
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst turnover analysis of water-oxidation catalyzed by Co-oxide nanoparticles†
Sergey
Koroidov‡
a,
Magnus F.
Anderlund
b,
Stenbjörn
Styring
b,
Anders
Thapper
b and
Johannes
Messinger
*a
aDepartment of Chemistry, Kemiskt Biologiskt Centrum (KBC), Umeå University, S-90187 Umeå, Sweden. E-mail: johannes.messinger@umu.se
bMolecular Biomimetics, Department of Chemistry – Ångström Laboratory, Uppsala University, S-75120 Uppsala, Sweden
First published on 2nd July 2015
Abstract
Co-oxides are promising water oxidation catalysts for artificial photosynthesis devices. Presently, several different proposals exist for how they catalyze O2 formation from water. Knowledge about this process at molecular detail will be required for their further improvement. Here we present time-resolved 18O-labelling isotope-ratio membrane-inlet mass spectrometry (MIMS) experiments to study the mechanism of water oxidation in Co/methylenediphosphonate (Co/M2P) oxide nanoparticles using [Ru(bpy)3]3+ (bpy = 2,2′-bipyridine) as chemical oxidant. We show that 16O–Co/M2P-oxide nanoparticles produce 16O2 during their first turnover after simultaneous addition of H218O and [Ru(bpy)3]3+, while sequential addition with a delay of 3 s yields oxygen reflecting bulk water 18O-enrichment. This result is interpreted to show that the O–O bond formation in Co/M2P-oxide nanoparticles occurs via intramolecular oxygen coupling between two terminal Co–OHn ligands that are readily exchangeable with bulk water in the resting state of the catalyst. Importantly, our data allow the determination of the number of catalytic sites within this amorphous nanoparticular material, to calculate the TOF per catalytic site and to derive the number of holes needed for the production of the first O2 molecule per catalytic site. We propose that the mechanism of O–O bond formation during bulk catalysis in amorphous Co-oxides may differ from that taking place at the surface of crystalline materials.
Broader contextOne of the grand challenges of mankind is providing enough free energy to the growing population and to simultaneously preserve nature for generations to come. Solar energy is the most abundant energy available on Earth, but it is dilute and cannot be used directly by mankind. Converting light into storable and transportable fuels via ‘artificial photosynthesis’ using earth-abundant elements is therefore intensely studied. Impressive progress has been achieved during recent years, but mechanistic understanding for all processes involved will be required for further rational improvements. Here we present a detailed study on how water is split by amorphous Co-oxides to form molecular oxygen. This renewable process delivers electrons and protons for fuel production. This fuel can be either molecular hydrogen or low chain alcohols, if coupled to CO2 reduction. |
1. Introduction
Developing technology for generating energy carriers from renewable resources is pivotal for the future of mankind.1,2 One promising approach is mimicking nature by constructing artificial photosynthetic systems that store the energy of solar light in solar fuels such as hydrogen, methanol or ethanol.3–10 Such devices generally comprise a light-absorber promoting vectorial charge separation, one catalyst for reducing protons or CO2, and another catalyst that oxidatively splits water into electrons, protons and molecular oxygen. These components may be either integrated into one ‘artificial leaf’ unit,3,11 or the photovoltaic and (photo)electrochemical units may be wired together.12–16 For large scale implementation of artificial photosynthesis devices it will be crucial that all parts are made of earth-abundant, non-toxic materials, and that the devices operate with high rates and efficiencies for long periods of time.Cobalt oxides, especially in amorphous form (CoPi-oxides), have attracted much interest recently as possible catalysts for large scale water oxidation.11,17–28 They self-assemble on electrode surfaces, when an oxidizing potential of ≥1 V vs. NHE (normal hydrogen electrode) is applied in presence of Co2+ and phosphate (Pi) in solution,29,30 they work well at neutral pH,31 and they have the ability to undergo self-repair.32 X-ray absorption spectroscopy studies revealed that CoPi-oxides are structurally similar to CoO(OH); both form layered structures with edge-sharing CoO6 octahedral units (Fig. 1A–C). However, for CoPi oxides the resulting layers are not continuous, but consist instead of molecular fragments containing 7–16 Co atoms.30 Two subsequent in situ X-ray scattering studies suggest a relatively narrow size distribution dominated by fragments with 13–14 Co atoms or 19 Co atoms, respectively.33,34 The presence of corner sharing complete and incomplete Co4O4 cubanes was also discussed, but this appears to be a less common motif.30,33,35 Depending on the deposition conditions, CoPi-oxide films consist of several stacked layers that are separated by water molecules, buffer ions (e.g. Pi) and other counterions (Fig. 1A).34,36–38 Overall, the CoPi-oxide films appear to be similar in water-splitting activity to earlier studied CoO(OH) and Co3O4; interestingly, all these Co-forms convert into each other over time depending on the applied potentials.39–45
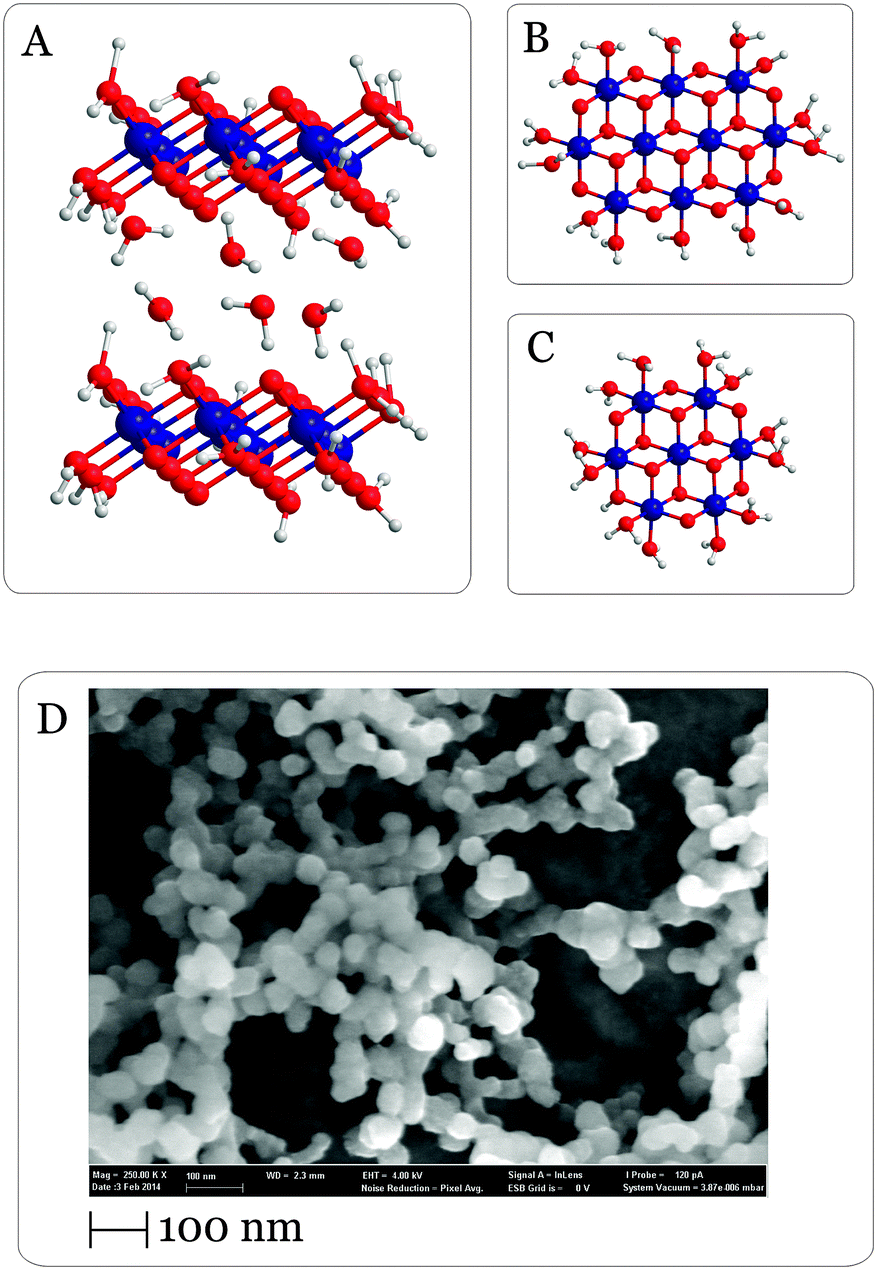 | ||
| Fig. 1 (A) Site view of the proposed layered structures of CoPi and Co/M2P-oxides.30,34,36–38,52 Between the Co-oxide layers there are likely buffer molecules (Pi), counterions and water molecules (only water is schematically shown). (B and C) Top view onto Co-oxide fragments containing either 10 (B) or 7 (C) Co atoms. Blue spheres, cobalt ions; red spheres, oxygen atoms; white spheres, hydrogen atoms. (D) SEM micrograph of Co/M2P-oxide NP formed in this study by addition of 76 μM [Ru(bpy)3]3+ into 10 μM Co(ClO4)2, 14 μM M2P and 20 mM Pi (pH 7). The SEM micrograph was taken after vacuum drying the sample on a grid. For an alternative structure with three M2P ligands see Fig. S4†. | ||
An alternative way for forming Co-oxide water-splitting catalysts was reported recently.24 In this approach, a colloidal suspension of Co-oxide nanoparticles (NP) with high oxygen-evolving capacity was formed by chemical oxidation of dissolved Co2+ in the presence of the phosphate derivate methylenediphosphonate (referred to in the following as M2P, independent of protonation state). The oxidative formation and subsequent (photochemical) water-splitting is promoted either by using the well-established [Ru(bpy)3]2+/S2O82− system, in which [Ru(bpy)3]2+ upon photoexcitation transfers an electron to the sacrificial acceptor S2O82− and subsequently oxidizes Co, or by adding the chemically preformed one-electron oxidant [Ru(bpy)3]3+.39,46–51 Structural and elemental information on the Co/M2P-oxide NP is available from an EXAFS study,52 from XRD and XPS (Text S1 and Fig. S1–S3†), light scattering24 and electron microscopy experiments (Fig. 1D). Collectively, these data show that the Co/M2P procedure produces NP with diameters of about 30–100 nm that are composed of fragments of Co-oxide layers that are structurally identical to the above described CoPi-oxides (Fig. 1).30,33 The role of M2P appears to be to limit the growth of the Co/M2P-oxide NP, since particles up to 2500 nm were obtained in presence of only Pi instead of M2P and Pi. In contrast, the O2 evolution activity of Co/M2P-oxide NP and CoPi-oxide NP was found to be identical.24
The protonation state of the terminal oxygen atoms of the molecular layer fragments in Co-oxide fragments has not yet been determined experimentally, and the possibility of Pi or M2P ligation instead of terminal water-derived ligands (Fig. 1A–C) is also discussed.30,33,35,52 The effect of M2P on the size of the Co/M2P-oxide NP suggests that there are at least unspecific interactions between Co and M2P. This is further supported by the finding of 3 M2P per 7 Co ions in XPS-measurements (Text S1 and Fig. S3†) of precipitated Co/M2P-NP that were prepared following the procedure developed by Risch and coworkers.52 EXAFS data of this solid material were consistent with Co–P interactions; however, a separate M2P phase could not be excluded.52 Importantly, 31P NMR measurements on CoPi-oxides suspensions showed that Pi is mobile within CoPi-oxides.37 It is thus an open question if in aqueous suspensions M2P is a terminal ligand to Co or not. In the resting state, which the CoPi-oxides attain a few minutes after catalysis, the Co atoms are predominantly in the oxidation state CoIII,30,35,53,54 but also some CoII and CoIV ions were detected by EPR spectroscopy.36,55 Prolonged storage in absence of applied potential bias leads to the release of CoII into solution.3,32
The mechanism of water oxidation by Co-oxide catalysts has been studied in great detail by electrochemistry, EPR, in situ X-ray spectroscopy, FTIR and H218O isotope labelling mass spectrometry, but no consensus has been reached.30,36,54–57 Kinetic, EPR and electrochemical studies revealed that CoIV needs to be formed before oxygen evolution commences,36,41–43,50,54,55 and that the rate of O2 formation (turn over frequency, TOF) is limited by a reversible step directly before the O–O bond formation that involves a proton coupled electron transfer (PCET).56 The isotope-labelling experiments in the same study raised the possibility that oxo-bridges act as substrate and form O2via intramolecular (direct) oxygen coupling (IMOC) with a terminal oxo/hydroxo group.3,56 In contrast, a recent FTIR study on crystalline Co3O4 concluded that the O–O bond formation occurs via bulk water nucleophilic attack (BWNA) onto a surface bound terminal CoIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species. This mechanistic proposal is based on the isotopic composition of a Co-bound superoxide intermediate formed after a brief charge injection.57
O species. This mechanistic proposal is based on the isotopic composition of a Co-bound superoxide intermediate formed after a brief charge injection.57
In addition, three theoretical studies have been performed on various fragments of CoPi-oxide layers, suggesting either BWNA or IMOC as dominating catalytic pathway for dioxygen formation.58–60 In the two latest reports, the formation of a CoIV–O˙ (formal CoVO) state was found to be a pre-requisite for O–O bond formation.58,59
In this work we employ highly sensitive membrane inlet isotope-ratio mass spectrometry with online sampling61–65 to study the first turnover of the Co/M2P-oxide NP after a sudden jump in H218O enrichment. The advantage of the Co/M2P-oxide NP for the present study is that their small size allows forming a colloidal suspension and thus fast mixing of the catalyst with H218O. Mathematical modelling of the data reveals the basic mechanism of O–O bond formation, the number of Co centers per catalytic site and the number of electrons that need to be removed from each catalytic site for the production of the first O2 molecule. On the basis of these results, published mechanistic proposals are discussed and amended.
2. Experimental
2.1 Materials
Methylenediphosphonic acid and [Ru(bpy)3](ClO4)3 were prepared as reported previously.24,66 [Ru(bpy)3](ClO4)2, Co(ClO4)2·6H2O (99.999% metals basis) and Na2S2O8 were purchased from Alfa Aesar or Aldrich and used as received. Labeled water (98% H218O) was obtained from Medical Isotopes, Inc. USA. All other chemicals were purchased in analytical grade quality and used as received. Photosystem II (PSII) membrane fragments were prepared as described elsewhere.67,682.2 Oxygen evolution measurements by membrane-inlet mass spectrometry (MIMS)
All oxygen evolution measurements in this study were done with an isotope-ratio mass spectrometer62,65 (ThermoFinnigan Delta plus XP) that was connected via a cooling trap (liquid N2) to a cylindrical, thermostated MIMS cell (modified Hansatech Clark electrode cell). The reaction volume of 600 μl was separated from the high vacuum of the mass spectrometer by a thin silicon membrane (25 μm, Mempro; MEM-213) resting on a porous plastic support, and isolated from the air by a lid that contained a narrow injection port. The experiments were performed at 20 °C under constant stirring. Before the measurements, the samples were degased in the MIMS cell for about 30 min to reach stable base lines. The O2 formed during the measurements was recorded simultaneously as non-labeled 16O2, mixed labeled 16,18O2 and double-labeled 18O2 species.The injections of [Ru(bpy)3]3+ solutions into the degased sample suspensions in the MIMS cell caused injection artifacts due to dissolved natural abundance O2, a small fraction of 36O2 arising from water oxidation by [Ru(bpy)3]3+ (this fraction is incubation time dependent; see results and Fig. S5†) and Argon (36Ar). The amplitudes caused by dissolved oxygen from the air were largest for the 16O2 (about 3000 mV); for comparison, the O2 signals arising from water oxidation by Co-oxides had amplitudes of 150–800 mV. Thus, careful background subtractions were required. For this, the injection amplitudes were determined repeatedly during the day by injections of [Ru(bpy)3]3+ solutions (90% H218O) into the reaction mixtures lacking Co2+. These ‘blank’ injections were found to yield reproducible amplitudes, and the average blank injections were subtracted from the signals collected in presence of Co2+ to obtain the traces displayed in the figures. Error bars in the figures are based on 2–3 independent data collections and take into account the deviations arising from the slight variations in the blank injections.
2.3 Calculation of the fraction of 18O in O2
From the experimental 16O2, 16,18O2 and 18O2 signals the total fraction of 18O atoms in the product, 18α, was calculated according to eqn (1):69,70 | (1) |
2.4 Modelling of isotope ratios
The dependence of the isotope ratios and total oxygen on the [Ru(bpy)3]3+ concentration was modelled within Microsoft Excel. The sum of the squared errors between the data points and the fits was minimized using the Generalized Reduced Gradient, GRG, algorithm in the Solver function. The equations used are presented in the main text.2.5 Calibration of O2 signals
The calibration of the O2 signals was performed by the injection of various known amounts of air-saturated water into the MIMS cell containing degased phosphate buffer. The calibration curve obtained was used to determine the absolute amount of O2 produced by the Co-oxide NP.2.6 Electron microscopy
Scanning electron microscopy (SEM) was performed using a Zeiss Merlin FEG-SEM instrument. For forming active Co/M2P-oxide NP 76 μM [Ru(bpy)3]3+ was added to a solution containing 10 μM Co(ClO4)2 and 14 μM M2P in 20 mM Pi buffer (pH 7). A few drops of suspension containing preformed Co/M2P-oxide NP were cast onto a Formvar (a polyvinyl-based thermoplastic resin) coated copper grid. The specimen was mounted onto a grid holder. The surface morphology was examined by SEM using an in-lens secondary electron detector operating at a beam accelerating voltage of 4 kV and probe current of 120 pA.3. Results and discussion
3.1 Steady state analysis (SSA)
The red line in Fig. 2 shows the dependence of the 18O-fraction of the O2 evolved by Co/M2P-oxide NP on the time of illumination in presence of [Ru(bpy)3]2+ and S2O82−. The 18α-fraction (eqn (1)) was calculated from the 16O2, 16,18O2 and 18O2 mass peaks measured simultaneously with an isotope ratio mass spectrometer via membrane-inlet sampling.62,65 After 75 s of illumination a stable value of 18α = 5% was reached, which corresponded to the 18O-enrichment of the water.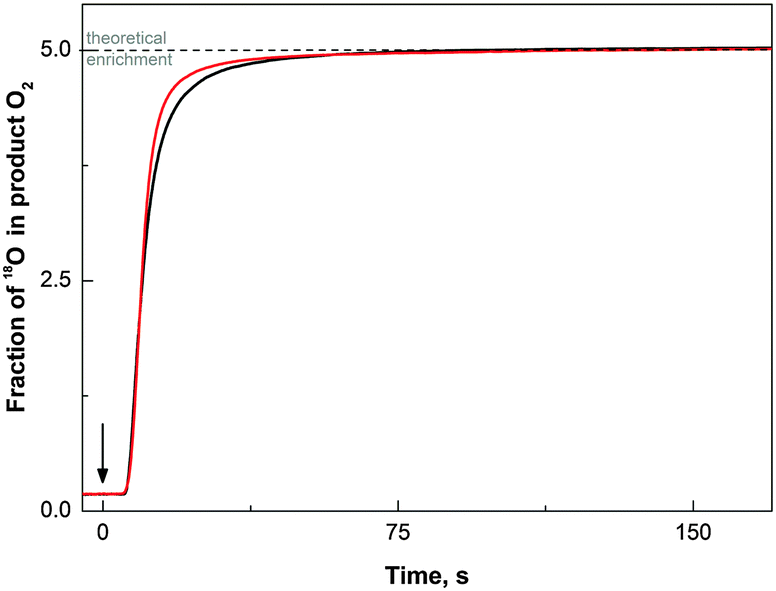 | ||
| Fig. 2 18O-fraction (18α) of molecular oxygen formed by the reaction of Co/M2P-oxide NP with photo-generated [Ru(bpy)3]3+ (red line), and by illumination of spinach PSII membrane fragments (black line). The traces were obtained using eqn (1) from the 16O2, 16,18O2 and 18O2 data recorded with a membrane-inlet isotope-ratio mass spectrometer. In case of the Co/M2P-oxide NP, the reaction mixture contained 100 mM [Ru(bpy)3]2+, 1 mM S2O82−, 10 μM Co(ClO4)2, 14 μM M2P and 5% H218O in 20 mM Pi buffer, pH 7.0. After a stable base line was reached, the sample was illuminated with white light from a 500 W slide projector (the arrow marks the start of illumination). PSII membrane fragments (0.03 mg Chl ml−1) were illuminated at pH 6.5 with the same light source in a buffered solution containing 40 mM MES, 5 mM MgCl2, 15 mM CaCl2, 15 mM NaCl, 400 mM sucrose, 5% H218O, and 2 mM K3[Fe(CN)6] plus 0.6 mM PPBQ as exogenous electron acceptors. | ||
The black line in Fig. 2 shows that illumination of a photosystem II (PSII) sample, which contained the natural water-splitting enzyme complex, gave essentially the same trace. Since PSII is known to have two rapidly exchanging substrate waters,71,72 this comparison demonstrates that the initial delay after the start of the illumination (arrow) and the slow rise of the 18α-fraction were caused by the slight difference in the pervaporation rate of the isotopologues and that the correct ratio can be obtained only after a steady state production is reached.63,64 The good agreement of the steady state 18α fraction of the O2 produced by Co/M2P-oxide NP with that found for PSII, i.e. with the value expected for the level of H218O enrichment in the buffer, shows that water is the ultimate substrate for molecular oxygen formation. This means that any contribution from non-exchangeable, pre-bound oxygen species such as oxo-bridges is negligible under steady state conditions. Similar results were reported previously for the CoPi catalysts31 and CaMn-oxides.63
While this information is important, steady state O2 production does not reveal any information on the mechanism of its formation from water, unless careful calibrations are performed to reveal kinetic isotope effects.73,74
3.2 First turnover analysis (FTA)
A direct path towards unravelling the basic mechanism of water oxidation is to study the isotopic composition of the O2 produced by the first turnover after a sudden jump in the H218O concentration. In order to achieve rapid oxidation of the Co/M2P-oxide NP after H218O addition and for controlling the number of turnovers, we injected in these experiments dissolved [Ru(bpy)3]3+ rather than forming [Ru(bpy)3]3+in situ by illumination. Since [Ru(bpy)3]3+ is not stable in Pi buffer at pH 7.0, for each injection a fresh aliquot was dissolved and injected into the preformed 16O–Co/M2P-oxide NP after 45 s. The concentration of [Ru(bpy)3]3+ at time of injection was determined by UV/Vis spectroscopy, taking the 675 nm absorption of a [Ru(bpy)3]3+ solution in 4 M H2SO4 as 100% value (see Fig. S6†), since [Ru(bpy)3]3+ is known to have a long halftime (>25 h) at this high acid concentration.50,75 In the following, we always refer to this effective [Ru(bpy)3]3+ concentration, which under our conditions was 38% of the initial concentration.For each measurement in these FTA experiments, the active 16O–Co/M2P-oxide NP were formed in situ by the injection of a 76 μM [Ru(bpy)3]3+ into the MIMS cell containing a solution of 10 μM Co2+ and 14 μM M2P in 20 mM Pi buffer. For this step, all chemicals were dissolved in natural abundance water. As shown on the left sides of Fig. 3A and B, a reproducible amount of natural abundance O2 was formed during this procedure (note: the 16,18O2 and 18O2 signals are displayed at 100- and 1000-fold amplification, respectively, relative to the 16O2 signal).
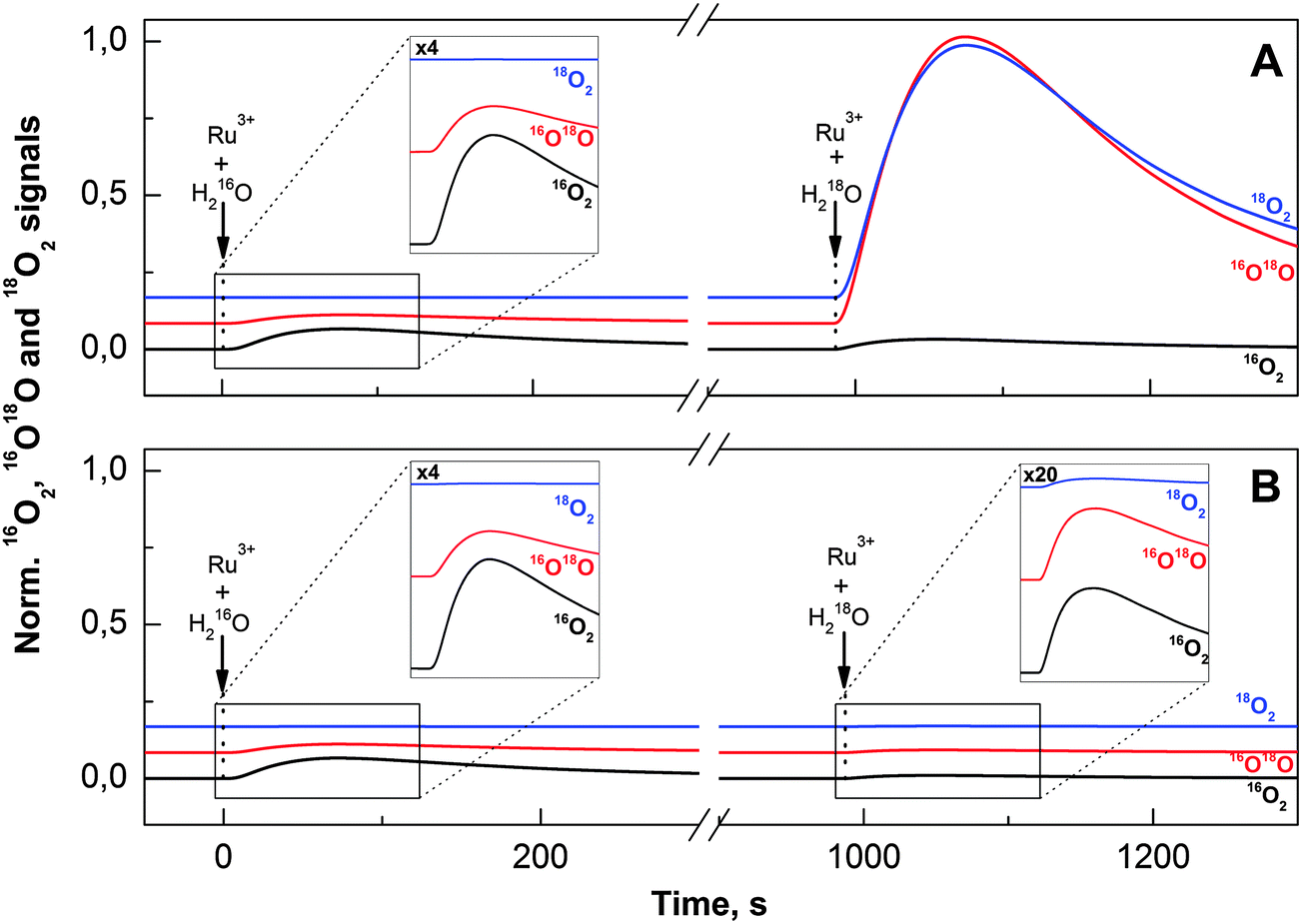 | ||
| Fig. 3 Isotopologues of O2 formed during the FTA experiments. The 16,18O2 and 18O2 signals are shown in 100- and 1000-fold amplification, respectively, relative to the 16O2 signal. At time zero 10 μl of [Ru(bpy)3]3+ (76 μM final concentration) dissolved in natural abundance water was injected into the reaction mixture (600 μl) containing 10 μM Co(ClO4)2, 14 μM M2P and 20 mM Pi (pH 7) to form active Co/M2P-oxide NP having 16O-labelled bridges and terminal ligands. This was followed by a second injection of various [Ru(bpy)3]3+ concentrations in 100 μl H218O (90%); 76 μM and 7.6 μM for panels A and B, respectively. The final H218O enrichment was in all cases 15%. The formation of molecular oxygen was monitored by MIMS, and the separately determined injection artifacts were subtracted. To allow easy comparison between panels A and B the amplified traces were normalized to the amplitude of the 16,18O2 signal obtained after injection of 76 μM [Ru(bpy)3]3+ (in 90% H218O) into the preformed Co/M2P-oxide NP. | ||
During the subsequent waiting time of 25 min all signals returned to their baselines. Then a second portion of [Ru(bpy)3]3+ was injected into the 16O–Co/M2P-oxide NP suspension; however, this time [Ru(bpy)3]3+ was dissolved in 90% H218O to give a final 18O-enrichment of 15%. While the volume of this second injection was kept constant, so that always the same final 18O-enrichment was reached in the buffer, the concentration of [Ru(bpy)3]3+ was successively reduced during the course of the FTA experiments (Table 1). The results obtained for the largest [Ru(bpy)3]3+ concentration (76 μM) and the lowest [Ru(bpy)3]3+ concentration (7.6 μM) are displayed on the right sides of Fig. 3A and B, respectively.
| [Ru(bpy)3]3+ μM | Experimental ratios | |
|---|---|---|
| 16O2/16,18O2 | 16,18O2/18O2 | |
| a The final 18O-enrichment was 15% and the final Co concentration was 8.16 μM. | ||
| 7.6 | 118 ± 10 | 76 ± 10 |
| 11.4 | 40 ± 5 | 12 ± 3 |
| 13.3 | 20 ± 2 | 11.7 ± 0.3 |
| 15.2 | 12 ± 1 | 11 ± 3 |
| 19 | 9 ± 1 | 11 ± 1.5 |
| 38 | 5 ± 2 | 11 ± 0.6 |
| 76 | 4.8 ± 0.1 | 11 ± 0.4 |
The isotopic composition of the O2 obtained at these two [Ru(bpy)3]3+ concentrations was markedly different. While the addition of 76 μM [Ru(bpy)3]3+/H218O induced the formation of labelled O2, the oxygen produced after the injection of 7.6 μM [Ru(bpy)3]3+ had nearly natural enrichment (compare to the injections of [Ru(bpy)3]3+ dissolved in H216O; left side). The experimental 16O2/16,18O2 and 16,18O2/18O2 ratios obtained for all seven [Ru(bpy)3]3+ concentrations used in this study are listed in Table 1. These data show that the change in the isotopic composition of O2 occurred gradually over the studied [Ru(bpy)3]3+ concentration range.
If the 16O–Co/M2P-oxide NP generated during the pre-formation and subsequent waiting time would not bind any substrate, or if both substrate oxygen atoms would exchange faster with bulk water than they would be oxidized by [Ru(bpy)3]3+, then no [Ru(bpy)3]3+ concentration dependence of the isotopic ratios would be observed. The data in Fig. 3 and Table 1 thus excluded these two possibilities, demonstrating that at least one relatively slowly exchanging substrate is pre-bound to the catalytic site. This leads to a distinct isotopic composition of the O2 produced in the first turnover, which in turn allows deriving the reaction mechanism and the determination of the number of catalytic sites.
3.3 Modelling of the FTA data
The main purpose of modelling the isotope ratios as a function of [Ru(bpy)3]3+ is to discern whether the O–O bond formation occurs via BWNA or IMOC. The latter could involve either a nucleophilic attack of a bound terminal water/hydroxo onto a Co-oxo species, or radical coupling. These options are summarized in Scheme 1.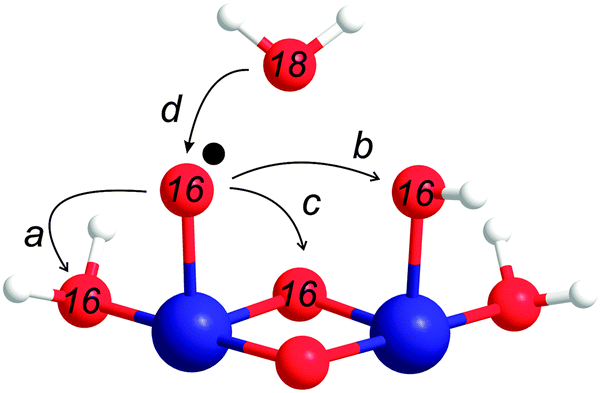 | ||
| Scheme 1 O–O bond formation through intramolecular oxygen coupling (IMOC) of two pre-bound oxygen atoms (a–c) or bulk water nucleophilic attack (BWNA) onto a pre-bound oxygen atom (d). The Co–O˙ group may be replaced by CoO. For the FTA experiments in this study, the oxygen atoms (red spheres) ligating Co (blue spheres) are of natural abundance, while the bulk water is enriched in H218O. Hydrogen atoms are shown as white spheres. The coordination sphere of Co is not completed for simplicity of presentation. | ||
Assuming that the O–O bond formation occurs via BWNA (reaction d in Scheme 1), the O2 evolved by the first turnover of each catalytic site will contain one pre-bound oxygen atom with natural enrichment, and a second oxygen atom originating from bulk water, i.e. with 15% 18O-enrichment. Therefore, if the effective [Ru(bpy)3]3+ concentration is smaller than the concentration of catalytic sites, [cat], the relative ratios of the oxygen isotopologues formed by water oxidation can be calculated, in absence of isotope effects, from the binomial distribution of 16O and 18O in the bulk water and the pre-bound substrate at the catalytic site, respectively. The absolute concentrations of these isotopologues are then obtained by multiplying these fractions by the concentration of catalytic sites that reacted with [Ru(bpy)3]3+ to form O2:
 | (2) |
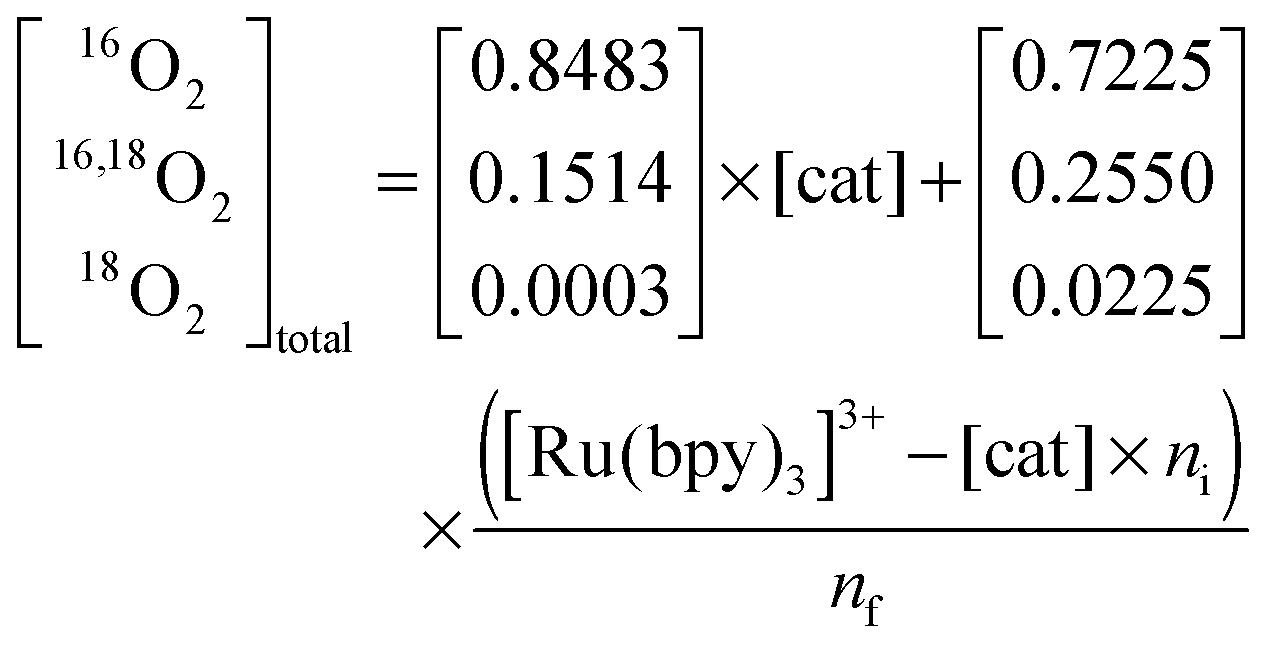 | (3) |
In contrast, the first turnover of a coupling mechanism (Scheme 1a–c) involves two pre-bound oxygen species, and thus O2 with natural abundance will be observed if the oxidation is faster than the exchange with bulk water:
 | (4) |
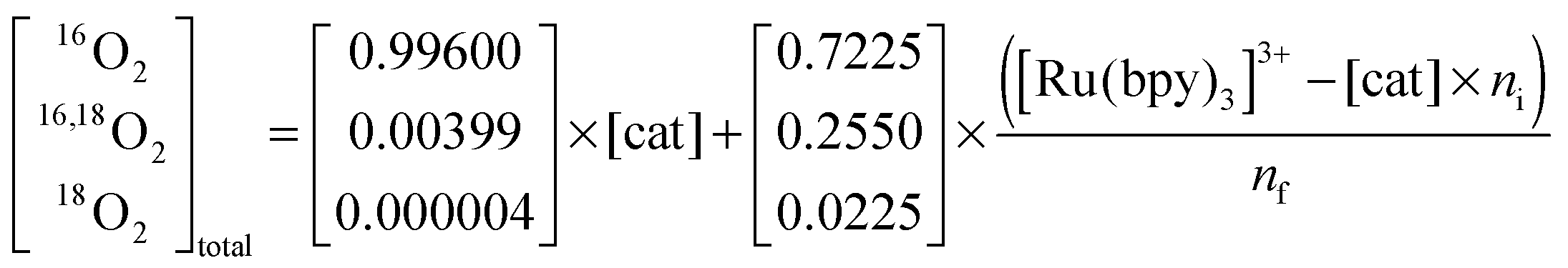 | (5) |
Eqn (2)–(5) contain implicitly a number of simplifying assumptions: (i) no catalytic site makes a second turnover before all catalytic sites have released their first O2 molecule (no double turnovers), (ii) the number of catalytic sites remains constant during the experiments and (iii) each catalytic site operates independently from all others, i.e. there is no exchange of substrate oxygen atoms between catalytic sites until all sites have completed the first turnover. Fig. 4 shows that despite these simplifications, the isotope ratios calculated for the intramolecular coupling mechanism (eqn (4) and (5)) reproduce the experimental values rather closely: only at the lowest [Ru(bpy)3]3+ concentration a significant deviation is observed (black line in Fig. 4A and B). This fit was obtained with [cat] = 3.6 μM and ni = 2.9. By contrast, for the nucleophilic attack mechanism (eqn (2) and (3)) the best fit showed only at the two largest [Ru(bpy)3]3+ concentrations an acceptable agreement with the experimental 16O2/16,18O2 ratio (blue line in Fig. 4A and B; see also Table S1† for an independent fit and Fig. S7† for an independent data set). It is noted that according to eqn (2) the 16O2/16,18O2 ratio cannot exceed a value of 5.6 (0.84/0.15) for BWNA under our experimental conditions. Similarly, clear deviations from the 16,18O2/18O2 ratio are observed for most [Ru(bpy)3]3+ concentrations.
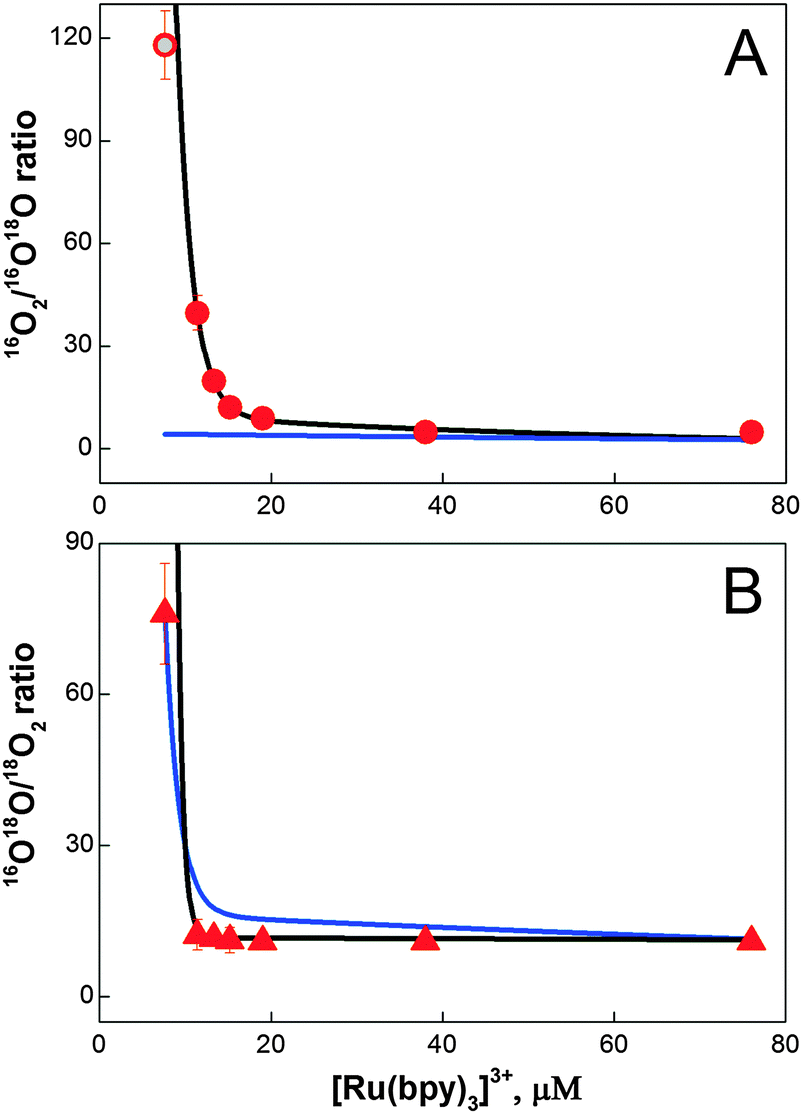 | ||
| Fig. 4 Simulation of the 16O2/16O18O (A, circles) and 16O18O/18O2 (B, triangles) ratios obtained during the FTA experiments for different [Ru(bpy)3]3+ concentrations (Fig. 3 and Table 1). Lines represent best fits to eqn (2) and (3) (blue line; BWNA, [cat] = 1.75 μM and ni = 4.0) and eqn (4) and (5) (black line; IMOC, [cat] = 3.6 μM and ni = 2.9), respectively. The grey dot represents the best fit of the isotopologue ratio measured at 7.6 mM [Ru(bpy)3]3+ obtained by an unrestricted FTA analysis assuming either 1.7% double hits or 3% BWNA in addition to IMOC (for details see eqn (S2) and (S3) and Table S2†). | ||
This simple analytical description of the data indicates strongly that the O–O bond is formed at the Co/M2P-oxide NP by IMOC, i.e. between two pre-bound substrate ligands. To further study both the deviations from the IMOC model at low [Ru(bpy)3]3+ concentrations and the possible effects of the implicit assumptions, we analyzed the data also by freely varying the fractions for the isotopologue distributions of molecular oxygen produced in the respective first and subsequent turnovers of the BWNA and IMOC mechanisms (for details see Text S2†). The fit results displayed in Table S1 and S2† demonstrate that the discrepancy observed for 7.6 μM [Ru(bpy)3]3+ in the IMOC (Fig. 4) can be equally well explained with either about 2% of double turnovers or 3% of BWNA in addition to IMOC (gray circle in Fig. 4A). They further show that less than 10% double turnovers/BWNA occurs until all centers have produced the first O2 molecule. As expected, also this flexible fit approach is unable to describe the data by assuming that the BWNA mechanism is predominant (Table S1†).
3.4 Analysis of the total oxygen yields (TOY)
In Fig. 5 the total oxygen yield (sum of all three isotopologues) as a function of [Ru(bpy)3]3+ concentration is presented. Two phases can be discerned, which were fit using eqn (4) and (5) by summing up all isotopologues. The best fit (black line in Fig. 5) revealed that in the first phase the O2-yield increased linearly with a slope of about 1 O2 per 3 [Ru(bpy)3]3+, while at higher [Ru(bpy)3]3+ concentrations the slope was about 1 O2 per 6 [Ru(bpy)3]3+. Both slopes thus deviate from the theoretical value of 1 O2 per 4 [Ru(bpy)3]3+. The lower slope at higher [Ru(bpy)3]3+ concentrations is consistent with known side reactions of [Ru(bpy)3]3+,75,76 while the steeper slope at low [Ru(bpy)3]3+ shows that the resting state of Co/M2P-oxide NP attained 25 min after their formation does not correspond to the lowest oxidation state within the catalytic cycle. To account for the small differences in [cat] and nf as compared to the FTA analysis, a simultaneous fit of the FTA and the TOY data was performed (blue line Fig. 5). The best parameters found were: ni = 3.1, [cat] = 3.2, and nf = 5.1. The value for ni, i.e. the number of [Ru(bpy)3]3+ required per catalytic site for the production of the first O2 molecule, is very robust and essentially identical in all three fits (see also Fig. S7†). Since it is unknown if the percentage of side reactions of [Ru(bpy)3]3+ is concentration dependent or not, this number is consistent with mechanisms in which the resting state is by 1 or 2 electrons more oxidized than the lowest oxidation state of the catalytic site during catalysis.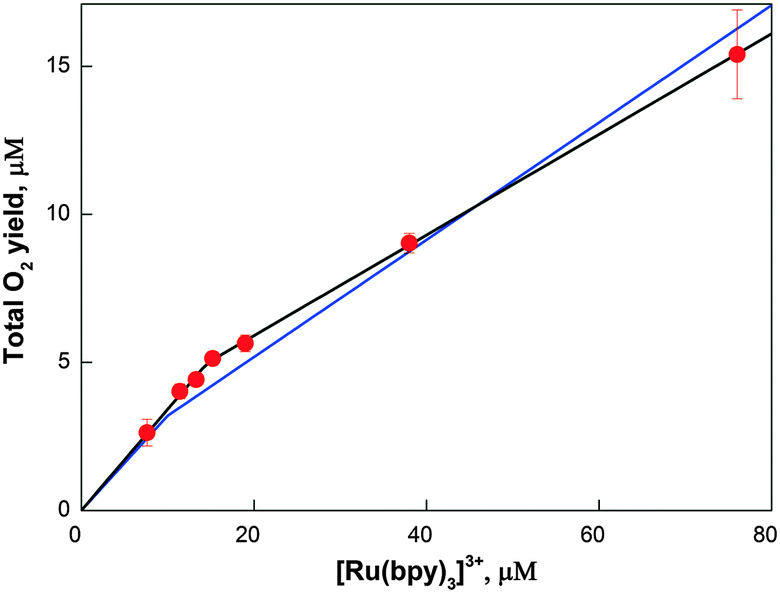 | ||
| Fig. 5 Total oxygen as a function of [Ru(bpy)3]3+ concentration. Symbols: experimental values. Black line: best fit using the parameters: ni = 2.9, [cat] = 5.0, and nf = 5.9. Blue line: simultaneous fit of FTA and TOY data: ni = 3.1, [cat] = 3.2, and nf = 5.1. | ||
3.5 Number of Co atoms and TOF per catalytic site
Given the good agreement between the TOY and FTA analyses and that only few double turnovers are required to make the IMOC approach fully consistent with the experimental data, the values derived for the concentration of catalytic sites, which corresponds to the amount of naturally labelled molecular oxygen released during the first turnover, can be viewed as rather reliable: [cat] = 4 ± 1 μM. Using the known concentration of Co2+ in the suspension (8.16 μM after the two injections) we can calculate the number of Co atoms per catalytic site to be 2.1 ± 0.5. This surprisingly low number indicates that each catalytic site is formed by two Co atoms plus possibly a small number of structural or charge storing Co centers. This is fully consistent with the molecular nature of small Co-oxide layer fragments (Fig. 1B and C) and the assumption that their edges form the catalytic sites (see also below). It also strongly supports recent reports showing that in amorphous Co-oxides the whole volume and not only the surface is catalytically active.45,56,77 A much larger number of structural Co centers were assumed to be present in electrodeposited CoPi-oxide films.56 This may be consistent with the 100-times lower turnover frequency of CoPi-oxide films as compared to Co/M2P-oxide NP.24,31,56 We note, however, that it is not straight forward to compare electrochemical experiments with the chemical oxidant approach employed here.The TOF per Co ion was determined recently for Co/M2P-oxide NP under essentially identical conditions to be TOF/Co = 0.26 s−1.24 Using the above estimate for the number of Co per catalytic site, the TOF per catalytic site can be calculated to be TOF/cat ≈ 0.5 s−1. This value is nearly three orders of magnitude lower than for the oxygen evolving complex in PSII.
3.6 Exchange rates and mode of substrate binding
Both terminal hydroxo/oxo ligands and μ-oxo bridges have been proposed as substrates for O2 formation during water oxidation by Co-oxides.3,36,56,59 Since bridging oxo groups in Co-oxide materials were shown to be practically non-exchangeable,56,57 observation of isotopic exchange of one or both substrate water(s) may favor terminal ligation of the respective substrate(s). While water exchange of terminal ligands on CoII complexes occurs generally with characteristic time constants in the order of 1 μs,78–80 significantly slower exchange is expected for terminal ligands on CoIII, which is the dominant oxidation state in the resting state of Co-oxide catalysts.30,35,52,54 The retarded ligand exchange of CoIII is due to the fact that it is a low spin diamagnetic t2g system.78 Characteristic times published for molecular complexes vary with ligand type and span a range of 10 days to 100 ms.81 To the best of our knowledge, no values are published so far for terminal water or hydroxo ligands on Co-oxides or Co(oxo)hydroxides.Our FTA data show that both substrate oxygen atoms are bound to the resting state of the 16O–Co/M2P-oxide catalyst and that they exchange slower with the bulk water than they react with [Ru(bpy)3]3+ to form O2. This is also seen in Fig. 6A where we repeated the FTA experiments at a [Ru(bpy)3]3+ concentration that induced just over 1 turnover per catalytic site (the initial formation of the Co/M2P in H216O is not shown). In full agreement with the data displayed in Fig. 3 and Table 1, the simultaneous addition of [Ru(bpy)3]3+ and H218O resulted in a large percentage of unlabeled 16O2, and the 16,18O2 and 18O2 yields are far below those expected for the H218O enrichment of 15%.
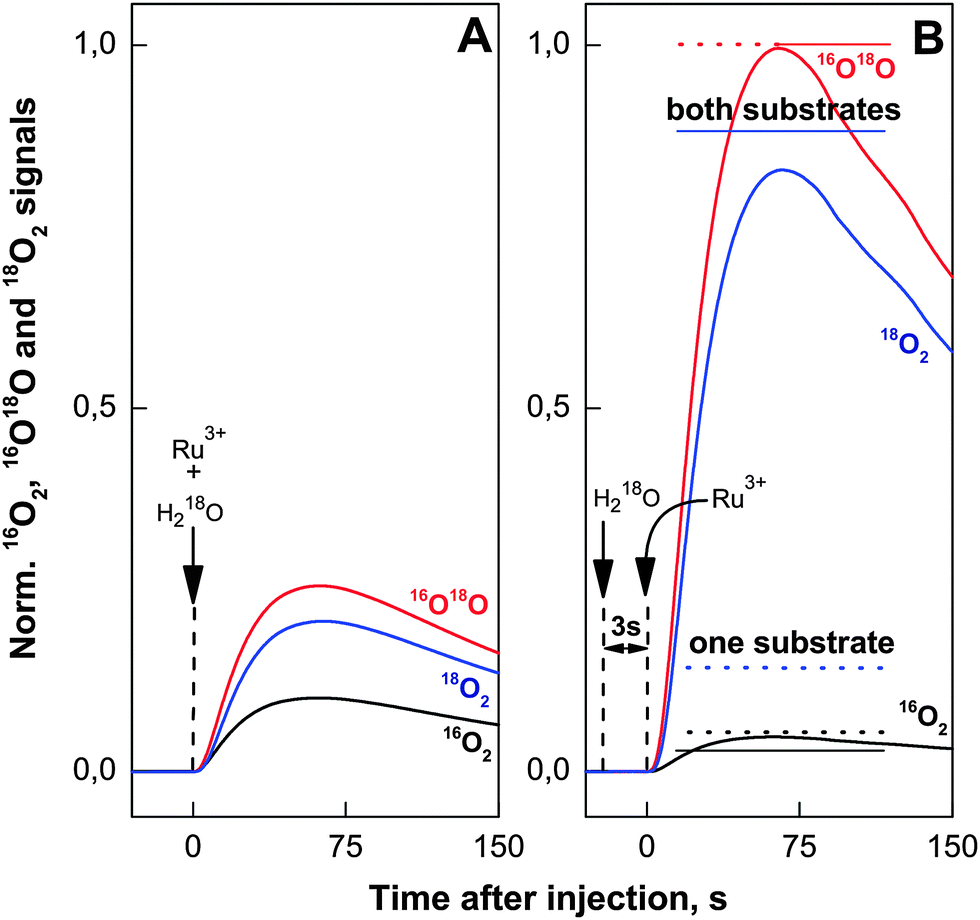 | ||
| Fig. 6 Evolution of O2 isotopologues after injection of 11.4 μM [Ru(bpy)3]3+ and labeled H218O into pre-formed 16O–Co/M2P-oxide NP suspensions giving final enrichments of 15% H218O and 8.16 μM Co. Panel A shows the result of a simultaneous injection (arrow) of [Ru(bpy)3]3+ and H218O, while the data in panel B were obtained by injecting H218O 3 s before the addition of [Ru(bpy)3]3+ (dissolved in 15% H218O). The horizontal bars in panel B show the calculated oxygen yields of the three O2 isotopologues for either complete exchange of both substrate water molecules (solid bars) or of just one substrate (dashed bars). In both cases 1.1 turnovers of the catalytic site were assumed. The signals for m/z 34 and m/z 36 are shown in 100- and 1000-fold amplification, respectively. All experimental and calculated (bars) O2 signals were normalized to the amplitude of the 16,18O2 signal in panel B. | ||
To probe the exchange of the substrates with bulk water we injected then the H218O (final enrichment 15%) 3 s before the addition of [Ru(bpy)3]3+ (Fig. 6B). To avoid any further change of the isotopic composition of the reaction mixture, [Ru(bpy)3]3+ was dissolved in 15% H218O in this case. The drop in the percentage of the 16O2 isotopologue and the corresponding increase of the 16,18O2 and 18O2 signals suggests that in almost all catalytic sites both substrate oxygen atoms have exchanged during the 3 s incubation time with H218O. The dashed and solid bars in Fig. 6B indicate the isotopologue ratios calculated for 1.1 turnovers assuming the exchange of only one substrate (dashed bars) or after exchange of both substrates per catalytic sites (solid bars). The much better agreement of the data with the latter scenario, especially for the 18O2 signal (blue line and bar in Fig. 6) favors O–O bond formation between two terminal ligands. This reinforces that water oxidation occurs on the outer boundaries of the layered Co-oxide fragments. Together with our finding that 2.1 ± 0.5 Co atoms form a catalytic site this argues for a dominance of small layer fragments with about 7–13 Co atoms that contain no more than 3 rows of cobalt atoms (Fig. 1B and C). As already mentioned in the introduction, such structures were suggested previously on the basis of EXAFS spectroscopy and X-ray scattering.30,33,52
3.7 A slowly exchangeable water pool or different reactivity of crystalline Co3O4vs. amorphous Co/M2P-oxide NP?
As noted above, most terminal water and hydroxo ligands on molecular CoIII complexes exchange orders of magnitudes slower than the exchange observed in the experiments displayed in Fig. 6.81 Therefore, also one alternative interpretation must be considered. Due to the layered structure of the Co/M2P-oxide NP (Fig. 1), it may be possible that the data displayed in Fig. 6 do not report on the exchange of the bound substrate molecules per se, but instead reflect the slow exchange of a pool of water molecules inside the interlayer spaces or of strongly ligated surface water molecules. The likelihood of this possibility is considered in the following.Detailed electrochemical studies have shown that mass transport of water and O2 is not rate limiting for the water-splitting catalysis in amorphous CoPi-oxide layers on electrode surfaces of thicknesses of up to at least 100 nm, the maximum size of our Co/M2P-oxide NP (Fig. 1).56,77 Similarly, the same TOF/Co was found earlier for Co/M2P-oxide NP with size distributions of either 10–60 nm or 200–2500 nm.24 This is understandable on the basis of reports that the interlayer spaces in CoPi-oxides are relatively large,34 allowing even Pi ions to move freely and to be exchanged.37,38 Since water molecules buried deep inside of proteins have residence times of usually much less than 1 ms,82 it appears unlikely that the exchange of the interlayer waters would occur at timescales longer than that. Since 1 ms is short as compared to the speed of mixing and the TOF/cat for Co/M2P-oxide NP (0.5 s−1), we think that it is more likely that the exchange observed in Fig. 6 reflects the equilibration of terminal water ligands on CoIII ions than that of trapped interlayer or surface water molecules. However, experiments far beyond the scope of the present study are required to clarify this point.
Thus, the question becomes if slow interlayer water exchange, or that of specifically bound surface water molecules, could mask a BWNA mechanism, thereby leading to an erroneous assignment to IMOC. This question is especially important, since recently a mixed labelled superoxide was identified by FTIR spectroscopy to be an intermediate during the cycle of water-oxidation by crystalline Co3O4 NP. Such a species can only arise by BWNA onto a non-exchangeable, Co-bound substrate oxygen.57 It is noted that for these FTIR experiments the Co3O4 nanocrystals were exposed to highly-enriched H218O for much longer times than the 3 s incubation used here for full exchange (Fig. 6), demonstrating the presence of a very slowly exchanging substrate oxygen in the Co3O4 sample. In contrast, as discussed above, for Co/M2P-oxide NP the significant increase of both the 16,18O2 and 18O2 isotopologues after 3 s of incubation in H218O (Fig. 6) excludes the participation of a very slowly exchanging substrate in O2 formation, and thus in agreement with the FTA analysis also the BWNA mechanism. However, if the number of catalytic sites would be significantly lower than determined by our FTA method, then the employed amount of [Ru(bpy)3]3+ would induce multiple turnovers and a distinction between BWNA and IMOC mechanisms may no longer be possible. To be consistent with the FTA data this scenario would require that after the first turnover the vacant substrate site would be filled for at least one subsequent cycle almost exclusively from a non-exchangeable pool of water molecules. That this can happen homogenously at all catalytic sites within an amorphous and as such heterogeneous sample appears rather unlikely, especially given the fast water exchange observed in the experiments presented in Fig. 6.
On the basis of these considerations we propose that O2 formation on the surface of Co3O4 nanocrystals occurs with a different mechanism (BWNA) than in the bulk catalysis supported by amorphous Co/M2P-oxide NP (IMOC). This hypothesis is supported by the recent finding that the few catalytic sites on the surface of crystalline Co-oxide samples show a higher site specific TOF than the many catalytic sites within the amorphous Co-oxides these crystalline samples convert into after prolonged catalysis (the total O2 evolution is higher in the amorphous materials due to the much larger number of catalytic sites).45 This result appears fully consistent with a change in the mechanism of O–O bond formation during this transformation. Similarly, for molecular Ru2-catalysts either BWNA or IMOC has been observed depending on constraints imposed by their ligands.74,83
3.8 Comparison to earlier isotope labelling mass spectrometry studies
The rapid exchange found here for the substrate oxygen atoms and the conclusion that the O–O bond is formed between two terminal water-derived ligands is consistent with two previous 18O isotope labelling mass spectrometry experiments on water-oxidation by Co-oxide materials.56,57 Nocera and coworkers prepared a thin 18O–CoPi-oxide film by electrodeposition of CoII dissolved in H218O. Subsequent catalysis in H216O water showed no initial burst of labelled O2, but instead a slow release of the 18O-lable into the product; over a period of 12 hours of continuous O2 production 22% of the 18O contained in the oxide appeared in the product.56,84–86 In accordance with the original interpretation by Nocera and coauthors (see ref. 3 for an alternative interpretation), we take this to show that the participation of oxo-bridges in O–O bond formation is a minor side reaction that reflects either a low probability for intramolecular coupling between terminal ligands and μ-oxo bridges or structural changes of the amorphous CoPi-oxides during catalysis.56Similarly, Frei and coworkers found neither an initial burst of 16,16O2 nor of 16,18O2 evolution during photo-chemical water oxidation ([Ru(bpy)3]2+/S2O82− system) catalyzed by Co316O4 nanocrystals that were equilibrated in H218O buffer.57 This is likely due to the lower sensitivity of the mass spec technique employed, which made it necessary to accumulate the O2 produced during 10 min of photocatalysis before the isotopologue ratios could be determined. The large number of turnovers induced by this procedure makes it extremely difficult to discern the isotopic signature of the first turnover. Thus, no distinction between IMOC and BWNA can be made on the basis of their mass spectrometric data.
3.9 Mechanism of water oxidation by Co/M2P-oxide NP
Below we derive a mechanism for water oxidation by Co/M2P-oxide NP on the basis of previously proposed mechanism and the results obtained in this study. While we employ a layer fragment size as structural model that is possibly more abundant in Co/M2P-oxide NP than in other Co-oxide materials, the overall similarities of the molecular Co-oxide fragments in Co/M2P-oxide NP with those in CoPi-oxides suggest that the proposed mechanism will be the same for both types of materials. Since our data appear to be incompatible with BWNA mechanisms, we concentrate below on IMOC mechanisms.For developing a mechanism, we first need a structural model for the resting state that includes the protonation state of the terminal ligands and the Co-oxidation states of the Co/M2P-oxide catalyst. Our data show that for each catalytic site there are no more than 2.6 Co atoms in the molecular fragment, and that the O–O bond is formed by terminal rather than bridging oxygen's. Therefore, Co-oxide layer fragments containing 7, 10 or 13 Co atoms arranged in 3 rows are the best structural models for further considerations (Fig. 1B and C), since the corresponding ratios of total Co to catalytic sites are 2.3, 2.5 and 2.6, respectively; here we assume that each catalytic site requires two peripheral Co atoms. Such fragments are in full agreement with models proposed earlier on the basis of EXAFS spectroscopy and X-ray scattering.30,33,34,52 For simplicity, we focus below on Co7 fragments. These Co7 fragments contain 12 oxo-bridges. Since EPR and XANES experiments are best consistent with CoIII as dominant oxidation state in the catalyst's resting state, the total charge of such fragments would be −3 in the absence of terminal ligands. Adding hydroxides or phosphates as terminal ligands to the 12 vacant terminal binding sites would increase the negative charge of the Co7 fragment up to −15. Such a high charge appears unrealistic, and it seems unlikely that such highly charged fragments could build layered structures such as those proposed for CoPi films or Co/M2P-oxide NP, even if partially screened by counterions (Fig. 1). We thus propose, in line with recent theoretical work,58,59 that the individual fragments shall be (nearly) neutral in charge. As such, the terminal ligands are likely fully protonated water molecules, and even three of the six outer μ2-oxo bridges may be protonated in the resting state. This proposed resting state structure of a Co7-fragment is shown in the center of Fig. 7. Incidentally, the mechanism proposed in Fig. 7 would not be affected if up to 50% of the terminal Co binding sites would be occupied by M2P or Pi ligands instead of H2O (see Fig. S4† for a possible structural model). While there are three catalytic sites per Co7 fragment, we will concentrate our further analysis on just one dimeric unit (dashed oval in Fig. 7). The structure labelled C1 in the outer ring of Fig. 7 marks the resting state of this catalytic site.
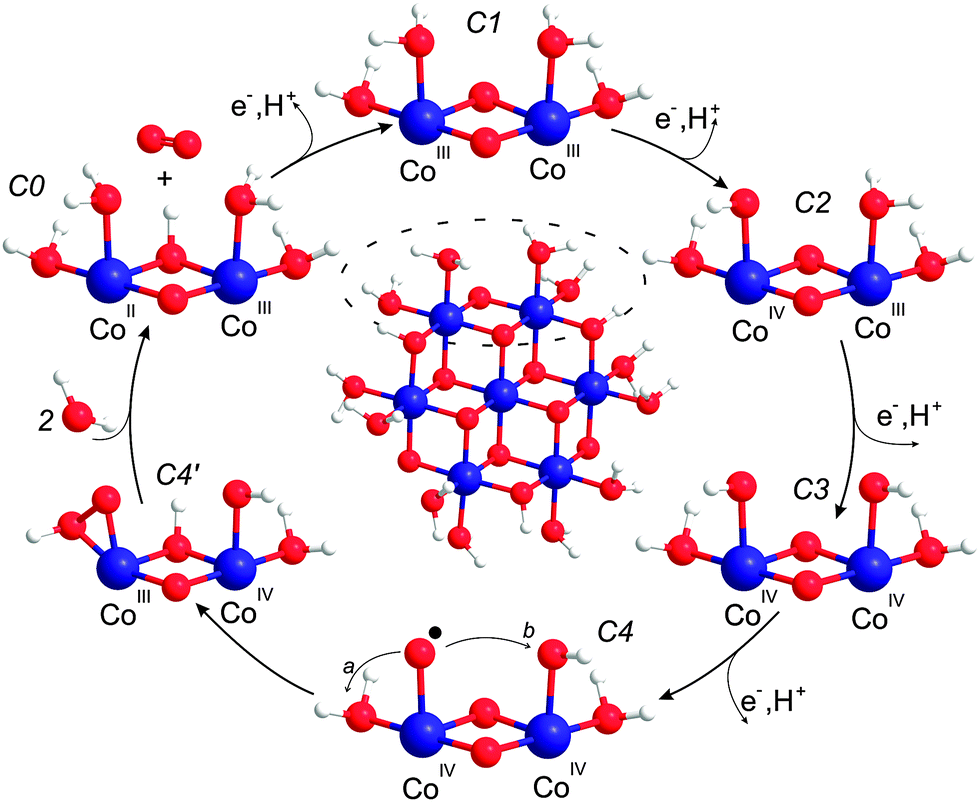 | ||
| Fig. 7 Mechanistic proposal. Center: structural model for the resting state of the Co-oxide fragments catalyzing water oxidation in Co/M2P-oxide NP. The outer circle shows the proposed reaction cycle for one catalytic site. C1 is the dominant ‘resting’ state, while C4 and C4′ are transient species during O–O bond formation. The mechanism builds on previous suggestions by Nocera and coworkers,3,30,56 Dau and coworkers54 and Guidoni and coworkers.59 Cobalt, blue; oxygen, red; hydrogen, white. | ||
In many published mechanistic proposals O–O bond formation is suggested to occur on the level of CoIVO, either by BWNA or by IMOC with another CoIVO or CoIII/IV–OH group.3,36,40,54,56,57,60 These suggestions are based on the findings that (i) CoIV formation is required before O–O bond formation can occur,36,55 (ii) that a reversible PCET step is rate limiting and occurs directly before O–O bond formation,56 and (iii) the assumption that the terminal ligands of the Co-oxide layer fragments are hydroxide groups. If, by contrast, one starts with a CoIII–OH2 in the resting state, also the CoIV–O˙ state becomes an option without violating restriction (ii).59 This idea is also in agreement with the finding by Gerken et al. that the formation of CoIV alone is insufficient for O2 formation, which commences at clearly higher potentials than the first CoIV formation.36 Indeed, both Siegbahn and coworkers58 and Guidoni and coworkers59 concluded in recent theoretical calculations that the formation of CoIV–O˙ is a prerequisite for O–O bond formation catalyzed by various small Co-oxides fragments. While Siegbahn used a cuboidal Co4O4 cluster (and some variations thereof with higher nuclearity) as structural model to propose nucleophilic attack of bulk water onto CoIV–O˙, Guidoni suggested, using a structural model similar to that displayed in Fig. 7 and one corner sharing Co6 cuboidal variant, that geminal coupling between CoIV–O˙ and a terminal OH group is the dominant pathway leading to O2 evolution.58,59 Siegbahn noted that the activation energy (31.6 kcal) for his mechanism is still significantly higher than the one he estimated from experimental data for CoPi oxide films (21.8 kcal).58 By contrast, the activation energy reported by Mattioli et al. for the intramolecular coupling pathway (16 kcal) is somewhat below that estimate.59
Our experiments showed that starting from the resting state the first O2 molecule is evolved after the removal of two or three electrons from the binuclear catalytic site. The CoIV–O˙ state can thus be reached from the resting C1 state by two sequential oxidation and deprotonation steps of one CoIII–OH2 group. However, we find it more probable that in between the adjacent CoIII–OH2 is oxidized and deprotonated to CoIV–OH. These steps are illustrated in Fig. 7 by structures C2 to C4. In state C4, O–O bond formation via IMOC then predominantly occurs either via germinal coupling of two terminal ligands (pathway a in Fig. 7, state C4) or via coupling with an OH group at the neighboring Co center (pathway b in Fig. 7, state C4). In contrast, we consider on the basis of our exchange data (Fig. 6) coupling between a terminal and a bridging ligand to be a clearly less frequent event.
Since our data do not allow deciding between pathways a and b, we give in Fig. 7 preference to the theoretically calculated geminal coupling pathway,59 in which the side-on hydroperoxo intermediate C4′ is formed under proton transfer from water to the μ-oxo bridge. This intermediate transfers two more electrons and a proton to the catalytic site. O2 then leaves the catalytic site while two water molecules from the bulk fill the empty coordination sites on cobalt. This leads to the formation of state C0, which is the lowest redox state in the reaction cycle, one electron reduced as compared to C1. The assignment of CoIICoIII in the C0 state is consistent with the observation of some CoII in the resting state of CoPi-oxide films, which may have fewer catalytic sites as compared to Co/M2P-oxides.3,36,54,56 The proposed reaction cycle is closed by the oxidation of CoII and the deprotonation of the μ-hydroxo bridge.
4. Conclusions
Our FTA approach resulted in unique information about the mechanism of water oxidation by Co/M2P-oxide NP. These include the number of Co atoms and the TOF per catalytic site, and the number of electrons that need to be removed from the resting state for the release of the first O2 molecule per site. Importantly, our results demonstrate that O2 formation from water occurs in Co/M2P-oxide NP almost exclusively via IMOC between two terminal ligands to Co. Our findings thus suggest that the mechanism by which the O–O bond is formed at the surface of Co3O4 (ref. 57) is different from that in amorphous bulk materials such as Co/M2P-oxide NP.Acknowledgements
The authors acknowledge discussions with Heinz Frei (Lawrence Berkeley National Laboratory) and Holger Dau (FU Berlin). This work was supported by the Strong Research Environment Solar Fuels (Umeå University), the Artificial Leaf Project Umeå (Knut & Alice Wallenberg foundation) and Energimyndigheten to JM, and by grants from the Swedish Energy Agency and the Knut & Alice Wallenberg Foundation to MFA, SS and AT. The authors acknowledge the facilities and technical assistance of the Umeå Core Facility for Electron Microscopy (UCEM), Umeå University, and the support by the COST Action CM1205 CARISMA (Catalytic Routines for Small Molecule Activation). XPS measurements and interpretation was done by Andrey Shchukarev. We thank Wai Ling Kwong for performing the XRD measurements and Anurag Kawde for the DLS experiment and the synthesis of additional M2P and [Ru(bpy)3]3+.References
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. deGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. G. Nocera, D. M. Tiede, H. Dau, W. Hillier, L. Z. Wang and R. Amal, Energy Environ. Sci., 2013, 6, 1074–1076 Search PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 CAS.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjö, S. Styring, V. Sundström and L. Hammarström, Acc. Chem. Res., 2009, 42, 1899–1909 CrossRef CAS PubMed.
- Y. Izumi, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef CAS.
- S. Rani, N. Z. Bao and S. C. Roy, Appl. Surf. Sci., 2014, 289, 203–208 CrossRef CAS.
- J. G. Yu, J. X. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- P. Zhou, J. G. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS PubMed.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed.
- C. R. Cox, J. Z. Lee, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14057–14061 CrossRef CAS PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- T. J. Jacobsson, V. Fjällström, M. Sahlberg, M. Edoff and T. Edvinsson, Energy Environ. Sci., 2013, 6, 3676–3683 CAS.
- J. H. Baxendale and C. F. Wells, J. Chem. Soc., Faraday Trans., 1957, 53, 800–812 RSC.
- M. Anbar and I. Pecht, J. Am. Chem. Soc., 1967, 89, 2553–2556 CrossRef CAS.
- V. Y. Shafirovich and V. V. Strelets, New J. Chem., 1978, 2, 199–201 CAS.
- V. Y. Shafirovich, N. K. Khannanov and V. V. Strelets, New J. Chem., 1980, 4, 81–84 CAS.
- G. L. Elizarova, L. G. Matvienko, N. V. Lozhkina, V. N. Parmon and K. I. Zamaraev, React. Kinet. Catal. Lett., 1981, 16, 191–194 CrossRef CAS.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841–1844 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018–1027 CAS.
- D. Shevchenko, M. F. Anderlund, A. Thapper and S. Styring, Energy Environ. Sci., 2011, 4, 1284–1287 CAS.
- G. P. Gardner, Y. B. Go, D. M. Robinson, P. F. Smith, J. Hadermann, A. Abakumov, M. Greenblatt and G. C. Dismukes, Angew. Chem., Int. Ed., 2012, 51, 1616–1619 CrossRef CAS PubMed.
- F. Song and X. L. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- X. H. Deng and H. Tüysüz, ACS Catal., 2014, 4, 3701–3714 CrossRef CAS.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- Y. Surendranath, M. Dincă and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 2615–2620 CrossRef CAS PubMed.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dincă, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- D. Lutterman, Y. Surendranath and D. Nocera, J. Am. Chem. Soc., 2009, 131, 3838–3839 CrossRef CAS PubMed.
- P. Du, O. Kokhan, K. W. Chapman, P. J. Chupas and D. M. Tiede, J. Am. Chem. Soc., 2012, 134, 11096–11099 CrossRef CAS PubMed.
- C. L. Farrow, D. K. Bediako, Y. Surendranath, D. G. Nocera and S. J. L. Billinge, J. Am. Chem. Soc., 2013, 135, 6403–6406 CrossRef CAS PubMed.
- M. Risch, V. Khare, I. Zaharieva, L. Gerencser, P. Chernev and H. Dau, J. Am. Chem. Soc., 2009, 131, 6936 CrossRef CAS PubMed.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- S. J. Harley, H. E. Mason, J. G. McAlpin, R. D. Britt and W. H. Casey, Chem. – Eur. J., 2012, 18, 10476–10479 CrossRef CAS PubMed.
- Y. Liu and D. G. Nocera, J. Phys. Chem. C, 2014, 118, 17060–17066 CAS.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795–2806 RSC.
- G. L. Elizarova, G. M. Zhidomirov and V. N. Parmon, Catal. Today, 2000, 58, 71–88 CrossRef CAS.
- R. Boggio, A. Carugati and S. Trasatti, J. Appl. Electrochem., 1987, 17, 828–840 CrossRef CAS.
- R. N. Singh, M. Hamdani, J. F. Koenig, G. Poillerat, J. L. Gautier and P. Chartier, J. Appl. Electrochem., 1990, 20, 442–446 CrossRef CAS.
- F. Švegl, B. Orel, M. G. Hutchins and K. Kalcher, J. Electrochem. Soc., 1996, 143, 1532–1539 CrossRef.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- D. Gonzales-Flores, I. Sanchez, I. Zaharieva, K. Klingan, J. Heidkamp, P. Chernev, P. W. Menezes, M. Driess, H. Dau and M. L. Montero, Angew. Chem., Int. Ed., 2015, 54, 2472 CrossRef PubMed.
- Y. V. Geletii, Q. S. Yin, Y. Hou, Z. Q. Huang, H. Y. Ma, J. Song, C. Besson, Z. Luo, R. Cao, K. P. O'Halloran, G. B. Zhu, C. C. Zhao, J. W. Vickers, Y. Ding, S. Mohebbi, A. E. Kuznetsov, D. G. Musaev, T. Q. Lian and C. L. Hill, Isr. J. Chem., 2011, 51, 238–246 CrossRef CAS.
- L. L. Duan, Y. H. Xu, P. Zhang, M. Wang and L. C. Sun, Inorg. Chem., 2010, 49, 209–215 CrossRef CAS PubMed.
- V. Y. Shafirovich, N. K. Khannanov and A. E. Shilov, J. Inorg. Biochem., 1981, 15, 113–129 CrossRef CAS.
- N. K. Khannanov, A. V. Khramov, A. P. Moravskii and V. Y. Shafirovich, Kinet. Catal., 1983, 24, 726–732 Search PubMed.
- P. K. Ghosh, B. S. Brunschwig, M. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1984, 106, 4772–4783 CrossRef CAS.
- H. J. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kogerler, P. F. Zhuk, J. Bacsa, G. B. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef CAS PubMed.
- M. Risch, D. Shevchenko, M. F. Anderlund, S. Styring, J. Heidkamp, K. M. Lange, A. Thapper and I. Zaharieva, Int. J. Hydrogen Energy, 2012, 37, 8878–8888 CrossRef CAS.
- H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2015, 137, 612–615 CrossRef CAS PubMed.
- M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Energy Environ. Sci., 2015, 8, 661–674 CAS.
- J. McAlpin, Y. Surendranath, M. Dincǎ, T. Stich, S. Stoian, W. Casey, D. Nocera and R. D. Britt, J. Am. Chem. Soc., 2010, 132, 6882–6883 CrossRef CAS PubMed.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- X. Li and P. E. M. Siegbahn, J. Am. Chem. Soc., 2013, 135, 13804–13813 CrossRef CAS PubMed.
- G. Mattioli, P. Giannozzi, B. A. Amore and L. Guidoni, J. Am. Chem. Soc., 2013, 135, 15353–15363 CrossRef CAS PubMed.
- L.-P. Wang and T. Van Voorhis, J. Phys. Chem. Lett., 2011, 2, 2200–2204 CrossRef CAS.
- K. Beckmann, H. Uchtenhagen, G. Berggren, M. F. Anderlund, A. Thapper, J. Messinger, S. Styring and P. Kurz, Energy Environ. Sci., 2008, 1, 668–676 CAS.
- D. Shevela and J. Messinger, Front. Plant Sci., 2013, 4, 473 Search PubMed.
- D. Shevela, S. Koroidov, M. M. Najafpour, J. Messinger and P. Kurz, Chem. – Eur. J., 2011, 17, 5415–5423 CrossRef CAS PubMed.
- L. Konermann, J. Messinger and W. Hillier, in Biophysical Techniques in Photosynthesis (Volume II), ed. T. J. Aartsma and J. Matysik, Springer, Dordrecht, 2008, vol. 26, pp. 167–190 Search PubMed.
- K. Beckmann, J. Messinger, M. R. Badger, T. Wydrzynski and W. Hillier, Photosynth. Res., 2009, 102, 511–522 CrossRef CAS PubMed.
- S. Mohamady and D. L. Jakeman, J. Org. Chem., 2005, 70, 10588–10591 CrossRef CAS PubMed.
- D. A. Berthold, G. T. Babcock and C. F. Yocum, FEBS Lett., 1981, 134, 231–234 CrossRef CAS.
- T. Kuwabara and N. Murata, Plant Cell Physiol., 1982, 23, 533–539 CAS.
- D. N. Silverman, Methods Enzymol., 1982, 87, 732–752 CAS.
- G. A. Mills and H. C. Urey, J. Am. Chem. Soc., 1940, 62, 1019–1026 CrossRef CAS.
- J. Messinger, M. Badger and T. Wydrzynski, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3209–3213 CrossRef CAS.
- N. Cox and J. Messinger, Biochim. Biophys. Acta, 2013, 1827, 1020–1030 CrossRef CAS PubMed.
- C. Ledesma, J. Yang, D. Chen and A. Holmen, ACS Catal., 2014, 4, 4527–4547 CrossRef CAS.
- A. M. Angeles-Boza, M. Z. Ertem, R. Sarma, C. H. Ibanez, S. Maji, A. Llobet, C. J. Cramer and J. P. Roth, Chem. Sci., 2014, 5, 1141–1152 RSC.
- R. D. Gerardi, N. W. Barnett and P. Jones, Anal. Chim. Acta, 1999, 388, 1–10 CrossRef CAS.
- A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247–2252 RSC.
- K. Klingan, F. Ringleb, I. Zaharieva, J. Heidkamp, P. Chernev, D. Gonzalez-Flores, M. Risch, A. Fischer and H. Dau, ChemSusChem, 2014, 7, 1301–1310 CrossRef CAS PubMed.
- D. T. Richens, Chem. Rev., 2005, 105, 1961–2002 CrossRef CAS PubMed.
- D. Lieb, A. Zahl, E. F. Wilson, C. Streb, L. C. Nye, K. Meyer and I. Ivanovic-Burmazovic, Inorg. Chem., 2011, 50, 9053–9058 CrossRef CAS PubMed.
- C. A. Ohlin, S. J. Harley, J. G. McAlpin, R. K. Hocking, B. Q. Mercado, R. L. Johnson, E. M. Villa, M. K. Fidler, M. M. Olmstead, L. Spiccia, R. D. Britt and W. H. Casey, Chem. – Eur. J., 2011, 17, 4408–4417 CrossRef CAS PubMed.
- N. E. Brasch, D. A. Buckingham, C. R. Clark and A. J. Rogers, Inorg. Chem., 1998, 37, 4865–4871 CrossRef CAS PubMed.
- B. Halle, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 1207–1224 CrossRef CAS PubMed.
- S. Romain, L. Vigara and A. Llobet, Acc. Chem. Res., 2009, 42, 1944–1953 CrossRef CAS PubMed.
- D. B. Hibbert, J. Chem. Soc., Chem. Commun., 1980, 202–203 RSC.
- D. B. Hibbert and C. R. Churchill, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 1965–1975 RSC.
- M. Wohlfahrt-Mehrens and J. Heitbaum, J. Electroanal. Chem. Interfacial Electrochem., 1987, 237, 251–260 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS, XRD and DLS measurements of precipitated Co/M2P particles; O2 production by [Ru(bpy)3]3+ in H218O; VIS absorption study of [Ru(bpy)3]3+ stability in phosphate buffer pH 7; additional FTA experiment; unrestricted FTA analysis. See DOI: 10.1039/c5ee00700c |
| ‡ Current address: PULSE Institute, SLAC National Accelerator Laboratory, Stanford University, Stanford, California 94305, USA. |
| This journal is © The Royal Society of Chemistry 2015 |