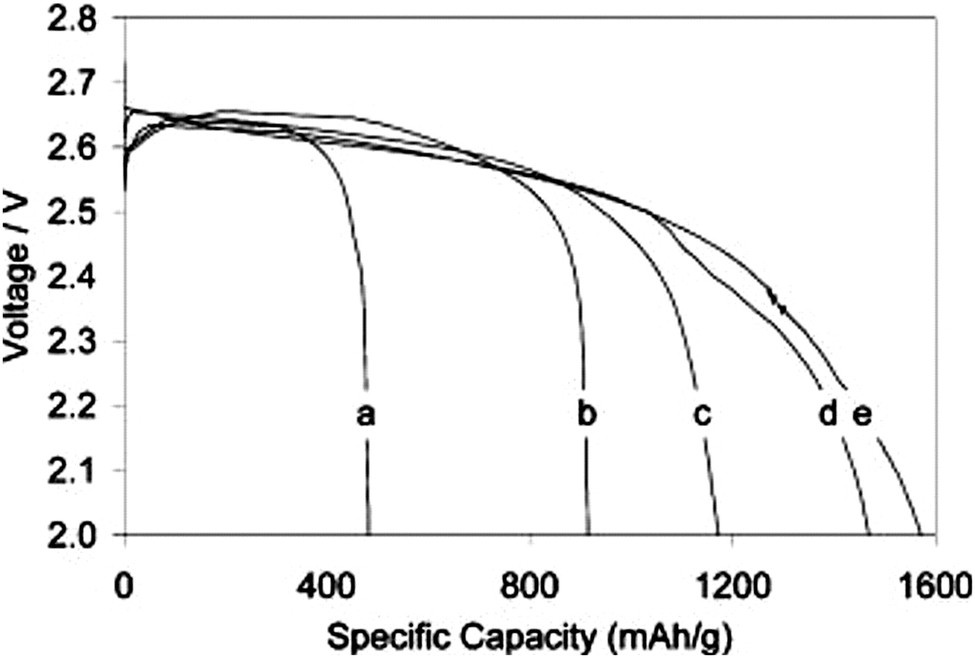 | ||
| Fig. 6 Discharge curves of Li–O2 cells using 1 M of: (a) LiBETI, (b) LiTFSI, (c) LiTf, and (d, e) LiBr dissolved in DME:DOL (1:1).72 | ||
Early studies on the Li–O2 battery utilized the most common LIBs salt i.e. the LiPF6. Subsequent investigations revealed that LiPF6 salt is unstable in Li–O2 batteries156–163 and in particular X-ray photoelectron spectroscopy (XPS) studies have revealed that it decomposes to LiF, LixPFyOz, and other P–O containing compounds during cell cycling.156–159 FT-IR, NMR, X-ray diffraction (XRD), and non-resonant inelastic X-ray scattering (NIXS) results indicate similar findings.158–160 LiF and other degradation products of LiPF6 can remain on the surface of cathode, and consequently, passivate the surfaces and/or block the pores. These results highlight the challenges of designing an electrolyte for Li–O2 cells despite all these decomposition reactions occurring at potentials (∼2.5–2.7 V vs. Li+/Li°) well within the electrochemical stability window of the electrolyte.
The degradation of the LiPF6 salt can be attributed to parasitic reactions with (i) intermediate and/or (ii) final ORR products. The former include degradation by O2− and LiO2, while the latter refers to degradation of the LiPF6 salt by Li2O2.140,161,162,164 Another common source of parasitic reactions is trace water, which has shown to substantially alter performance of the Li–O2 cells.75,96 The presence of water contamination commonly leads to an increase in the first discharge capacity, but also the formation of LiOH.96,157 In the presence of water, LiPF6 readily forms HF, which consequently degrade cell components.
The degradation of LiPF6 guided researchers to study other Li-salts, often previously used in LIBs. LiTFSI has been widely used in Li–O2 cells, but the TFSI anion has been shown to be unstable vs. ORR and OER in ether- and sulfone-based electrolyte solvents.157,159,164–166 LiTf has been used in several studies indicating high re-chargeability, however, the salt stability is still under debate.159,167 A unique feature of LiTf is the very high DN of the Tf anion (DN = 16.9) compared to all other anions employed (see Table 2). This could be favorable for Li–O2 cells since it has been shown that a high DN of electrolytes results in increase higher solubility of LiO2 as well as the discharge capacity of Li–O2 cells.74–76 Degradation of LiTFSI and LiTf both result in the formation of LiF and –CF3.157,159,164–166 Similarly, LiF has been detected as a degradation product of the LiBF4 salt.157,159,161
Taking into account that LiF always forms when any of the aforementioned Li-salts are used, a fluorine-free anion could arguably improve the performance. Hence, LiBison was used,168 but here the B–C bonds were found to break and form B2O3 during cycling. For LiBison, the solvent chemistry also influences the degradation.168 Another non-fluorinated Li-salt is LiBOB, which has also been shown to decompose.159,169–172 Nazar et al. proposed a decomposition mechanism by superoxide radicals resulting in formation of lithium oxalate and lithium triborate (Fig. 7).170 Although it has, and will have, no industrial application, due to severe safety issues, LiClO4 has been used in several academic studies reporting high re-chargeability of Li–O2 cells.173,174 The relatively higher stability of LiClO4 suggests it to continue to be suitable for the purpose of tests and development at the research level.157,159,169
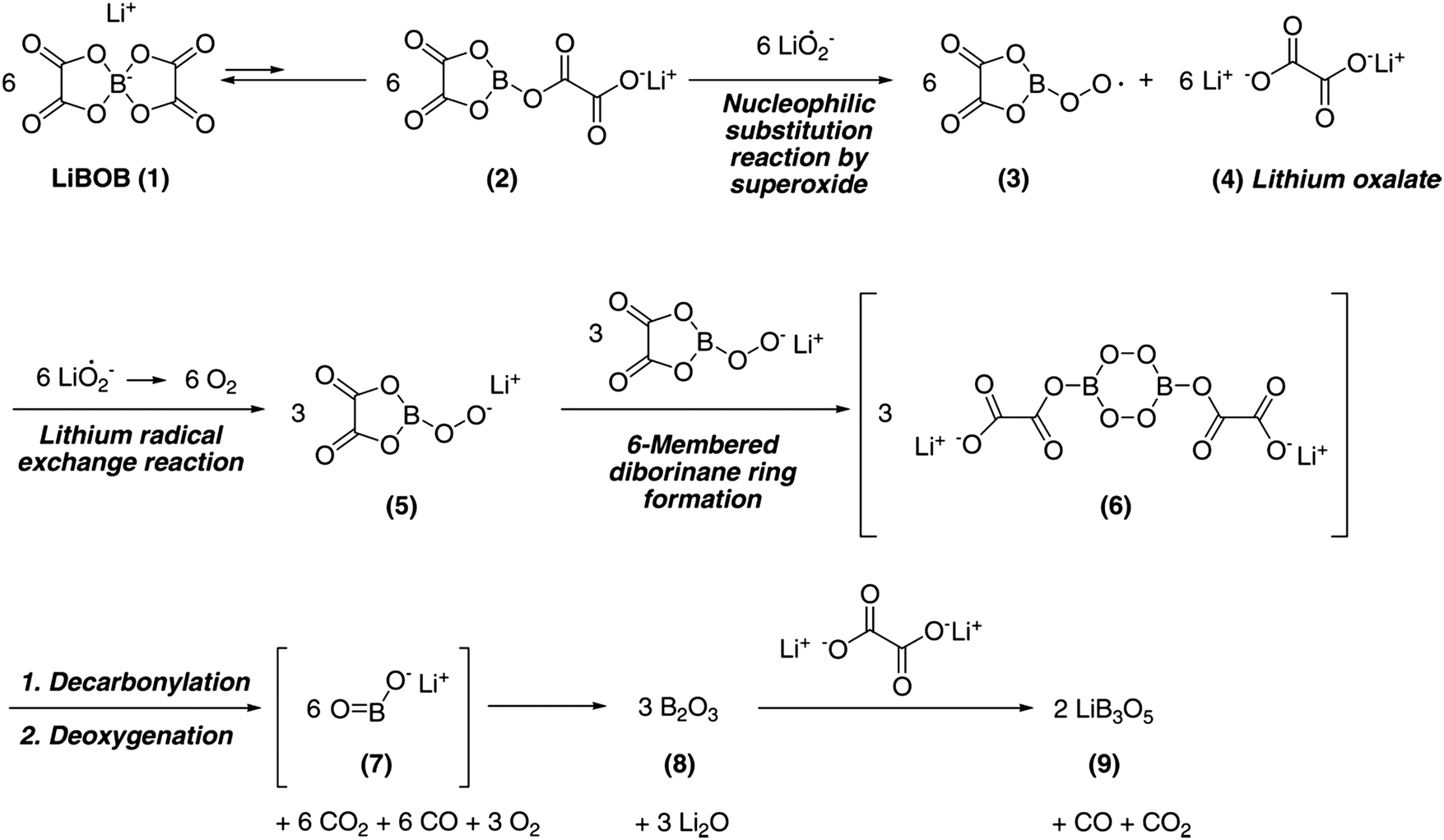 | ||
| Fig. 7 Proposed mechanism for decomposition of LiBOB by superoxide radicals, obtained from Hyoung Oh et al.170 | ||
Alongside the “super- concentrated” electrolyte approach for Li–metal and Li–S batteries, two studies have recently showed that a relatively high concentration of Li-salt, 3–5 M, can also improve the performance of Li–O2 batteries.175,176 These studies suggested the mechanism of protection to be that the superoxide radical now has a higher probability to react with solvated Li+ than with solvent molecules.
Alongside practical cell studies, the stability of various Li-salts has also been addressed by quantum chemical computational methods. Due to the vast computational cost associated with analyzing reactions within the Li–O2 system properly as high level accuracy methods are needed to describe, e.g. interactions of the salt with the redoxactive molecule or solvent and the solvation structure of salt in solution,177 only a limited set of studies have been performed thus far. Du et al. combined ab initio molecular dynamics (AIMD) and density functional theory (DFT) calculations at the B3LYP level to investigate the hydrolytic stability of LiTFSI, LiTf, and LiPF6 in tri(ethylene glycol)-substituted trimethylsilane (1NM3) and tetraglyme, respectively.158 LiPF6 was shown to react with even small amounts of H2O in 1NM3, whereas LiTf showed a much higher stability. All three salts were, however, found to be compatible with TEGDME. It should be noted that the investigated system sizes are small and the time-scales short, so additional verification is needed. Subsequent work by the Lau et al. found that LiBOB leads to the formation lithium oxalate during discharge and, according to DFT calculations, the formation of lithium oxalate is exothermic and thermodynamically favorable why LiBOB probably is not a suitable salt for the Li–O2 battery.171
6. Li-salts for Li–S batteries
The Li–S battery concept has the promise of both very high energy densities (2567 W h kg−1 and 2800 W h l−1) and the use of, in principle, an extremely in-expensive cathode material – sulfur.9 The overwhelming part of the total number of Li–S battery studies has, in similarity to Li–O2 batteries, been performed during the last decade and thus most Li-salts cited above have been available. Just as for Li–O2 batteries, there is currently no Li-salt which has been specially designed for Li–S batteries, why the most commonly chosen Li-salts differs. In order to understand the specific choices made, the special Li–S battery demands on the electrolyte (and the Li-salt) must first be properly understood.The overall possible chemistry choices for Li–S non-aqueous organic liquid electrolytes (solvents, salts, additives) as well as other types of electrolytes (ionic liquid based and non-liquid concepts) have recently been reviewed.77 The Li–S battery has one strong common feature with the Li–O2 battery – the use of a Li–metal anode, which indeed is meta-stable vs. the prevailing electrolytes used and is why additives like LiNO3 have been employed; although with rather unsatisfactory results.178 The other special situation is the need for the electrolyte to be stable vs. the various poly-sulfides (PS) created during cell cycling. Another aspect of the same feature is whether the choice of Li-salt can affect the solubility of the PS in total and/or selectively (short-chain vs. long-chain PS). To date, many studies have focused on the solvent choice(s) with respect to affecting the PS solubility, but only a few reports on the influence of the choice of Li-salt.
Yet another notable difference is the rather limited ESW needed for the Li–S concept: max ca. 2.5 V (even including a proper safety margin) – which expands the choice of Li-salts much beyond the “usual suspects” from LIB studies i.e. LiTFSI is for Li–S cells a real contender to LiPF6. Another candidate is LiTf – both these Li-salts are not viable for LIBs as Al (current collector) corrosion starts at ca. 2.8 V vs. Li+/Li°.179 Indeed, during the last decade, these two salts, LiTFSI and LiTf, both at ∼1 M concentration,14 totally dominate the Li–S literature. Their popularity is mainly based on their high thermal stabilities and compatibilities with the ether solvents often applied,180,181 and, especially for LiTFSI, a high dissociation ability.182,183 There is thus a renewed interest in LiTf, but despite its lower cost, it is not as popular as LiTFSI. This is because by employing LiTFSI the electrolyte conductivity approximately doubles compared to using LiTf; e.g. 1 M LiX in DME:PC (1:1) show conductivities of 11.2 and 5.9 mS cm−1 at 20 °C, respectively.35,184
Along with the overall increase in Li–S research activities, electrolyte development has accelerated, based on variations of either the Li-salt concentration or other components being added to the electrolyte, as well as novel design concepts.69,185 In addition, there are some Li-salts that we can foresee to be employed for Li–S batteries in the near future. As one example, the LiFSI salt could be employed just as well as the LiTFSI salt, and result in electrolytes of even higher conductivities (but with less stability), and we also propose that the LiTDI salt giving intermediate anodic stability could be employed – but there are yet some question marks concerning also this salt's stability vs. Li–metal anodes.59,61 As a very novel salt alternative the Li[B(OCH2CF3)4] borate salt was recently launched; stable up to 4.8 V vs. Li+/Li° and tested in a Li–S cell set-up,186 whilst providing Coulombic efficiencies of 97% for each cycle for the 40 cycles shown. Notable is the use of a 0.2 M salt concentration in DME:DOL 1:1 wt% in this study, despite no clear motivation for this rather low concentration – as electrolytes based on this salt were easily made up to 0.8 M.
Having outlined the requirements needed for Li–S electrolytes and the Li-salt candidates most likely to fulfil these, there is a need for controlled studies in order to analyse the unique rôle of each choice of Li-salt for the Li–S performance or other specific behaviour. In most studies employing several different Li-salts concomitant changes in the solvent composition hinder any unambiguous analysis.187,188 However, there is a rather limited number of studies where the Li-salt is the only variable and all other parameters constant. Kim et al.189 studied four Li-salts in the same solvent; LiPF6, LiTFSI, LiTf, and LiBETI and Gao et al.190 did the same for three salts: LiTf, LiPF6, and LiClO4. Both these studies used standard Li–S battery electrolyte solvents DME/DOL (4:1) and tetraglyme, respectively, and a salt concentration of 1 M.
In the work of Kim et al.,189 the Li-salts were ranked by the initial capacity and capacity decay of the Li–S cell – the latter taken as the discharge capacity at the 50th cycle: LiTFSI (770 mA h g−1) > LiBETI (730 mA h g−1) > LiPF6 (620 mA h g−1) > LiTf (560 mA h g−1) (Fig. 8). The film forming properties at the lithium anode surface, in the field of Li–S batteries an often neglected factor in comparison to the ability to handle PS solubility, was suggested as a partial explanation for the markedly better performance of the LiTFSI and LiBETI based electrolytes, as also observed earlier with PC as the solvent.120
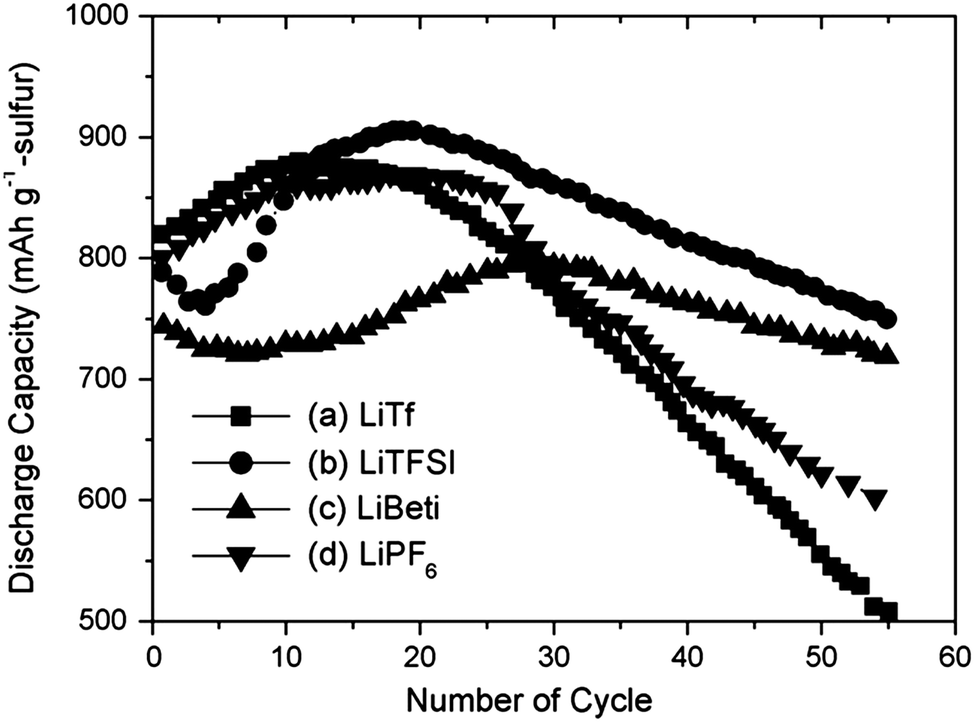 | ||
| Fig. 8 Cycling performance of Li–S cells using DME/DOL (4:1, v:v) solvent mixture with a 1 M concentration of different salts: (a) LiTf; (b) LiTFSI; (c) LiBETI; (d) LiPF6. Modified from Kim et al.189 | ||
In contrast, Gao et al.190 claimed to find no significant effect on Li–S cell performance by altering the Li-salt, despite the data contained in the publication which clearly show that the cells with the LiClO4 salt demonstrated a quite significant stabilization (for the limited 10 charge–discharge cycles made).
Another path forward in the research on Li–S battery electrolytes is what has been coined “quasi ionic liquids” or more precisely equimolar mixtures of a glyme solvent Gn (e.g. triglyme, G3) and a Li-salt to create [Li(Gn)]X species.191 This is a quite intriguing concept, with the salt concentration totally dependent on how to obtain the 1:1 Li:Gn ratio. It has been applied for LiBETI, LiTFSI, LiTf, LiNO3, and LiBF4, and dramatic differences in cell performance have been reported.191 The PS solubility was much higher in [Li(G3)]NO3 and [Li(G3)]Tf as compared to the remaining electrolytes, resulting in a poor Li–S cell performance. Poor cell performance was also found for [Li(G3)]BF4, and BF4− was also suggested to decompose in the presence of PS. Ueno et al.191 also made one of very few attempts to at the molecular level explain the poor performance observed at a macroscopic level; a stronger interaction of the Tf and NO3− anions with Li+ was suggested and as a consequence slightly less stable [Li(G3)]+ complexes resulting. Thus, also for the quasi ionic liquid based electrolytes, LiTFSI and LiBETI are the two best Li-salts tested.
Another line of work is that of studying the rôle of the salt concentration. Using both LiClO4 and LiTf, Kolosnitsyn et al.192 studied how the salt concentration affects the viscosity, the conductivity, and the PS solubility of TMS based electrolytes. They also found the viscosity to increase with salt basicity, LiClO4 > LiTf, and at higher LiClO4 concentrations, the viscosity rapidly increased – likely due to dynamic cross-linking of PS chains by the ‘free’ Li+ ions present. While most studies, as mentioned previously, use a 1 M standard – the Li2S6 PS solubility was found to have a minimum at ∼0.3 M, while the conductivity had a maximum at 0.5 M. Two single salt studies of salt concentration are Chang et al.,193 using LiTf to study the PS solubility, and Barchasz et al.194,195 using LiTFSI between 0.1–2 M, the latter concluding that, in order to balance cost and performance, 1 M is still to be the recommended salt concentration. Very recently also the solubility of elemental sulfur (S) was quantitatively determined – using electrolytes with LiTFSI, LiClO4, LiBF4, and LiTf dissolved in both organic solvents and ionic liquids – combined creating no less than 22 electrolytes.196 The choice of Li-salt was by chromatography found to be of much less importance than the choice of solvent and the salt concentration (either 0.1 or 1 M) – for the former the solubility varied by maximum ca. 25% (at 1 M) while the choice of solvent could indeed alter the S solubility by orders of magnitude.
Even higher salt concentrations have recently attracted a lot of interest. Shin et al.197 studied LiTFSI based electrolytes up to 5 M concentration – obtaining less dissolution of PS at higher concentrations and also a decreased overcharge and an improved Coulombic efficiency. Moving to even higher LiTFSI concentrations in the “solvent-in-salt” (≤7 M) electrolytes by Suo et al.,69 where the salt becomes the dominant component, PS dissolution was negligible and uniform Li–metal anode plating and stripping was demonstrated at a salt concentration of 7 M – close to the saturation limit. This resulted in high initial capacity, excellent rate capability (≤3 C) (Fig. 9) – a property usually quite limiting for Li–S cells (in general cycling at C/20 is commonly needed to get sustainable performance), a high capacity retention, and a high Coulombic efficiency – close to 100% for 100 cycles. The much higher viscosity compared to having a 1 M system resulted in only a slightly increased polarization.
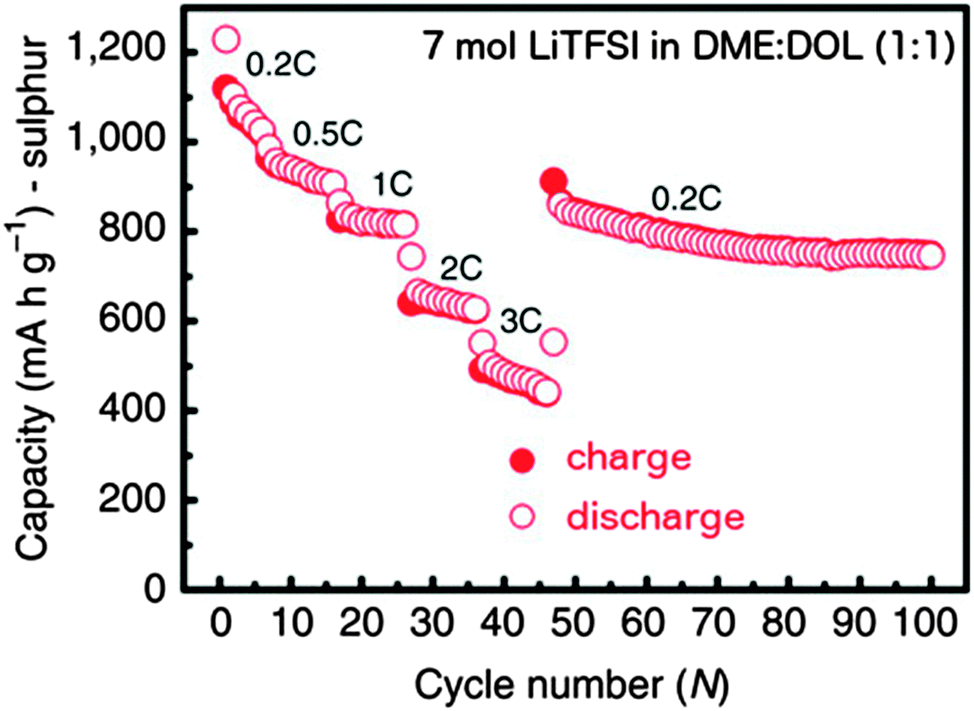 | ||
| Fig. 9 Rate capability of a Li–S battery with 7 M LiTFSI in DME/DOL (1:1). Modified from Suo et al.69 | ||
While the concept of very high salt concentrations seems promising from a performance point of view – the main concern is cost; a large amount of a costly Li-salt may prove detrimental to the otherwise formally inexpensive Li–S battery design.198 In this respect, the balance of performance and cost is delicate, but if a truly excellently working cell can be demonstrated, we do not believe that the cost carried by the Li-salt will be an obstacle to commercialization.
Overall, due to the rather different demands in terms of PS solubility and the limited ESW needed, the main problems of Li–S batteries as of today do not in any way originate in the Li-salt employed. On the contrary, the right salt (currently LiTFSI seems to be the main candidate) at the right concentration (rather much higher than 1 M it seems – in the light of both the quasi ionic liquid systems and the studies of Shin et al. and Suo et al.) can indeed enhance the performance of the cells.
7. Conclusions
The development of advanced batteries such as Li–metal, Li–O2, and Li–S batteries requires new electrolyte chemistries, which could (i) remain preserved during cell cycling and in contact with intermediate and final reaction products, and (ii) provide high Coulombic efficiency with a Li–metal anode. Though several Li-salts have been studied, none has provided a package of appropriated properties that LiPF6 provides for LIBs with graphite anode and moderate voltage cathodes (3–4 V).To date, virtually all of the studies focused on forming a stable SEI layer in carbonate solvent on Li–metal have failed to produce a robust electrode, which cycles with sufficient Coulombic efficiency and without dendrite formation. The examples highlighted in Section 4 provide evidence that significant advances can be realized with the right electrolyte chemistry, although the performance of Li anodes does not match traditional graphite materials. There are promising results coming out for IL-based and solid-state electrolytes, but these materials are still many years away from production. For those continuing to work on liquid electrolytes and the formation of passivation layers in contact with Li–metal, we recommend focusing on anions capable of forming polymerized and flexible structures, while optimizing the ionic transport. Indeed as noted by Xu, “an electrolyte with higher bulk ionic conductivity usually results in an SEI of lower impedance”.14
The Li–O2 battery mainly suffers from degradation of electrolytes. Along with different solvents, several Li-salts including LiPF6, LiTFSI, LiBF4, LiTf, LiBOB, Li[B(CN)4], and LiClO4 have been employed, but they all degrade in cells. Therefore, a key step in the development of metal–oxygen batteries is to design Li-salts and electrolyte solvents which are electrochemically and chemically stable in presence of oxygen species such as superoxides and peroxides, when ORR and OER occur at the cathode. Such an electrolyte should also provide sufficient ionic conductivity, oxygen solubility, and anodic stability at high surface area cathodes. As a high DN of electrolyte solvents has been shown to increase the solubility of LiO2 as well as the discharge capacity of Li–O2 cells, LiTf could be a preferred Li-salt to be used together with high DN solvents. However, stability of LiTf in contact with all reactive species in long term cycling as well as its compatibility with Li–metal anode need to be more comprehensively explored. The complexity of oxygen batteries declines feasibility of achieving commercial Li–O2 batteries in near future, but we believe that further research on metal–oxygen batteries will assist the battery community to develop new chemistries for electrochemical energy storage and conversion systems.
As for the employment of salts for Li–S cells, the LiTFSI and LiTf salts are commonly used and can assist in controlling the PS solubility, but only given a suitable solvent. Contrary to many other Li based battery concepts, the Li–S battery is not particularly limited by the choice of Li-salt (apart from the common issue of passivating layers for the Li–metal electrode). Exciting developments are those using very high salt concentrations and employing glymes in equimolar concentrations to the salt – both resulting in better cell cyclabilities.
Acknowledgements
The authors acknowledge support of this work from the ReLiable project (project nr. 11-116792/0603-00462B) funded by the Danish Council for Strategic Research Programme Commission on Sustainable Energy and Environment under the Danish Innovation Foundation. A portion of this work (GMV) was supported by U.S. Department of Energy's Office of Basic Energy Science (DOE-BES), Division of Materials Sciences and Engineering, under contract with UT-Battelle, LLC. In addition, the UU/KTH StandUp for Energy as well as several of Chalmers Areas of Advance: Materials Science, Energy and Transport, the Swedish Foundation for Strategic Research (SSF) within the project Road to load and the Battery Fond administrated by the Swedish Energy Agency, are all acknowledged for support.References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed
.
- F. T. Wagner, B. Lakshmanan and M. F. Mathias, J. Phys. Chem. Lett., 2010, 1, 2204–2219 CrossRef CAS
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2012, 159, R1–R30 CrossRef CAS PubMed
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750–768 CAS
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2012, 46, 1125–1134 CrossRef PubMed
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed
-
D. Aurbach, Nonaqueous Electrochemistry, Marcel Dek., New York, 1999 Search PubMed
-
G.-A. Nazri and G. Pistoia, Lithium Batteries: Science and Technology, Springer, 2003 Search PubMed
-
R. A. Huggins, Advanced Batteries, Springer US, Boston, MA, 2009 Search PubMed
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS
-
W. A. Henderson, Electrolytes for Lithium and Lithium-Ion Batteries, Springer New York, NY, 2014, vol. 58 Search PubMed
-
H. J. Gores, J. Barthel, S. Zugmann and D. Moosbauer, Handbook of Battery Materials, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed
- W. Tang, L. Liu, Y. Zhu, H. Sun, Y. Wu and K. Zhu, Energy Environ. Sci., 2012, 5, 6909–6913 CAS
- X. Wang, Q. Qu, Y. Hou, F. Wang and Y. Wu, Chem. Commun., 2013, 49, 6179–6181 RSC
- Z. Chang, X. Wang, Y. Yang, J. Gao, M. Li, L. Liu and Y. Wu, J. Mater. Chem. A, 2014, 2, 19444–19450 CAS
- S. J. Visco, V. Y. Nimon, A. Petrov, K. Pridatko, N. Goncharenko, E. Nimon, L. De Jonghe, Y. M. Volfkovich and D. a. Bograchev, J. Solid State Electrochem., 2014, 18, 1443–1456 CAS
- V. Aravindan, J. Gnanaraj, S. Madhavi and H.-K. Liu, Chem. – Eur. J., 2011, 17, 14326–14346 CrossRef CAS PubMed
- R. W. Schmitz, P. Murmann, R. Schmitz, R. Müller, L. Krämer, J. Kasnatscheew, P. Isken, P. Niehoff, S. Nowak, G.-V. Röschenthaler, N. Ignatiev, P. Sartori, S. Passerini, M. Kunze, A. Lex-Balducci, C. Schreiner, I. Cekic-Laskovic and M. Winter, Prog. Solid State Chem., 2014, 42, 65–84 CrossRef CAS PubMed
- T. Böttcher, B. Duda, N. Kalinovich, O. Kazakova, M. Ponomarenko, K. Vlasov, M. Winter and G.-V. Röschenthaler, Prog. Solid State Chem., 2014, 42, 202–217 CrossRef PubMed
- M. Marcinek, J. Syzdek, M. Marczewski, M. Piszcz, L. Niedzicki, M. Kalita, A. Plewa-Marczewska, A. Bitner, P. Wieczorek, T. Trzeciak, M. Kasprzyk, P. Łżak, Z. Zukowska, A. Zalewska and W. Wieczorek, Solid State Ionics, 2015, 276, 107–126 CrossRef CAS PubMed
- G. Pistoia, J. Electrochem. Soc., 1971, 118, 153–158 CrossRef CAS PubMed
- C. D. Desjardins, T. G. Cadger, R. S. Salter, G. Donaldson and E. J. Caser, J. Electrochem. Soc., 1985, 132, 529–533 CrossRef CAS PubMed
- J. O. Besenhard and G. Eichinger, J. Electroanal. Chem. Interfacial Electrochem., 1976, 68, 1–18 CrossRef CAS
- V. R. Koch, J. Power Sources, 1981, 6, 357–370 CrossRef CAS
- A. M. Andersson, M. Herstedt, A. G. Bishop and K. Edström, Electrochim. Acta, 2002, 47, 1885–1898 CrossRef CAS
- R. Fong, U. von Sacken and J. R. Dahn, J. Electrochem. Soc., 1990, 137, 2009–2013 CrossRef CAS PubMed
- Y. Ein-Eli, B. Markovsky, D. Aurbach, Y. Carmeli, H. Yamin and S. Luski, Electrochim. Acta, 1994, 39, 2559–2569 CrossRef CAS
- A. M. Andersson, K. Edström and J. O. Thomas, J. Power Sources, 1999, 81–82, 8–12 CrossRef CAS
- Z. Ogumi and M. Inaba, Bull. Chem. Soc. Jpn., 1998, 71, 521–534 CrossRef CAS
- E. Peled, D. Golodnitsky, C. Menachem and D. Bar-Tow, J. Electrochem. Soc., 1998, 145, 3482–3486 CrossRef CAS PubMed
- J. T. Dudley, D. P. Wilkinson, G. Thomas, R. LeVae, S. Woo, H. Blom, C. Horvath, M. W. Juzkow, B. Denis, P. Juric, P. Aghakian and J. R. Dahn, J. Power Sources, 1991, 35, 59–82 CrossRef CAS
- M. Schmidt, U. Heider, A. Kuehner, R. Oesten, M. Jungnitz, N. Ignat'ev and P. Sartori, J. Power Sources, 2001, 97–98, 557–560 CrossRef CAS
- S. E. Sloop, J. K. Pugh, S. Wang, J. B. Kerr and K. Kinoshita, Electrochem. Solid-State Lett., 2001, 4, A42–A44 CrossRef CAS PubMed
- A. M. Andersson and K. Edström, J. Electrochem. Soc., 2001, 148, A1100–A1109 CrossRef CAS PubMed
- S. S. Zhang, K. Xu and T. R. Jow, J. Electrochem. Soc., 2002, 149, A586–A590 CrossRef CAS PubMed
- N. Takami, T. Ohsaki, H. Hasebe and M. Yamamoto, J. Electrochem. Soc., 2002, 149, A9–A12 CrossRef CAS PubMed
- S. Zhang, K. Xu and T. Jow, J. Solid State Electrochem., 2003, 7, 147–151 CrossRef CAS PubMed
- W. Gorecki, M. Jeannin, E. Belorizky, C. Roux and M. Armand, J. Phys.: Condens. Matter, 1995, 7, 6823–6832 CrossRef CAS
- K. Xu, S. Zhang, T. R. Jow, W. Xu and C. A. Angell, Electrochem. Solid-State Lett., 2002, 5, A26–A29 CrossRef CAS PubMed
- B. Fahys and M. Herlem, J. Power Sources, 1991, 34, 183–188 CrossRef CAS
- J. Foropoulos and D. D. DesMarteau, Inorg. Chem., 1984, 23, 3720–3723 CrossRef CAS
- J. S. Gnanaraj, M. D. Levi, Y. Gofer, D. Aurbach and M. Schmidt, J. Electrochem. Soc., 2003, 150, A445–A454 CrossRef CAS PubMed
- D. Di Censo, I. Exnar and M. Graetzel, Electrochem. Commun., 2005, 7, 1000–1006 CrossRef CAS PubMed
- P. Murmann, P. Niehoff, R. Schmitz, S. Nowak, H. Gores, N. Ignatiev, P. Sartori, M. Winter and R. Schmitz, Electrochim. Acta, 2013, 114, 658–666 CrossRef CAS PubMed
- M. Ue, T. Fujii, Z.-B. Zhou, M. Takeda and S. Kinoshita, Solid State Ionics, 2006, 177, 323–331 CrossRef CAS PubMed
-
S. Tsujioka, H. Takase, M. Takahashi and Y. Isono, US Pat., 6849752 B2, 2005 Search PubMed
- T. Küppers, E. Bernhardt, H. Willner, H. W. Rohm and M. Köckerling, Inorg. Chem., 2005, 44, 1015–1022 CrossRef
- J. Scheers, P. Johansson and P. Jacobsson, J. Electrochem. Soc., 2008, 155, A628–A634 CrossRef CAS PubMed
- J. Scheers, J. Pitawala, F. Thebault, J.-K. Kim, J.-H. Ahn, A. Matic, P. Johansson and P. Jacobsson, Phys. Chem. Chem. Phys., 2011, 13, 14953–14959 RSC
- J. Scheers, D.-H. Lim, J.-K. Kim, E. Paillard, W. A. Henderson, P. Johansson, J.-H. Ahn and P. Jacobsson, J. Power Sources, 2014, 251, 451–458 CrossRef CAS PubMed
- M. Egashira, B. Scrosati, M. Armand, S. Béranger and C. Michot, Electrochem. Solid-State Lett., 2003, 6, A71–A73 CrossRef CAS PubMed
- C. Herriot, S. Khatun, E. T. Fox, P. Judeinstein, M. Armand, W. A. Henderson and S. Greenbaum, J. Phys. Chem. Lett., 2012, 3, 441–444 CrossRef CAS
- C. Liao, K. S. Han, L. Baggetto, D. A. Hillesheim, R. Custelcean, E.-S. Lee, B. Guo, Z. Bi, D. Jiang, G. M. Veith, E. W. Hagaman, G. M. Brown, C. Bridges, M. P. Paranthaman, A. Manthiram, S. Dai and X.-G. Sun, Adv. Energy Mater., 2014, 4, 1301368 Search PubMed
- L. Niedzicki, S. Grugeon, S. Laruelle, P. Judeinstein, M. Bukowska, J. Prejzner, P. Szczeciñski, W. Wieczorek and M. Armand, J. Power Sources, 2011, 196, 8696–8700 CrossRef CAS PubMed
- J. Scheers, P. Johansson, P. Szczeciński, W. Wieczorek, M. Armand and P. Jacobsson, J. Power Sources, 2010, 195, 6081–6087 CrossRef CAS PubMed
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed
- P. Johansson, H. Nilsson, P. Jacobsson and M. Armand, Phys. Chem. Chem. Phys., 2004, 6, 895–899 RSC
- P. Johansson, Phys. Chem. Chem. Phys., 2007, 9, 1493–1498 RSC
- M. Armand and P. Johansson, J. Power Sources, 2008, 178, 821–825 CrossRef CAS PubMed
- E. Jónsson and P. Johansson, Phys. Chem. Chem. Phys., 2012, 14, 10774–10779 RSC
- M. Carboni, R. Spezia and S. Brutti, J. Phys. Chem. C, 2014, 118, 24221–24230 CAS
- J. Scheers, E. Jónsson, P. Jacobsson and P. Johansson, Electrochemistry, 2012, 80, 18–25 CrossRef CAS PubMed
- H. P. Chen, J. W. Fergus and B. Z. Jang, J. Electrochem. Soc., 2000, 147, 399–406 CrossRef CAS PubMed
- S.-K. Jeong, H.-Y. Seo, D.-H. Kim, H.-K. Han, J.-G. Kim, Y. B. Lee, Y. Iriyama, T. Abe and Z. Ogumi, Electrochem. Commun., 2008, 10, 635–638 CrossRef CAS PubMed
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed
- H.-S. Kim and C.-S. Jeong, Bull. Korean Chem. Soc., 2011, 32, 3682–3686 CrossRef CAS
- W. Xu and C. A. Angell, Electrochem. Solid-State Lett., 2001, 4, L3 CrossRef CAS PubMed
- J. Read, J. Electrochem. Soc., 2006, 153, A96–A100 CrossRef CAS PubMed
- V. Gutmann, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef CAS
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, J.-M. Tarascon, P. C. Ashok, B. B. Praveen, K. Dholakia and P. G. Bruce, Nat. Chem., 2014, 6, 1–9 Search PubMed
- N. B. Aetukuri, B. D. McCloskey, J. M. García, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2015, 7, 50–56 CrossRef CAS PubMed
- K. M. Abraham, J. Electrochem. Soc., 2015, 162, A3021–A3031 CrossRef CAS PubMed
- J. Scheers, S. Fantini and P. Johansson, J. Power Sources, 2014, 255, 204–218 CrossRef CAS PubMed
- W. Linert, A. Camard, M. Armand and C. Michot, Coord. Chem. Rev., 2002, 226, 137–141 CrossRef CAS
- P. Johansson, J. Phys. Chem. A, 2006, 110, 12077–12080 CrossRef CAS PubMed
- E. Jónsson and P. Johansson, Phys. Chem. Chem. Phys., 2015, 17, 3697–3703 RSC
- K. Edström, T. Gustafsson and J. O. Thomas, Electrochim. Acta, 2004, 50, 397–403 CrossRef PubMed
- S.-T. Myung, Y. Hitoshi and Y.-K. Sun, J. Mater. Chem., 2011, 21, 9891–9911 RSC
- A. Hofmann, M. Schulz, V. Winkler and T. Hanemann, J. Electrochem. Soc., 2014, 161, A431–A438 CrossRef CAS PubMed
- S. Wilken, M. Treskow, J. Scheers, P. Johansson and P. Jacobsson, RSC Adv., 2013, 3, 16359–16364 RSC
- B. G. Nolan and S. H. Strauss, J. Electrochem. Soc., 2003, 150, A1726–A1734 CrossRef CAS PubMed
- J. Jiang, H. Fortier, J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 2004, 151, A609–A613 CrossRef CAS PubMed
- U. von Sacken, E. Nodwell, A. Sundler and J. R. Dahn, Solid State Ionics, 1994, 69, 284–290 CrossRef CAS
- K. Sato, Solid State Ionics, 2002, 148, 463–466 CrossRef CAS
- G. V. Zhuang, H. Yang, P. N. Ross, K. Xu and T. R. Jow, Electrochem. Solid-State Lett., 2006, 9, A64–A68 CrossRef CAS PubMed
- L. Yang, C. Smith, C. Patrissi, C. R. Schumacher and B. L. Lucht, J. Power Sources, 2008, 185, 1359–1366 CrossRef CAS PubMed
- Sigma-Aldrich, http://www.sigmaaldrich.com/materialsscience/material-science-products.html?TablePage=19295337.
- L. Yang, H. Zhang, P. F. Driscoll, B. Lucht and J. B. Kerr, ECS Trans., 2011, 33, 57–69 CAS
- H.-B. Han, S.-S. Zhou, D.-J. Zhang, S.-W. Feng, L.-F. Li, K. Liu, W.-F. Feng, J. Nie, H. Li, X.-J. Huang, M. Armand and Z.-B. Zhou, J. Power Sources, 2011, 196, 3623–3632 CrossRef CAS PubMed
- Y. Hu, H. Li, X. Huang and L. Chen, Electrochem. Commun., 2004, 6, 28–32 CrossRef CAS PubMed
- K. Tasaki, K. Kanda, S. Nakamura and M. Ue, J. Electrochem. Soc., 2003, 150, A1628–A1636 CrossRef CAS PubMed
- S. Meini, M. Piana, N. Tsiouvaras, A. Garsuch and H. A. Gasteiger, Electrochem. Solid-State Lett., 2012, 15, A45–A48 CrossRef CAS PubMed
-
M. J. Trahan, PhD thesis, Northeastern University, US, 2014
- R. D. W. Kemmitt, D. R. Russell and D. W. A. Sharp, J. Chem. Soc., 1963, 4408 RSC
-
W. N. Smith, US Pat., 3607020, 1970 Search PubMed
-
R. A. Wiesboeck, US Pat., 3654330, 1972 Search PubMed
-
Fluorinated Materials for Energy Conversion, ed. T. Nakajima and H. Groult, Elsevier, 1st edn, 2005 Search PubMed
-
D. J. Salmon, W. Barnette and R. A. Baenett, US Pat., 5378445, 1995 Search PubMed
-
H.-J. Belt, D. Seffer and W. Rudolph, US Pat., 5866093, 1999 Search PubMed
-
P. Bonnet, S. Perdrieux and S. G. Schon, US Pat., 5935541, 1999 Search PubMed
-
A. Subramaniam, T. Vasudevan and R. Gangadharan, US Pat., 6824754 B2, 2004 Search PubMed
- J.-H. Kim, H. Umeda, M. Ohe, S. Yonezawa and M. Takashima, Chem. Lett., 2011, 40, 360–361 CrossRef CAS
- H. Kim, G. Jeong, Y.-U. Kim, J.-H. Kim, C.-M. Park and H.-J. Sohn, Chem. Soc. Rev., 2013, 42, 9011–9034 RSC
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS
- A. Ferrese and J. Newman, J. Electrochem. Soc., 2014, 161, A1350–A1359 CrossRef CAS PubMed
- C. Monroe and J. Newman, J. Electrochem. Soc., 2005, 152, A396–A404 CrossRef CAS PubMed
- S. Kalnaus, A. S. Sabau, W. E. Tenhaeff, N. J. Dudney and C. Daniel, J. Power Sources, 2012, 201, 280–287 CrossRef CAS PubMed
- D. Blanchard, A. Nale, D. Sveinbjörnsson, T. M. Eggenhuisen, M. H. W. Verkuijlen, Suwarno, T. Vegge, A. P. M. Kentgens and P. E. de Jongh, Adv. Funct. Mater., 2015, 25, 184–192 CrossRef CAS PubMed
- J. B. Bates, N. J. Dudney, D. C. Lubben, G. R. Gruzalski, B. S. Kwak, X. Yu and R. A. Zuhr, J. Power Sources, 1995, 54, 58–62 CrossRef CAS
- N. J. Dudney, J. Power Sources, 2000, 89, 176–179 CrossRef CAS
- S. Neuhold, D. J. Schroeder and J. T. Vaughey, J. Power Sources, 2012, 206, 295–300 CrossRef CAS PubMed
- S. Shui Zhang, Electrochem. Commun., 2006, 8, 1423–1428 CrossRef PubMed
- R. Herr, Electrochim. Acta, 1990, 35, 1257–1265 CrossRef CAS
- D. Aurbach, Solid State Ionics, 2002, 148, 405–416 CrossRef CAS
- G. E. Blomgren, J. Power Sources, 1999, 81–82, 112–118 CrossRef CAS
- K. Naoi, M. Mori, Y. Naruoka, W. M. Lamanna and R. Atanasoski, J. Electrochem. Soc., 1999, 146, 462–469 CrossRef CAS PubMed
- M. Odziemkowski and D. E. Irish, J. Electrochem. Soc., 1992, 139, 3063–3074 CrossRef CAS PubMed
- M. Odziemkowski and D. E. Irish, J. Electrochem. Soc., 1993, 140, 1546–1555 CrossRef CAS PubMed
- D. Rahner, J. Power Sources, 1999, 81–82, 358–361 CrossRef CAS
- K. Nishikawa, Y. Fukunaka, T. Sakka, Y. H. Ogata and J. R. Selman, J. Electrochem. Soc., 2007, 154, A943–A948 CrossRef CAS PubMed
- D. Aurbach and M. Moshkovich, J. Electrochem. Soc., 1998, 145, 2629–2639 CrossRef CAS PubMed
- K. Nishikawa, T. Mori, T. Nishida, Y. Fukunaka and M. Rosso, J. Electroanal. Chem., 2011, 661, 84–89 CrossRef CAS PubMed
- X.-G. Sun, C. Liao, L. Baggetto, B. Guo, R. R. Unocic, G. M. Veith and S. Dai, J. Mater. Chem. A, 2014, 2, 7606–7614 CAS
- T. Schedlbauer, B. Hoffmann, S. Krüger, H. Gores and M. Winter, Energies, 2013, 6, 3481–3505 CrossRef CAS PubMed
- T. Schedlbauer, S. Krüger, R. Schmitz, R. W. Schmitz, C. Schreiner, H. J. Gores, S. Passerini and M. Winter, Electrochim. Acta, 2013, 92, 102–107 CrossRef CAS PubMed
- T. Schedlbauer, U. C. Rodehorst, C. Schreiner, H. J. Gores and M. Winter, Electrochim. Acta, 2013, 107, 26–32 CrossRef CAS PubMed
- M. Wu, Z. Wen, Y. Liu, X. Wang and L. Huang, J. Power Sources, 2011, 196, 8091–8097 CrossRef CAS PubMed
- K. Chung, J.-D. Lee, E.-J. Kim, W.-S. Kim, J.-H. Cho and Y.-K. Choi, Microchem. J., 2003, 75, 71–77 CrossRef CAS
- W. Walker, V. Giordani, J. Uddin, V. S. Bryantsev, G. V Chase and D. Addison, J. Am. Chem. Soc., 2013, 135, 2076–2079 CrossRef CAS PubMed
- M. Roberts, R. Younesi, W. Richardson, J. Liu, T. Gustafsson, J. Zhu and K. Edström, ECS Electrochem. Lett., 2014, 3, A62–A65 CrossRef CAS PubMed
- V. Giordani, W. Walker, V. S. Bryantsev, J. Uddin, G. V Chase and D. Addison, J. Electrochem. Soc., 2013, 160, A1544–A1550 CrossRef CAS PubMed
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V Sushko, J. Liu and J.-G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed
- J. Qian, W. a. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef PubMed
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed
- Y.-C. Lu, H. a. Gasteiger, M. C. Parent, V. Chiloyan and Y. Shao-Horn, Electrochem. Solid-State Lett., 2010, 13, A69–A72 CrossRef CAS PubMed
- C. O. Laoire, S. Mukerjee, K. M. Abraham, E. J. Plichta and M. A. Hendrickson, J. Phys. Chem. C, 2009, 113, 20127–20134 CAS
-
C. M. Ó. Laoire, PhD thesis, Northeastern University, US, 2010
-
R. Younesi, PhD thesis, Uppsala University, Sweden, 2012
- N. Garcia-Araez and P. Novák, J. Solid State Electrochem., 2013, 17, 1793–1807 CrossRef CAS PubMed
- M. Balaish, A. Kraytsberg and Y. Ein-Eli, Phys. Chem. Chem. Phys., 2014, 16, 2801–2822 RSC
- R. Younesi, P. Norby and T. Vegge, ECS Electrochem. Lett., 2014, 3, A15–A18 CrossRef CAS PubMed
- M. M. Storm, R. E. Johnsen, R. Younesi and P. Norby, J. Mater. Chem. A, 2015, 3, 3113–3119 CAS
- Y. S. Mekonnen, K. B. Knudsen, J. S. G. Mýrdal, R. Younesi, J. Højberg, J. Hjelm, P. Norby and T. Vegge, J. Chem. Phys., 2014, 140, 121101 CrossRef PubMed
- J. Chen, J. S. Hummelshøj, K. S. Thygesen, J. S. G. Myrdal, J. K. Nørskov and T. Vegge, Catal. Today, 2011, 165, 2–9 CrossRef CAS PubMed
- J. M. Garcia-Lastra, J. S. G. Myrdal, R. Christensen, K. S. Thygesen and T. Vegge, J. Phys. Chem. C, 2013, 117, 5568–5577 CAS
- J. Højberg, K. B. Knudsen, J. Hjelm and T. Vegge, ECS Electrochem. Lett., 2015, 4, A63–A66 CrossRef PubMed
- J. Liu, R. Younesi, T. Gustafsson, K. Edström and J. Zhu, Nano Energy, 2014, 10, 19–27 CrossRef CAS PubMed
- D. Aurbach, M. Daroux, P. Faguy and E. Yeager, J. Electroanal. Chem. Interfacial Electrochem., 1991, 297, 225–244 CrossRef CAS
- W. Xu, J. Xiao, D. Wang, J. Zhang and J.-G. Zhang, J. Electrochem. Soc., 2010, 157, A219–A224 CrossRef CAS PubMed
- G. A. Elia, J.-B. Park, Y.-K. Sun, B. Scrosati and J. Hassoun, ChemElectroChem, 2014, 1, 47–50 CrossRef CAS PubMed
- R. Younesi, M. Hahlin, M. Roberts and K. Edström, J. Power Sources, 2013, 225, 40–45 CrossRef CAS PubMed
- R. Younesi, S. Urbonaite, K. Edström and M. Hahlin, J. Phys. Chem. C, 2012, 116, 20673–20680 CAS
- G. M. Veith, J. Nanda, L. H. Delmau and N. J. Dudney, J. Phys. Chem. Lett., 2012, 3, 1242–1247 CrossRef CAS
- P. Du, J. Lu, K. C. Lau, X. Luo, J. Bareno, X. Zhang, Y. Ren, Z. Zhang, L. A. Curtiss, Y.-K. Sun and K. Amine, Phys. Chem. Chem. Phys., 2013, 15, 5572–5581 RSC
- E. Nasybulin, W. Xu, M. H. Engelhard, Z. Nie, S. D. Burton, L. Cosimbescu, M. E. Gross and J.-G. Zhang, J. Phys. Chem. C, 2013, 117, 2635–2645 CAS
- N. K. Karan, M. Balasubramanian, T. T. Fister, A. K. Burrell and P. Du, J. Phys. Chem. C, 2012, 116, 18132–18138 CAS
- R. Younesi, M. Hahlin, F. Björefors, P. Johansson and K. Edström, Chem. Mater., 2013, 25, 77–84 CrossRef CAS
- D. Chalasani and B. L. Lucht, ECS Electrochem. Lett., 2012, 1, A38–A42 CrossRef CAS PubMed
- G. M. Veith, N. J. Dudney, J. Howe and J. Nanda, J. Phys. Chem. C, 2011, 115, 14325–14333 CAS
- D. Sharon, V. Etacheri, A. Garsuch, M. Afri, A. A. Frimer and D. Aurbach, J. Phys. Chem. Lett., 2012, 4, 127–131 CrossRef
- B. D. McCloskey, A. Valery, A. C. Luntz, S. R. Gowda, G. M. Wallraff, J. M. Garcia, T. Mori and L. E. Krupp, J. Phys. Chem. Lett., 2013, 4, 2989–2993 CrossRef CAS
- W. Xu, J. Hu, M. H. Engelhard, S. A. Towne, J. S. Hardy, J. Xiao, J. Feng, M. Y. Hu, J. Zhang, F. Ding, M. E. Gross and J.-G. Zhang, J. Power Sources, 2012, 215, 240–247 CrossRef CAS PubMed
- I. Gunasekara, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. M. Abraham, J. Electrochem. Soc., 2014, 161, A381–A392 CrossRef CAS PubMed
- R. Younesi, M. Hahlin, M. Treskow, J. Scheers, P. Johansson and K. Edström, J. Phys. Chem. C, 2012, 116, 18597–18604 CAS
- R. Younesi, M. Hahlin and K. Edström, ACS Appl. Mater. Interfaces, 2013, 5, 1333–1341 CAS
- S. Hyoung Oh, T. Yim, E. Pomerantseva and L. F. Nazar, Electrochem. Solid-State Lett., 2011, 14, A185–A188 CrossRef PubMed
- K. C. Lau, J. Lu, J. Low, D. Peng, H. Wu, H. M. Albishri, D. A. Al-Hady, L. A. Curtiss and K. Amine, Energy Technol., 2014, 2, 348–354 CrossRef CAS PubMed
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, T. Mori, R. Scheffler, A. Speidel, M. Sherwood and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 3043–3047 CrossRef CAS
- Z. Peng, S. A. Freunberger, L. J. Hardwick, Y. Chen, V. Giordani, F. Bardé, P. Novák, D. Graham, J.-M. Tarascon and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 6351–6355 CrossRef CAS PubMed
- E. Yoo and H. Zhou, ACS Nano, 2011, 5, 3020–3026 CrossRef CAS PubMed
- F. Li, T. Zhang, Y. Yamada, A. Yamada and H. Zhou, Adv. Energy Mater., 2013, 3, 532–538 CrossRef CAS PubMed
- Y. Liu, L. M. Suo, H. Lin, W. Yang, Y. Fang, X. Liu, D. Wang, Y.-S. Hu, W. Han and L. Chen, J. Mater. Chem. A, 2014, 2, 9020–9024 CAS
- L. Cheng, R. S. Assary, X. Qu, A. Jain, S. P. Ong, N. N. Rajput, K. Persson and L. A. Curtiss, J. Phys. Chem. Lett., 2015, 6, 283–291 CrossRef CAS
-
Y. V. Mikhaylik, US Pat., US7354680 B2, 2008 Search PubMed
- L. J. Krause, W. Lamanna, J. Summerfield, M. Engle, G. Korba, R. Loch and R. Atanasoski, J. Power Sources, 1997, 68, 320–325 CrossRef CAS
- D.-J. Lee, M. Agostini, J.-W. Park, Y.-K. Sun, J. Hassoun and B. Scrosati, ChemSusChem, 2013, 6, 2245–2248 CrossRef CAS PubMed
- K. M. Abraham, Z. Jiang and B. Carroll, Chem. Mater., 1997, 9, 1978–1988 CrossRef CAS
- M. Ue, J. Electrochem. Soc., 1997, 144, 2684–2688 CrossRef CAS PubMed
- W. A. Henderson, J. Phys. Chem. B, 2006, 110, 13177–13183 CrossRef CAS PubMed
- K. Xu and C. A. Angell, J. Electrochem. Soc., 2002, 149, A920–A926 CrossRef CAS PubMed
- R. Demir-Cakan, M. Morcrette, Gangulibabu, A. Gueguen, R. Dedryvere and J.-M. Tarascon, Energy Environ. Sci., 2013, 6, 176–182 CAS
- M. Rohde, P. Eiden, V. Leppert, M. Schmidt, A. Garsuch, G. Semrau and I. Krossing, ChemPhysChem, 2015, 16, 666–675 CrossRef CAS PubMed
- J. S. Park, D. J. Kim, J. W. Park, H. S. Ryu, K. W. Kim, G. X. Wang and H. J. Ahn, J. Nanosci. Nanotechnol., 2012, 12, 5794–5798 CrossRef CAS PubMed
- S. Xin, L. Gu, N.-H. Zhao, Y.-X. Yin, L.-J. Zhou, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2012, 134, 18510–18513 CrossRef CAS PubMed
- S. Kim, Y. Jung and S.-J. Park, Electrochim. Acta, 2007, 52, 2116–2122 CrossRef CAS PubMed
- J. Gao, M. A. Lowe, Y. Kiya and H. D. Abruña, J. Phys. Chem. C, 2011, 115, 25132–25137 CAS
- K. Ueno, J.-W. Park, A. Yamazaki, T. Mandai, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2013, 117, 20509–20516 CAS
- V. Kolosnitsyn, E. Kuzmina and E. Karaseva, ECS Trans., 2009, 19, 25–30 CAS
- D.-R. Chang, S.-H. Lee, S.-W. Kim and H.-T. Kim, J. Power Sources, 2002, 112, 452–460 CrossRef CAS
- C. Barchasz, F. Mesguich, J. Dijon, J.-C. Leprêtre, S. Patoux and F. Alloin, J. Power Sources, 2012, 211, 19–26 CrossRef CAS PubMed
- C. Barchasz, J.-C. Lepretre, S. Patoux and F. Alloin, J. Electrochem. Soc., 2013, 160, A430–A436 CrossRef CAS PubMed
- D. Zheng, X. Zhang, C. Li, M. E. McKinnon, R. G. Sadok, D. Qu, X. Yu, H.-S. Lee, X.-Q. Yang and D. Qu, J. Electrochem. Soc., 2015, 162, A203–A206 CrossRef CAS PubMed
- E. S. Shin, K. Kim, S. H. Oh and W. Il Cho, Chem. Commun., 2013, 49, 2004–2006 RSC
- M. Hagen, S. Dörfler, P. Fanz, T. Berger, R. Speck, J. Tübke, H. Althues, M. J. Hoffmann, C. Scherr and S. Kaskel, J. Power Sources, 2013, 224, 260–268 CrossRef CAS PubMed
Footnote |
| † Current address: Visiting Professor at LRCS/CNRS UMR7314, Université de Picardie Jules Verne, 33 rue Saint Leu, 80039 Amiens, France. |
| This journal is © The Royal Society of Chemistry 2015 |