
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spatially-confined lithiation–delithiation in highly dense nanocomposite anodes towards advanced lithium-ion batteries†
Yinzhu
Jiang‡
*a,
Yong
Li‡
a,
Wenping
Sun
b,
Wei
Huang
a,
Jiabin
Liu
a,
Ben
Xu
c,
Chuanhong
Jin
a,
Tianyu
Ma
a,
Changzheng
Wu
*d and
Mi
Yan
*a
aState Key Laboratory of Silicon Materials, Key Laboratory of Advanced Materials and Applications for Batteries of Zhejiang Province, Key Laboratory of Novel Materials for Information Technology of Zhejiang Province, and School of Materials Science and Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, P. R. China. E-mail: yzjiang@zju.edu.cn; mse_yanmi@zju.edu.cn; Fax: +86-(0)-571-85038549; Fax: +86-(0)-571-87952730; Tel: +86-(0)-571-85038549 Tel: +86-(0)-571-87952730
bSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore
cLaboratory of Smart Materials and Surface, Mechanical Engineering, Faculty of Engineering and Environment, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK
dHefei National Laboratory for Physical Sciences at Microscale & Collaborative Innovation Center of Chemistry for Energy Materials, University of Science & Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: czwu@ustc.edu.cn; Fax: +86-(0)-551-63603134; Tel: +86-(0)-551-63603134
First published on 23rd March 2015
Abstract
Spatially-confined electrochemical reactions are firstly realized in a highly dense nanocomposite anode for high performance lithium ion batteries. The spatially-confined lithiation–delithiation effectively avoids inter-cluster migration and perfectly retains full structural integrity. Large reversible capacity, high rate capability and superior cycling stability are achieved simultaneously. This spatially-confined lithiation–delithiation offers novel insight to enhance cycling performance of high capacity anode materials.
Broader contextHigh capacity anode materials have been wandering around the corner of commercialization for advanced lithium ion batteries. Nanostructuring these electrochemically active materials through reserving large void spaces has been extensively utilized to extend the cycling stability in terms of the volume change view. However, the ultra-low volumetric capacity associated with the loosely-packed nano-sized materials is a critical disadvantage for the practical applications of high capacity materials. Here in a highly dense nanocomposite anode, we successfully eliminate the cycling failure of high capacity anodes by suppressing atom migration during lithiation–delithiation, demonstrating a novel approach of spatially-confined electrochemical reactions through which the atoms/clusters can be rapidly lithiated–delithiated at their original sites. The spatially-confined lithiation–delithiation effectively avoids inter-cluster migration and perfectly retains full structural integrity during prolonged cycling. Impressively, a volumetric capacity as high as 6034.5 mA h cm−3 (1206.9 mA h g−1, 86.4% of the first discharge capacity) can still be maintained after 200 stable cycles and 4704.0 mA h cm−3 (940.8 mA h g−1) is retained at an ultra-high current density of 20 A g−1. This spatially-confined lithiation–delithiation offers novel insight to enhance the cycling performance of high capacity anode materials. The successful demonstration of dense anodes may shed light on the practical applications of high capacity materials for the upgradation of lithium ion batteries. |
The coming decades will see an explosive demand for rechargeable lithium ion batteries (LIBs) with higher energy and stronger power, which greatly extends enormous usages in various electric vehicles (xEVs) and energy storage systems (ESSs).1 High capacity anode materials, classified by alloying or conversion reactions, are under intense research for potential upgradation of the state-of-the-art graphitic anode.2,3 Despite high gravimetric/volumetric capacity of these novel materials in bulk, they suffer from lithium-induced drastic volume change and micro-scale sluggish kinetics, resulting in serious capacity fading and poor rate capability (Fig. 1a).4 To address the huge volume change during lithiation–delithiation processes, there is a general strategy of reserving a large amount of void space by fabricating porous/nanostructured anode materials, including nanospheres,5 nanowires,6–8 yolk–shell nanoarchitectures,9–11 and nanosheets.12,13 They have been demonstrated to accommodate the volume change and shorten the diffusion length, which bring about longer cycling life and higher rate capability compared with bulk counterparts (Fig. 1a). Unsatisfactorily, the practical application of such nanoscale powder materials has been generally hindered by their relatively low packing density (packing factor: 0.2–0.3 or less),14,15 which severely lowers the volumetric capacity of electrodes (Table S1, ESI†) and has actually been long neglected in the research of LIBs.11
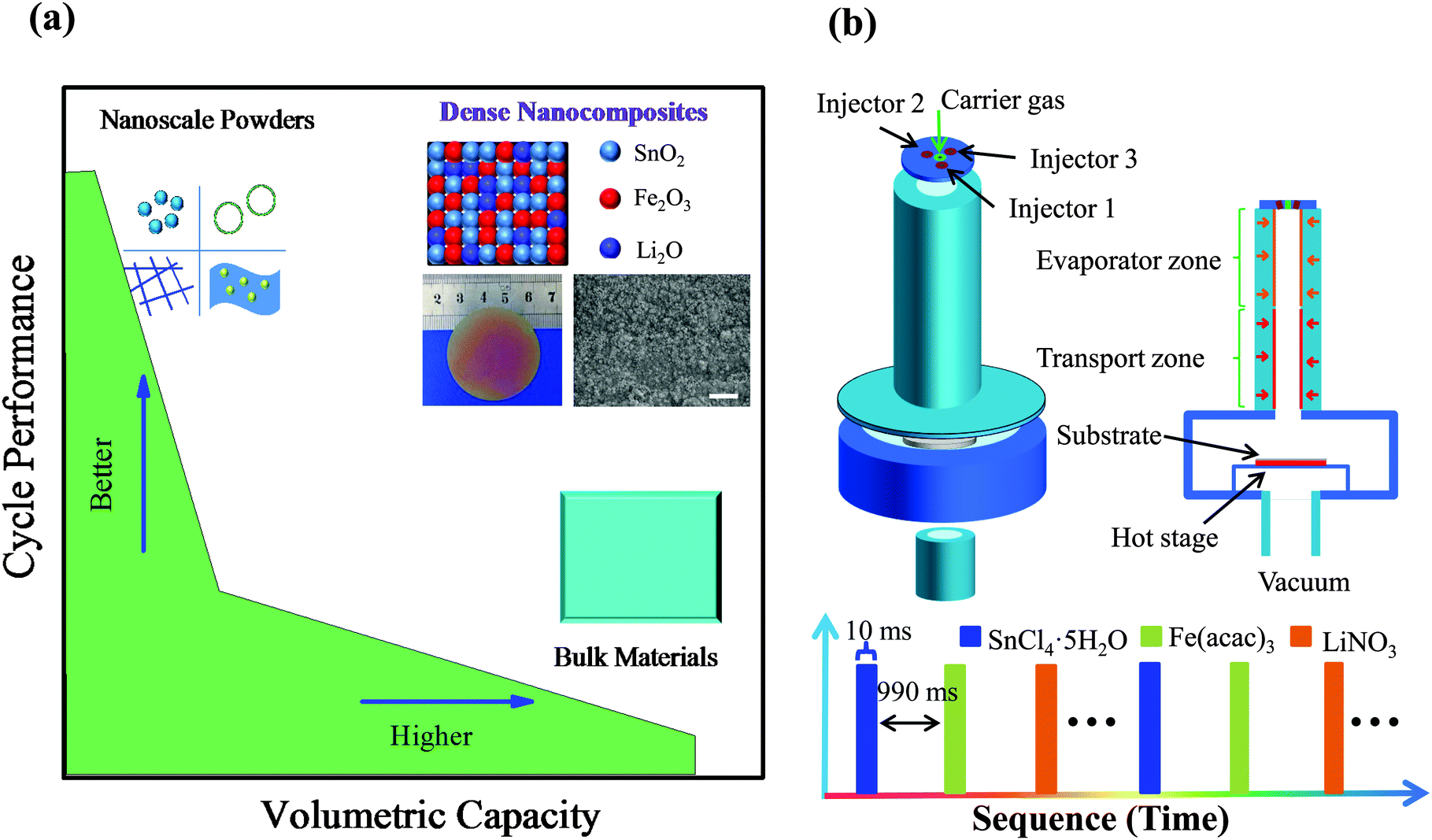 | ||
| Fig. 1 (a) Comparison of the volumetric capacity and cycle performance among the present dense nanocomposites and other materials. Insets (from left to right, top to down): illustration of the various nanoscale powders with much reserved void space, the schematic spatial distribution of dense SnO2–Fe2O3–Li2O nanocomposites, photograph of the nanocomposite film (uniform over a large area), SEM image of the deposited film (consisting of closely-packed nanoparticles, scale bar: 400 nm) and illustration of bulk materials. (b) Illustration of the deposition system (top) and the deposition sequence (time per pulse) used for the growth of dense nanocomposite films (down). | ||
Furthermore, if we deeply investigate the morphology evolution after long cycling in the reported literature, we will find that the electrode morphology after the cycling test greatly changed, by showing denser structure/particle aggregation instead of pristine porous/nanoscale structures.15–18 The study on phase evolution of NiO nanosheet electrodes clearly showed that the heterogeneous phase conversion prevails in charge reactions, which is likely caused by preferential nucleation at grain boundaries and inevitably results in atom migration from the interior of the grain.19 The de-alloying study of Li–Sn alloys also demonstrated the significant solid-state diffusive transport, leading to the remarkable morphology evolution.20 Fundamentally speaking, there is enormous spontaneous atom migration during lithiation–delithiation processes; in other words, the delithiation reaction sites are different from the lithiation ones, resulting in atom migration and hence electrode structure disintegration. In terms of structural integrity, atom migration during lithiation–delithiation processes might be the fundamental cause for the cycling failures of high capacity anodes.
Herein, from the point of atom migration and structural integrity view, we propose a novel approach of spatially-confined electrochemical reactions to enhance the cycling stability. During lithiation–delithiation processes, the atoms/clusters are largely confined at their original sites with little migration and therefore the electrode structural integrity is well retained, which is critical for the prolonged cycling behavior. A highly dense nanocomposite anode, composed of multi-oxide nanoclusters uniformly and alternately, is utilized to realize the proposed spatially-confined electrochemical reactions. Our designed anode architecture can undergo rapid lithiation–delithiation through local stepwise electrochemical reactions, leading to little migration of inter-nanoclusters and facilitated lithium kinetics. Based on the close-to-theoretical density, high volumetric capacity and superior electrochemical performance are achieved simultaneously for the first time (Fig. 1a). The dense SnO2–Fe2O3–Li2O nanocomposite anode exhibits an initial volumetric discharge capacity of 6984.9 mA h cm−3 (gravimetric capacity of 1396.8 mA h g−1), and maintains 6034.5 mA h cm−3 (1206.9 mA h g−1, 86.4% of the first discharge capacity) after 200 cycles, which has been the highest volumetric capacity value reported so far. Equally impressively, as high as 4704.0 mA h cm−3 (940.8 mA h g−1) is retained even when cycling at an ultra-large current density of 20 A g−1.
A novel strategy of pulsed spray evaporation chemical vapor deposition (PSE-CVD) was applied to fabricate such a highly dense nanocomposite anode of uniformly and alternately distributed multi-oxide nanoclusters, which is schematically presented in Fig. 1b.21,22 For the dense SnO2–Fe2O3–Li2O films, three different precursors dissolved in ethanol were injected as a fine spray into the reactor alternately with a spacing interval in between (see the Experimental section in the ESI†). The as-deposited nanocomposite film is uniform over a large area (Φ > 4 cm) and highly dense as evidenced in the scanning electron microscopy (SEM) image (Fig. 1a, insets). Furthermore, pure SnO2 films were also deposited via a similar process for comparison.
Fig. S1 (ESI†) shows that both pure SnO2 and SnO2–Fe2O3–Li2O films are composed of closely packed nanoparticles, resulting in high densities of ∼6.1 g cm−3 for pure SnO2 and ∼5.0 g cm−3 for SnO2–Fe2O3–Li2O. It is worth noting that such close-to-theoretical densities are crucial for acquiring high volumetric capacity of electrodes. X-ray photoelectron spectroscopy (XPS) investigation demonstrates that three components in the film are in the form of SnO2, Fe2O3 and Li2O (Fig. S2, ESI†),23–25 the weight ratio of which is around 59.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :36.6:4.4 based on inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis. X-ray diffraction (XRD) patterns (Fig. S3, ESI†) show that both films are X-ray amorphous, indicating the amorphous/nanocrystalline nature of as-deposited films. Transmission electron microscope (TEM) images and selected area electron diffraction (SAED) patterns (Fig. S4, ESI†) clearly demonstrate the densely packed morphology and amorphous/nanocrystalline structure of the SnO2–Fe2O3–Li2O film. In a further step, an advanced spherical aberration-corrected scanning transmission electron microscope (STEM) was used to identify the refined morphology, microstructure and elemental distribution. The high-angle annular dark field (HAADF) and energy dispersive X-ray spectroscope (EDS) mapping images shown in Fig. 2a clearly demonstrate that both Sn and Fe species are homogeneously distributed in a staggered manner over nanometer sized areas, which indicates that SnO2 and Fe2O3 are effectively separated from each other without any aggregation. Although light element Li cannot be detected by the EDS mapping, we can still infer that the distribution of Li is also homogeneous according to the overlay mapping images of Sn and O, and Fe and O (Fig. S5, ESI†). Additionally, the spatial distribution of these three components was further identified in detail by high resolution TEM (HRTEM) as shown in Fig. 2b–d, where three characteristic areas were chosen for investigations. Interestingly, Fig. 2b demonstrates the existence of SnO2–Fe2O3 nano heterostructures, where the lattice spacings of 0.33 nm and 0.27 nm are in good agreement with the (110) planes of SnO2 and the (104) planes of α-Fe2O3, respectively.26 A similar heterostructure has also been reported previously for other SnO2–Fe2O3 composites.26,27 Another typical feature is observed in Fig. 2c in which a 10 nm particle consists of several 2–5 nm continuous nanoclusters. Supported by the fast Fourier transformation (FFT) patterns, it can be inferred that these nanoclusters belong to different phases with a d-spacing of 0.333 nm corresponding to the (110) planes of SnO2 and of 0.273 nm to the (104) planes of α-Fe2O3.27 Similarly, at the edge of the selected area shown in Fig. 2d, a d-spacing of 0.264 nm can be assigned to the (101) plane of SnO2 and of 0.253 nm to the (110) plane of α-Fe2O3.26,27 However, no lattice fringes of Li2O can be found probably due to the noncrystalline nature of low temperature deposition.18,28 All of the above analyses reveal advantageous features that the 2–10 nm nanoclusters of SnO2, Fe2O3, and Li2O distribute alternately with special interfacial relation among them (e.g. coherent boundary), which exhibits a uniform and dense structure of anodes. Fig. S6 (ESI†) shows TEM images of SnO2 films for comparison. Similarly, the SAED pattern (diffuse halos) implies the amorphous nature of pure SnO2 films (Fig. S6a and b, ESI†). HRTEM images (Fig. S6c and d, ESI†) illustrate that ultra-small nanoparticles (∼5 nm) are densely packed, which is consistent with the SEM image (Fig. S1, ESI†). Corresponding to the SAED result, the deposited film is largely disordered with a small part of regions which show poor crystallinity (Fig. S6c and d, ESI†).
:36.6:4.4 based on inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis. X-ray diffraction (XRD) patterns (Fig. S3, ESI†) show that both films are X-ray amorphous, indicating the amorphous/nanocrystalline nature of as-deposited films. Transmission electron microscope (TEM) images and selected area electron diffraction (SAED) patterns (Fig. S4, ESI†) clearly demonstrate the densely packed morphology and amorphous/nanocrystalline structure of the SnO2–Fe2O3–Li2O film. In a further step, an advanced spherical aberration-corrected scanning transmission electron microscope (STEM) was used to identify the refined morphology, microstructure and elemental distribution. The high-angle annular dark field (HAADF) and energy dispersive X-ray spectroscope (EDS) mapping images shown in Fig. 2a clearly demonstrate that both Sn and Fe species are homogeneously distributed in a staggered manner over nanometer sized areas, which indicates that SnO2 and Fe2O3 are effectively separated from each other without any aggregation. Although light element Li cannot be detected by the EDS mapping, we can still infer that the distribution of Li is also homogeneous according to the overlay mapping images of Sn and O, and Fe and O (Fig. S5, ESI†). Additionally, the spatial distribution of these three components was further identified in detail by high resolution TEM (HRTEM) as shown in Fig. 2b–d, where three characteristic areas were chosen for investigations. Interestingly, Fig. 2b demonstrates the existence of SnO2–Fe2O3 nano heterostructures, where the lattice spacings of 0.33 nm and 0.27 nm are in good agreement with the (110) planes of SnO2 and the (104) planes of α-Fe2O3, respectively.26 A similar heterostructure has also been reported previously for other SnO2–Fe2O3 composites.26,27 Another typical feature is observed in Fig. 2c in which a 10 nm particle consists of several 2–5 nm continuous nanoclusters. Supported by the fast Fourier transformation (FFT) patterns, it can be inferred that these nanoclusters belong to different phases with a d-spacing of 0.333 nm corresponding to the (110) planes of SnO2 and of 0.273 nm to the (104) planes of α-Fe2O3.27 Similarly, at the edge of the selected area shown in Fig. 2d, a d-spacing of 0.264 nm can be assigned to the (101) plane of SnO2 and of 0.253 nm to the (110) plane of α-Fe2O3.26,27 However, no lattice fringes of Li2O can be found probably due to the noncrystalline nature of low temperature deposition.18,28 All of the above analyses reveal advantageous features that the 2–10 nm nanoclusters of SnO2, Fe2O3, and Li2O distribute alternately with special interfacial relation among them (e.g. coherent boundary), which exhibits a uniform and dense structure of anodes. Fig. S6 (ESI†) shows TEM images of SnO2 films for comparison. Similarly, the SAED pattern (diffuse halos) implies the amorphous nature of pure SnO2 films (Fig. S6a and b, ESI†). HRTEM images (Fig. S6c and d, ESI†) illustrate that ultra-small nanoparticles (∼5 nm) are densely packed, which is consistent with the SEM image (Fig. S1, ESI†). Corresponding to the SAED result, the deposited film is largely disordered with a small part of regions which show poor crystallinity (Fig. S6c and d, ESI†).
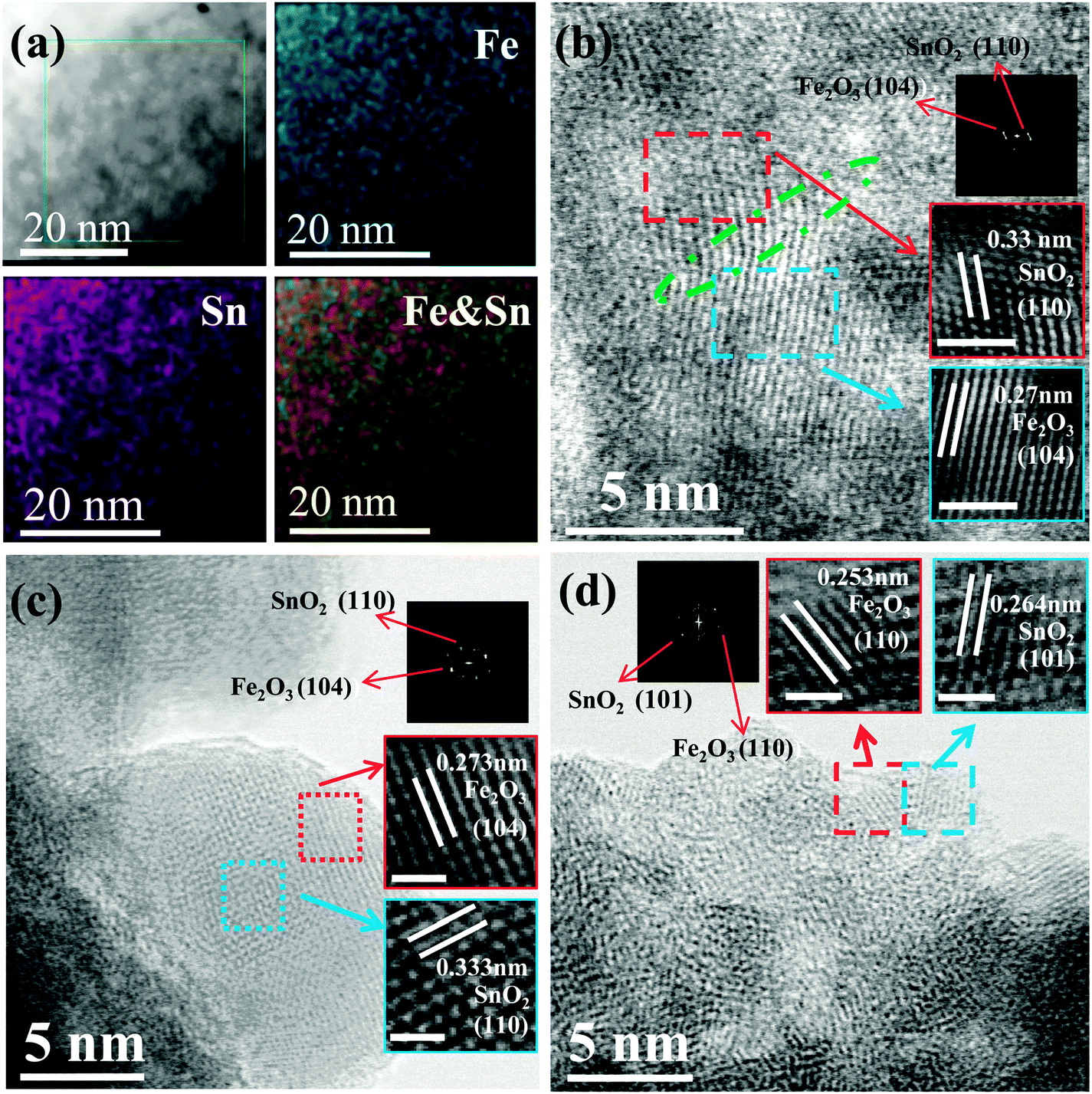 | ||
| Fig. 2 (a) Elemental mapping images of SnO2–Fe2O3–Li2O nanocomposites. (b–d) Characteristic HR-TEM and FFT, IFFT (inset) images of SnO2–Fe2O3–Li2O: (b) the special nanoheterostructure between SnO2 and α-Fe2O3; (c) a typical 10 nm scale nanoparticle consisting of many smaller (e.g. 2–5 nm) grains, some of which are identified as SnO2 and α-Fe2O3, distributed in a staggered manner; (d) demonstration of two ultra-small (2 nm scale) grains locate closely, which are proved to be SnO2 and α-Fe2O3, respectively. | ||
The as-deposited dense films, as working electrodes directly without using conductive carbon and a binder, were subsequently assembled into 2025-type coin cells. Cyclic voltammetry (CV, Fig. 3a) was firstly performed to characterize the electrochemical properties of the dense SnO2–Fe2O3–Li2O electrode in the voltage range of 0.005–3 V (versus Li/Li+) at a scan rate of 0.10 mV s−1. During the initial sweeping at 5 mV, four well-defined reduction peaks are observed at 1.21 V, 0.78 V, 0.49 V and 0.15 V, respectively. The peak at 1.21 V can be attributed to the reduction of SnO2 to metal Sn and the intercalation of lithium into Fe2O3 (Li2Fe2O3).8,29 The second peak at 0.78 V corresponds to the further reduction of Li2Fe2O3 to metal Fe and the formation of a solid electrolyte interphase (SEI) layer.8,25 The other two peaks are associated with Li–Sn alloying.29 When sweeping back to 3 V, the peak centered at 0.48 V appears due to the dealloying of LixSn, while the oxidation peak at 1.28 V is most likely due to the reversible oxidation from Sn to SnO.30 The strong wide peak at around 1.82 V can be ascribed to the further oxidation of SnO and Fe0.25,29 Additionally, the CV peaks reappear well during the subsequent cycles, indicating good reversibility of the electrochemical reactions. The CV curves clearly show that SnO2 and Fe2O3 are electrochemically reacted at different potentials; in other words, when one metal oxide anode is electrochemically engaged, the surrounding one is inactive and acts as a buffering matrix.31,32 In accordance with the multi-peaks observed during CV scans, the voltage profiles of the SnO2–Fe2O3–Li2O electrode present sectional sloping lines during both charge and discharge processes, as displayed in Fig. 3b. The discharge and charge capacity for the first cycle is 1396.8 and 1146.8 mA h g−1, respectively, representing an ultra-high Coulombic efficiency (CE) of around 82.1%. In comparison, only 64.4% (CE) inserted lithium ions are extracted reversibly for the pure SnO2 electrode (Fig. S7, ESI†). Such multi-oxide configuration and nanocomposite nature of the SnO2–Fe2O3–Li2O electrode can effectively facilitate the reversible electrochemical reactions and reduce the lithium consumption in the irreversible formation of the SEI layer.18 More impressively, benefited from the close-to-theoretical density of the SnO2–Fe2O3–Li2O electrode, it exhibits an initial volumetric discharge and charge capacity of 6984.9 and 5734.6 mA h cm−3.
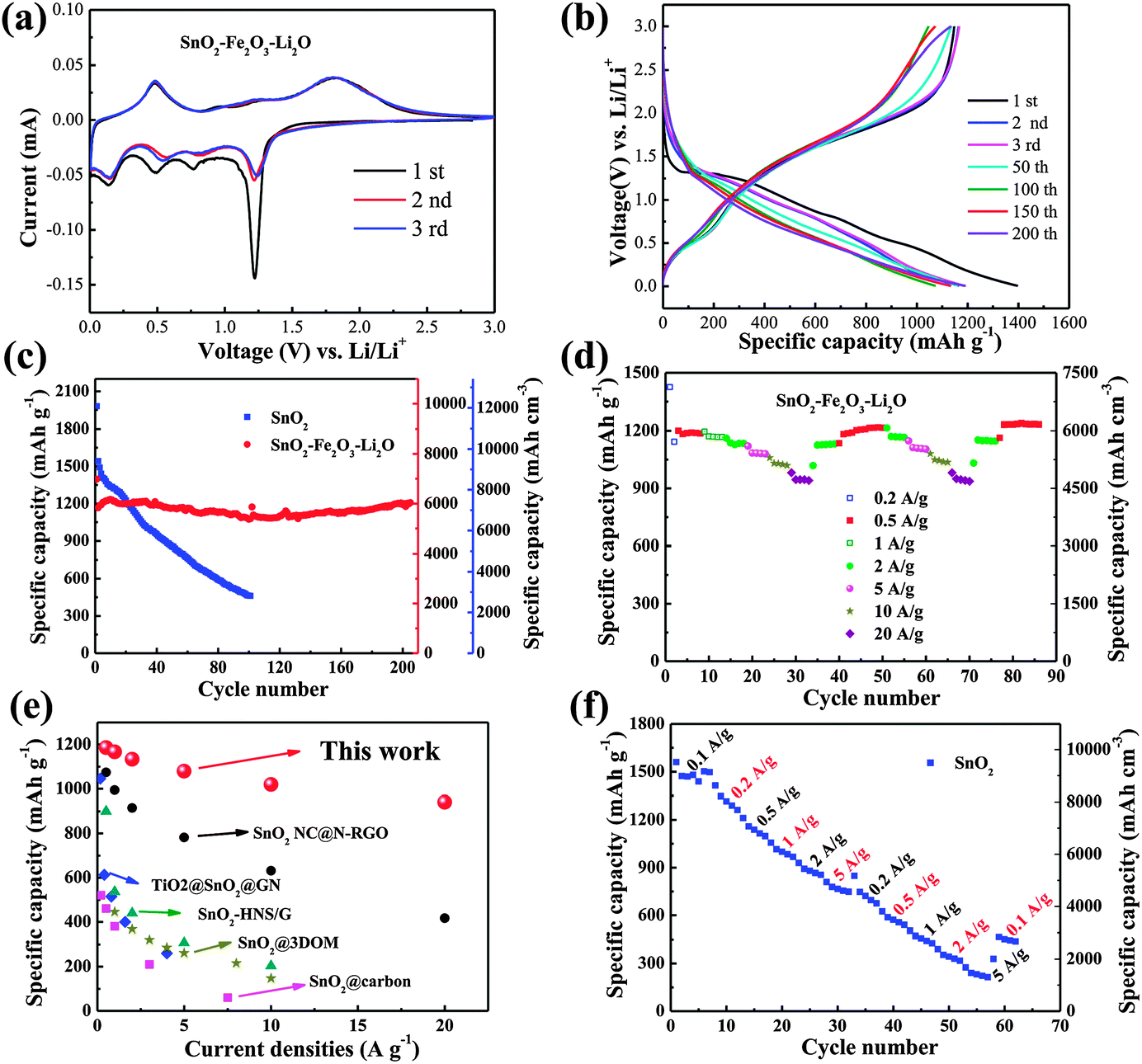 | ||
| Fig. 3 (a) CV curves of the SnO2–Fe2O3–Li2O. (b) Galvanostatic discharge–charge profiles of SnO2–Fe2O3–Li2O at a constant current density of 200 mA g−1. (c) Capacities vs. cycle number of SnO2 and SnO2–Fe2O3–Li2O at 0.2 A g−1. (d) Cycling performance at various current rates of SnO2–Fe2O3–Li2O (0.2–20 A g−1). Impressively, the capacity at 20 A g−1 can still be maintained at 940.8 mA h g−1, which is as high as 82.4% of the capacity at 0.2 A g−1. (e) Comparison of the rate capability among the reported materials (SnO2 NC@N-RGO,33 TiO2@SnO2@GN,44 SnO2-HNS/G,54 SnO2@carbon,51 SnO2@3DOM55) and SnO2–Fe2O3–Li2O in this work. (f) Rate capability of pure SnO2. | ||
Fig. 3c presents the cycling performance of the two electrodes cycled at 200 mA g−1 in the range of 0.005–3 V versus Li/Li+. In order to make the measurements more accurate, a bare steel substrate was also tested as shown in Fig. S8 (ESI†), the capacity of which is quite low and ignorable. After 200 stable cycles, as high as 6034.5 mA h cm−3 (1206.9 mA h g−1) representing 86.4% capacity retention is sustained for the SnO2–Fe2O3–Li2O nanocomposite electrode. To the best of our knowledge, the present SnO2–Fe2O3–Li2O anode exhibits the highest reversible volumetric capacity, nearly sixteen times of the commercial graphite anodes, among the reported LIB anodes as shown in Table S1 (ESI†). In contrast, the pure SnO2 anode suffers from rapid capacity decay, from the ultrahigh initial capacity of 12169.5 mA h cm−3 (1995 mA h g−1) to only 2814.5 mA h cm−3 (461.4 mA h g−1) in the 100th cycle. The electrochemical performances of Fe2O3, SnO2–Fe2O3 and SnO2–Li2O electrodes are demonstrated in Fig. S9 (ESI†). Similar to pure SnO2, pure Fe2O3 also suffers from poor cycling stability due to large volume change and unavoidable aggregation (Fig. S9a, ESI†). The SnO2–Fe2O3 composite demonstrates improved cycling stability with high reversible capacity compared to bare SnO2 or Fe2O3 (Fig. S9b, ESI†). Nevertheless, as huge volume change occurs in both SnO2 and Fe2O3 anodes during charge–discharge processes, the improvement of cycling stability in the SnO2–Fe2O3 electrode is still limited. Although there is a great improvement in cycling stability in the reported SnO2–graphene and SnO2–carbon composite electrodes, the incorporation of Li2O is compulsory as both the Li-ion diffusion pathway and the buffering matrix in such a dense composite system.33,34
Besides the stirring volumetric capacity and cycling behaviors, the SnO2–Fe2O3–Li2O anode also possesses outstanding rate capability, as shown in Fig. 3d. At high current densities of 5, 10 and even 20 A g−1, the SnO2–Fe2O3–Li2O anode can retain high reversible capacities of 5398.5, 5097.0 and 4704.0 mA h cm−3 (1079.7, 1019.4 and 940.8 mA h g−1), respectively. All of these values are much higher than the theoretical capacity of the commonly used graphite anode material (837 mA h cm−3, or 372 mA h g−1). Moreover, when the SnO2–Fe2O3–Li2O composite electrodes were tested at high current densities directly, large capacities of 5754.0 mA h cm−3 (1150.8 mA h g−1) at 0.5 A g−1 after 100 cycles, 4791.5 mA h cm−3 (958.3 mA h g−1) at 1 A g−1 after 100 cycles, 4971.5 mA h cm−3 (994.3 mA h g−1) at 5 A g−1 after 50 cycles and 4530.0 mA h cm−3 (906.0 mA h g−1) at 10 A g−1 after 50 cycles can still be maintained (Fig. S10, ESI†). Such a remarkable high-rate performance has never been observed before for carbon-free SnO2-based anode materials, which is even much better than that of SnO2–CNT composite or SnO2–graphene composite electrodes (Fig. 3e).13,35–44 Contrarily, for the pure SnO2 anode, the capacity drops rapidly upon increasing the current density, dropping to 4559.8 mA h cm−3 (747.5 mA h g−1) at 5 A g−1, which is only about 47.9% of the capacity at 0.1 A g−1 (Fig. 3f). Moreover, the discharge capacity of the pure SnO2 anode could not recover when the current density was reduced from 5 to 0.2 A g−1. It is worthy of note that the present SnO2–Fe2O3–Li2O (59.0:36.6:4.4 of weight ratio) demonstrates the most satisfying results in view of the cycling stability and specific capacity. Upon increasing the content of SnO2, the SnO2–Fe2O3–Li2O (70.0:25.6:4.4) electrode shows worse cycling stability though the capacity improved (Fig. S11a, ESI†). However, the specific capacity decreases to only 1000.0 mA h g−1 when the content of Li2O is increased (the weight ratio of SnO2, Fe2O3 and Li2O is 50.2:31.1:18.7), even though better cycling stability can be maintained (Fig. S11b, ESI†).
The electrode micro-morphology was further characterized to understand the difference of local mechanical behaviors between dense nanocomposites and pure SnO2, after cyclic lithiation–delithiation processes. The SEM observations present a state of aggregated nanoparticles with surface cracks after 100 cycles for the pure SnO2 film (Fig. 4a and b), whilst an improved rate capability and good cycling performance are found for SnO2–Fe2O3–Li2O without any notable surface fractures after 200 cycles (Fig. 4c and d). For the pure SnO2 electrode, a schematic diagram in Fig. 4e illustrates the generation and evolution of fractures. Sn particles firstly nucleate in the Li2O matrix during the lithiation process. When the lithiation process continues, the LixSn alloy is formed followed by the coarsening of LixSn and depletion of Sn, resulting in a wider size distribution of LixSn particles.45 As for the delithiation, the extraction of lithium occurs in energetically-favored sites (e.g. grain boundaries, surfaces) instead of the previous Sn/SnO2 sites, which leads to the microscopic transfer of Sn atoms in the whole electrode. Such a so-called ex situ delithiation process causes the aggregation of active materials and the disintegration of electrodes.46 The particle size of SnO2 increases during the cyclic lithiation–delithiation processes, due to the aggregation via viscous flow or diffusive bonding between SnO2 particles. Larger particle size increases the Li diffusion length and leads to higher diffusion-induced stress, therefore, the crack initializes easier. Furthermore, voids start to form during the initial delithiation process because of the large volumetric shrinkage and stresses generate in the big LixSn particles as a result of the nonsynchronous reactions between the interface and the center.19,47 After certain cycles, with a further increase in the number and size of the voids and continuous generation of stresses, pulverization and cracks occur inevitably, resulting in low capacity and poor stability.
 | ||
| Fig. 4 SEM images of samples after the battery test: (a), (b) SnO2 and (c), (d) SnO2–Fe2O3–Li2O. Schematic illustration of the lithiation–delithiation process: (e) SnO2 and (f) SnO2–Fe2O3–Li2O. (g) Elemental mapping images of SnO2–Fe2O3–Li2O after 200 cycles, which clearly demonstrates that Sn and Fe species are still homogeneously distributed in a staggered manner and similar to the pristine film. | ||
For the SnO2–Fe2O3–Li2O nanocomposite electrode as illustrated in Fig. 4f, on one hand, nanoclusters ranging from 2–10 nm (Fig. 2) can greatly reduce the diffusion length for Li-ions and accelerate the local lithiation–delithiation reaction even at high current densities.48 Meantime, highly uniformly distributed nanoclusters could effectively relieve the stress generated in lithium insertion–extraction processes, keeping the electrode structure stable under long cycling.4 Recent study on silicon alloy anodes suggested a critical size (∼10 nm) for ensuring a stable structure for the electrode exhibiting the optimal electrochemical performance.49 On the other hand, a stepwise electro-chemical reaction could be induced by the different reactive potential of SnO2 and Fe2O3 as indicated in CV scans.27 With the alternatively ‘Chessboard’ staggered state of multi-oxide nanoclusters as shown in Fig. 1a, the nano-sized periodic lithiation–delithiation zone could be formed as the electrochemically engaged anode nanoclusters are surrounded by the temporarily inactivated ones, where they act as buffer matrices or isolators to prevent the direct chemical-contact between the active nanoclusters.31,32 Most importantly, the dense microstructures offer boundaries to limit the diffusion freedom of active atoms/clusters, leading to spatially-confined lithiation–delithiation reactions and full electrode integrity, thus improving the macroscopic structure stability by preventing the formation of voids. The elemental mapping (Fig. 4g) images (20 nm scale) after completing 200 cycles clearly verify that Sn and Fe species are still homogeneously distributed in a staggered manner and similar to the pristine film, which positively confirms the spatially-confined charge–discharge reactions during the long cycling process. Finally, it was reported that the maximum of the diffusion induced stress decreases with the increase of δ (shell thickness characterizes the degree of the buffering) when using a core–shell model for the diffusion-induced stress.50 Here, we noted that Li2O in the electrode facilitates spatially-confined electrochemical reactions between active nanoclusters and Li2O, enhancing the penetration of the Li-ion electrolyte into a dense electrode. It allows a shortened Li diffusion path which further reduces the diffusion-induced stress. This also results in the existence of residual Li concentration, eventually benefiting a decrease in the maximum diffusion-induced stress which is determined by the local Li concentration.51 The homogeneously distributed oxide nanoclusters induce a spatially-confined lithiation–delithiation state, benefit the release of the stored elastic energy and retard crack nucleation in the composite electrode.
In addition, although there is no conductive medium inside the nanocomposite films, the generated Fe metal (Fig. S12a, ESI†) during the conversion reaction of Fe2O3 at low voltages could provide a fast pathway for electron transport.19,52,53 Such an enhancement in conductivity contributes to the reversibility of Sn to SnO2 in majority (Fig. S12b, ESI†), which could improve the reversible capacity to some extent.18 Overall, benefited by the unique design of the nanostructure, the dense SnO2–Fe2O3–Li2O nanocomposite electrode can charge–discharge under space confinement with little migration and aggregation, which maintain sub 10 nm scale nanostructures, resulting in improved cycling stability and a faster charge–discharge rate in the current state of close-packed nanoparticles. Based on the above features, the dense SnO2–Fe2O3–Li2O nanocomposite can simultaneously achieve large volumetric capacity and high cycling stability (Fig. 5).
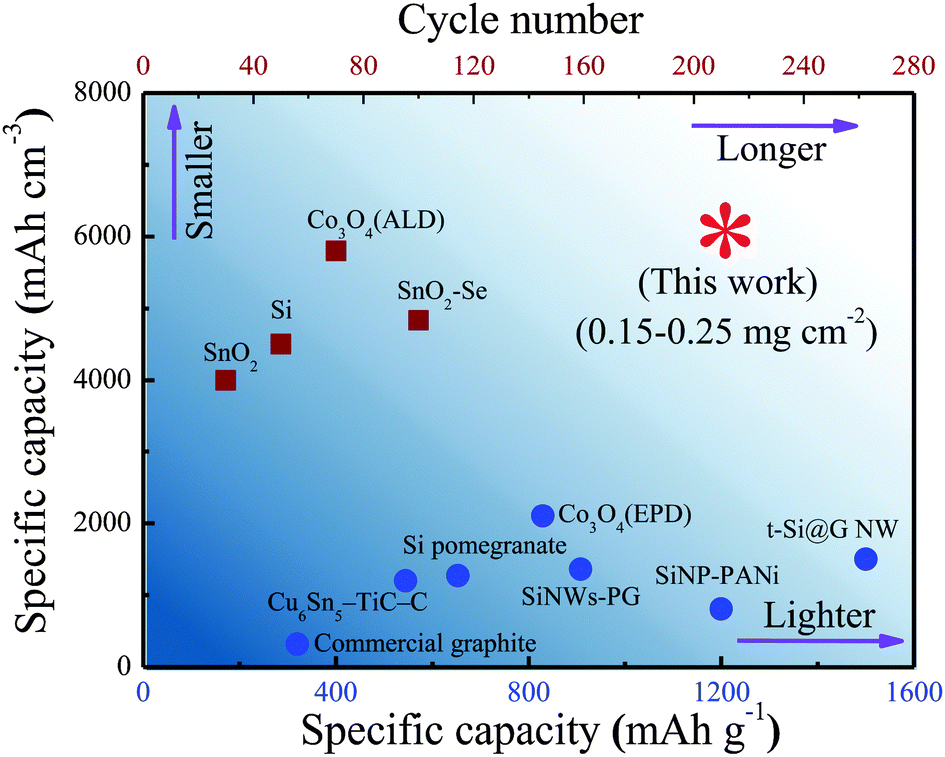 | ||
| Fig. 5 Performance comparison of various materials. Our dense nanocomposite anode shows the highest volumetric capacity with longer cycle life compared with other materials, such as commercial graphite, Cu6Sn5–TiC–C,56 SiNP-PANi (including the mass of PANi),57 Si pomegranate,15 SiNW-PG,17 Co3O4 (EPD),58 t-Si@G NW,59 SnO2,60 Si,61 Co3O4 (ALD)62 and SnO2–Se.63 | ||
To better understand the spatially-confined lithiation–delithiation mechanism, in situ TEM characterization was performed to directly observe the relationship between the electrochemical performance and microstructure of the active materials. The in situ TEM setup is based on previous studies and a half-cell LIB was constructed inside TEM using the SnO2–Fe2O3–Li2O nanocomposite as the working electrode and Li metal as the counter electrode.64 The vivid displays of lithiation–delithiation processes are presented in Movies S1–S6 (ESI†). TEM images at different charge–discharge stages were captured from these videos, as shown in Fig. 6 and Fig. S13 (ESI†). The time (such as 0 s, 5 s and 10 s) indicates the degree (state) of lithiation–delithiation. Fig. 6a demonstrates the TEM images of the time series of the lithiation process. Obvious volume expansion occurred and full lithiation can be completed in a short time of 10 s. As marked by the red dashed line, the nanoclusters expanded outward due to the volume change. After the lithiation process, a positive bias was applied to conduct the delithiation process as demonstrated in Fig. 6b. Contrary to the lithiation process, volume contraction occurs as the blue line implies. The subsequent second cycle repeated the processes of the first cycle (Fig. 6c and d). As expected, the superfine nanoparticles (black spots) are still confined at their original sites with little migration during the lithiation–delithiation processes. As demonstrated in Fig. 5, all the nanoparticles show synchronous movement without aggregation in the different states of charge–discharge processes, which is consistent with the illustration in Fig. 4f. Furthermore, the lithiation–delithiation processes can be completed in a short time without occurrence of cracks and pulverization, indicating that the nanoparticles can effectively accommodate the large stress associated with volume changes and accelerate the electrochemical reactions due to a shortened Li+ diffusion path. Fig. S13 (ESI†) displays morphology evolutions of the subsequent 3rd–6th cycles to further validate the cycling stability. Impressively, the nanoparticles are still confined at their original sites without migration and aggregation. Combined with the elemental mapping images showing that the homogeneous distribution of SnO2 and Fe2O3 is maintained during battery testing (Fig. 4g), spatially-confined electrochemical reactions can be realized, which maintain the integrity of the structure as illustrated in Fig. 4f.
 | ||
| Fig. 6 In situ TEM characterization. (a) Lithiation process and (b) delithiation process of the 1st cycle. (c) Lithiation process and (d) delithiation process of the 2nd cycle. | ||
In conclusion, a unique spatially-confined lithiation–delithiation reaction in a highly dense nanocomposite anode is proposed, successfully achieving the structural integrity during cycling through local, independent, rapid and step-by-step electrochemical reactions. Furthermore, the present design of architecture takes full advantages of nanomaterials in volume change/lithium ion diffusion and a highly dense structure in tap density, which assure superior cycling performance and ultra-high volumetric capacity simultaneously. The nanocomposite electrode maintains about 6034.5 mA h cm−3 (1206.9 mA h g−1, 86.4% of the first discharge capacity) after 200 extremely stable cycles, which has been the highest volumetric capacity even after long cycling so far. More inspiringly, the volumetric capacity can be as high as 5398.5, 5097.0 and 4704.0 mA h cm−3 (1079.7, 1019.4 and 940.8 mA h g−1) at high current densities of 5, 10 and 20 A g−1, respectively. We anticipate that spatially-confined lithiation–delithiation would lead to a rational design to address the cycling failures of high capacity materials and further development for harvesting high volumetric capacity anodes with superior lithium-storage performance.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC-21373184 and 51222202), the National Basic Research Program of China (2014CB932500), the Program for Innovative Research Team in University of Ministry of Education of China (IRT13037) and the Fundamental Research Funds for the Central Universities (2014XZZX003-07). Dr Yinzhu Jiang wishes to acknowledge for an equipment subsidy offered by the Alexander von Humboldt (AvH) Foundation. The work on microscopy was carried out at the Center of Electron Microscopy of Zhejiang University.Notes and references
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994 CrossRef CAS PubMed.
- Y. Idota, Science, 1997, 276, 1395 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS PubMed.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966 CrossRef CAS PubMed.
- J. M. Jeong, B. G. Choi, S. C. Lee, K. G. Lee, S. J. Chang, Y. K. Han, Y. B. Lee, H. U. Lee, S. Kwon, G. Lee, C. S. Lee and Y. S. Huh, Adv. Mater., 2013, 25, 6250 CrossRef CAS PubMed.
- P. Meduri, C. Pendyala, V. Kumar, G. U. Sumanasekera and M. K. Sunkara, Nano Lett., 2009, 9, 612 CrossRef CAS PubMed.
- C. Zhu, X. Xia, J. Liu, Z. Fan, D. Chao, H. Zhang and H. J. Fan, Nano Energy, 2014, 4, 105 CrossRef CAS PubMed.
- J. Luo, X. Xia, Y. Luo, C. Guan, J. Liu, X. Qi, C. F. Ng, T. Yu, H. Zhang and H. J. Fan, Adv. Energy Mater., 2013, 3, 737 CrossRef CAS.
- Z. W. Seh, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. McDowell, P. C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331 CrossRef PubMed.
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 309 Search PubMed.
- Y. J. Hong, M. Y. Son and Y. C. Kang, Adv. Mater., 2013, 25, 2279 CrossRef CAS PubMed.
- C. Wang, Y. Zhou, M. Y. Ge, X. B. Xu, Z. L. Zhang and J. Z. Jiang, J. Am. Chem. Soc., 2010, 132, 46 CrossRef CAS PubMed.
- J. Deng, C. Yan, L. Yang, S. Baunack, S. Oswald, H. Wendrock, Y. Mei and O. G. Schmidt, ACS Nano, 2013, 7, 6948 CrossRef CAS PubMed.
- F. Han, D. Li, W. C. Li, C. Lei, Q. Sun and A. H. Lu, Adv. Funct. Mater., 2013, 23, 1692 CrossRef CAS.
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187 CrossRef CAS PubMed.
- S. Xu, C. M. Hessel, H. Ren, R. Yu, Q. Jin, M. Yang, H. Zhao and D. Wang, Energy Environ. Sci., 2014, 7, 632 CAS.
- S. Jeong, J. P. Lee, M. Ko, G. Kim, S. Park and J. Cho, Nano Lett., 2013, 13, 3403 CrossRef CAS PubMed.
- Y. Yu, C. H. Chen and Y. Shi, Adv. Mater., 2007, 19, 993 CrossRef CAS.
- F. Lin, D. Nordlund, T. C. Weng, Y. Zhu, C. Ban, R. M. Richards and H. L. Xin, Nat. Commun., 2014, 5, 3358 Search PubMed.
- Q. Chen and K. Sieradzki, Nat. Mater., 2013, 12, 1102 CrossRef CAS PubMed.
- N. Bahlawane, K. K. Hoinghaus, P. A. Premkumar and D. Lenoble, Chem. Sci., 2012, 3, 9290 RSC.
- N. Bahlawane, K. K. Hoinghaus, T. Weimann, P. Hinze, S. Rohe and M. Baumer, Angew. Chem., Int. Ed., 2011, 50, 9957 CrossRef CAS PubMed.
- Y. Jiang, Y. Li, M. Yan and N. Bahlawane, J. Mater. Chem., 2012, 22, 16060 RSC.
- L. Martin, H. Martinez, D. Poinot, B. Pecquenard and F. Le Cras, J. Phys. Chem. C, 2013, 117, 4421 CAS.
- Y. Jiang, D. Zhang, Y. Li, T. Yuan, N. Bahlawane, C. Liang, W. Sun, Y. Lu and M. Yan, Nano Energy, 2014, 4, 23 CrossRef CAS PubMed.
- M. Niu, F. Huang, L. Cui, P. Huang, Y. Yu and Y. Wang, ACS Nano, 2010, 4, 681 CrossRef CAS PubMed.
- W. Zhou, C. Cheng, J. Liu, Y. Y. Tay, J. Jiang, X. Jia, J. Zhang, H. Gong, H. H. Hng, T. Yu and H. J. Fan, Adv. Funct. Mater., 2011, 21, 2439 CrossRef CAS.
- Y. Yu, C. H. Chen, J. L. Shui and S. Xie, Angew. Chem., Int. Ed., 2005, 44, 7085 CrossRef CAS PubMed.
- Y. Jiang, T. Yuan, W. Sun and M. Yan, ACS Appl. Mater. Interfaces, 2012, 4, 6216 CAS.
- C. Guan, X. Wang, Q. Zhang, Z. Fan, H. Zhang and H. J. Fan, Nano Lett., 2014, 14, 4852 CrossRef CAS PubMed.
- W. Zeng, F. Zheng, R. Li, Y. Zhan, Y. Li and J. Liu, Nanoscale, 2012, 4, 2760 RSC.
- X. J. Zhu, Z. P. Guo, P. Zhang, G. D. Du, R. Zeng, Z. X. Chen, S. Li and H. K. Liu, J. Mater. Chem., 2009, 19, 8360 RSC.
- X. Zhou, L. J. Wan and Y. G. Guo, Adv. Mater., 2013, 25, 2152 CrossRef CAS PubMed.
- X. Zhou, Z. Dai, S. Liu, J. Bao and Y. G. Guo, Adv. Mater., 2014, 26, 3943 CrossRef CAS PubMed.
- F. Y. Cheng, J. Liang, Z. L. Tao and J. Chen, Adv. Mater., 2011, 23, 1695 CrossRef CAS PubMed.
- Y. Wang, I. Djerdj, B. Smarsly and M. Antonietti, Chem. Mater., 2009, 21, 3202 CrossRef CAS.
- B. Zhang, Q. B. Zheng, Z. D. Huang, S. W. Oh and J. K. Kim, Carbon, 2011, 49, 4524 CrossRef CAS PubMed.
- L. Wang, D. Wang, Z. Dong, F. Zhang and J. Jin, Nano Lett., 2013, 13, 1711 CAS.
- Z. Chen, M. Zhou, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2012, 2, 95 CrossRef CAS.
- X. Wang, X. Cao, L. Bourgeois, H. Guan, S. Chen, Y. Zhong, D. M. Tang, H. Li, T. Zhai, L. Li, Y. Bando and D. Golberg, Adv. Funct. Mater., 2012, 22, 2682 CrossRef CAS.
- X. W. Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536 CrossRef CAS.
- Y. Su, S. Li, D. Wu, F. Zhang, H. Liang, P. Gao, C. Cheng and X. Feng, ACS Nano, 2012, 6, 8349 CrossRef CAS PubMed.
- J. Lin, Z. W. Peng, C. S. Xiang, G. D. Ruan, Z. Yan, D. Natelson and J. M. Tour, ACS Nano, 2013, 7, 6001 CrossRef CAS PubMed.
- Y. Tang, D. Wu, S. Chen, F. Zhang, J. Jia and X. Feng, Energy Environ. Sci., 2013, 6, 2447 CAS.
- C. M. Wang, W. Xu, J. Liu, J. G. Zhang, L. V. Saraf, B. W. Arey, D. Choi, Z. G. Yang, J. Xiao, S. Thevuthasan and D. R. Baer, Nano Lett., 2011, 11, 1874 CrossRef CAS PubMed.
- M. Gu, Y. Li, X. Li, S. Hu, X. Zhang, W. Xu, S. Thevuthasan, D. R. Baer, J. G. Zhang, J. Liu and C. Wang, ACS Nano, 2012, 6, 8439 CrossRef CAS PubMed.
- Y. Liu, N. S. Hudak, D. L. Huber, S. J. Limmer, J. P. Sullivan and J. Y. Huang, Nano Lett., 2011, 11, 4188 CrossRef CAS PubMed.
- Y. Zhao, L. Peng, B. Liu and G. Yu, Nano Lett., 2014, 14, 2849 CrossRef CAS PubMed; C. Kim, M. Noh, M. Choi, J. Cho and B. Park, Chem. Mater., 2005, 17, 3297 CrossRef.
- H. Kim, M. Seo, M. H. Park and J. Cho, Angew. Chem., Int. Ed., 2010, 49, 2146 CrossRef CAS PubMed.
- F. Hao and D. Fang, J. Electrochem. Soc., 2013, 160, A595 CrossRef CAS PubMed.
- Y. T. Cheng and M. W. Verbrugge, J. Power Sources, 2009, 190, 453 CrossRef CAS PubMed.
- F. Wang, R. Robert, N. A. Chernova, N. Pereira, F. Omenya, F. Badway, X. Hua, M. Ruotolo, R. Zhang, L. Wu, V. Volkov, D. Su, B. Key, M. S. Whittingham, C. P. Grey, G. G. Amatucci, Y. Zhu and J. Graetz, J. Am. Chem. Soc., 2011, 133, 18828 CrossRef CAS PubMed.
- F. Wang, H. C. Yu, M. H. Chen, L. Wu, N. Pereira, K. Thornton, A. Van der Ven, Y. Zhu, G. G. Amatucci and J. Graetz, Nat. Commun., 2012, 3, 1201 CrossRef PubMed.
- X. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, J. Mater. Chem., 2012, 22, 17456 RSC.
- X. Huang, J. Chen, Z. Lu, H. Yu, Q. Yan and H. H. Hng, Sci. Rep., 2013, 3, 2317 Search PubMed.
- D. Applestone and A. Manthiram, RSC Adv., 2012, 2, 5411 RSC.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed.
- D. H. Ha, M. A. Islam and R. D. Robinson, Nano Lett., 2012, 12, 5122 CrossRef CAS PubMed.
- B. Wang, X. Li, T. Qiu, B. Luo, J. Ning, J. Li, X. Zhang, M. Liang and L. Zhi, Nano Lett., 2013, 13, 5578 CrossRef CAS PubMed.
- Z. Cui, Y. Huang and X. Guo, Electrochim. Acta, 2012, 60, 7 CrossRef CAS PubMed.
- J. B. Kim, H. Y. Lee, K. S. Lee, S. H. Lim and S. M. Lee, Electrochem. Commun., 2003, 5, 544 CrossRef CAS.
- M. E. Donders, H. C. M. Knoops, W. M. M. Kessels and P. H. L. Notten, J. Power Sources, 2012, 203, 72 CrossRef CAS PubMed.
- X. L. Ding, Q. Sun, F. Lu and Z. W. Fu, J. Power Sources, 2012, 216, 117 CrossRef CAS PubMed.
- Q. Li, P. Wang, Q. Feng, M. Mao, J. Liu, S. X. Mao and H. Wang, Chem. Mater., 2014, 26, 4102 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and supplementary characterization including XPS, XRD, TEM, EDS mapping of the nanocomposite electrode and other electrochemical test results, and also comparison of volumetric capacities between references and the present work. See DOI: 10.1039/c5ee00314h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |