
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnassisted solar-driven photoelectrosynthetic HI splitting using membrane-embedded Si microwire arrays†
Shane
Ardo
a,
Sang Hee
Park
b,
Emily L.
Warren
a and
Nathan S.
Lewis
*acde
aDivision of Chemistry and Chemical Engineering, California Institute of Technology, 1200 E. California Blvd., Pasadena, California 91125, USA
bThomas J. Watson Laboratories of Applied Physics, California Institute of Technology, 1200 E. California Blvd., Pasadena, California 91125, USA
cJoint Center for Artificial Photosynthesis, Pasadena, California 91125, USA. E-mail: nslewis@caltech.edu; Fax: +1-626-395-8867; Tel: +1-626-395-6335
dBeckman Institute, California Institute of Technology, 1200 E. California Blvd., Pasadena, California 91125, USA
eKavli Nanoscience Institute, California Institute of Technology, 1200 E. California Blvd., Pasadena, California 91125, USA
First published on 1st April 2015
Abstract
Free-standing, membrane-embedded, Si microwire arrays have been used to affect the solar-driven, unassisted splitting of HI into H2 and I3−. The Si microwire arrays were grown by a chemical-vapor-deposition vapor–liquid–solid growth process using Cu growth catalysts, with a radial n+p junction then formed on each microwire. A Nafion proton-exchange membrane was introduced between the microwires and Pt electrocatalysts were then photoelectrochemically deposited on the microwires. The composite Si/Pt–Nafion membrane was mechanically removed from the growth substrate, and Pt electrocatalysts were then also deposited on the back side of the structure. The resulting membrane-bound Si microwire arrays spontaneously split concentrated HI into H2(g) and I3− under 1 Sun of simulated solar illumination. The reaction products (i.e. H2 and I3−) were confirmed by mass spectrometry and ultraviolet–visible electronic absorption spectroscopy.
Broader contextTechnologies that perform aqueous hydrohalic acid splitting can form the basis for a photoelectrochemical flow battery that combines solar energy conversion and storage into one integrated system. A low-cost, impure series of micron-sized semiconducting light absorbers can be incorporated into a low-cost membrane. Using the well-established principles of semiconductor photoelectrochemistry, the light-driven incipient photocurrent produced by this structure can directly charge the redox species of a flow battery. The approach allows for the use of inexpensive semiconducting materials while providing a unique, potentially transformational approach to obtaining integrated, scalable energy storage. The round-trip charge–discharge efficiency of the flow battery is over twice that achievable by the production of fuels or through use of a fuel cell. Furthermore, the balance-of-systems benefits offered by this integrated approach would enable very low-cost conversion of solar energy into stored electricity, in direct combination with the scalability features offered by flow batteries. The development of an integrated membrane-bound microwire array system for aqueous hydrohalic acid splitting is also a step along the technology development path toward a membrane-embedded tandem microwire array structure for solar-fuels production such as water splitting or sustainable CO2 reduction. |
I. Introduction
Arrays of semiconducting nanowires or microwires embedded in an ionically conducting polymer provide an interesting approach to the direct production of fuels from sunlight.1 The highly anisotropic geometry of the nanowire or microwire provides a long axis to achieve high light absorption, while maintaining a short, orthogonal distance in the radial direction of the wire to facilitate the effective collection and utilization of photogenerated charge carriers.2 For optimized spacings and wire diameters, sparse arrays of microwires can produce very efficient absorption of the illumination incident onto the array, over a variety of wavelengths and angles of illumination.3 Under constant illumination conditions, the high internal surface area of the microwire arrays results in a relatively low electron flux to the interfaces by comparison to a planar device, while also providing a high surface-area support for the strategically located adsorption of electrocatalysts, to thereby optimize internal light management as well as reactant access and product egress from the internal volume of the structure. In addition, the spacing between the microwires in an array produces porosity that readily facilitates an ionic flux of protons or hydroxide ions through the microwire array and accompanying ionophoric polymer support. Such an assembly allows effective, facile conduction of protons or hydroxide ions and thus neutralizes any pH gradients that might otherwise occur between the site of water oxidation and the site of fuel formation in the photosystem.4,5Several architectures have been proposed, evaluated, and constructed for a solar-fuels generator based on membrane-bound microwire arrays.5–7 One specific architecture utilizes an array of membrane-supported n-type semiconducting microwires, along with needed electrocatalysts, to oxidize water. An array of p-type semiconducting microwires is positioned on the other side of the membrane, and in conjunction with any necessary electrocatalysts, reduces H2O or CO2 to produce fuels.8,9 In this case, the membrane also must contain an intermediate, electronically and protonically conductive layer to facilitate trans-membrane ionic transport while providing ohmic electrical contact between the two sets of wire arrays on either side of the membrane.10 Another architecture involves the use of a core–shell configuration, in which the wire arrays act as a voltage-generating scaffold for a self-aligned, conformally deposited photoanode (or photocathode) material, with the microwires providing contact to the cathode (or anode) in the system.7 In this architecture, the microwires physically penetrate the membrane, obviating the need for an intermediate electrically conductive layer, and requiring instead an ohmic contact and/or tunnel junction between the shell and the core of each wire in the array. In the core–shell architecture, one electrocatalyst can be placed on the surfaces of the microwires, and the other electrocatalyst can be semi-continuous in nature on the other side of the membrane, essentially forming one element of a membrane–electrode assembly as are employed in fuel cells and electrolyzers.11 The “back side” catalyst can have a relatively high loading so that the electrocatalyst film reflects light back into the microwire array core–shell structure, where the light can then be absorbed more effectively and thus can lead to enhanced solar-driven fuel production.
To provide the photovoltage needed to split water and/or reduce CO2, while also oxidizing water to O2, both membrane-based architectures optimally use a tandem-type structure, with two light absorbers having complementary band gaps.12 The operational characteristics of such a system, however, can be demonstrated and evaluated for a simplified architecture that only consists of one wire array embedded in an ionically permeable membrane, provided that the absorber can provide sufficient photovoltage to drive the anodic and cathodic electrochemical half-reactions of interest. Specifically, the photovoltage requirement can be relaxed if the electron donor is not water but instead is a halide. Sunlight-driven splitting of aqueous hydrohalic acids, HX, results in H2 evolution with concomitant X2/X3− generation. At Si and group VI metal–dichalcogenide photoelectrodes, ∼10% solar-to-hydrogen (STH) efficiencies have been demonstrated for the direct conversion of solar energy to H2 through splitting of HI, HBr, or HCl (i.e. STH(I), STH(Br), or STH(Cl), respectively, where STH stands for solar-to-hydrogen and I/Br/Cl are abbreviations for the other component of the reagent).13–19 Stable sunlight-driven oxidation of Cl− has been achieved using MoS2, MoSe2, and metal-coated Si photoanodes,17,20 which is of interest because the oxidation product (chlorine) is thermodynamically more difficult to form than O2 and because chlorine is a top commodity chemical globally, with the process to generate Cl2 requiring >1% of global electricity.21 The products from the HX splitting reactions can additionally be utilized in redox flow batteries to generate electricity.22,23 Recent reports have demonstrated >90% energy efficiency at ∼1000 mA cm−2 current density from an H2/Br2 redox flow battery, due to the rapid catalysis of the H2 oxidation reaction and of the Br2 reduction reaction. In contrast, H2/air fuel cells, which would be coupled with water electrolyzers, typically exhibit ∼50% energy efficiency.17,24 The Texas Instruments Corporation demonstrated a ∼9% STH(Br) efficiency,16 which when used in conjunction with a redox flow battery, could yield an ∼8% overall solar-to-chemical-to-electricity efficiency with H2/Br2 intermediate storage.
We describe herein proof-of-concept, functional assemblies that consist of silicon microwire arrays partially embedded in a Nafion proton-exchange membrane, with catalysts on the microwire surfaces (Fig. 1). Each p-type silicon microwire was doped radially with an n+ emitter to form a buried junction, so that the photovoltage was independent of the energetics of the semiconductor–liquid contact. An oxide boot was fabricated at the base of each microwire to attenuate shunting across the Nafion membrane. Nafion was introduced between the microwires to support the microwires once they were mechanically removed from their underlying growth substrate, and to attenuate mixing of reaction products. Pt was deposited on both sides of the composite membrane to catalyze the faradaic reactions. The performance of each major component of the composite device, i.e. the Si microwire photoelectrodes, back-side electrodes, and Nafion membranes, respectively, was characterized and evaluated individually. The complete composite material performs unassisted solar-driven HI splitting, mitigates ion-transport losses, and is amenable to facile combination with another light-absorbing material.7,25
 | ||
| Fig. 1 Schematic depiction of the multiple-component device that is described, fabricated, characterized, and evaluated in this work, and with the predominant electron-transfer chemistries and ion-transport processes indicated in red. The composite membrane consisted of a p-type Si microwire array with an oxide boot base and a degenerately n+-doped radial emitter, and Pt electrocatalysts deposited on the tops, sides, and backs of the Si microwires, with the array partially embedded in a Nafion proton-exchange membrane. The inset depicts an approximate energy-band diagram for this structure when illuminated and located between aqueous electrolytes that contain I3− and I−, and H+ and H2. | ||
II. Experimental
Materials synthesis and processing
Crystalline Si microwire arrays were grown using previously reported procedures,2 and Pt was deposited photoelectrochemically and/or by electron-beam evaporation. Further details are available in the ESI.†Unassisted HI splitting electrochemical evaluation
Using the two electrodes and conditions described in the ESI,† the photocurrent at zero applied potential was measured over time using a two-electrode configuration in an H-cell arrangement with a commercial Nafion membrane separator without stirring and with air blown onto the illuminated cell.Free-standing composite membrane fabrication
Silicon microwires were embedded in a Nafion proton-exchange membrane and removed from the underlying growth substrate, via a procedure similar to that reported previously,10 and described in more detail in the ESI† (Fig. S1).Electrochemical evaluation
Each free-standing sample was used as the membrane in an H-cell configuration, with both compartments containing ∼7.6 M HI electrolyte with adventitious I3−. The current versus potential data were measured using a four-electrode setup with two SCE potential-sensing electrodes immersed in Luggin–Haber capillaries and positioned to within 1 mm of the membrane surface. The other two electrodes were Pt mesh current-carrying electrodes that were positioned far from the membrane surface. The potential reported at each current is the difference in potentials measured in the presence and absence of the membrane.Photoelectrochemical evaluation
Each large free-standing sample was cut into smaller samples (∼0.5 cm × 0.5 cm) using a Teflon-coated steel razor blade, and each of the smaller samples was sandwiched between two black Viton sheets (∼1 cm × ∼4.5 cm × ∼1 mm thick) each with a hole cut out (∼3 mm in diameter) so that the membrane could be contacted on either side by electrolyte. This material was then placed between two borosilicate glass cuvettes (a 10 mm path length cuvette on the catholyte side and a 1 mm path length cuvette on the anolyte side) each with a hole on the side of the cuvette that faced the sample (3 mm in diameter). The cuvettes were held together using a custom acrylic sample holder and were illuminated using a fiber optic attached to a 150 W EKE (3250 K) halogen lamp (ThorLabs Inc., OSL1 Fiber Illuminator). The light intensity was measured using a calibrated Si photodiode (ThorLabs, Inc., FDS100) by placing the photodiode at the same location of the membrane in the sample holder, with the incident light passing through the borosilicate glass windows used to hold the sample and electrolyte. The temperature was monitored externally using an infrared sensor (ThermoHAWK 200), which indicated that the temperature was ∼22 °C under ambient light and ∼23 °C under steady-state 1 Sun illumination with air blown onto the cell. A quadrupole mass spectrometer coupled to a Faraday cup continuously recorded m/z = 2 (H2), and m/z = 18 (H2O) signals to verify that the m/z = 2 signal was from H2 as opposed to the fragment ions of water. A Pt wire was inserted into the silicone septum of each cuvette so that the potential difference between the electrolytes could be measured in situ using cyclic voltammetry. These electrodes were also used at the end of the experiment to generate H2(g) as an internal standard. A galvanostatic current density of 3 mA cm−2 between the Pt wires, based on the projected geometric area of the composite-material membrane in contact with electrolyte, resulted in a steady-state m/z = 2 signal of 1.9 × 10−9 Torr. The absorption spectrum of the electrolyte in the 1 mm thin-path length cuvette was recorded once per minute using a custom LabView program (National Instruments, Inc.) that controlled and communicated with an HP 8452A diode-array spectrophotometer, and was converted into an apparent current density using the known molar decadic absorption coefficients of I3−.26 Apparent current densities were referenced to the geometric area of the composite-membrane–electrolyte contact (∼7 mm2). Additional experimental details can be found in the ESI.†III. Results
Three-electrode photoelectrochemical characterization of Si microwire array electrodes
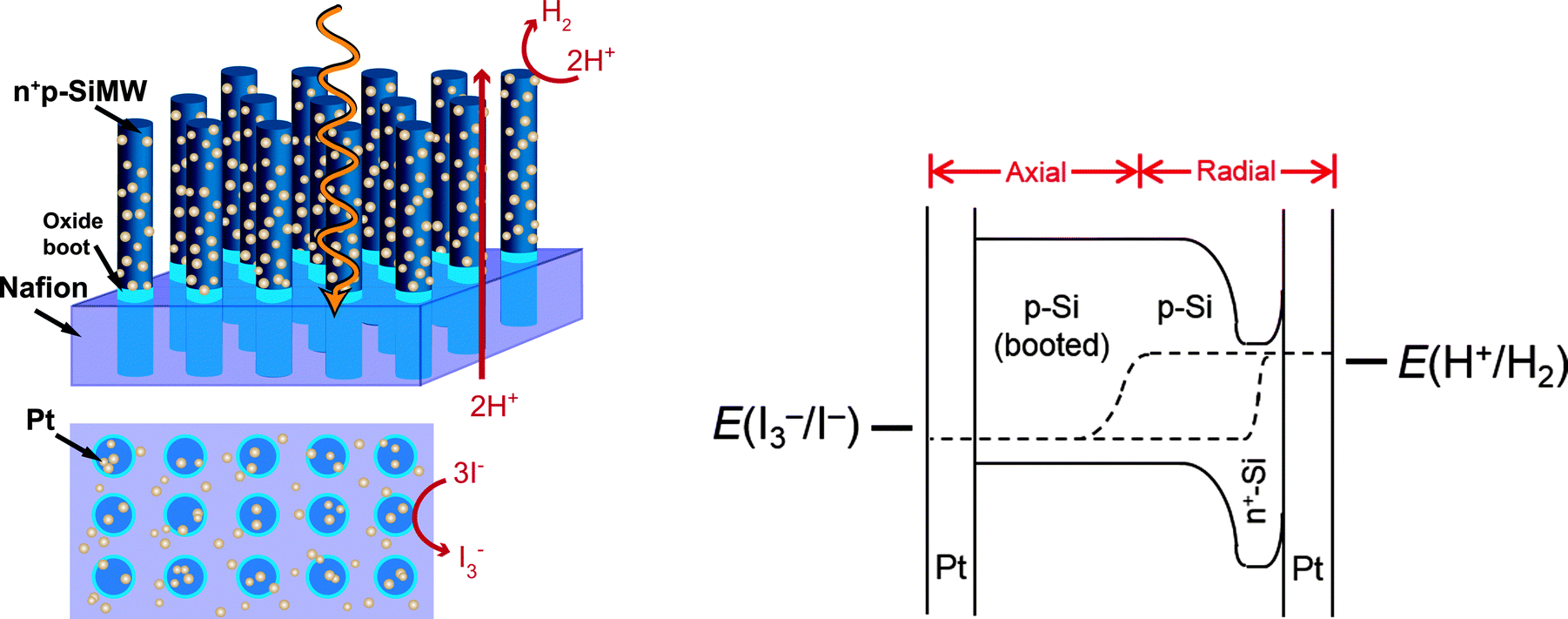
Fig. 2 shows the current–density versus potential (J–E) behavior in H2(g)-saturated, ∼12 M HCl(aq) of arrays of n+p-Si microwires (∼65 μm in height) with photoelectrochemically deposited Pt (Fig. S1c, ESI†). Under 100 mW cm−2 of simulated solar illumination, the photoelectrode exhibited an open-circuit potential, Eoc, of 470 mV vs. RHE, and exhibited a photocurrent density of J = −4.9 mA cm−2 at E = 0 V vs. RHE for the photoinduced evolution of H2 (Fig. 2), and a light-limited current density of ∼−5.1 mA cm−2.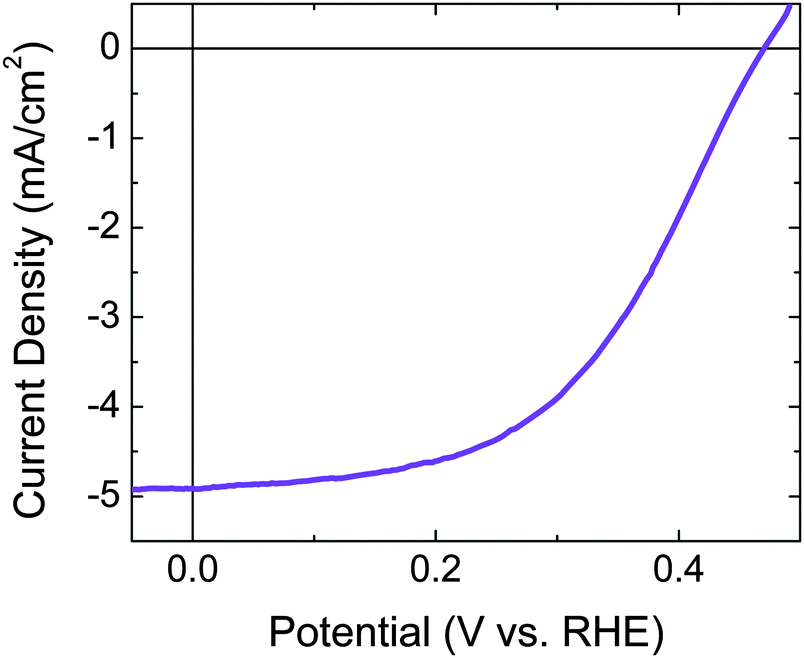 | ||
| Fig. 2 Three-electrode current–density versus potential data recorded under 1 Sun simulated solar illumination for a p-type Si microwire array, with a degenerate n+-doped radial emitter. | ||
Fig. 3a shows a typical scanning-electron micrograph (SEM) image of the back side of a free-standing device that was fabricated by infilling an n+p-Si–Pt microwire array with a polymer, mechanically removing the polymer-embedded microwire arrays from the underlying growth substrate, and depositing Pt electrocatalysts on the back side of the microwire–polymer membrane. Energy-dispersive spectroscopy (EDS) mapping measurements confirmed that Pt was present on the back side of the microwires as well as on the surface of the Nafion. Physical deposition methods readily permitted the deposition of Pt on the back side of the free-standing microwire–Nafion materials.
 | ||
| Fig. 3 (a) SEM image of the back side of an array of oxide-booted p-type Si microwires without a radial n+-doped emitter and partially embedded in PDMS with 5 nm planar-equivalent Pt on the back side of the microwire–PDMS membrane. (b) Three-electrode current–density versus potential data recorded in the dark for the electrode from panel a immersed in ∼7.6 M HI(aq) electrolyte containing adventitious I3−. | ||
Fig. 3b shows the J–E behavior in the dark for the electrode shown in Fig. 3a immersed in ∼7.6 M HI(aq) that contained adventitious I3−. The data indicate that Pt formed a low-resistance contact with the I3−/I− redox system in HI(aq), and exhibited a Tafel slope of ∼80 mV per decade for iodide oxidation over the potential range of ∼30 mV to ∼90 mV.
Four-electrode photoelectrochemical characterization of Nafion-embedded Si microwire arrays
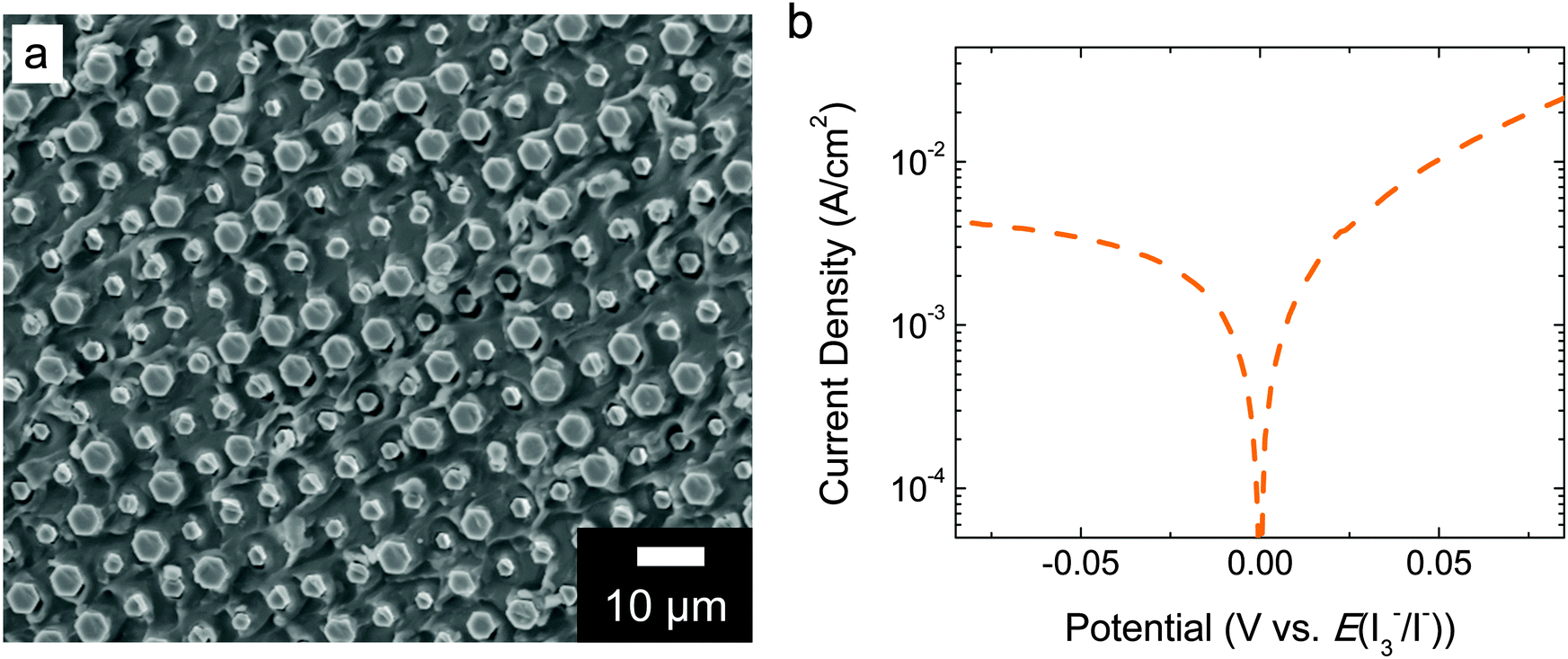
Pt evaporated to a 5 nm planar equivalent thickness on the back side of the microwire–Nafion membrane did not conformally coat the Nafion surface, and afforded rapid proton conduction through the Nafion, with an area-specific resistance of ∼0.3 Ohm cm2 (Fig. 4). In contrast, Pt at a thickness of 10 nm (planar equivalent) produced a large attenuation of the protonic conduction.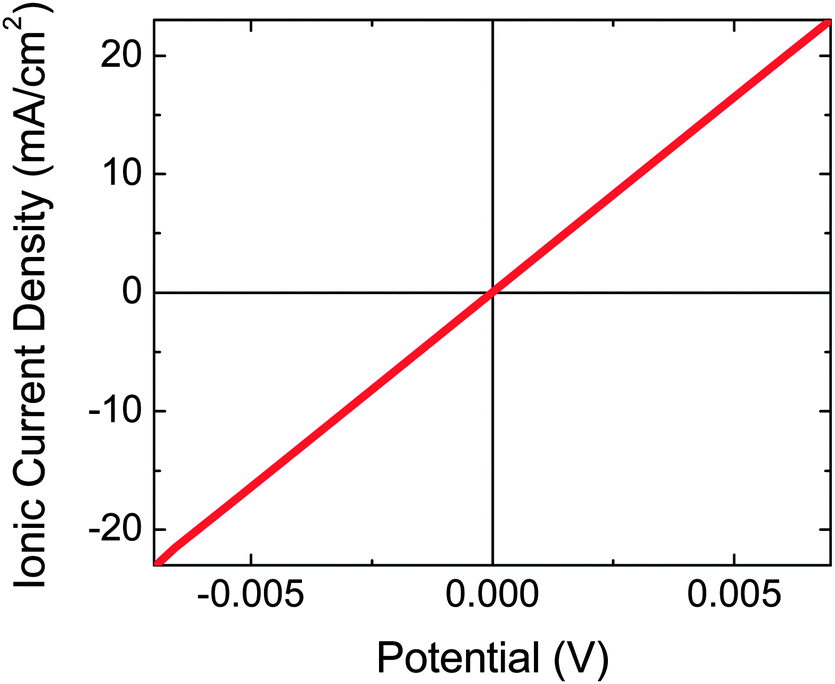 | ||
| Fig. 4 Four-electrode ionic current–density versus potential data for a composite material membrane consisting of an array of Si microwires partially embedded in Nafion and with 5 nm planar-equivalent Pt deposited on the microwire–Nafion back side and in contact with ∼7.6 M HI(aq) electrolyte containing adventitious I3− on both sides of the membrane. | ||
Two-electrode photoelectrochemical characterization of Nafion-embedded Si microwire arrays
A load-line analysis (Fig. 5a) was conducted using the data in Fig. 2 and 3b, which, when coupled with the observation of negligible potential loss due to ionic resistance in the Si–Nafion–Pt membrane (Fig. 4), indicated that a free-standing device should operate near its maximum power point. A measurement at an applied potential bias of 0 V performed in an H-cell using a wired two-electrode arrangement between the front-side electrode from Fig. 2 and the back-side electrode from Fig. 3 confirmed the predicted operating point (Fig. 5b).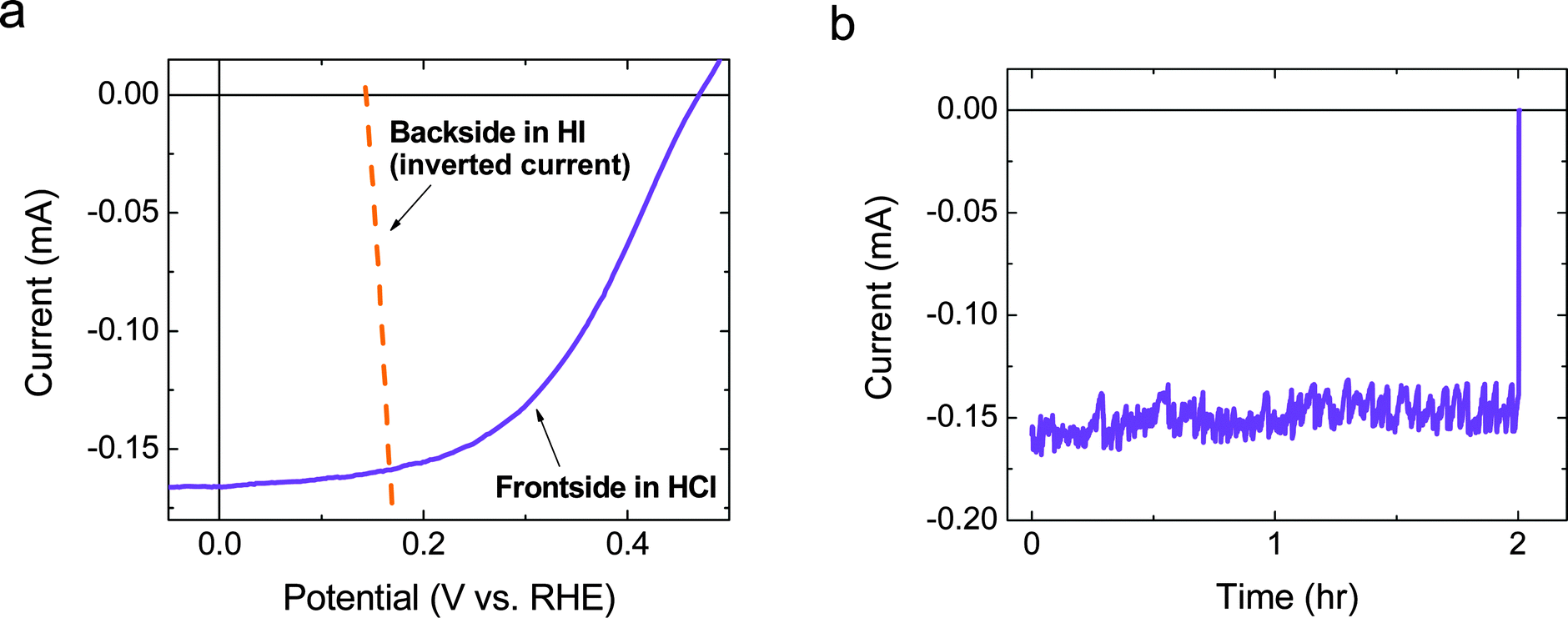 | ||
| Fig. 5 (a) Load-line analysis for the current versus potential data for the electrodes from Fig. 2 and 3b (inverted current). The area of the photocathode was 3.675 mm2 and the anode area was 4.296 mm2. (b) Chronoamperometry data recorded under 1 Sun simulated solar illumination and at an applied potential bias of 0 V between the electrodes used for the data in panel a. | ||
Fig. 5b shows the stability of the two electrodes over a 2 h period when the electrodes were illuminated and shorted together through a potentiostat that was used as a low-impedance ammeter. After ∼2 h, the illumination was stopped, and the concomitant drop in current verified that the observed current was due to the illumination. The initial observed photocurrent was within 5% of that predicted by the load-line analysis (Fig. 5a). Based on the current at the intersection of the two curves, which predicted the operating current for the device, i.e. −0.159 mA (∼4.3 mA cm−2, based on the area of the photocathode), as well as the potential at zero current measured electrochemically, i.e. ∼140 mV, the solar-to-hydrogen-and-triiodide energy-conversion efficiency (ηSTH(I)) was determined to be ∼0.60%. After continuously operating the electrode for 2 h, ηSTH(I) decreased slightly to ∼0.56%.
Product analysis for Nafion-embedded Si microwire arrays
Initially, HI splitting was assessed qualitatively and was successfully observed for ∼80% of the composite membranes fabricated, quantifying the reproducibility of the deposition techniques. For direct and quantitative detection of the products of the putative reaction, free-standing devices were evaluated in an H-cell that consisted of two custom borosilicate-glass cuvettes held together with a custom acrylic sample holder and containing ∼7.6 M HI(aq) (Fig. S2, ESI†). I3− was detected in situ and in operando using visible-absorption spectroscopy, while H2 was detected in operando using an in-line mass spectrometer (Fig. 6). Under simulated solar illumination, bubbles were clearly observed on the cathode side of the membrane, and were confirmed to be H2 via mass spectrometry. No visual evidence suggested that H2 or I3− transported through the membrane at an appreciable rate. Upon illumination, an induction period of ∼10 min was observed before H2 was detected by mass spectrometry. Over the course of ∼15 min, the mass spectroscopic signal then rose to the H2 production rate expected from a current density of ∼0.35 mA cm−2. Between 15.5 and 19.5 min into the experiment, a large bubble detached from the composite material membrane surface (Fig. S3a, ESI†), and this detachment resulted in a >10-fold increase in the intensity of the m/z = 2 mass spectroscopic signal, to a maximum apparent H2 production current density of approximately −4.5 mA cm−2. An analogous event occurred at ∼35 min into the experiment, and the system then transitioned to a situation in which large bubbles were no longer observed to form on the membrane surface but instead small bubbles released continually from the membrane surface (Fig. S3b, ESI†). Fig. 6 also shows the rate of I3− generation due to reactions in the anolyte. This responded much more rapidly to starting and stopping the illumination which occurred at 0 h and 3.5 h, and 2.5 h, respectively. Using two-electrode cyclic voltammetry, the potential difference between the two compartments, each containing a Pt electrode, was determined to be ∼200 mV. Using this potential difference and the data in Fig. 6, ηSTH(I) of the free-standing device was determined to be ∼0.5%.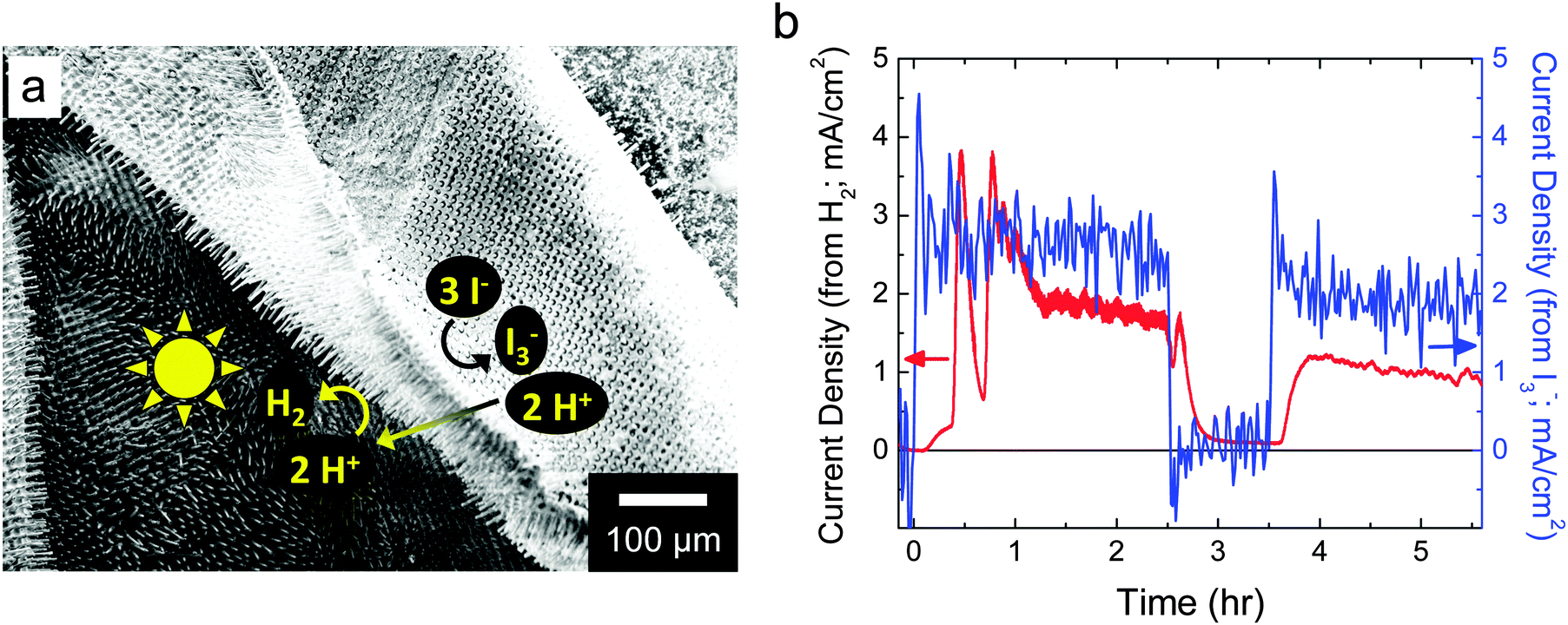 | ||
| Fig. 6 (a) SEM image of a free-standing device that is partially folded over so that both the front side and the back side of the microwires are visible. The general redox reactions and ion transport processes that occurred are indicated on the SEM image. (b) Rates (reported as current densities) for the evolution of H2 and generation of I3− as a function of time, under 1 Sun simulated solar illumination, except for 2.5–3.5 h which was in the dark. | ||
IV. Discussion
Component characterization
Prior to fabricating and operating the freestanding polymer-embedded composite material to affect the solar-driven splitting of HI, each of the system components was fabricated and individually evaluated using three-electrode and/or four-electrode electrochemical techniques. This independent component evaluation afforded rapid assessment of specific characteristics and provided predictive capabilities for the overall device function. Ultimately each component was modelled or physically assembled electrically in series at matched current densities to mimic the complete freestanding structure, so that insight into the overall device operation could be obtained. This procedure was informative, because the electrochemical behavior of the complete freestanding structure cannot easily be controlled, and chemical product detection, which is the most straightforward and accepted means to evaluate the performance of a wireless structure, is laborious. Also, measurement of the reaction products does not provide significant fundamental rationale for the underlying principles that govern the ultimate device performance.Silicon microwires have been shown to effectively collect photogenerated majority charge carriers at the bases of such microwires.27,28 Electrodes based on arrays of these microwires have demonstrated open-circuit photovoltages in excess of 500 mV when immersed in aqueous or non-aqueous electrolytes that contain outer-sphere one-electron-transfer redox couples.29,30 Deposition of Pt by electron-beam evaporation onto on the microwires has enabled the effective production of H2(g) from 0.5 M H2SO4(aq).30 Similar H2-evolution behavior was obtained herein from microwire array electrodes that contained photoelectrochemically deposited Pt and were immersed in ∼7.6 M HI(aq) (Fig. 2). The photoelectrochemical deposition procedure was chosen because it resulted in the most reproducible performance. The light-limited current density in Fig. 2 is about half of the value reported previously for state-of-the-art Si microwire arrays.29,30 However, the photocurrent density from Si microwire arrays is highly dependent on the length of the microwires, the angle of incidence of incoming illumination with respect to the long microwire axis, and the presence of light-scattering particles or other optical components (e.g. Ag back reflector) as part of the electrode.3,30,31 The microwires used in the electrodes shown in Fig. 2 were intentionally shorter than often reported in the literature. These microwires were chosen because short microwires facilitated the introduction of a compact Nafion proton-exchange membrane between the microwires, which is important for structural stability and product separation in free-standing devices. In a more optimized device structure, back reflectors, scattering particles, and an off-normal axis alignment for the microwires would be used to increase the photocurrent. The fill factor in Fig. 2 is also less than that observed previously. This behavior can be rationalized based on the photoelectrochemical deposition protocol used herein relative to the electron-beam evaporation process used previously.30 In addition, halides poison Pt and increase the required overpotential for catalysis of the H2 evolution reaction,18,32 which would manifest itself as a decreased fill factor. Future efforts will focus on replacing Pt with earth-abundant materials such as carbon or conducting polymers such as poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate), which are known to be stable and efficient electrocatalysts for the oxidation of halides.23,33,34
This architecture is advantageous with respect to prior demonstrations of ∼10% STH efficiencies using planar materials or systems without orthogonalization of light absorption and charge-carrier collection, because in theory inexpensive and impure semiconductor materials can be used, ionic conduction distances approach the smallest possible therefore minimizing any ionic resistance losses, and the single composite integrated structure could be amenable to inexpensive large-scale production. A core–shell architecture for solar-driven water splitting has been reported previously in an electrode form factor.7,35 If core–shell materials were partially embedded in an ionophoric polymer support and mechanically removed from the growth substrate, the electrocatalyst layer on the back side of the composite material would likely possess two characteristics: it would be deposited by physical deposition methods and it would be semi-continuous to allow facile ion transport. Once devices are peeled, physical methods are most amenable to catalyst deposition in lieu of electrochemical methods, which require electrical contact to each microwire. The Pt loading used herein on the back side of the microwire–Nafion membrane was 5 nm planar equivalent by electron-beam evaporation, which was optimized for efficient iodide oxidation electrocatalysis (Fig. 3b) and rapid proton conduction (Fig. 4). The electrocatalyst layer was determined to be discontinuous because a small ionic resistance was measured when steady-state current was passed through the membrane. If the Pt electrocatalyst layer had been conformal, ions would not have been able to migrate through the membrane and, under illumination, after rapid capacitive charging between both sides of the device, the device would cease to pass current. Ultimately, the amount of Pt that was deposited was limited to that which still afforded rapid conduction of protons through Nafion. If the planar equivalent thickness of Pt was ≥10 nm, protons were not able to conduct through the Nafion membrane, likely due to a conformal coating of Pt on the Nafion surface. Physical removal of some Pt resulted in efficient proton conduction, suggesting that the Pt had not irreversibly altered the membrane. EDS mapping only showed a small signal above background for Pt, and the noise precluded identification of isolated Pt regions of the back side of the array. Pt particles could not be seen in the SEM images.
The results from the three electrochemical measurements described above (illuminated photocathode; anode in the dark; membrane in the dark) suggest that a free-standing membrane-bound material without an external bias can affect the splitting of HI, accompanied by robust separation of the reaction products. Because the area-specific resistance of the membrane was small, the potential losses at solar photon fluxes would be negligible (<2 mV), and inclusion of the membrane resistance was thus not required in the load-line analysis for accurate prediction of the operating current. The load-line analysis indicated that the device would operate at ∼4.5 mA cm−2, which was also supported by two-electrode measurements that mimicked the overall free-standing device structure (Fig. 5).
Free-standing device analysis
Free-standing devices were fabricated using the protocols from the individual component analyses described above. Silicon microwires, containing radial n+p junctions and oxide boots, were embedded in a Nafion proton-exchange membrane and removed from the underlying growth substrate followed by deposition of electrocatalysts on both sides of this composite membrane. One electrocatalyst (photoelectrochemically deposited Pt) was deposited on the microwire sidewalls, and the other electrocatalyst (evaporated Pt) was deposited as a semi-continuous material on the back side of the membrane-embedded microwires. The oxide boots attenuated shunts between the two sides of the Nafion membrane. The back-side catalyst can have a relatively high loading because in this location the catalyst will not attenuate light absorption in the microwire array. However, the back-side catalyst must not be continuous, so that proton conduction through the Nafion membrane remains rapid. The ultimate device structure resembled that of a membrane–electrode assembly akin to those utilized in fuel cells and electrolyzers, except the bipolar plates and external wires to deliver/collect charge were replaced by internal semiconducting Si microwires that delivered charge and potential upon solar illumination.Most of the composite membranes leaked. Crossover of red triiodide from the anolyte into the clear catholyte was observed visually. However, even these leaky composite membranes could be qualitatively evaluated for their ability to evolve H2(g) and generate I3− when immersed in ∼7.6 M HI(aq) and illuminated using simulated sunlight. Under illumination, bubble formation and release was clearly observed from the microwire side of ∼80% of the composite membranes studied, and a darkening of the electrolyte near the back side of the composite membrane was apparent over time, collectively suggesting that the Si–Pt materials drove both H2 evolution and I3− generation irrespective of tears in the membrane. Previously, Nafion-embedded Si microwire arrays have been shown to significantly attenuate H2 crossover.10 The tears in the materials reported herein would allow H2 (and I3−) crossover and were likely generated due to utilization of thin Nafion membranes (∼15 μm) and/or poor adhesion of Nafion to the H-terminated Si or oxide-booted microwire surfaces. Thus, to remove the array from its growth substrate while keeping the membrane intact, a custom home-built microwire cutter and composite membrane peel-off tool was used instead of a razor blade.36 This approach resulted in devices that did not tear based on visual inspection of leakage into the catholyte, even when under ∼3 cm of pressure from aqueous HI, although this does not completely exclude the possibility of small rates of I3− crossover.
Fig. 6b compares the rates of H2 evolution and I3− generation observed herein. The maximum rates for H2 evolution and I3− generation are consistent with calculations using data obtained from two-electrode and three-electrode on-wafer (photo)electrochemical measurements (Fig. 5). H2 had to equilibrate between the electrolyte and headspace and only then could the evolved gas flow from the electrochemical cell to the mass spectrometer. Initial mass spectroscopic signals due to H2 bubbles were observed within one min after their release from the membrane surface, indicating that mass transport of H2 to the detector was not slow. Similarly, the electrolyte containing I3− was constantly, but slowly, stirred and hence I3− generation was observed immediately after optical excitation was initiated. The rate of I3− generation was approximately independent of time, while that for the detection of H2 evolution was not, which suggests that bubbles may have altered the photoelectrosynthesis. However, optical effects from the interaction of light with bubbles did not result in a substantial net change in the rate of I3− generation. Moreover, bubbles did not physically occlude the electrocatalytic reaction sites or ion-conducting pathways in the membrane because that would have resulted in a slowed rate of I3− formation, which was not observed.
The active area of the composite membranes was circular, with an approximate diameter of 3 mm. SEM images suggested that these composite membranes, which consist of autonomous HI-splitting silicon microwire units that were ∼2.5 μm in diameter, were homogeneous on this size scale. It is therefore reasonable to assume that larger samples will operate at very similar efficiency. Qualitatively, by visual inspection of bubble formation, the rate of H2 evolution for a larger sample immersed in HI was reasonably homogeneous on the 3 × 3 mm2 scale. The non-linear optical properties of the microwire array arise from effects on the <100 μm scale, so samples of the size evaluated, or larger, will not be affected by additional non-linear optical effects.
Although Cl2 is highly corrosive and the electrolytes are strongly acidic, these chemistries can nevertheless form the basis for scalable technologies, as evidenced by large-scale electrochemical technologies used in the chloralkali process (Cl2 and strong base) and state-of-the-art electrolyzers and fuel cells (strongly acidic local environments in the ionically conductive membrane). These conditions would obviously require corrosion-resistant cells and gaskets, as well as stringent safety protocols, if such a system were deployed technologically.
A benefit of microstructured ordered arrays of light absorbers is the ease by which they can be embedded in solid polymer electrolytes. This approach highlights an advantage inherent to arrays of autonomous fuel-forming units, which differs from a design consisting of wired photoelectrochemical electrodes or a design comprising a photovoltaic cell coupled to a separate electrolyzer. The demonstration reported herein is a model system for future photoelectrosynthetic devices that drive other chemistries, such as water splitting. This proof-of-concept demonstrates that a “solar carpet” can be fabricated and used to store the energy in sunlight in chemical bonds via unassisted solar HI splitting. These photoelectrosynthetic reactions are unassisted because the chemical biases utilized, i.e. the H2/H+ electrochemical bias that can reduce I3−/I−, thermodynamically opposes the light-driven reactions that form H2 and I3−. Future research will focus on using a tandem light-absorber device architecture so that overall solar-driven water splitting can be performed using self-contained, membrane-embedded solar reactors.
V. Conclusions
Arrays of silicon microwires grown by a chemical-vapor-deposition vapor–liquid–solid growth process were fabricated and processed into radial n+p junctions. When Pt electrocatalysts were photoelectrochemically deposited on the tops and sides of the microwires, microwire-array electrodes exhibited Eoc ≈ 470 mV for H2 evolution from concentrated acid. Nafion was spin cast into the microwire array and cured, and subsequently the composite Si–Nafion membrane was mechanically removed from the underlying growth substrate. Pt electrocatalysts were also deposited on the back side of the microwires and were found to create a low-resistance contact to the p-type Si microwire core. Peeled, free-standing arrays of partially Nafion-embedded Si microwires with Pt deposited on their back sides exhibited efficient proton conduction when immersed in ∼7.6 M HI(aq), and were shown to split HI into H2 and I3− under simulated solar illumination with a solar-to-hydrogen-and-triiodide energy-conversion efficiency of ∼0.5%. These devices thus provide a first demonstration of photoelectrosynthesis from a free-standing composite membrane that is isotropic on the millimeter scale, for storage of sunlight in the bonds of H2.Acknowledgements
This material is based upon work performed by the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award Number DE-SC0004993. S.A. acknowledges support from a U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy (EERE) Postdoctoral Research Award under the EERE Fuel Cell Technologies Program. The authors would like to thank Dan Turner–Evans, Morgan Putnam, and Mike Kelzenberg for their generous assistance and donation of Si microwire array samples for some of the preliminary studies; Jacob Good for his assistance with the mass spectrometer; Rick P. Gerhart from the Caltech Glassblowing Shop for fabricating custom borosilicate electrochemical cells; Steve Olson and Mike Roy from the Caltech Machine Shop for fabricating custom free-standing device holders and a sample holder for the absorption spectrometer; and Harry Gray, Harry Atwater, and Bruce Brunschwig for useful discussions and guidance.References
- E. L. Warren, H. A. Atwater and N. S. Lewis, Silicon Microwire Arrays for Solar Energy-Conversion Applications, J. Phys. Chem. C, 2014, 118, 747–759 CAS.
- B. M. Kayes, H. A. Atwater and N. S. Lewis, Comparison of the Device Physics Principles of Planar and Radial p-n Junction Nanorod Solar Cells, J. Appl. Phys., 2005, 97, 114302 CrossRef PubMed.
- M. D. Kelzenberg, S. W. Boettcher, J. A. Petykiewicz, D. B. Turner-Evans, M. C. Putnam, E. L. Warren, J. M. Spurgeon, R. M. Briggs, N. S. Lewis and H. A. Atwater, Enhanced Absorption and Carrier Collection in Si Wire Arrays for Photovoltaic Applications, Nat. Mater., 2010, 9, 239–244 CrossRef CAS.
- J. Jin, K. Walczak, M. R. Singh, C. Karp, N. S. Lewis and C. Xiang, An Experimental and Modeling/simulation-Based Evaluation of the Efficiency and Operational Performance Characteristics of an Integrated, Membrane-Free, Neutral pH Solar-Driven Water-Splitting System, Energy Environ. Sci., 2014, 7, 3371–3380 CAS.
- J. R. McKone, N. S. Lewis and H. B. Gray, Will Solar-Driven Water-Splitting Devices See the Light of Day?, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- S. Ardo, M. Shaner, R. Coridan, N. C. Strandwitz, J. R. McKone, K. Fountaine, H. A. Atwater and N. S. Lewis, Semiconductor Structures for Fuel Generation, US20130269761 A1, 2013 Search PubMed.
- M. R. Shaner, K. T. Fountaine, S. Ardo, R. H. Coridan, H. A. Atwater and N. S. Lewis, Photoelectrochemistry of Core–shell Tandem Junction n–p+-Si/n-WO3 Microwire Array Photoelectrodes, Energy Environ. Sci., 2014, 7, 779–790 CAS.
- N. S. Lewis and D. G. Nocera, Powering the Planet: Chemical Challenges in Solar Energy Utilization, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- N. S. Lewis, An Integrated, Systems Approach to the Development of Solar Fuel Generators, Electrochem. Soc. Interface, 2013, 22, 43–49 CAS.
- J. M. Spurgeon, M. G. Walter, J. Zhou, P. A. Kohl and N. S. Lewis, Electrical Conductivity, Ionic Conductivity, Optical Absorption, and Gas Separation Properties of Ionically Conductive Polymer Membranes Embedded with Si Microwire Arrays, Energy Environ. Sci., 2011, 4, 1772–1780 CAS.
- H. Zhang and P. K. Shen, Recent Development of Polymer Electrolyte Membranes for Fuel Cells, Chem. Rev., 2012, 112, 2780–2832 CrossRef CAS PubMed.
- S. Hu, C. Xiang, S. Haussener, A. D. Berger and N. S. Lewis, An Analysis of the Optimal Band Gaps of Light Absorbers in Integrated Tandem Photoelectrochemical Water-Splitting Systems, Energy Environ. Sci., 2013, 6, 2984–2993 CAS.
- F. F. Fan, H. S. White, B. L. Wheeler and A. J. Bard, Semicondcutor Electrodes. 31. Photoelectrochemistry and Photovoltaic Systems with n- and p-Type WSe2 in Aqueous Solution, J. Am. Chem. Soc., 1980, 102, 5142–5148 CrossRef CAS.
- E. L. (Pete) Johnson, The TI Solar Energy System Development, IEEE Trans. Acoust., Speech, Signal Process., 1981, 81, 2–6 Search PubMed.
- C. Levy-Clement, A. Heller, W. A. Bonner and B. A. Parkinson, Spontaneous Photoelectrolysis of HBr and HI, J. Electrochem. Soc., 1981, 129, 1701–1705 CrossRef PubMed.
- J. D. Luttmer and I. Trachtenberg, Performance Predictions for Solar-Chemical Converters Based on Photoelectrochemical I–V Curves, J. Electrochem. Soc., 1985, 132, 1312–1315 CrossRef CAS PubMed.
- J. R. White, F.-R. F. Fan and A. J. Bard, Semiconductor Electrodes. LVI. Principles of Multijunction Electrodes and Photoelectrosynthesis at Texas Instruments' p/n-Si Solar Arrays, J. Electrochem. Soc., 1985, 132, 544–550 CrossRef CAS PubMed.
- Y. Nakato, Y. Egi, M. Hiramoto and H. Tsubomura, Hydrogen Evolution and Iodine Reduction on an Illuminated n-p Junction Silicon Electrode and Its Application to Efficient Solar Photoelectrolysis of Hydrogen Iodide, J. Phys. Chem., 1984, 88, 4218–4222 CrossRef CAS.
- Y. Nakato, M. Yoshimura, M. Hiramoto, A. Tsumura, T. Murahashi and H. Tsubomura, p-n Junction Silicon Electrode Coated with Noble Metal for Efficient Solar Photoelectrolysis of Hydrogen Iodide, Bull. Chem. Soc. Jpn., 1984, 57, 355–360 CrossRef CAS.
- C. P. Kubiak, L. F. Schneemeyer and M. S. Wrighton, Visible Light Driven Generation of Chlorine and Bromine. Photooxidation of Chloride and Bromide in Aqueous Solution at Illuminataed n-Type Semiconducting Molybdenum Diselenide and Molybdenum Disulfide Electrodes, J. Am. Chem. Soc., 1980, 102, 6898–6900 CrossRef CAS.
- United States Department of Energy, Energy Information Administration, http://www.eia.doe.gov/.
- B. Huskinson, J. Rugolo, S. K. Mondal and M. J. Aziz, A High Power Density, High Efficiency Hydrogen–Chlorine Regenerative Fuel Cell with a Low Precious Metal Content Catalyst, Energy Environ. Sci., 2012, 5, 8690–8698 CAS.
- K. T. Cho, M. C. Tucker, M. Ding, P. Ridgway, V. S. Battaglia, V. Srinivasan and A. Z. Weber, Cyclic Performance Analysis of Hydrogen/Bromine Flow Batteries for Grid-Scale Energy Storage, ChemPlusChem, 2014, 80, 402–411 CrossRef PubMed.
- V. Livshits, A. Ulus and E. Peled, High-Power H2/Br2 Fuel Cell, Electrochem. Commun., 2006, 8, 1358–1362 CrossRef CAS PubMed.
- S. Haussener, C. Xiang, J. M. Spurgeon, S. Ardo, N. S. Lewis and A. Z. Weber, Modeling, Simulation, and Design Criteria for Photoelectrochemical Water-Splitting Systems, Energy Environ. Sci., 2012, 5, 9922–9935 CAS.
- A. D. Awtrey and R. E. Connick, The Absorption Spectra of I2, I3−, I−, IO3−, S4O6= and S2O3=. Heat of the Reaction I3− = I2 + I−, J. Am. Chem. Soc., 1951, 73, 1842–1843 CrossRef CAS.
- M. C. Putnam, D. B. Turner-Evans, M. D. Kelzenberg, S. W. Boettcher, N. S. Lewis and H. A. Atwater, 10 μm Minority-Carrier Diffusion Lengths in Si Wires Synthesized by Cu-Catalyzed Vapor-Liquid-Solid Growth, Appl. Phys. Lett., 2009, 95, 163116 CrossRef PubMed.
- M. D. Kelzenberg, D. B. Turner-Evans, M. C. Putnam, S. W. Boettcher, R. M. Briggs, J. Y. Baek, N. S. Lewis and H. A. Atwater, High-Performance Si Microwire Photovoltaics, Energy Environ. Sci., 2011, 4, 866–871 CAS.
- S. W. Boettcher, J. M. Spurgeon, M. C. Putnam, E. L. Warren, D. B. Turner-Evans, M. D. Kelzenberg, J. R. Maiolo, H. A. Atwater and N. S. Lewis, Energy-Conversion Properties of Vapor-Liquid-Solid-Grown Silicon Wire-Array Photocathodes, Science, 2010, 327, 185–187 CrossRef CAS PubMed.
- S. W. Boettcher, E. L. Warren, M. C. Putnam, E. A. Santori, D. Turner-Evans, M. D. Kelzenberg, M. G. Walter, J. R. McKone, B. S. Brunschwig, H. A. Atwater and N. S. Lewis, Photoelectrochemical Hydrogen Evolution Using Si Microwire Arrays, J. Am. Chem. Soc., 2011, 133, 1216–1219 CrossRef CAS PubMed.
- E. A. Santori, J. R. Maiolo III, M. J. Bierman, N. C. Strandwitz, M. D. Kelzenberg, B. S. Brunschwig, H. A. Atwater and N. S. Lewis, Photoanodic Behavior of Vapor–Liquid–Solid–Grown, Lightly Doped, Crystalline Si Microwire Arrays, Energy Environ. Sci., 2012, 5, 6867–6871 CAS.
- J. D. Luttmer, D. Konrad and I. Trachtenberg, Electrode Materials for Hydrobromic Acid Electrolysis in Texas Instruments' Solar Chemical Converter, J. Electrochem. Soc., 1985, 132, 1054–1058 CrossRef CAS PubMed.
- R. A. Simon and M. S. Wrighton, Stabilization of n-Type Silicon Photoanodes against Photoanodic Decomposition with Thin Films of Polyacetylene, Appl. Phys. Lett., 1984, 44, 930–932 CrossRef CAS PubMed.
- S. Mubeen, J. Lee, N. Singh, M. Moskovits and E. W. McFarland, Stabilizing Inorganic Photoelectrodes for Efficient Solar-to-Chemical Energy Conversion, Energy Environ. Sci., 2013, 6, 1633–1639 CAS.
- C. Liu, J. Tang, H. M. Chen, B. Liu and P. Yang, A Fully Integrated Nanosystem of Semiconductor Nanowires for Direct Solar Water Splitting, Nano Lett., 2013, 13, 2989–2992 CrossRef CAS PubMed.
- A. C. Tamboli, C. T. Chen, E. L. Warren, D. B. Turner-Evans, M. D. Kelzenberg, N. S. Lewis and H. A. Atwater, Wafer-Scale Growth of Silicon Microwire Arrays for Photovoltaics and Solar Fuel Generation, IEEE J. Photovoltaics, 2012, 2, 294–297 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee00227c |
| This journal is © The Royal Society of Chemistry 2015 |