
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heat-to-current conversion of low-grade heat from a thermocapacitive cycle by supercapacitors†
Andreas
Härtel‡
*a,
Mathijs
Janssen
a,
Daniel
Weingarth
b,
Volker
Presser
bc and
René
van Roij
a
aInstitute for Theoretical Physics, Center for Extreme Matter and Emergent Phenomena, Utrecht University, Leuvenlaan 4, 3584 CE Utrecht, The Netherlands
bINM – Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany
cSaarland University, Campus D2 2, 66123 Saarbrücken, Germany
First published on 13th July 2015
Abstract
Thermal energy is abundantly available, and especially low-grade heat is often wasted in industrial processes as a by-product. Tapping into this vast energy reservoir with cost-attractive technologies may become a key element for the transition to an energy-sustainable economy and society. We propose a novel heat-to-current converter which is based on the temperature dependence of the cell voltage of charged supercapacitors. Using a commercially available supercapacitor, we observed a thermal cell-voltage rise of around 0.6 mV K−1 over a temperature window of 0 °C to 65 °C. Within our theoretical model, this can be used to operate a Stirling-like charge–voltage cycle whose efficiency is competitive to the most-efficient thermoelectric (Seebeck) engines. Our proposed heat-to-current converter is built from cheap materials, contains no moving parts, and could operate with a plethora of electrolytes which can be chosen for optimal performance at specific working temperatures. Therefore, this heat-to-current converter is interesting for small-scale, domestic, and industrial applications.
Broader contextIn times of ever-increasing worldwide energy demand, innovative renewable energy sources must be developed. One highly attractive candidate is exploiting thermal energy, as it is abundantly available for instance in the form of waste heat. Thermocapacitive devices have the potential to harvest low-grade heat and to convert it directly into electric energy. They can be constructed from cheap and sustainable materials and are highly tunable to their specific field of application. Especially at small temperature differences, they reach efficiencies close to the most efficient Carnot efficiency and could compete with thermoelectric engines of ZT ≥ 20. Our work also impacts on the field of large-scale energy storage, where the thermal voltage rise can be used to easily increase the already installed storage efficiency or even harvest energy directly from the storage conversion. |
Introduction
With ever-increasing worldwide energy demand, together with the depletion of fossil fuels, there is an urgent call for innovative renewable energy sources.1 One highly attractive candidate is thermal energy, as it is abundantly available for instance in industry, power plants, data centers, combustion engines, or as solar or geothermal heat in nature. Yet, the efficient conversion of (low-grade) heat into electric energy is still a great technological challenge.2In a straightforward conversion, a heat engine converts heat into mechanical energy, which is subsequently used to drive an electric generator. More direct heat-to-current converters (HTCCs) avoid the intermediate mechanical step and directly convert thermal into electric energy. In recent years, continuous power-generating thermoelectric engines have been intensely studied.3–6 These devices work on the temperature difference between two heat reservoirs and they often exploit the Seebeck effect, the generation of a potential difference due to a temperature gradient.3 Other thermoelectric engines are based on the temperature-dependence of electrochemical cells, called the thermogalvanic effect,4 on the thermoionic, and on the thermoelectronic effect.5 Recently even a single ion heat engine7 and a thermoelectric engine with ultracold atoms6 attracted interest. However, thermoelectric engines are typically expensive and flawed with a poor efficiency.8
Contrary to continuous power generation, the thermogalvanic effect has also been used in thermally regenerative electrochemical cycles,9 where the connection to only one of two heat reservoirs at the same time is alternated in a cyclic fashion. The conversion of heat into electricity while operating in the cyclic charging–cooling–discharging–heating mode can even be performed without a membrane10 or without external voltage source.11 This cycle resembles the Stirling and Carnot cycle of a heat engine that generates mechanical work via a heating–expansion–cooling–compression cycle.12–15
Recently, in two new HTCC designs, nanoporous carbon electrodes with an internal surface area exceeding >1000 m2 g−1 were used: in a first design, supercapacitors coated with ion-exchange membranes exploit the so-called thermal membrane potential, the voltage across an ion-exchange membrane subject to a temperature gradient;16 a second design used a thermally driven distiller to create a difference in salt concentration between two solutions to subsequently feed them in a capacitive mixing (Capmix) engine.17 Such capacitive devices perform a cyclic charging/discharging cycle to harvest the mixing free energy of the involved solutions at different concentrations.18,19 This energy would dissipate if both solutions would mix spontaneously. Crucial to the working of these Capmix engines is the ion concentration dependence of the electrostatic potential of an electrified surface with an adjacent electrolyte. In particular, with decreasing salt concentration a higher electrostatic potential is required to attract counter-ionic charges which form the electric double layer and screen the charge on the electrode. Moreover, the electrostatic potential also shows a near-linear increase with temperature, because a temperature rise leads to increased thermal motion of the ions, which, at fixed electrode charge, again must be compensated for by an increased cell potential. This thermal voltage rise (TVR) can significantly enhance the Capmix performance.20,21
Interestingly, the TVR can be isolated to create a new HTCC for capacitive thermal energy extraction. Similar to the thermally regenerative electrochemical cycles (TREC),9 the new HTCC approach exploits the temperature dependence of the electrostatic potential in a thermocapacitive charging–heating–discharging–cooling cycle to convert thermal into electric energy (Fig. 1(a)). Note that whereas TREC uses the thermogalvanic effect, capacitive thermal energy extraction is based on increased thermal motion and a related entropic change in the electric double layer as sketched in Fig. 1(b). Apart from local rearrangements, no current flows during the heating/cooling steps, which significantly differs from thermoelectric (Seebeck) engines.
 | ||
| Fig. 1 (a) Schematic drawing of the charging–heating–discharging–cooling cycle of the proposed heat-to-current converter. The inset shows the supercapacitor immersed in hot water. (b) The supercapacitor is composed of an electrolyte reservoir and two porous electrodes, each modeled as a parallel plate capacitor. Furthermore, a section of the positive electrode and its electric double layer is sketched for all four states of the proposed cycle ABCDA at high (H) and low (L) temperatures T and charges Q of the device. (c) Canonical charging curves predicted by our theoretical model supercapacitor at different temperatures. In addition, the operation cycle ABCDA is indicated as sketched in (a), where A and D are positioned at zero charge. The enclosed area of this thermocapacitive cycle indicates the available work. The corresponding efficiency η of the cycle is shown in the inset for fixed TL = 0 °C and three different recuperation efficiencies ηrec together with the Carnot efficiency ηC and the approximative eqn (3). For further details see ESI.† | ||
Using (classical) density functional theory,22 well established in previous work,15,20 we predict a charge–potential cycle which uses the TVR to convert heat into electric work. Moreover, we experimentally demonstrate the facile nature of harvesting thermal energy using the TVR with readily available system technology.
Materials and methods
All measurements were done using a commercially available 10 Farad supercapacitor (YEC Pl series) because it has a very low cell resistance compared to laboratory cells. We attached a Pt-100 temperature sensor and welded together both parts in a piece of plastic (Fig. 1(a)) to avoid any shortcut in the water bath used for temperature regulation. The cold bath consists of an ice/water mixture at constant temperature TL = 0 °C; the second water bath was heated to a certain temperature TH using a laboratory heater (IKA). The cell was completely charged in galvanostatic mode with potential limitation to 2.5 V cell voltage at a temperature of 0 °C for two days in order to have a fully charged system. After charging, the system was put to open circuit potential mode and the cell voltage was recorded while the cell was transferred to the heated reservoir. Here, we always started from TL = 0 °C and increased the temperature to several values (up to 70 °C) for TH, listed in the ESI,† Table S1. To avoid any degradation of the electrolyte at elevated temperatures we preferred a cell voltage of 2.5 V over the maximum specified one of 2.7 V.Similar to previous work,15,20 we modeled the porous carbon supercapacitor with an electrolyte of tetraethylammonium tetrafluoroborate (TEA-BF4) solvated in acetonitrile (ACN) by a dielectric solvent within infinite parallel-plate pores, see Fig. 1(b). We determined a mean plate-separation of H = 1 nm from gas-sorption measurements (shown in ESI,† Fig. S2) and ensured that the description with just one mean pore size approximates a more detailed description with a set of differently sized pores within a few percent. Additionally, we connected the pores to a finite-size reservoir of three times the total pore volume of the device to take account of large pores and inter-electrode space. Within our restricted primitive model (charged hard spheres), the TEA+ and BF4− ions at a bulk reference concentration c∞ = 1 mol L−1 carry unit charges ±e; for simplicity, we chose the same effective solvated ion diameter of d = 0.6 nm for both species.23 Furthermore, we incorporated the acetonitrile solvent by a dielectric constant ε = 35, neglecting its temperature dependence24 (see ESI†).
We calculated concentration profiles and the electrostatic potential Ψ(Q,T) as a function of temperature T and electrode charge ±Q within a fundamental measure density functional theory involving the mean spherical approximation.15 This theory excellently describes the complex interplay between electrostatic interactions and packing effects due to finite ion sizes. To achieve charging curves, we fixed the number of ions at zero electrode potential and, for each given potential Ψ, adjusted the reference concentration c∞ that enters our grand canonical theory.
Results and discussion
Working principle
The new thermocapacitive engine is based on the rise of the electrostatic potential Ψ with increasing temperature, which is attributable to the increased thermal motion of ions in the electric double layer. This thermal voltage rise (TVR) is characterized by a pseudo-Seebeck coefficient , which, next to the temperature T, depends on the charge Q stored on each electrode of the thermocapacitive HTCC.
, which, next to the temperature T, depends on the charge Q stored on each electrode of the thermocapacitive HTCC.
The electric potential Ψ(Q,T) enters the thermodynamic internal energy state function U(Q,S), which describes the system. Its differential dU = ΨdQ + TdS, involving the entropy S of the capacitive system, describes the flow of energy during the operation of the HTCC: while the electrodes charge up, a positive amount ΨdQ of work is done on the device by its surrounding, such that the work W delivered by the engine is given by the differential dW = −ΨdQ. In a reversible (closed) cycle as depicted in Fig. 1 this differential integrates to the total amount of work
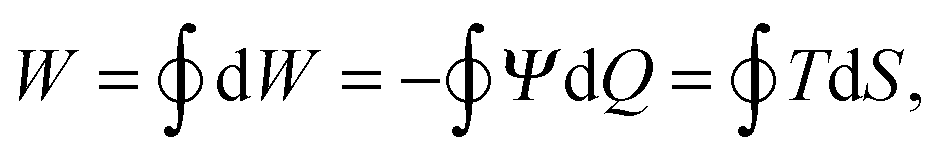 | (1) |
 . To characterize the performance of this cycle, its efficiency η is measured as the relation between the amount of delivered work W and consumed heat,
. To characterize the performance of this cycle, its efficiency η is measured as the relation between the amount of delivered work W and consumed heat,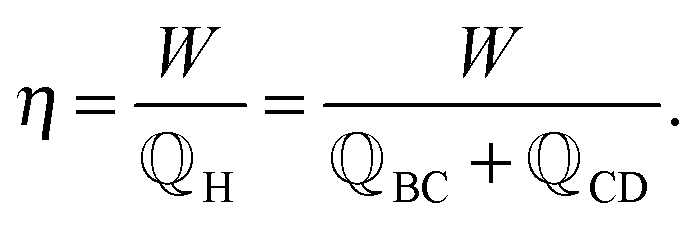 | (2) |
![[Doublestruck Q]](https://www.rsc.org/images/entities/char_e174.gif) H is the total heat flow from the reservoir at temperature TH to the device and consists of the heat flows during the dissipative heating step BC and the isothermal discharging step CD, as illustrated in Fig. 1(a).
H is the total heat flow from the reservoir at temperature TH to the device and consists of the heat flows during the dissipative heating step BC and the isothermal discharging step CD, as illustrated in Fig. 1(a).
Fig. 1(c) shows a cycle ABCDA as calculated within our restricted primitive model. It shows the HTCCs temperature-dependent charging behavior from which the pseudo-Seebeck coefficient Π(Q,T) can be estimated (see ESI,† inset of Fig. S4). Furthermore, eqn (1) and (2) allow to determine the cycle efficiency, shown in the inset of Fig. 1(c).
The HTCCs efficiency can be improved by recuperating heat from the cooling process (mainly step DA) back to the heating process (inset of Fig. 1(b)). In this case, the heat flow BC into the engine is (partially) fed with a fraction ηrec of the heat flow DA out of the engine such that the amount of consumed heat from the reservoir diminishes by the amount rec = ηrecDA of recuperated heat. This affects the working efficiency in eqn (2), where the denominator must be replaced by H − rec. Currently, ηrec = 80% can be realized experimentally.9
For a better understanding of the TVR-based cycle, the efficiency η can be well approximated (as shown in the inset of Fig. 1(c)) by (for a derivation see ESI,† eqn (S13))
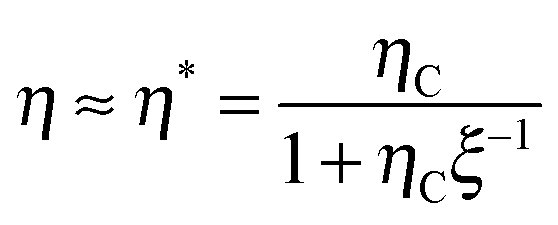 | (3) |
![[Doublestruck C]](https://www.rsc.org/images/entities/char_e166.gif) Ψ−1Π(Q,TH), in terms of the uptaken charge ΔQ = QC − QD, the heat capacity
Ψ−1Π(Q,TH), in terms of the uptaken charge ΔQ = QC − QD, the heat capacity 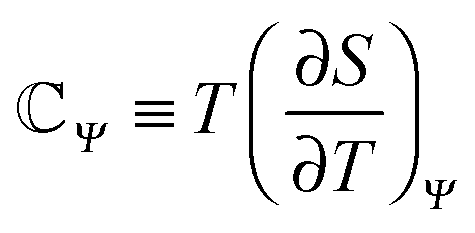 of the device material, and the system-dependent pseudo-Seebeck coefficient Π(Q,T). The figure of merit ξ is the ratio of the change in electrostatic energy due to varied temperature and the heat capacity of the engine and must be maximized in order to achieve a high efficiency. Thus, it could be used as a selection guideline for materials with low heat capacity, high pseudo-Seebeck sensitivity, and large charge capacity. Furthermore, the efficiency in eqn (3) increases when the amount of charge ΔQ involved in a cycle rises. For this reason, we have chosen points A and D in Fig. 1(c) to be located at vanishing charge Q = 0. The figure of merit associated to the cycle ABCDA is ξ = 0.066. Note that ξ is a quality factor which cannot be compared directly to the figure of merit ZT known from thermoelectric devices.25 However, efficiencies are comparable and a thermocapacitive engine with ξ = 0.066 reaches the same efficiency in the investigated range of temperatures as a thermoelectric device with ZT ≈ 2 (ηrec = 0) or even ZT ≈ 20 (ηrec = 80%) (see ESI,† Fig. S6).
of the device material, and the system-dependent pseudo-Seebeck coefficient Π(Q,T). The figure of merit ξ is the ratio of the change in electrostatic energy due to varied temperature and the heat capacity of the engine and must be maximized in order to achieve a high efficiency. Thus, it could be used as a selection guideline for materials with low heat capacity, high pseudo-Seebeck sensitivity, and large charge capacity. Furthermore, the efficiency in eqn (3) increases when the amount of charge ΔQ involved in a cycle rises. For this reason, we have chosen points A and D in Fig. 1(c) to be located at vanishing charge Q = 0. The figure of merit associated to the cycle ABCDA is ξ = 0.066. Note that ξ is a quality factor which cannot be compared directly to the figure of merit ZT known from thermoelectric devices.25 However, efficiencies are comparable and a thermocapacitive engine with ξ = 0.066 reaches the same efficiency in the investigated range of temperatures as a thermoelectric device with ZT ≈ 2 (ηrec = 0) or even ZT ≈ 20 (ηrec = 80%) (see ESI,† Fig. S6).
Experimental results
To gain insight into the supercapacitor, we recorded a scanning electron micrograph (ESI,† Fig. S1(f)) which shows common activated carbon consolidated by polymer binder. We derived the carbon's specific surface area of ca. 1200 m2 g−1 by applying the Brunauer–Emmett–Teller equation on data from gas-sorption measurements (ESI,† Fig. S2). After fully charging the system to an equilibrium state at TL = 0 °C, the cell voltage of 2.5 V remained essentially unchanged within 0.2 mV as seen from the open circuit potential measurement depicted in Fig. 2(a). This is a unique feature at low temperatures since at higher temperatures we see a nearly linear decay of the cell voltage over time. This decay is well-known for supercapacitors and an even stronger (exponential) decay of the open circuit potential has been observed for low-charged systems.26 The decay can be related to either charge loss via self-discharge or ion redistribution but, since our system was fully charged at low temperature, the decay of cell voltage is dominated by ion redistribution.27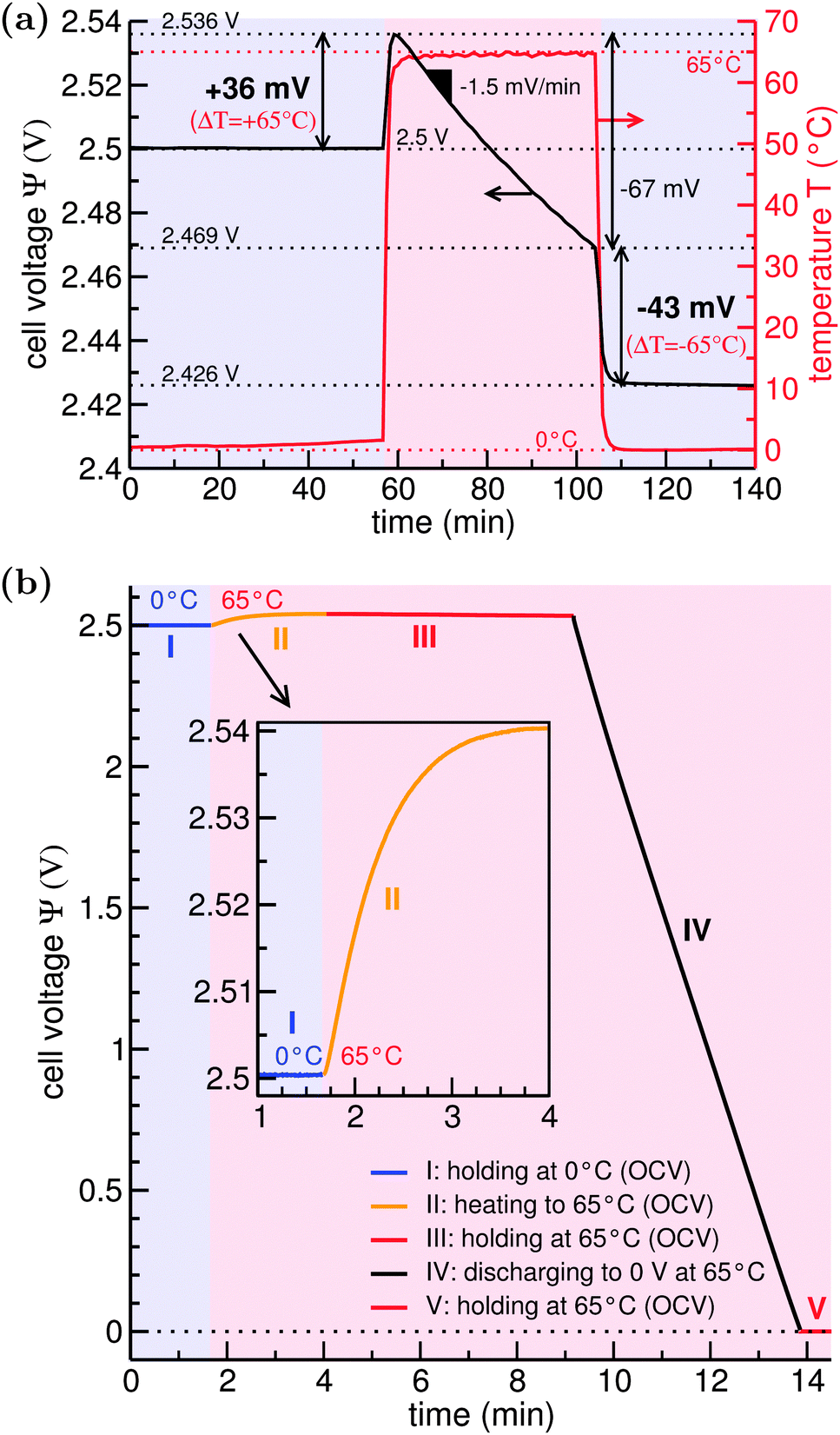 | ||
| Fig. 2 (a) Open circuit potential measurements when cycling the fully charged cell between 0 °C and 65 °C. We kept the device at each temperature for about 40 minutes to clearly present the effect of self-discharge at the respective temperatures (almost zero at T = 0 °C and around −1.5 mV min−1 at T = 65 °C). (b) Full half-cycle measurement of the thermal voltage rise (I → III) and discharging to 0 V (III → V). Again, steps I, III, and V just show that the open circuit potential stays almost constant in the respective states. For application this time can be reduced to a minimum. | ||
Most noticeably in Fig. 2, when increasing the temperature from 0 °C to 65 °C by placing the device in a hot water reservoir, the TVR instantaneously sets in, reaching a maximum of +36 mV after 2 min. This increase, however, does not reflect the maximum increase caused by thermal effects on the electric double layer, as we see the discussed subsequent decay of the cell voltage in the hot cell. By keeping the system in the hot state for about 40 min, we determined a decay rate of ca. −1.5 mV min−1 from the measured overall decay of 67 mV. Considering the period of ca. 2 min of heating to observe the TVR, the voltage decay lowers the TVR by around 3 mV such that a TVR of 39 mV seems to be more reasonable to assume for ΔT = 65 °C (when starting at 0 °C). Thus, the decay rate gives a timescale in which experiments should be performed. We furthermore measured the TVR as a function of the high temperature TH with a fixed TL = 0 °C and listed results in Table S1 (see ESI†). Visualized in Fig. S5 (ESI†), the data show a corresponding pseudo-Seebeck coefficient Π0°C→65°C = 0.6 mV K−1.
The increase in cell voltage with an increase of temperature is mirrored by a decrease in cell voltage when the system is cooled again (Fig. 2(a)). We measured a drop of the open circuit potential by 43 mV. When we correct for the 2 min cooling period again with the voltage decay of ca. −1.5 mV min−1, we obtain a TVR of ∼40 mV. This is in perfect agreement with the initial cell voltage increase such that Π65°C→0°C = Π0°C→65°C.
In a follow-up experiment with a faster heating rate, shown in Fig. 2(b), we corroborated a TVR of ∼40 mV for ΔT = 65 °C. Furthermore, Fig. 2(b) illustrates a half cycle BCD (see Fig. 1) for thermocapacitive energy extraction: the system at ΨB = 2.5 V at 0 °C in point B is rapidly heated to 65 °C in open circuit potential mode and then discharged to 0 V. Considering the voltage change ΔΨ of 40 mV, we estimate the energy-harvesting ability of the commercial 10 F supercapacitor device from (for a derivation see ESI,† eqn (S3))
| W = ½CHΨBΔΨ | (4) |
In order to compare the experimental findings with our theoretical data as shown in Fig. 1(c), we determined the electrode charge density of the charged 10 F supercapacitor at 2.5 V: charged with around 25 C (also measured from the integrated current during charging) and with a surface area of 440–600 m2 (see ESI†), the surface charge density is 0.0417–0.0568 C m−2 = 0.260–0.355 e nm−2, a factor 7 lower than our theoretical model predicts. These values lead to a corresponding experimental figure of merit ξexp = 0.0039 (for a cycle between ΨA = 0 V and ΨB = 2.5 V), which is one order of magnitude lower than those of the theoretical model, ξmodel = 0.066, obtained from a TVR of ∼1.7 mV K−1 (ESI,† inset of Fig. S4). Even though structural effects due to finite ion sizes are included in our model, there are several features missing in our theory. For example, polarization28,29 and (de)solvation of ions30,31 could play a major role inside the narrow pores of the electrodes material, leading to highly inhomogeneous dielectric permittivities. Furthermore, the description of image charges in the pore walls32 and the complex geometry of the pores could be necessary to describe the properties of the porous electrodes in more detail. Finally, a dynamical description of the cycle is required to optimize its power.
The biggest obstacle to overcome for the facile exploitation of the TVR will be cell design and operation optimization. In particular, our data clearly establishes that the TVR can only be maximized for a supercapacitor near charged equilibrium at given Q. Incidentally, having the system initially at low temperatures with corresponding low ion mobility helps, since the notorious issue of voltage decrease (“self-discharge”) is negligible. Yet, the target must be to rapidly heat and subsequently discharge the system because the voltage drop over time rivals the TVR effect.
Towards application
We have shown, both theoretically and experimentally, that common supercapacitors can be used to construct a new type of HTCC. The proposed engine effectively isolates the “thermal voltage rise” (TVR), introduced in earlier work to boost the work output of capacitive mixing engines.20,21 Compared to a method which combines Capmix and a distiller,17 our thermocapacitive HTCC device has a less complex lay-out, since it consists solely of the supercapacitor. The engines proposed in this study can work with (but are not restricted to) very low-temperature heat, which makes them especially interesting for the use of non-combustion waste heat of, for example, data centers, geothermal reservoirs, bio-mass heat or ocean thermal energy. Moreover, in our HTCC no solvents or electrolytes must be exchanged, as necessary in Capmix.21 Instead, the whole sealed device at once is successively connected to heat reservoirs at different temperatures. The resulting cyclic change of the ionic entropy, and hence of the double layer capacitance, causes a change in the open circuit potential of the charged device (this is the TVR) and represents the driving mechanism of the thermocapacitive HTCC.Other thermoelectric HTCCs operating in the low-grade heat regime typically reach efficiencies not higher than a few percent.3–6,8–11 Even though in this first study we are not yet able to surpass these values, the experimental figure of merit ξexp = 0.0039 leads to an efficiency of the same order of magnitude with the possibility to be optimized in future work; the efficiency would for instance benefit greatly from reducing the dead-space reservoir volume which is heated and cooled without contributing to the performed work. In comparison to the experimentally found value, the figure of merit extracted from the theoretical model (results shown in Fig. 1(c)) is an order of magnitude higher with ξmodel = 0.066 and has an associated efficiency of η ≈ 5%. For low-grade thermocapacitive heat conversion, this value compares to the figure of merit ZT ≈ 2 of a thermoelectric device. Note that an even higher efficiency of around 13% (ZT ≈ 20) is possible if the heat flowing out of the system during dissipative cooling is recuperated by 80% (see ESI,† Fig. S6). In fact, even Carnot efficiency could be achieved just by construction, when instead of the dissipative heating step an adiabatic heating by charging step is considered.20,33 Finally, the TVR will also appear in flowing-electrodes, which could provide continuous power generation.34,35
In summary, the proposed thermocapacitive HTCC operates without chemical reactions or moving parts and can be optimized by choosing ideal and cheap electrode and electrolyte materials, including sodium-chloride in water, organic solvents, lithium salts, multivalent ions, or ionic liquids. By this, the system is tunable to work within a wide range of different temperatures, including the commercially interesting low-grade heat regime as well as temperatures around 200 °C.36 Using a supercapacitor designed for high power application employing a different carbon material might lead to faster cycle times, because the equilibration time to have a fully charged system is much smaller (reduced time for period “I”, cf.Fig. 2). Facilitating a modified cell design with high outer surface area37 and, hence, very fast heat transfer in addition would lead to shorter cycle times and more cycles per hour, meaning higher power output. In both cases, the time for period “II” in Fig. 2 can be largely reduced and the voltage loss in that period might get negligible. Most importantly, the supercapacitor used in the experiments is cost-attractive and readily available compared to typical thermoelectric devices. Thus, scaling our thermocapacitive approach is facile. It is this simplicity, along with the low cost of all components, that makes the TVR-based HTCC a candidate for a sustainable-energy future.
Acknowledgements
This work is part of the D-ITP consortium, a program of the Netherlands Organisation for Scientific Research (NWO) that is funded by the Dutch Ministry of Education, Culture and Science (OCW). AH, MJ and RvR acknowledge financial support from a NWO-VICI grant. DW and VP thank Prof. Eduard Arzt (INM) for his continuing support, Dr Ingrid Grobelsek for SEM, and acknowledge funding from the German Federal Ministry for Research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- A. S. Rattner and S. Garimella, Energy, 2011, 36, 6172–6183 CrossRef.
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105–114 CrossRef CAS PubMed.
- T. I. Quickenden and Y. Mua, J. Electrochem. Soc., 1995, 142, 3985–3994 CrossRef CAS.
- S. Meir, C. Stephanos, T. H. Geballe and J. Mannhart, J. Renewable Sustainable Energy, 2013, 5, 043127 CrossRef.
- J.-P. Brantut, C. Grenier, J. Meineke, D. Stadler, S. Krinner, C. Kollath, T. Esslinger and A. Georges, Science, 2013, 342, 713–715 CrossRef CAS PubMed.
- O. Abah, J. Roßnagel, G. Jacob, S. Deffner, F. Schmidt-Kaler, K. Singer and E. Lutz, Phys. Rev. Lett., 2012, 109, 203006 CrossRef CAS PubMed.
- C. B. Vining, Nat. Mater., 2009, 8, 83–85 CrossRef CAS PubMed.
- S. W. Lee, Y. Yang, H.-W. Lee, H. Ghasemi, D. Kraemer, G. Chen and Y. Cui, Nat. Commun., 2014, 5, 3942 CAS.
- Y. Yang, J. Loomis, H. Ghasemi, S. W. Lee, Y. J. Wang, Y. Cui and G. Chen, Nano Lett., 2014, 14, 6578–6583 CrossRef CAS PubMed.
- Y. Yang, S. W. Lee, H. Ghasemi, J. Loomis, X. Li, D. Kraemer, G. Zheng, Y. Cui and G. Chen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17011–17016 CrossRef CAS PubMed.
- H. B. Callen, Thermodynamics, John Wiley and Sons, New York, London, 1960 Search PubMed.
- N. Boon and R. van Roij, Mol. Phys., 2011, 109, 1229–1241 CrossRef CAS.
- R. van Roij, in Electrostatics of soft and disordered matter, ed. D. S. Dean, J. Dobnikar, A. Naji and R. Podgornik, Pan Stanford Publishing, Singapore, 2012 Search PubMed.
- A. Härtel, M. Janssen, S. Samin and R. van Roij, J. Phys.: Condens. Matter, 2015, 27, 194129 CrossRef PubMed.
- B. B. Sales, O. S. Burheim, S. Porada, V. Presser, C. J. N. Buisman and H. V. M. Hamelers, Environ. Sci. Technol. Lett., 2014, 1, 356–360 CrossRef CAS.
- M. Marino, L. Misuri, A. Carati and D. Brogioli, Energies, 2014, 7, 3664–3683 CrossRef; A. Carati, M. Marino, D. Brogioli, arXiv:1309.3643v3 [physics.chem-ph], 2014.
- D. Brogioli, Phys. Rev. Lett., 2009, 103, 058501 CrossRef PubMed.
- H. V. M. Hamelers, O. Schaetzle, J. M. Paz-García, P. M. Biesheuvel and C. J. N. Buisman, Environ. Sci. Technol. Lett., 2014, 1, 31–35 CrossRef CAS.
- M. Janssen, A. Härtel and R. van Roij, Phys. Rev. Lett., 2014, 113, 268501 CrossRef PubMed.
- S. Ahualli, M. M. Fernández, G. Iglesias, Á. V. Delgado and M. L. Jiménez, Environ. Sci. Technol., 2014, 48, 12378–12385 CrossRef CAS PubMed.
- R. Evans, Adv. Phys., 1979, 28, 143–200 CrossRef CAS.
- J. Varghese, H. Wang and L. Pilon, J. Electrochem. Soc., 2011, 158, A1106–A1114 CrossRef CAS.
- L. G. Gagliardi, C. B. Castells, C. Ràfols, M. Rosés and E. Bosch, J. Chem. Eng. Data, 2007, 52, 1103–1107 CrossRef CAS.
- D. Nemir and J. Beck, J. Electron. Mater., 2010, 39, 1897 CrossRef CAS.
- M. Kaus, J. Kowal and D. U. Sauer, Electrochim. Acta, 2010, 55, 7516–7523 CrossRef CAS.
- H. A. Andreas, J. Electrochem. Soc., 2015, 162, A5047–A5053 CrossRef CAS.
- D. Jiang and J. Wu, J. Phys. Chem. Lett., 2013, 4, 1260–1267 CrossRef CAS PubMed.
- A. Abrashkin, D. Andelman and H. Orland, Phys. Rev. Lett., 2007, 99, 077801 CrossRef PubMed.
- A. Levy, D. Andelman and H. Orland, Phys. Rev. Lett., 2012, 108, 227801 CrossRef PubMed.
- A. Bankura, V. Carnevale and M. L. Klein, J. Chem. Phys., 2013, 138, 014501 CrossRef PubMed.
- P. M. Biesheuvel, S. Porada, M. Levi and M. Z. Bazant, J. Solid State Electrochem., 2014, 18, 1365 CrossRef CAS.
- J. Schiffer, D. Linzen and D. U. Sauer, J. Power Sources, 2006, 160, 765–772 CrossRef CAS.
- S.-I. Jeon, H.-R. Park, J.-G. Yeo, S. Yang, C. H. Cho, M. H. Han and D. K. Kim, Energy Environ. Sci., 2013, 6, 1471–1475 CAS.
- S. Porada, D. Weingarth, H. V. M. Hamelers, M. Bryjak, V. Presser and P. M. Biesheuvel, J. Mater. Chem. A, 2014, 2, 9313–9321 CAS.
- R. S. Borges, A. L. M. Reddy, M.-T. F. Rodrigues, H. Gullapalli, K. Balakrishnan, G. G. Silva and P. M. Ajayan, Sci. Rep., 2013, 3, 2572 Search PubMed.
- D. Pech, M. Brunet, H. Durou, P. Huang, V. Mochalin, Y. Gogotsi, P.-L. Taberna and P. Simon, Nat. Nanotechnol., 2010, 5, 651 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of modelling a supercapacitor, about the efficiency of the Stirling-like cycle, and a comparison with a thermoelectric device, including: experimental setup and scanning electron microscopy image of the supercapacitor device (Fig. S1), gas-sorption data (Fig. S2), sketch of the model supercapacitor (Fig. S3), full temperature-dependent charge–potential curves including a discussion of the dielectric constant (Fig. S4), the measured thermal voltage rise (Table S1 and Fig. S5), the derivation of eqn (3) and (4), and efficiencies of the thermocapacitive device compared to a thermoelectric one (Fig. S6). See DOI: 10.1039/c5ee01192b |
| ‡ Current address: KOMET 331, Institute of Physics, Johannes Gutenberg University Mainz, 55099 Mainz, Germany. E-mail: AnHaerte@uni-mainz.de; Tel: +49 (0)6131 39-20498. |
| This journal is © The Royal Society of Chemistry 2015 |